

Abstract
Dominant mutations in the RNA/DNA-binding protein TDP-43 have been linked to amyotrophic lateral sclerosis (ALS). Here, we screened genomic DNA extracted from spinal cord specimens of sporadic ALS patients for mutations in the TARDBP gene and identified a patient specimen with previously reported Q331K mutation. The patient spinal cord tissue with Q331K mutation showed accumulation of higher levels of DNA strand breaks and the DNA double-strand break (DSB) marker γH2AX, compared to age-matched controls, suggesting a role of the Q331K mutation in genome-damage accumulation. Using conditional SH-SY5Y lines ectopically expressing wild-type (WT) or Q331K-mutant TDP-43, we confirmed the increased cytosolic sequestration of the poly-ubiquitinated and aggregated form of mutant TDP-43, which correlated with increased genomic DNA strand breaks, activation of the DNA damage response factors phospho-ataxia-telangiectasia mutated (ATM), phospho-53BP1, γH2AX and neuronal apoptosis. We recently reported the involvement of WT TDP-43 in non-homologous end joining (NHEJ)-mediated DSB repair, where it acts as a scaffold for the recruitment of XRCC4-DNA ligase 4 complex. Here, the mutant TDP-43, due to its reduced interaction and enhanced cytosolic mislocalization, prevented the nuclear translocation of XRCC4-DNA ligase 4. Consistently, the mutant cells showed significantly reduced DNA strand break sealing activity and were sensitized to DNA-damaging drugs. In addition, the mutant cells showed elevated levels of reactive oxygen species, suggesting both dominant negative and loss-of-function effects of the mutation. Together, our study uncovered an association of sporadic Q331K mutation with persistent genome damage accumulation due to both damage induction and repair defects.
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disease characterized by a complex pathology. A major subgroup of ALS patients show cytoplasmic mislocalization and aggregation of RNA-binding protein TDP-43 in spinal motor neurons (1). Since the identification of TDP-43 in 2006 as the ubiquitinated protein in inclusion bodies found in ALS spinal cord neurons, TDP-43 inclusions have also been reported in other neurodegenerative diseases, including frontotemporal dementia and Alzheimer’s disease-related dementia (2–5). TDP-43 inclusions in ALS are composed of hyper-phosphorylated, poly-ubiquitinated and fragmented forms of TDP-43 (6). TDP-43 protein encoded by the TARDBP gene belongs to the family of heterogeneous nuclear ribonucleoproteins with a highly conserved RNA/DNA-binding domain. TDP-43 contains two RNA recognition motifs (RRM1 and RRM2) and a glycine-rich C-terminus. The RRMs are necessary for nucleic acid binding, whereas the C-terminus provides a binding interface for its protein partners. The C-terminus contains the majority of the mutational hotspots for several dozen point mutations identified in both familial and sporadic cases of ALS (1,7). In mature neurons, TDP-43 is predominantly present in the nucleus but also shuttles to other cellular compartments. A unique feature of TDP-43 is its autoregulation property that maintains its level in neuronal cells by binding to its transcript, initiating alternative splicing of intron 7 in the TARDBP 3′ UTR region that leads to the destruction of the TDP-43 mRNA (8).
Several familial and sporadic missense point mutations in the TARDBP gene have been identified in ALS patients, many of which are linked to early disease onset and enhanced disease progression (1). How these mutants affect the function and toxicity of TDP-43 is not completely understood. In 2008, Sreedharan et al. (7) identified the Q331K mutation in sporadic ALS patients, which was later shown to affect its autoregulation in a transgenic mouse, leading to the mutant protein’s progressive accumulation (9). The C-terminus of TDP-43 is intrinsically disordered with propensity to assemble into beta-sheet-rich dynamic oligomers. Furthermore, studies have suggested abnormal nucleic acid binding by the Q331K mutant, which results in an increased aggregation rate of the protein (9).
We recently discovered that TDP-43 is a critical component of DNA damage response (DDR) in motor neurons and plays a role in non-homologous end joining (NHEJ)-mediated DNA double-strand break (DSB) repair (10). In healthy neurons, TDP-43 facilitates the optimal recruitment of XRCC4-DNA ligase 4 complex at DSB sites and regulates DSB ligation and repair. In ALS, nuclear loss of function of TDP-43 causes deficient repair of DSBs, leading to DSB accumulation, and persistent DDR activation, contributing to neurodegeneration. It is intriguing to elucidate how mutant forms of TDP-43, which exhibit both loss-of-function and gain-of-toxicity, impact its genome maintenance role, which we address in this manuscript with respect to the Q331K mutation.
We demonstrate that expression of Q331K mutation not only affects DSB repair by preventing the nuclear translocation of XRCC4 but also contributes to oxidative genome damage accumulation via increased reactive oxygen species (ROS) generation. The cumulative effect of the persistent DNA-damage accumulation and DDR activation correlated with neuronal apoptosis.
Results
Elevated genome damage in the spinal cord tissue of an ALS patient with acquired TDP-43 Q331K mutation
ALS-linked mutations in TDP-43 are clustered in exon 6 of the TARDBP gene that codes for the nuclear export sequence (NES)-containing C-terminal domain. We performed Sanger sequencing of exon 6 of the TARDBP gene from genomic DNA isolated from spinal cord (cervical) tissue specimens from 10 sporadic ALS patients obtained from the Veterans Affairs Biorepository Brain Bank, USA. A Q331K mutation was identified in one specimen from a 83-year-old Caucasian male 0028 (Fig. 1A). This mutation was not present in genomic DNA isolated from the occipital lobe of the same patient, ruling out a familial linkage. Immunoblotting analysis of mutant spinal cord tissue extracts detected decreased levels of the monomeric form of TDP-43 due to its apparent degradation/aggregation, compared to age-matched control spinal cord extracts. The mutant spinal cord also showed increased levels of the DSB marker γH2AX (Fig. 1B). Long-amplicon polymerase chain reaction (LA-PCR) analysis of genomic DNA isolated from the mutant spinal cord showed significantly (~2-fold) decreased amplification of the LA from the OCT3/4, NANOG, Polβ and HPRT gene regions compared to the controls indicating the presence of higher levels of DNA strand breaks in the ALS spinal cord (Fig. 1C and D). There was no change in the amplification of 200 bp short amplicon, which served as control. Immunohistochemistry (IHC) analysis showed typical TDP-43 pathology with increased cytoplasmic accumulation, which also correlated with higher staining for γH2AX and TUNEL staining compared to control spinal cord sections (Fig. 1E and F). These data suggest that the Q331K mutation may be linked to increased DNA damage in neuronal genomes.
Figure 1.
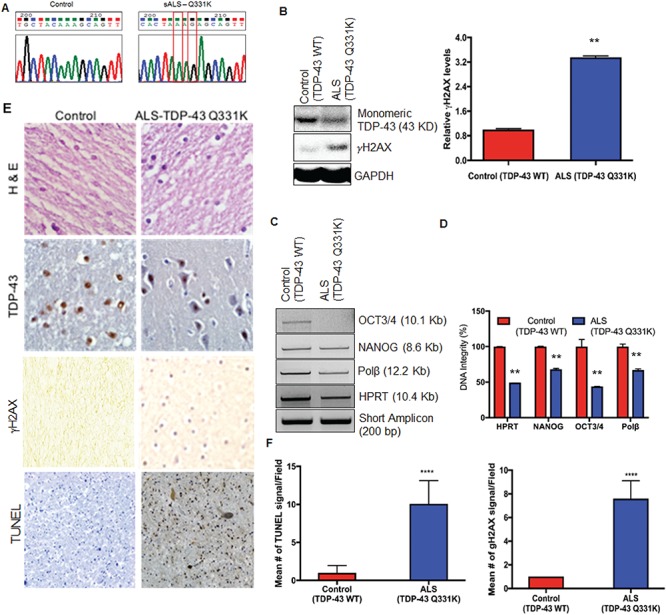
Targeted sequencing of exon-6 of the TARDBP gene in spinal cord specimens from ALS patients. (A) Chromatogram showing detection of the Q331K mutation in genomic DNA isolated from spinal cord tissue from an ALS patient. (B) Immunoblots probed with TDP-43 antibody and γH2AX antibody show reduced monomeric TDP-43 protein and increased γH2AX in an ALS specimen carrying the Q331K mutation compared to an age-matched control sample. GAPDH served as the loading control. (C) Analysis of DNA integrity by LA-PCR. Representative agarose gel images indicate reduced DNA amplification of OCT3/4, NONOG, Polβ and HPRT gene segments in TDP-43-Q331K spinal cord compared to control. The 200 bp short amplicon served as control.( D) Quantitation of PCR products by Pico Green-based DNA quantitation from triplicate experiments. (E) IHC of spinal cord from an ALS patient carrying a Q331K mutation for TDP-43, γH2AX and TUNEL staining. Representative images acquired at 20× magnification. (F) Quantitation of mean γH2AX signal and mean TUNEL-positive cells per field in ALS spinal cord sample and age-matched controls. **P<0.05; ****P<0.0001.
Enhanced cytosolic sequestration of the TDP-43 Q331K mutant
To examine the cellular distribution of mutant TDP-43, we transiently transfected neuroblastoma SH-SY5Y cells with either empty vector control or FLAG-tagged TDP-43 wild-type (WT) or Q331K mutant plasmid (Fig. 2A). After confirming FLAG expression by immunoblotting (Fig. 2B), we first evaluated the effect of mutant expression on cell proliferation. In a time-lapse cell proliferation assay, we observed moderately decreased proliferation rate in Q331K-expressing cells compared to WT as well as control cells (Fig. 2C). Immunofluorescence (IF) analysis with anti-TDP-43 antibody at 96 h post-transfection showed increased cytosolic TDP-43 in mutant-expressing cells compared to WT cells (Fig. 2D). The nuclei and cytosol were stained with DAPI and phalladoin, respectively. Furthermore, the mutant cells also showed higher levels of γH2AX, compared to WT-expressing cells (Fig. 2B).
