

Abstract
Decades of research have elucidated the critical role of Akt isoforms in cancer as pro-tumorigenic and metastatic regulators through their specific effects on the cancer cells, tumor endothelial cells and the stromal cells. The pro-cancerous role of Akt isoforms through enhanced cell proliferation and suppression of apoptosis in cancer cells and the cells in the tumor microenvironment is considered a dogma. Intriguingly, studies also indicate that the Akt pathway is essential to protect the endothelial-barrier and prevent aberrant vascular permeability, which is also integral to tumor perfusion and metastasis. To complicate this further, a flurry of recent reports strongly indicates the metastasis suppressive role of Akt, Akt1 in particular in various cancer types. These reports emanated from different laboratories have elegantly demonstrated the paradoxical effect of Akt1 on cancer cell epithelial-to-mesenchymal transition, invasion, tumor endothelial-barrier disruption, and cancer metastasis. Here, we emphasize on the specific role of Akt1 in mediating tumor cell-vasculature reciprocity during the advanced stages of cancers and discuss how Akt1 differentially regulates cancer metastasis through mechanisms distinct from its pro-tumorigenic effects. Since Akt is integral for insulin signaling, endothelial function, and metabolic regulation, we also attempt to shed some light on the specific effects of diabetes in modulating Akt pathway in the promotion of tumor growth and metastasis.
Keywords: Akt1, cancer, diabetes, metastasis, tumor endothelium
Graphical Abstract

Introduction
In the advanced stages, cancer cells become highly invasive and eventually spread to distant organs, resisting treatments and risking the patients’ lives (1, 2). Once tumor cells acquire the ability to invade the surrounding tissues, the process of metastasis is instigated, and the cells enter the circulation through the lymphatic or vascular networks (3). Loss of cell-cell adhesion and acquisition of the migratory features allow malignant tumor cells to dissociate from the primary tumor, break the cell-matrix interactions and disintegrate the extracellular matrix (ECM) network that enables their invasion to the surrounding areas (4). Upon reaching a congenial microenvironment, these cells settle and adhere to a new location, start to colonize and profusely proliferate to generate the life-threatening secondary tumors (1, 5). Akin to the primary tumors, secondary tumors must also re-initiate angiogenesis in order for their growth to exceed 1–2 mm3 in size. Without angiogenesis, these metastasized tumors are deprived of oxygen and nutrients delivered through diffusion and thereby fail to develop further (2). Indeed, these events demonstrate the importance of vascular networks in cancer metastasis. Thus, cooperation between the tumor and vascular compartments ensures the overall growth of tumors, their trans-endothelial migration, invasion as well as metastasis and colonization in distant organs (6, 7).
Although the cross-talk between the tumor and vascular compartments is crucial in the regulation of tumor growth and metastasis, less attention has been given to the mechanisms by which tumor and vascular cells reciprocate with each other within the tumor microenvironment (8). In a recent review, we outlined the importance of Src family of kinases (SFKs) in the regulation of tumor vascular permeability, endothelial-barrier regulation, tumor growth and metastasis (9). In addition to SFKs, another important molecule that mediates such a cross-talk is protein kinase B (PKB or Akt), a serine-threonine kinase that exists in three isoforms namely Akt1, Akt2 and Akt3 (10). Akts are known to elicit isoform-, cell-and context-specific effects (11–14). In this review, we will shed the light on the molecular aspect of Akt1 to understand how it orchestrates the interaction between the tumor cells and the vascular compartment in the advanced stages of cancer. Here, we also present a molecular comparison between the specific effects of Akt1 activity modulation in tumor endothelial cells and the diabetes-related effects on endothelial cells on cancer metastasis.
Phosphoinositide-3-Kinase and Akt signaling pathway
The family of PI3Kinase (PI3K) consists of a group of lipid kinases that belong to three different classes (class I, II and III) and have the ability to phosphorylate hydroxyl group of inositol ring in the membranous inositol phospholipids (1–3). Class I PI3Ks are heterodimers that have the catalytic subunit (p110) linked to one of the regulatory subunits (p55, p65, p85 or p101). This class has been well documented in human cancers and can be further divided into 2 subclasses; subclass la and lb. Subclass la comprises 3 isoforms (PI3Kα, PI3Kβ, and PI3Kδ) with the catalytic domain (p110) combined to the regulatory domain (p55, p65 or p85, respectively), while subclass ib has γ isoform (PI3Kγ) with p110 catalytic subunit combined to the p101 regulatory subunit. The catalytic subunit is responsible for adding phosphate group to phosphatidylinositol 4,5-bisphosphate (PIP2) and producing phosphatidylinositol 3,4,5-trisphosphate (PIP3), an important second messenger functioning as a binding site in the inner cellular membrane for many proteins that contain pleckstrin homology (PH) domain, such as Phosphoinositide-Dependent Kinase-1 (PDK-1), Akt and other serine/threonine kinases. When Akt interacts with PIP3, it transiently localizes to the inner membrane, so PDK-1 can phosphorylate Akt at threonine residue and activate it. Currently, less is known about class II PI3K enzymes, namely PI3K-C2α, PI3K-C2β and PI3K-C2γ, and Class III PI3K member PI3K-C3, also known as vacuolar protein sorting 34 (Vps34).