Figure 2.
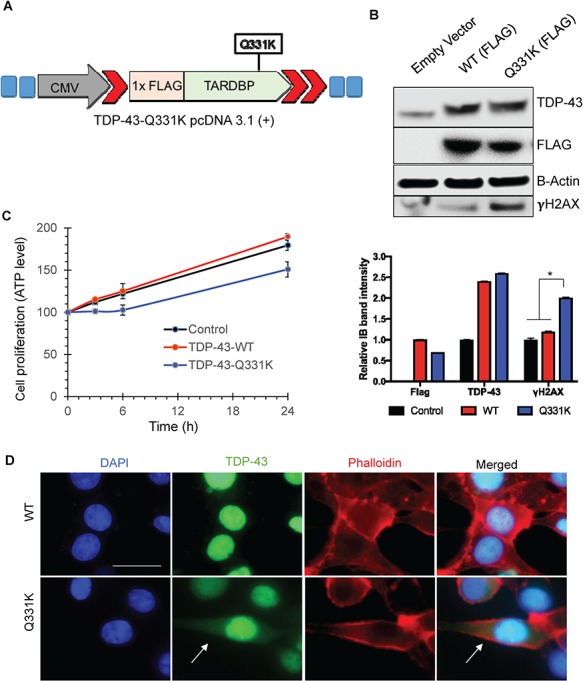
Ectopic transient expression of TDP-43-Q331K shows increased cytoplasmic accumulation in SH-SY5Y cells. (A) Vector construct expressing 1× FLAG and TDP-43-Q331K mutation. (B) Immunoblots of extracts from transiently transfected cells at 48 h, probed with FLAG, TDP-43 and γH2AX antibodies, show increased γH2AX, in cells expressing Q331K. β-Actin is shown as a loading control. The histogram shows quantitation of band intensity from three independent experiments. *P<0.01. (C) Cell-proliferation assay reveals reduced cell proliferation in Q331K-expressing cells. (D) IF of Q331K cells shows increased cytosolic accumulation of TDP-43 in cells expressing the Q331K mutant compared to WT. Left panel shows DAPI staining. Second from the left panel shows TDP-43 staining. Third panel shows Phalloidin staining as a cytoplasmic marker. Right panel show the merged image. Arrows indicate cytoplasmic localization of TDP-43 in Q331K-expressing cells. Scale bar represents 10 μm.
Generation and characterization of doxycycline-inducible TDP-43 Q331K mutant cell line
The transient transfection of WT or Q331K described in Figure 2 presented difficulties in obtaining reproducible and comparable levels of FLAG, particularly after the neuronal differentiation of SH-SY5Y cells. To overcome this limitation, we first generated a stable cell line constitutively expressing WT and Q331K. These stable lines, however, only expressed the ectopic TDP-43 up to 2–3 cell passages, then gradually showed large variability in FLAG expression, likely due to the autoregulatory activity of TDP-43 (8). We then designed doxycycline (Dox)-inducible vectors, pCW-TDP-43 Q331K (Fig. 3A) and pCW-TDP-43 WT, and isolated stable SH-SY5Y clones with comparable FLAG expression for WT and mutant TDP-43 after Dox induction (Fig. 3B). The conditional cell lines were differentiated with retinoic acid prior to Dox induction. Neuronal differentiation prior to induction was critical to avoid effects of Q331K on differentiation, because it has been reported that Q331K expression affects axonal growth and growth cones (11). Furthermore, to avoid robust overexpression, we optimized the WT and mutant lines for Dox induction for 24 h, followed by their culture in media-lacking Dox for 72 h, to obtain a comparable level of FLAG-TDP-43 with endogenous TDP-43 (Fig. 3B). This condition was used for most experiments unless otherwise indicated. The IF with anti-FLAG antibody in these lines showed increased cytoplasmic localization of Q331K, consistent with our observation in transiently transfected cells (Fig. 3C). Cytosolic translocation of mutant TDP-43 was further confirmed by co-IF with anti-FLAG antibody and cytosolic marker, phalloidin, which showed an increased cytoplasmic localization and nuclear loss of the mutant TDP-43 compared to WT (Fig. 3D). We also performed fractionation of nuclear and cytosolic extracts. Immunoblotting of the nuclear extracts with anti-TDP-43 and anti-FLAG antibodies confirm significant loss of nuclear Q331K compared to WT TDP-43 (Fig. 3E). The ectopic mutant was mostly excluded from the nucleus as indicated by FLAG levels. In addition, there is a decrease in endogenous TDP-43 in mutant-expressing cells, likely caused by autoregulation activity. However, we could not detect corresponding increase in monomeric mutant TDP-43 in the cytosol. This was perhaps expected due to aggregation/hyper-ubiquitination of the cytosolic forms as shown in Figure 4. We further confirmed nuclear exclusion of the mutant TDP-43 by a modified proximity ligation assay (PLA) using anti-FLAG versus anti-TDP-43 antibody, which detects the induced WT or mutant TDP-43. These data clearly showed an ~2-fold increase in cytosolic PLA signals for Q331K compared to WT TDP-43 (Fig. 3F).
Figure 3.
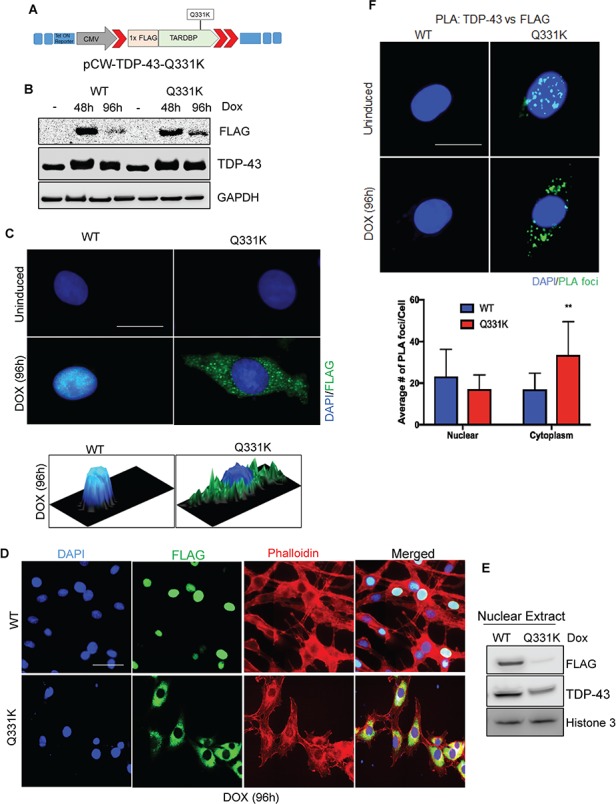
Characterization of differentiated SH-SY5Y cells with inducible expression of TDP-43-Q331K. (A) pCW-TDP-43-Q331K vector construct containing Dox-inducible 1× FLAG-tagged WT or 1× FLAG-tagged Q331K mutation. (B) Immunoblots confirming comparable FLAG and TDP-43 expression after Dox induction. GAPDH served as loading control. (C) IF with FLAG antibody 96 h after induction shows substantially increased cytoplasmic localization of Q331K in comparison to WT (Scale bar, 10 μm). The 2.5-dimensional view of the localization is shown in the image below. (D) IF of staining with FLAG antibody shows increased cytosolic accumulation of mutant TDP-43 in cells expressing the Q331K compared to WT. Left panel shows DAPI staining. Second from the left panel shows FLAG staining. Third panel shows Phalloidin staining as a cytoplasmic marker. Right panel shows merged image (Scale bar, 10 μm). (E) Immunoblots confirming nuclear exclusion of mutant TDP-43 as compared to WT. Histone 3 served as a nuclear loading control. (F) PLA using TDP-43 and FLAG antibody and foci quantitation showed the accumulation of cytosolic foci in cells expressing Q331K compared to WT cells (Scale bar, 10 μm). **P<0.05.
Figure 4.
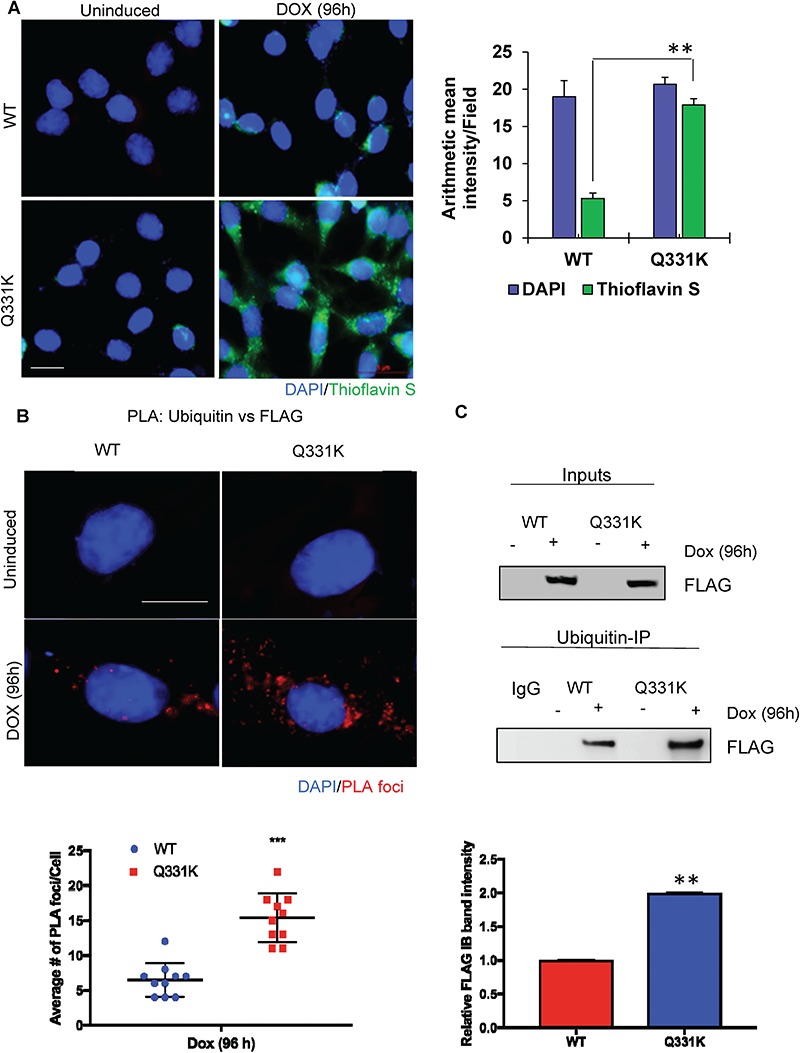
Increased poly-ubiquitination and aggregation of Q331K in neuronal cytosol. (A) IF with Thioflavin S shows increased formation of aggregates in the cytoplasm of Q331K-expressing differentiated neurons (Scale bar, 5 μm). Histogram indicates quantitation of Thioflavin S aggregates. (B) Detection of PLA foci between ubiquitin and FLAG antibodies and quantitation of PLA foci from 25 cells (Scale bar, 10 μm). (C) Immunoblots of ubiquitin Co-IP from neuronal cells expressing TDP-43-Q331K, using FLAG antibody, show increased ubiquitinated TDP-43-Q331K compared to WT. Histogram represents quantitation of mean band intensity from three independent experiments. **P<0.05; ***P<0.0005.
Hyper-ubiquitination of mutant TDP-43 correlates with its increased cytosolic aggregation
Cytoplasmic aggregates of TDP-43 in spinal motor neurons have been associated with ALS (2,6), and Q331K has been shown to be more aggregation-prone in vitro than WT (12). To test the cytosolic aggregation propensity of the Q331K mutant and WT, cells were stained with Thioflavin S after Dox induction, a dye that preferentially binds with protein aggregates to emit green fluorescence at 426 nm. Figure 4A shows significantly increased Thioflavin S fluorescence in mutant cells compared to WT, confirming its aggregated state. The aggregates appear both in perinuclear region and as large plaque-like cytoplasmic aggregates. Mitchell et al. (13) previously showed the presence of ubiquitin inclusions in an 8-week-old human Q331K-expressing mouse model. Here, using PLA of anti-ubiquitin versus anti-FLAG antibodies, we observed an ~2-fold higher level of ubiquitination in the Q331K mutant compared to WT (Fig. 4B). Hyper-ubiquitination of TDP-43 Q331K was further confirmed by immunoprecipitation (IP) of total cell extracts with anti-ubiquitin antibody, followed by detection of FLAG in the immunoblots of the IP eluate (Fig. 4C). Together, these data demonstrate an increased rate of cytosolic translocation, aggregation and ubiquitination of the Q331K mutant compared to WT TDP-43.
Accumulation of genomic DNA strand breaks in TDP-43-Q331K-expressing cells
Following on our studies that showed that TDP-43 silencing leads to genome damage accumulation in neurons (10), we tested the effect of mutant TDP-43 on genome integrity. Amyloidogenic protein aggregates in neurodegenerative disease have been linked to increased ROS production (14–16). To test whether the Q331K mutation with enhanced cytosolic aggregate formation elevates oxidative stress, we quantitated intracellular ROS levels in the WT and mutant cell lines, using a Cellular Reactive Oxygen Species Detection Assay system (17). We observed increased levels of ROS formation in the Q331K mutant cells compared to WT or uninduced control (Fig. 5A). We further confirmed elevated ROS levels by fluorometric microplate cellular ROS detection assay (Fig. 5B) and observed a significant increase in cellular ROS levels in Q331K cells compared to WT and uninduced control. Increased levels of ROS in cells can cause damages to cellular macromolecules including genomic DNA, which can contribute to neurodegenerative diseases (18). We tested genome integrity by LA-PCR and single-cell comet analysis in differentiated SH-SY5Y cells after WT and Q331K induction. LA-PCR analysis of genomic DNA isolated from the Q331K-expressing cells showed an ~50–80% reduction in DNA amplification of the ~12–8 kb genomic regions of NANOG, Polβ and HPRT genes compared to the WT control (Fig. 5C). Quantitation of LA-PCR products using the PicoGreen dsDNA assay (19) confirmed significantly reduced amplification products in the Q331K mutant compared to WT cells (Fig. 5D). To investigate the accumulation of DNA strand breaks, we performed single-cell electrophoresis and compared the comet tail moments under alkaline conditions. The alkaline comet assay is a measure of total alkali-labile sites in genomic DNA, including single-strand breaks (SSBs) and DSBs (20). We observed significantly increased tail moments in Q331K-expressing cells, compared to WT or uninduced control cells under alkaline conditions, indicating the accumulation of DNA strand breaks (Fig. 5E). These data cumulatively reveal that the Q331K mutation induces genomic instability in neurons by both accumulating DNA damage and inducing oxidative genome damage via ROS induction.
Figure 5.
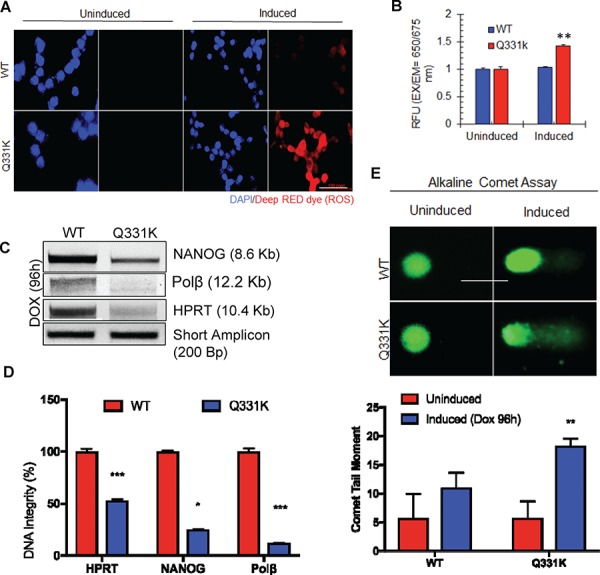
Q331K expression induces ROS stress and accumulation DNA strand breaks in neurons. (A) Cellular Reactive Oxygen Species Detection Assay by IF microscopy. Upper panels represent WT-expressing cells; lower panels represent Q331K-expressing cells. The two panels on the left represent uninduced cells. Panels to the right represent cells after Dox induction. Deep RED dye staining indicates the presence of ROS (Scale bar, 10 μm). (B) Quantitation of cellular ROS using a microplate fluorescence reader. (C) LA-PCR analysis of genomic DNA isolated from TDP-43-Q331K neurons shows reduced DNA integrity. Representative agarose gel images of amplified DNA products. (D) Quantification of PCR products by Pico Green-based DNA quantitation from triplicate experiments. (E) Alkaline comet analysis of differentiated SH-SY5Y cells expressing WT or Q331K (lower panel). Quantitation of mean tail moment before and after Dox induction in 25–50 cells reveals an ~5-fold increase in DNA damage in Q331K cells (Scale bar, 10 μm). *P<0.01; **P<0.05; ***P<0.0005.
The TDP-43 Q331K mutation induces DDR
We next tested for DDR markers γH2AX and phospho Ser1981-ataxia-telangiectasia mutated (ATM) activation in Q331K-expressing cells. Dox induction of the WT and mutant lines for 24 h followed by DDR analysis at 72 h by immunoblotting showed the presence of increased levels of γH2AX and p-ATM in Q331K compared to WT or uninduced cells (Fig. 6A). IF data confirmed the presence of a higher number of average foci for γH2AX, p-ATM and p-53BP1 in Q331K cells (Fig. 6B). Although WT cells also showed a small increase (~1.5–2-fold) in the levels of activated DDR factors, consistent with a moderate increase in cytosolic aggregation, the increase was significantly profound (>4–6-fold) in Q331K cells. Taken together, these data link the accumulation of unrepaired, steady-state levels of DNA strand breaks and DDR activation with Q331K mutant expression in neurons. DSBs may be produced by a variety of exogenous or endogenous insults and in general by cell stress due to abnormal physiological conditions. ROS can cause secondary DSBs, because single-stranded lesions caused by ROS are converted to DSBs (21,22). To test the nature of these DNA strand breaks, we investigated the accumulation of DSBs and performed neutral comet assay, which exclusively visualizes DSBs (Fig. 6C) (20). We observed a significantly increased tail moment in Q331K-expressing cells compared to WT or uninduced control cells, indicating the accumulation of DSBs.
Figure 6.
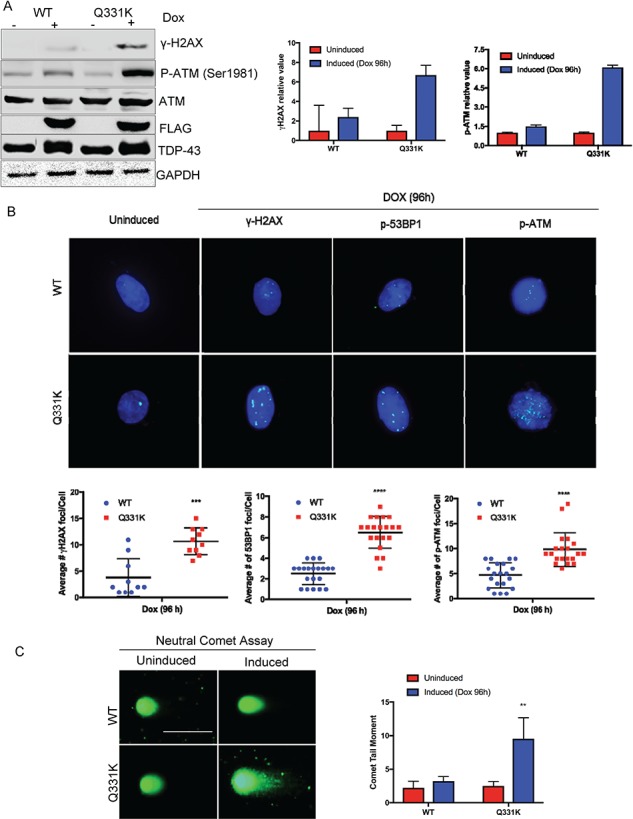
Activation of DDR signaling in Q331K-expressing neurons. (A) Immunoblots of extracts from inducible Q331K cells before and after Dox induction, probed with γH2AX and p-ATM antibodies. GAPDH served as loading control. Histogram represents quantitation of mean band intensity from three independent experiments. (B) Representative IF image of γH2AX, p53BP1 and p-ATM foci and quantitation of foci (Scale bar, 10 μm). (C) Neutral comet analysis of differentiated SH-SY5Y cells expressing WT or Q331K (lower panel). Quantitation of mean tail moment before and after Dox induction in 25–50 cells reveals an ~3-fold increase in DNA DSBs in Q331K cells (Scale bar, 10 μm). **P<0.05; ***P<0.0005; ****P<0.0001.
Q331K mutation affects kinetics of DSB repair
To test the effect of the Q331K mutation in DSB repair, we performed ionizing radiation (IR)-induced γH2AX foci disappearance analysis (Fig. 7A). Differentiated SH-SY5Y cells were first induced with Dox for 24 h and allowed to grow for an additional 72 h as before. The cells were treated with IR (3 Gy), and γH2AX foci were counted at 0.5, 3 and 6 h post-irradiation by IF microscopy. Quantitation of the average number of foci per cell, counted in at least 25 cells, showed that while the foci gradually disappeared in WT cells at 3 and 6 h, the mutant cells retained a significant number of foci even at 6 h. This suggested delayed kinetics of DSB repair in Q331K mutant cells. These results were further supported by comet analysis at 0.5 and 6 h after damage induction by IR (Fig. 7B). The data showed persistent comet tails at 6 h after IR treatment in mutant cells, whereas most of the induced damage was repaired in the WT cells (Fig. 7B).
Figure 7.
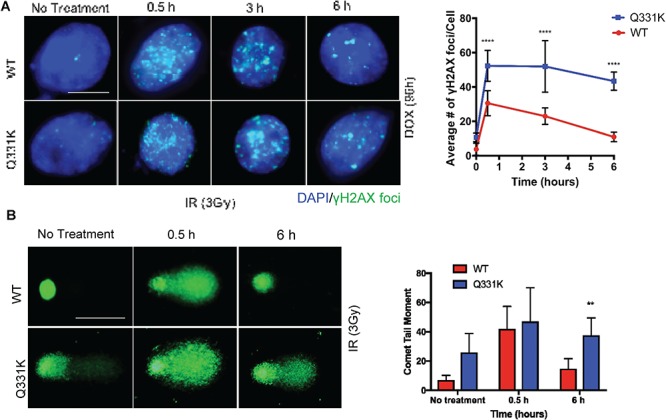
Q331K inhibits DNA DSB repair. (A) Analysis of γH2AX foci kinetics after IR (3 Gy) treatment by IF microscopy. γH2AX foci are shown in green (Scale bar, 5 μm). Quantitation of the average number of foci per cell shown in histogram. (B) Time course comet analysis of cells treated with 3 Gy radiation. Quantitation of mean comet tail moment in 50 randomly selected cells (Scale bar, 10 μm). **P<0.05 ; ****P<0.0001.
The TDP-43 Q331K mutation induces neuronal apoptosis in the presence of DNA-damaging drugs
Next, we examined neuronal apoptosis using the TUNEL assay in Q331K cells, with or without the presence of various DNA-damaging drugs. At steady state, the Q331K cells showed significant TUNEL positivity compared to WT following Dox induction (Fig. 8A). The cells were treated 72 h after Dox induction with a DNA DSB-inducing topoisomerase II inhibitor etoposide (10 μM), a DNA SSB-inducing topoisomerase I inhibitor camptothecin (CPT) (5μm), or ROS-inducing glucose oxidase (100 ng/mL). After treatment with DNA-damaging drugs, the cells were allowed to grow for additional 24 h, before the TUNEL analysis was performed. The data showed a significantly higher number of TUNEL-positive cells in the Q331K-expressing line compared to WT cells (Fig. 8A). An MTT-based cell viability measurement under similar conditions confirmed the enhanced sensitivity of mutant cells to DNA-damaging drugs (Fig. 8B). Immunoblotting of total cell extracts with antibodies specific to apoptosis markers showed the presence of cleaved-caspase 3 and cleaved-PARP1 products in Q331K cells (Fig. 8C). Taken together, these data suggest enhanced sensitivity of the neurons with Q331K mutation to DNA damage stress.
Figure 8.
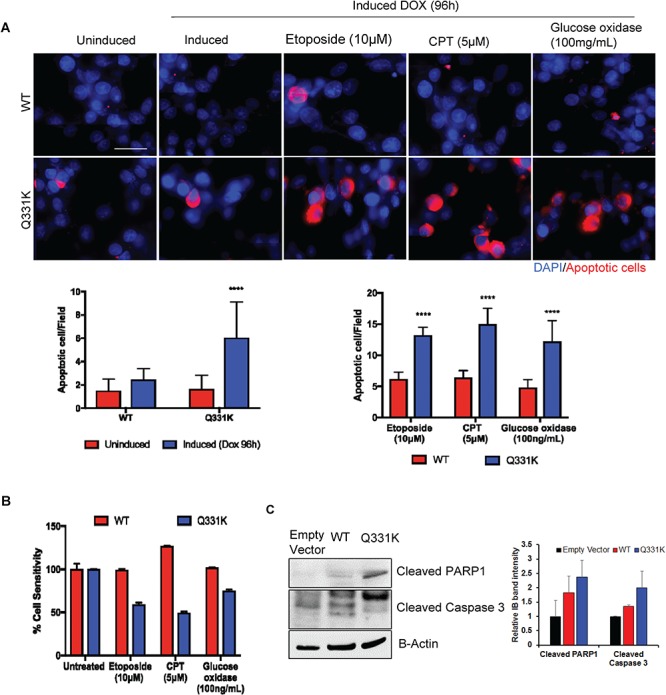
Apoptosis and sensitivity of Q331K neurons to DNA damage. (A) TUNEL analysis by IF microscopy. Upper panels represent differentiated SH-SY5Y cells expressing WT while lower panels represent cells expressing Q331K mutation. Cells were induced with Dox for 24 h and cultured for an additional 72 h before treating with DNA damage-inducing drugs. The cells were then allowed to recover for 24 h before TUNEL analysis. Red staining indicates TUNEL-positive cells. Histogram represents the quantitation of TUNEL-positive cells per field from three independent experiments. (B) MTT assay for measuring the viability of cells after treatment with DNA-damaging agents. The viability was measured at 24 h of recovery after damage induction. (C) Immunoblots of cell extracts were probed for apoptotic markers with antibodies against cleaved PARP1 and cleaved Caspase 3. β-Actin is shown as a loading control.****P<0.0001.
Mutant TDP-43 prevents nuclear translocation of the XRCC4-DNA ligase 4 complex and inhibits DSB ligation activity
Cytosolic XRCC4 is translocated to the nucleus in response to genome damage, and this translocation requires its interaction with DNA ligase 4 (23). The formation of the XRCC4-DNA ligase 4 complex is critical for efficient recruitment and stability of DNA ligase 4 at DNA damage sites (24). Our recent studies showed that TDP-43 interacts with the XRCC4-DNA ligase 4 complex to regulate its recruitment at DSBs in the neuronal genome (10). Here we investigated how the Q331K mutation affects DNA damage-dependent nuclear translocation of XRCC4 and DNA ligase 4. IF microscopy with anti-XRCC4 and anti-DNA ligase 4 antibodies in irradiated WT- or Q331K-expressing cells showed that both DNA ligase 4 (Fig. 9A) and XRCC4 (Fig. 9B) were predominantly localized in the nucleus (stained with DAPI) of WT cells. However, significant immunoreactivity was observed in the cytosol for XRCC4 and DNA ligase 4 in Q331K cells. Immunostaining with anti-FLAG antibody confirmed increased nucleocytoplasmic mislocalization of the Q331K mutant. These data suggest that mutant TDP-43 prevents the DNA damage-dependent nuclear translocation of XRCC4 and DNA ligase 4. We next confirmed the cytoplasmic sequestration of XRCC4 and DNA ligase 4 using PLA for anti-FLAG versus anti-XRCC4 or anti-DNA ligase 4 antibodies and observed more cytoplasmic localization in the Q331K mutant compared to WT (Fig. 9C). We next isolated the FLAG IP complex from FLAG-tagged WT- and Q331K mutant-expressing cells and performed DNA-ligation analysis using a 5′Cy3-labeled nicked oligonucleotide substrate (Fig. 10A). The ligation activity was markedly reduced in the IP complex from mutant cells compared to the WT cells. Substrate alone, and T4 DNA ligase-mediated ligation product, served as size controls. This inhibition in DNA ligase activity was due to the reduced association of the Q331K mutant with DNA ligase 4 as observed in immunoblotting analysis of WT and Q331K IP (Fig. 10B). Here, while FLAG levels in WT and mutant IPs were comparable, the level of DNA ligase 4 in the mutant co-IP complex was significantly reduced compared to WT, both from unstressed and irradiated cells. To further confirm the reduced interaction of mutant TDP-43 with DNA ligase 4 and XRCC4, we performed an in vitro His-affinity co-elution (pulldown) experiment using purified proteins. Immunoblotting of the co-eluted proteins confirmed reduced binding of mutant TDP-43 to the XRCC4-DNA ligase 4 complex, in the absence of DNA (Fig. 10C). Together, these results demonstrate that the Q331K mutation inhibits DNA ligase 4 activity by preventing its nuclear translocation in neuronal cells.
Figure 9.
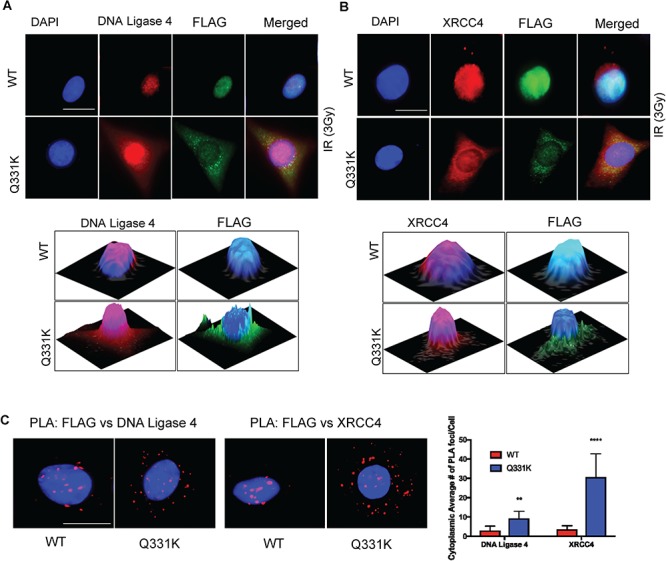
The Q331K mutation affects the nuclear translocation of XRCC4-DNA ligase 4. (A) IF of DNA ligase 4 in WT or Q331K cells shows increased cytoplasmic presence in mutant cells (Scale bar, 10 μm). The 2.5-dimensional view of the localization is shown in the image below. (B) IF of XRCC4 in WT or Q331K cells shows their reduced nuclear presence in mutant cells (Scale bar, 10 μm). The 2.5-dimensional view of the localization is shown in the image below. (C) PLA of FLAG versus DNA ligase 4 and FLAG versus XRCC4 in cells expressing WT and mutant TDP-43 (Scale bar, 10 μm). The higher number of foci in the cytoplasm of Q331K-expressing cells compared to WT-expressing cells indicates the increased interaction of XRCC4-DNA ligase 4 after DNA-damage induction by IR (3 Gy). **P<0.05 ; ****P<0.0001.
Figure 10.
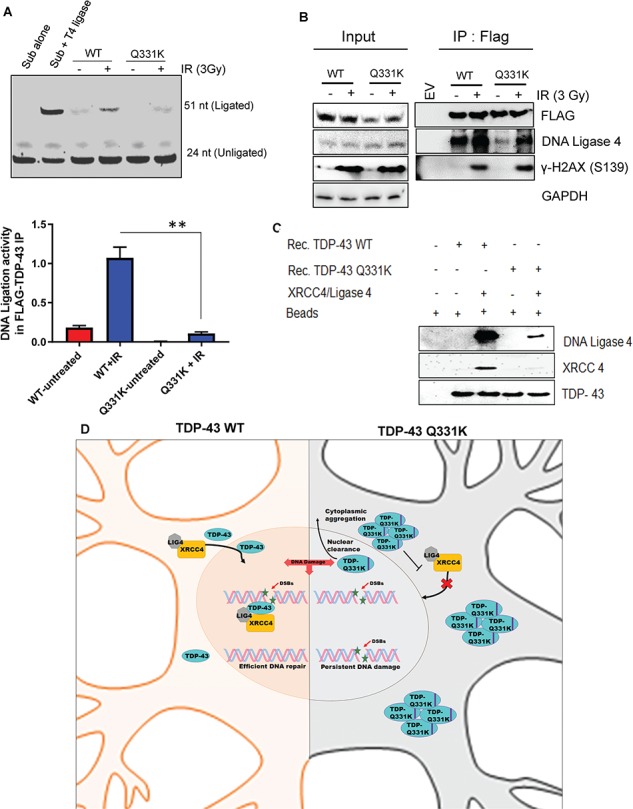
The Q331K mutation affects DNA-ligation activity. (A) In vitro DNA-ligation assay using extracts of neurons expressing WT or mutant TDP-43 before and after DNA damage induction by IR (3 Gy). Quantitation of DNA-ligation activity from three independent experiments expressed as fold change in the histogram. (B) IP of WT- and Q331K-expressing cells with anti-FLAG antibody or IgG (mouse) before and after DNA-damage induction by IR (3 Gy) shows reduce DNA ligase 4 interaction in Q331K mutant cells. (C) His-affinity pulldown assay using recombinant TDP-43-WT and TDP-43-Q331K shows reduced association of the Q331K protein with XRCC4-DNA ligase 4 complex in vitro. (D) A model showing TDP-43 WT facilitates XRCC4-Ligase4 complex nuclear translocation in healthy neuronal cells leading to efficient DNA repair. ALS-linked Q331K mutation nuclear clearance and cytoplasmic aggregation impairs XRCC4-Ligase4 complex nuclear translocation leading to persistent DNA damage accumulation. **P<0.05.
Discussion
Since the discovery that implicated TDP-43 toxicity in ALS in 2006 (2,6), genetic-screening studies have identified several missense mutations in the TARDBP gene that encodes the TDP-43 protein in both familial and sporadic ALS, which firmly established a causal role for TDP-43 in neurodegeneration. Although several TDP-43 mutations have been identified in ALS patients, how mutant TDP-43 triggers neuronal dysfunction is unclear. A common feature of TDP-43 pathology is its nucleo-cytoplasmic mislocalization in spinal motor neurons, leading to both nuclear clearance and cytoplasmic sequestration of poly-ubiquitinated aggregates of TDP-43. The lack of nuclear TDP-43 is consistently observed in inclusion-bearing neurons (25–28) in the spinal cord in ALS patients. Most of the mutations are localized in the C-terminal glycine-rich region encoded by exon 6 (7,28–30). Cell culture expression of disease-linked mutations replicates a biochemical profile similar to pathological TDP-43, including its hallmark nuclear clearance (2,31).
The RNA-binding functions of TDP-43 are well characterized and involve global regulation of gene expression, including regulation of transcription and multiple aspects of RNA processing such as splicing, stability, transport, translation and microRNA maturation (32–35). Although TDP-43 also binds DNA, its DNA-binding functions and implications in ALS were not investigated until our recent report on the involvement of TDP-43 in the NHEJ-mediated DSB repair pathway (10). TDP-43 regulates the recruitment and activity of the DSB break-sealing ligation complex XRCC4-DNA ligase 4 in healthy neurons, and its functional loss leads to DSB repair defects. The resulting persistent accumulation of DSBs and activation of DDR factors contribute to neurodegeneration. These studies raised the question of whether TDP-43 mutations affect its DNA repair functions.
In this study, we investigated the role of the Q331K mutation in neuronal genome instability. The Q331K mutation, first identified in sporadic ALS patients (7), was shown to progressively aggregate in transgenic mice (9). Furthermore, studies have suggested abnormal nucleic acid binding by the Q331K mutant that results in an increased aggregation rate of the protein (9). The effect of this mutation on DNA binding by TDP-43 suggested its likely impact on the DNA repair functions of this protein, providing the rationale for our study.
We observed significant accumulation of DNA strand breaks, both in genomic DNA isolated from ALS patient spinal cord tissue harboring the Q331K mutation and in neuronal cells ectopically expressing Q331K. Furthermore, the levels of DDR markers γH2AX, p-53BP1 and p-ATM were markedly activated in patient spinal cord tissue and in cell lines. This suggested a possible role for mutant TDP-43 in promoting DNA damage or affecting their repair.
It is important to mention that the mutation was not present in the genomic DNA isolated from occipital lobe tissue of the same patient. Furthermore, the mutation was present in ~10–20% of total genomic DNA isolated from the patient spinal cord. This suggests that the mutation is acquired sporadically, consistent with previous observations (7).
To gain molecular insights into the effect of Q331K mutation on the integrity of the neuronal genome, we first generated a conditional neuroblastoma cell line that expresses WT or mutant TDP-43 upon Dox induction. Although our initial efforts using transiently expressing cell lines strongly linked the mutation to DNA damage accumulation, the reproducibility of comparable transfection efficiency and expression of WT and mutant TDP-43 in differentiated neurons was a challenge. We then generated a stable SH-SY5Y line constitutively expressing mutant TDP-43, which unexpectedly lost ectopic protein expression after 2–3 passages. This is likely due to the autoregulatory activity of TDP-43 and the expansion of surviving cells with reduced mutant expression.
We finally generated a Dox-inducible neuronal line, which allowed us to induce controlled expression of WT or mutant TDP-43. Dox induction for 24 h, followed by cell culture for 72 h reproducibly contained comparable levels of ectopic and endogenous TDP-43, which is critical to prevent possible robust toxicity of overexpression. In addition, the conditional line allowed for induction of TDP-43 expression after neuronal differentiation, thus preventing any impact of mutant TDP-43 on the neuronal differentiation process. We recently generated and characterized a similar cell line model for α-synuclein to study Parkinson’s disease (36). We believe that conditional and controlled expression of amyloidogenic proteins is very critical for physiologically modeling neurodegenerative disease.
Using this conditional cell line model, we demonstrate that mutant TDP-43 not only affects DSB repair but also contributes to damage induction by promoting ROS stress in neurons. Previous studies have shown generation of ROS by amyloid aggregates by gain of toxicity. It is likely that TDP-43 aggregates in the neuronal cytosol contribute to ROS accumulation. Consistently, mutant cells showed significant sensitization when exposed to a variety of DNA-damaging drugs, including DSB-inducing IR or etoposide, and SSB-inducing CPT or ROS stress. These DNA-damaging drugs induced neuronal apoptosis in mutant TDP-43 cells. TDP-43 has recently been shown to localize in neuronal mitochondria (37,38). Mutant TDP-43 may thus affect mitochondrial function, thereby increasing ROS stress, which needs to be investigated. The DSB repair defects and damage accumulation in mutant cells strongly correlates with the nuclear clearance and cytosolic sequestration of poly-ubiquitinated forms of TDP-43.
Kinetic analysis of repair of DNA strand breaks induced by IR showed that mutant TDP-43 expression significantly delays the repair, compared to WT cells. To elucidate the molecular mechanism of delayed repair in Q331K cells, we examined the functions of the XRCC4-DNA ligase 4 complex, a key factor in sealing DSBs via NHEJ in neurons. As neurons lack replication-associated homologous recombination, NHEJ is the predominant DSB repair pathway. It was previously shown that XRCC4, a scaffold for DNA ligase 4, is largely present in the cytosol in unstressed cells and is translocated to the nucleus to localize in damaged chromatin after damage induction (23). This damage-dependent translocation requires the binding of XRCC4 to DNA ligase 4, as shown by its reduced translocation in cells lacking DNA ligase 4 (23). Interestingly, the mutant TDP-43, which also mislocalizes to the cytosol, inhibits the nuclear translocation of XRCC4. This was shown by two independent methods: IF and immunoblotting analysis. Our in vitro interaction analysis showed that while WT TDP-43 binds the XRCC4-DNA ligase 4 complex, the mutant shows significantly reduced interaction. Thus, it appears that nuclear TDP-43 is important for the damage-dependent nuclear translocation of the XRCC4-DNA ligase 4 complex. Consistent with this, DNA-ligation activity was markedly reduced in nuclear extracts from Q331K cells compared to those from WT cells. These data provided an important molecular insight into how mutant TDP-43 impedes DSB repair by inhibiting the nuclear translocation of XRCC4-DNA ligase 4 complex. Moreover, the role of TDP-43 in regulating the function of DSB sealing XRCC4-DNA ligase 4 complex appears to be specific. A related RNA-binding protein FUS, which is primarily linked to familial ALS, is associated with XRCC1-DNA ligase 3 functions but not XRCC4-DNA ligase 4 (39).
Our studies thus unravel the role of nuclear TDP-43 in facilitating the nuclear translocation of the XRCC4-DNA ligase 4 complex for the repair of DSBs via NHEJ and the effect of the Q331K mutation in disrupting this process, causing defective DSB repair, which may play a critical role in neurodegeneration (Fig. 10D).
Materials and Methods
Expression plasmids
The FLAG-TDP-43 WT pcDNA 3.1(+) expression plasmid was a kind gift from Prof. Paul Taylor (St. Jude Children’s Research Hospital; Memphis, TN). pCW-Cas9 was a gift from Eric Lander and David Sabatini (Addgene plasmid # 50661). The TDP-43 coding DNA sequence (CDS) along with the N-terminal 1X FLAG tag sequence was PCR-amplified from FLAG-TDP-43 pcDNA 3.1 vector using high-fidelity Deep Vent DNA polymerase (# M0258, NEB, Ipswich, MA). The following primers were used for PCR amplification: Fl-TDP43-F: 5′-CGGCGCTAGCATGGACTACAAAGACGATGACGACAAGTCTGAATATATTCGGGTAAC-3′ and Fl-TDP43-R: 5′-CAACGGATCCCTACATTCCCCAGCCAGAAGACTTAGAATCCA-3′.
Thermal profile: the cycle included an initial incubation at 95°C for 2 min, followed by 35 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 90 s and a final elongation at 72°C for 10 min.
Both the pCW-Cas9 vector and TDP-43 CDS were double digested with NheI (#R0131, NEB) and BamHI (#R0136, NEB) restriction enzymes, and subsequently, 3× FLAG-Cas9 CDS was replaced with 1× FLAG-TDP-43 by ligation using T4 DNA ligase (#EL0011, Thermo Scientific, Waltham, MA) to generate Dox-inducible pCW-FLAG-TDP43 mammalian expression vector.
For the construction of the pCW-TDP-43 Q331K mutant, the Q331K mutation was introduced in the pcDNA-TDP-43 plasmid using QuickChange II XL site-Directed Mutagenesis kit (#200521, Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. The following primers were used for Q331K mutation: TDP43-QK-Sense: 5′-GCCATGATGGCTGCCGCCCAGGCAGCACTAAAGAGC AGTTGGGGTATGATGGG-3′ and TDP43-QK-Antisense: 5′-CCCAT CATACCCCAACTGCTCTTTAGTGCTGCCTGGGCGGCAGCCATCATGGC-3′.
1× FLAG-TDP-43 Q331K mutant CDS was then PCR amplified, double digested and cloned into the pCW vector similar to WT TDP-43 to generate the pCW-TDP-43 Q331K expression plasmid.
Cell culture
SH-SY5Y cells were cultured in DMEM/F12 (#11320-033, Gibco, Waltham, MA) medium supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin in a humidified chamber at 37°C with 5% CO2. SH-SY5Y cells were differentiated using 10 μM retinoic acid (#R2625, Sigma, St. Louis, MO) and brain-derived neurotrophic factor (#B3795, Sigma) (50 ng/mL) for 6 days in DMEM/F12 medium supplemented with 1% FBS and 1% PS.
Generation of stable inducible cell line
First, SH-SY5Y cells were transfected with 2 μg of either pCW-TDP-43 WT or Q331K plasmid DNA using lipofectamine 2000 reagent (#11668-019, Invitrogen, Carlsbad, CA) and opti-MEM (#31985-070, Gibco), per standard protocol in 60–70% confluent cells. Cells were subjected to antibiotic selection by puromycin (#ant-pr, Invivogen, San Diego, CA) 24 h after transfection at a concentration of 5 μg/ml. Following selection, individual discrete colonies were isolated and cultured individually as independent clones. Finally, each of clones were induced with Dox (#D9891, Sigma) (5 μg/ml) for 48 h, and FLAG-TDP-43 expression was analyzed by immunoblotting.
Human tissue
Sporadic ALS spinal cord tissue specimens and the respective age-matched controls were obtained from the Department of Veterans Affairs Biorepository, as de-identified, frozen specimens. Studies on human tissues were conducted in accordance with the ethics board standards at the Department of Veteran’s Affairs and the institutional review boards at the Houston Methodist Research Institute (Houston, Texas).
Protein extraction
Cells transiently or stably transfected with expression plasmids were washed with Dulbecco’s phosphate-buffered saline (DPBS) and harvested with cell scrapers. Cells were pelleted at 1500 rpm at 4°C for 5 min, lysed with whole cell lysis buffer (50 mm Tris-HCl pH 7.5, 150 mm NaCl, 1 mm EDTA and protease inhibitor cocktail) for 15 min on ice and centrifuged for 10 min at 10 000 rpm at 4°C to obtain clear cell lysates. For protein extraction from tissues, tissues were homogenized in RIPA buffer supplied with cocktail protease inhibitors (#A32955, Thermo Scientific) and phosphatase inhibitors (#5870, Cell Signaling Tech, Danvers, MA) (20 mm Tris-HCl, pH 7.5; 150 mm NaCl; 1 mm EDTA, 0.5 EGTA; 1% Sodium deoxycholate; 1% Triton-X-100 and 0.1% SDS). Tissue lysates were sonicated 6–7× at 8 amplitude for 10 s each time, with 2 min intervals between 2 consecutive pulses, followed by centrifugation 2× at 13 000 rpm for 10 min at 4°C. Protein concentrations were measured by the Bradford assay method.
Antibodies
The antibodies used in this study were as follows: polyclonal anti-TDP-43 (ProteinTech, Rosemont, IL, 10782-2-AP), FLAG M2 (Sigma, A8592, F1804), pSer1981-ATM (Abcam, United Kingdom, ab81292-100), phospho-H2AX (Cell Signaling, 9718s), ATM (Abcam, ab32420), phospho-53BP1 (Cell Signaling, 2675), phospho-di-TDP-43 (Sigma, SAB4200225), Apoptosis Cocktail (pro/p17-caspase 3, cleaved-PARP, muscle actin) (Abcam, ab136812), Glyceraldehyde phosphate dehydrogenase (GAPDH) (ProteinTech, 6004-1-IG), Amersham ECL rabbit IgG HRP-linked (GE Healthcare Life Sciences, Chicago, IL, NA934), Amersham ECL mouse IgG HRP-linked (GE Healthcare Life Sciences, NA931), Anti-Mouse IgG Texas Red-X conjugate (Thermo Scientific, T-862) and Anti-Rabbit IgG Alexa Fluor® 488 conjugate (Thermo Scientific, A-11008).
Immunoblotting
Proteins were separated on a NuPAGE 4–12% Bis-Tris Gel (#NP0335, Novex), and gel electrophoresis were carried out in 1× NuPAGE running buffer (#NP0002-02, Novex). Proteins were electrotransferred onto a 0.45 μm pore size nitrocellulose membrane (#1620115, Bio-Rad) in 1× NuPAGE transfer buffer (#NP0006-1, Novex). After protein transfer, membranes were blocked in 5% skimmed milk (Millipore, Burlington, MA) solution in 1% Tris-Buffered saline with Tween-20 buffer (TBST) (#IBB-775, Boston BioProducts, Ashland, MA). Membranes were immunoblotted with appropriate primary and secondary antibodies in 1% skimmed milk and washed three times with 1% TBST.
Co-IP
Endogenous ubiquitin co-IP was performed using Protein A/G PLUS agarose beads (#sc-2003, Santa Cruz Biotechnology, Dallas, TX). Cells were harvested and lysed with whole-cell lysis buffer (0.2% NP-40, 150 mm NaCl, 50 mm Tris-HCl pH 7.5, 1 mm EDTA and protease inhibitor). Protein A/G beads were incubated with the appropriate primary antibody for 30 min at 4°C. Lysate was pre-cleared with 1 μg IgG/mg of total protein and incubated with A/G beads at 4°C for 30 min. The supernatant was incubated with the antibody-bound beads overnight at 4°C. After washing, the protein complex was eluted from the beads with 2× LDS sample buffer (#NP0008, Novex).
For FLAG co-IP, cells were harvested and lysed as previously described. Lysate was incubated with FLAG-M2 agarose beads (#M8823, Sigma) overnight at 4°C. After three washes with Tris-buffered saline (TBS), the protein complex was eluted from the beads as before.
IF Microscopy
Conditionally expressing WT TDP-43 and TDP-43 Q331K mutant SH-SY5Y cell lines were cultured in 8-well chamber slides (Millicell EZ slides, Millipore), fixed in 4% paraformaldehyde in 1× PBS for 20 min and permeabilized with 0.2% Tween-20 in PBS for 20 min at room temperature. Blocking was performed with 3% BSA solution in 1× PBS for 30 min at room temperature. Probing was done by incubating with appropriate primary antibody overnight at 4°C in 1% BSA solution and fluorescent secondary antibodies for 1 h at 37°C in 1% BSA solution. Slides were washed three times and counterstained with DAPI. For Figure 2D and 3D, slides were incubated with the cytoplasmic marker Phalloidin (2:100 in PBS, Life Technologies, Carlsbad, CA, A22282) for 25 min at room temperature before washing. Images were captured using an AXIO Observer inverted microscope (Carl Zeiss, Germany). The 2.5-dimensional images showed in Figures 3C and 9A and B were created by ZEN 2.3 software (Carl Zeiss Microscopy GmbH). Angle X: 53. Angle Y: 41. Z Scaling: 1.
PLA
In cellulo protein–protein association was analyzed using PLA (Duolink) according to the manufacturer’s instructions. PLA is an antibody-based method that detects molecules in close proximity (approximately 16 nm) and is a standard experiment used in our lab (40). SH-SY5Y cells were cultured in 8-well chamber slides (Millicell EZ slides, Millipore), fixed in 4% paraformaldehyde in PBS for 20 min and permeabilized with 0.2% Tween-20 in PBS for 20 min at room temperature before overnight incubation with primary antibodies. Secondary antibody probing and subsequent proximity ligation signal amplification were performed according to the manufacturer’s protocol. Images were analyzed using an AXIO Observer inverted microscope (Carl Zeiss).
MTT assay
Conditionally expressing WT TDP-43 and TDP-43 Q331K mutant SH-SY5Y cell lines were seeded in a 96-well ELISA plate (Corning, NY) in triplicate. After 4 days of induction with 10 μg/mL Dox, MTT assay (#4890-25-K, Trevigen, Gaithersburg, MD) was performed following the manufacturer’s protocol. In summary, 10 μl of MTT reagent was added to each well and incubated for 2–4 h until the purple dye was visible, then 100 μl of detergent reagent was added. After 2–4 h of incubation, the absorbance at 570 nm was measured using a microplate reader (Bio-Rad, 680 XR).
Single-cell gel electrophoresis (comet) assay
Alkaline and neutral Comet assays were performed in differentiated SH-SY5Y cells after 4 days of induction with 10 μg/mL Dox, according to the manufacturer’s protocol (Trevigen). Briefly, SH-SY5Y cells were mixed with low-melt agarose (#4250-050-02, Trevigen) and placed in Comet Assay Slides (#4250-050-03, Trevigen). Agarose-embedded cells were lysed with lysis solution (#4250-010-01, Trevigen) in a coupling jar overnight at 4°C, followed by incubation in the recommended freshly prepared alkaline buffer. Comet assay slides were subjected to electrophoresis at 21 V for 30 min at 4°C, whereas for the neutral comet assay, the slides were subjected to electrophoresis at 21 V for 45 min at 4°C, followed by 70% ethanol fixation and staining with SYBR® Green. Images were analyzed using an AXIO Observer inverted fluorescence microscope (Carl Zeiss).
TUNEL staining
Apoptotic cells in adherent condition were detected using the in situ BrdU-Red DNA Fragmentation (TUNEL) Assay kit (#ab66110, Abcam) following the manufacturer’s protocol. Briefly, cells were fixed in 4% paraformaldehyde and washed with washing buffer supplied with the kit. DNA labeling solution and anti-BrdU-Red antibody were incubated for 1 h at 37°C under humidified conditions and washed three times with ddH2O. Cells were counterstained with DAPI and images were analyzed using an AXIO Observer inverted fluorescence microscope (Carl Zeiss).
Quantitation of DNA strand breaks by LA-PCR assay
Genomic DNA was isolated from SH-SY5Y cells using the Qiagen Blood and Tissue kit (#69504, Qiagen, Germany), and DNA was quantified using a nanodrop spectrophotometer (DeNovix DS-11). To estimate the accumulation of strand breaks using LongAmp Taq DNA polymerase (#M0323, New England Biolabs), four distinct gene segments (10--12kb) of genomic DNA were amplified using appropriate primer pairs (Table 1).
Table 1.
List of primers used for LA-PCR analysis and their sequence
| Gene target | Primer pair |
|---|---|
| HPRT (10.4 kb) | 5′-TGGGAT TACACGTGTGAACCAACC-3′ |
| 5′-GCTCTACCCTGTCCTCTACCGTCC-3′ | |
| Polβ (12.2 kb) | 5′-CAT GTC ACC ACT GGA CTC TGC AC-3′ |
| 5′-CCT GGA GTA GGA ACA AAA ATT GCT G-3′ | |
| NANOG (8.6 kb) | 5′-CTCCGGAATGGTAGTCTGAGAA GAA-3′ |
| 5′-ATTTAGGGCAGGCACAAGATGG-3′ | |
| OCT ¾ (10.1 kb) | 5′-TCTGTGGCCTCACCCTATGA-3′ |
| 5′-CAGACCTGTGGCAGGTATTGAA-3′ |
To ensure the linearity of PCR amplification with respect to the number of cycles and DNA concentration, preliminary standardization was carried out (19,41). As a control, a shorter fragment (~200 bp length) was also amplified using suitable primers (forward: 5′-TGCTCGAGATGTGATGAAGG-3′; reverse: 5′-CTGCATTGTTTTGCCAGTGT-3′) (42). PCR products were separated on a suitable agarose gel and visualized using the Gel Logic 2200 imaging system (Kodak, Rochester, NY).
IHC of human tissues
Paraffin-embedded sections were deparaffinized and rehydrated by subsequent xylene immersions in different dilutions followed by ethanol and rinsing with water for 5 min. Slides were boiled in 10 mm sodium citrate buffer for 10 min and cooled for 20 min. A 3% hydrogen peroxide solution in methanol was used to quench endogenous peroxidase for 10 min at room temperature. Sections were rinsed three times in ddH2O and TBST for 5 min each time. Sections were perfused in 0.2% TritonX-100 in PBS and blocked in horse serum for 30 min at room temperature. Primary antibodies were diluted in Dako antibody diluent for 1 h at room temperature. Sections were incubated for 30 min with peroxidase-coupled secondary antibody at room temperature and washed three times before developing in the DAB-substrate chromogen. Sections were counterstained with hematoxylin for 30–60 s. Then, sections were dehydrated with ethanol and xylene before the slides were mounted with coverslips.
In vitro protein–protein interaction by His-affinity co-elution
In vitro His-affinity pulldown assay was carried as previously described (43). Briefly, purified XRCC4/Lig4 complex (100 pmole) was purified with His-tagged TDP-43 and His-tagged Q331K (40 pmole) pre-bound to HisPur magnetic nickel-nitrilotriacetic acid beads (Thermo Fisher). After 1 h of incubation at 4°C with continuous rotation in buffer containing 1× TBS, 5% BSA and 10% glycerol, beads were washed with TBS containing 0.1% Triton X-100 and 300 mm NaCl. Bound protein was eluted with SDS sample dye and subjected to western blot analysis.
DNA nick-ligation assay
DNA-ligation assay was performed via an endogenous XRRC4 IP elution using the stably expressing FLAG TDP-43 and FLAG Q331K cell line. For the rescue assay, endogenous XRRC4 IP from SHSY-5Y cell line was incubated with 100 fmole of purified TDP-43 and Q331K, respectively. Briefly, oligos for the nicked duplex DNA substrate were custom-synthesized by Sigma, as follows: p24-5′-Cy3-GGCACGGTCTACACGGCACACGAG-3′, p27-5′-TGTACATGATACGATTCCAAGCTAAGC-3′ and p51-5′-CCGTGCCAGATGTGCCGTGTGCTCACATGTACTATGCTAAGGTTCGATTCG-3′.
Following the annealing of 10 pmol of each oligomer with 50 mm NaCl in boiling water, annealed oligomers were mixed with IP products with or without recombinant TDP-43 and recombinant Q331K in 1× T4 ligation buffer, and the mixture was incubated in a water bath for 10 min at 30°C for ligation. Samples were then mixed with 2× TBE sample buffer, heated for 3 min at 100°C and, subsequently, cooled down on ice for 3 min before loading onto a denaturing urea polyacrylamide gel for electrophoresis. The band was detected by Typhoon FLA 7000 (GE Healthcare) for ligation kinetic analysis.
Statistical analysis
GraphPad Prism 6 or Microsoft Excel software was used for data analysis. Comparisons of groups were generated with two-way analysis of variance or Student’s t-test. P-values are indicated in the associated figure legends.
Acknowledgements
E.N.G. is thankful to IFHARU and SENACYT for doctoral fellowships. The authors thank the Pathology core facilities at the University of Texas Medical Branch at Galveston and Houston Methodist Research Institute for IHC services. Control and sporadic ALS spinal cord tissue specimens were provided by the Department of Veterans Affairs Biorepository (VA Merit Review BX002466).
Conflict of Interest statement. None declared.
Funding
National Institute of Neurological Disorders and Stroke (R01 NS088645); Muscular Dystrophy Association (MDA 294842); Houston Methodist Research Institute (M.L.H); Secretaria Nacional de Ciencia, Tecnolgía e Innovación (K.S.R.); Melo Brain Grant (K.S.R.).
References
- 1. Guerrero E.N., Wang H., Mitra J., Hegde P.M., Stowell S.E., Liachko N.F., Kraemer B.C., Garruto R.M., Rao K.S. and Hegde M.L. (2016) TDP-43/FUS in motor neuron disease: complexity and challenges. Prog. Neurobiol., 145–146, 78–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M. et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 3. Amador-Ortiz C., Lin W.L., Ahmed Z., Personett D., Davies P., Duara R., Graff-Radford N.R., Hutton M.L. and Dickson D.W. (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol., 61, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakashima-Yasuda H., Uryu K., Robinson J., Xie S.X., Hurtig H., Duda J.E., Arnold S.E., Siderowf A., Grossman M., Leverenz J.B. et al. (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol., 114, 221–229. [DOI] [PubMed] [Google Scholar]
- 5. Leverenz J.B., Yu C.E., Montine T.J., Steinbart E., Bekris L.M., Zabetian C., Kwong L.K., Lee V.M., Schellenberg G.D. and Bird T.D. (2007) A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain, 130, 1360–1374. [DOI] [PubMed] [Google Scholar]
- 6. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y. et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun., 351, 602–611. [DOI] [PubMed] [Google Scholar]
- 7. Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E. et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science, 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ayala Y.M., De Conti L., Avendano-Vazquez S.E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E. et al. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J., 30, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lim L., Wei Y., Lu Y. and Song J. (2016) ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol., 14, e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitra J, GE, Hegde PM, Liachko NF, Wang H, Vasquez V, Gao J, Pandey A, Taylor JP, Kraemer BC et al. (2019) Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-Strand break repair defects. Proc. Natl. Acad. Sci. U. S. A., 116, 4696–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baskaran P., Shaw C. and Guthrie S. (2018) TDP-43 causes neurotoxicity and cytoskeletal dysfunction in primary cortical neurons. PLoS One, 13, e0196528. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Johnson B.S., Snead D., Lee J.J., McCaffery J.M., Shorter J. and Gitler A.D. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem., 284, 20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitchell J.C., Constable R., So E., Vance C., Scotter E., Glover L., Hortobagyi T., Arnold E.S., Ling S.C., McAlonis M. et al. (2015) Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol. Commun., 3, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X., Guo C. and Kong J. (2012) Oxidative stress in neurodegenerative diseases. Neural Regen. Res., 7, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Butterfield D.A., Bader Lange M.L. and Sultana R. (2010) Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta, 1801, 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Wang W., Li L., Perry G., Lee H.G. and Zhu X. (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim. Biophys. Acta, 1842, 1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larasati Y.A., Yoneda-Kato N., Nakamae I., Yokoyama T., Meiyanto E. and Kato J.Y. (2018) Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci. Rep., 8, 2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rowe L.A., Degtyareva N. and Doetsch P.W. (2008) DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med., 45, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovalenko O.A. and Santos J.H. (2009) Analysis of oxidative damage by gene-specific quantitative PCR. Curr Protoc Hum Genet, Chapter 19, Unit 19 11. [DOI] [PubMed] [Google Scholar]
- 20. Collins A.R. (2004) The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol., 26, 249–261. [DOI] [PubMed] [Google Scholar]
- 21. De Zio D., Bordi M. and Cecconi F. (2012) Oxidative DNA damage in neurons: implication of ku in neuronal homeostasis and survival. Int J Cell Biol, 2012, 752420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martinez I., Garcia-Carpizo V., Guijarro T., Garcia-Gomez A., Navarro D., Aranda A. and Zambrano A. (2016) Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence, 7, 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Francis D.B., Kozlov M., Chavez J., Chu J., Malu S., Hanna M. and Cortes P. (2014) DNA ligase IV regulates XRCC4 nuclear localization. DNA Repair (Amst.), 21, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sibanda B.L., Critchlow S.E., Begun J., Pei X.Y., Jackson S.P., Blundell T.L. and Pellegrini L. (2001) Crystal structure of an Xrcc4-DNA ligase IV complex. Nat. Struct. Biol., 8, 1015–1019. [DOI] [PubMed] [Google Scholar]
- 25. Davidson Y., Kelley T., Mackenzie I.R., Pickering-Brown S., Du Plessis D., Neary D., Snowden J.S. and Mann D.M. (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol., 113, 521–533. [DOI] [PubMed] [Google Scholar]
- 26. Igaz L.M., Kwong L.K., Xu Y., Truax A.C., Uryu K., Neumann M., Clark C.M., Elman L.B., Miller B.L., Grossman M. et al. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol., 173, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickson D.W., Josephs K.A. and Amador-Ortiz C. (2007) TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol., 114, 71–79. [DOI] [PubMed] [Google Scholar]
- 28. Van Deerlin V.M., Leverenz J.B., Bekris L.M., Bird T.D., Yuan W., Elman L.B., Clay D., Wood E.M., Chen-Plotkin A.S., Martinez-Lage M. et al. (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol., 7, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gitcho M.A., Baloh R.H., Chakraverty S., Mayo K., Norton J.B., Levitch D., Hatanpaa K.J., White C.L. 3rd, Bigio E.H., Caselli R. et al. (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol., 63, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Vande Velde C., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F. et al. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet., 40, 572–574. [DOI] [PubMed] [Google Scholar]
- 31. Winton M.J., Igaz L.M., Wong M.M., Kwong L.K., Trojanowski J.Q. and Lee V.M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem., 283, 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buratti E. and Baralle F.E. (2010) The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol., 7, 420–429. [DOI] [PubMed] [Google Scholar]
- 33. Polymenidou M., Lagier-Tourenne C., Hutt K.R., Huelga S.C., Moran J., Liang T.Y., Ling S.C., Sun E., Wancewicz E., Mazur C. et al. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci., 14, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tollervey J.R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., Konig J., Hortobagyi T., Nishimura A.L., Zupunski V. et al. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci., 14, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawahara Y. and Mieda-Sato A. (2012) TDP-43 promotes microRNA biogenesis as a component of the Drosha and dicer complexes. Proc. Natl. Acad. Sci. U. S. A., 109, 3347–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vasquez V., Mitra J., Perry G., Rao K.S. and Hegde M.L. (2018) An inducible alpha-Synuclein expressing neuronal cell line model for Parkinson's Disease1. J. Alzheimers Dis., 66, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis S.A., Itaman S., Khalid-Janney C.M., Sherard J.A., Dowell J.A., Cairns N.J. and Gitcho M.A. (2018) TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett., 678, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang W., Wang L., Lu J., Siedlak S.L., Fujioka H., Liang J., Jiang S., Ma X., Jiang Z., da Rocha E.L. et al. (2016) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med., 22, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H., Guo W., Mitra J., Hegde P.M., Vandoorne T., Eckelmann B.J., Mitra S., Tomkinson A.E., Van Den Bosch L. and Hegde M.L. (2018) Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun., 9, 3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hegde M.L., Hegde P.M., Bellot L.J., Mandal S.M., Hazra T.K., Li G.M., Boldogh I., Tomkinson A.E. and Mitra S. (2013) Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U. S. A., 110, E3090–E3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chakraborty A., Tapryal N., Venkova T., Horikoshi N., Pandita R.K., Sarker A.H., Sarkar P.S., Pandita T.K. and Hazra T.K. (2016) Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun., 7, 13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sarker A.H., Chatterjee A., Williams M., Lin S., Havel C., Jacob P. 3rd, Boldogh I., Hazra T.K., Talbot P. and Hang B. (2014) NEIL2 protects against oxidative DNA damage induced by sidestream smoke in human cells. PLoS One, 9, e90261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dou H., Theriot C.A., Das A., Hegde M.L., Matsumoto Y., Boldogh I., Hazra T.K., Bhakat K.K. and Mitra S. (2008) Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem., 283, 3130–3140. [DOI] [PubMed] [Google Scholar]