The family of Akt consists of 3 different isoforms namely Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ, which are transcribed from three different chromosomes, and not through alternative splicing (15). These isoforms share structural resemblances to protein kinases A and C, hence called PKB. Although different genes encode these isoforms, they share ~80% homology in their structure and substrate specificities (16, 17). All Akt isoforms have a pleckstrin homology (PH) domain that is located at the N-terminal side and responsible for binding to PIP3. Catalytic domain of Akt has a threonine residue (Thr308, Thr309, and Thr305 in Akt1, Akt2, and Akt3, respectively) that is phosphorylated by PDK-1 for its activation. For complete activation, Akt requires additional phosphorylation at a serine residue (Ser473, Ser474 and 472 in Akt1, Akt2, and Akt3, respectively). Although a significant structural similarity is present among all the isoforms as mentioned previously, their functions in physiology and pathology do not appear to be redundant, which could be attributed to its tightly regulated subcellular localization (18).
Akt1 in cell survival, proliferation, and tumor development
The Akt pathway has a distinguished role in different cellular processes such as metabolism, proliferation, growth and cell death, and due to its integral role in promoting cell survival and inhibition of apoptosis, it is known as the “survival kinase” (1, 2). Akt mediates cell survival proliferation mainly by inhibiting the Bcl2 and MDM2 pathways, which otherwise promotes apoptosis (Figure 1). Although PI3K/Akt pathway is tightly regulated in normal cells, it gets deregulated in cancer cells leading to enhanced proliferation, growth and survival, and resistance to apoptosis (19, 20). Deregulation of Akt pathway builds the elements that are necessary for oncogenic transformation (21), promotes tumor growth (22, 23) and recruitment of inflammatory cells that are required in the tumor microenvironment (3). Mouse gene knockout studies focused on the Akt isoforms revealed their tissue and stage-specific expression and function (5, 24). Moreover, the fact that different genes encode different Akt isoforms and certain isoforms are hyper-activated in specific tumors supports the notion that Akt-isoform specificity determines tumorigenesis and cancer progression differently in various cancers. Since Akt1 is the predominantly expressed and the best-characterized isoform in many cancers, owing to its tissue versatility and context-specific effects, we will focus on the recent advances on its role in various stages of tumor growth with an emphasis on the cross-talk between the tumor and vascular compartments. Figure 1 summarizes the Akt1 targeted genes and their biological functions such as cell survival, proliferation, metabolism, and growth, all of which are essential for the overall tumor growth.
Figure 1. Pro-tumorigenic effects of Akt in early stages of cancer.
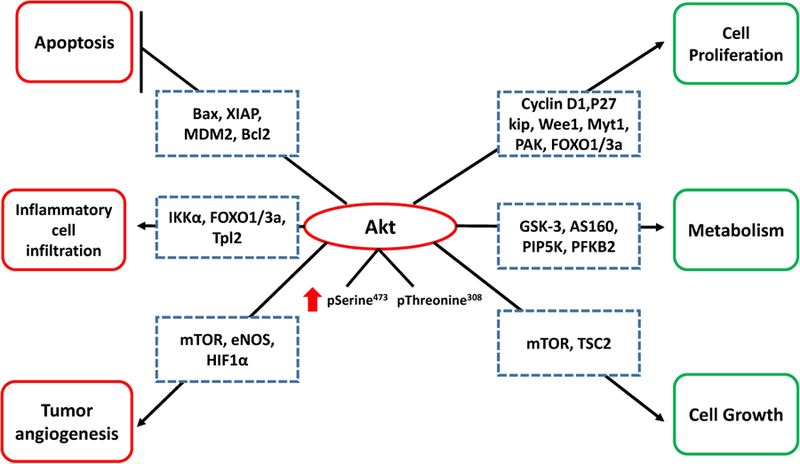
Enhanced Akt expression/activity in cancer cells results in enhanced proliferation, metabolism and cell cycle through various downstream targets including mTOR, GSK3α, GSK3β, FoxO transcription factors, MDM2, BAD, p27KIP1, etc. In addition, inhibition of cellular apoptosis and induction of tumor promoting cytokines, which favors inflammatory cell infiltration and tumor angiogenesis, further support cancer cell survival and tumor growth. Arrowhead indicates enhanced cell function and the flat arrow indicates inhibition of function. Green: Direct effect of Akt activity within cancer cell; Red: Indirect effects of increased Akt activity to promote tumor growth.
Bax- Bcl-2-associated X protein, XIAP- X-linked inhibitor of apoptosis protein, MDM2- mouse double minute 2 homolog, Bcl2- B-cell lymphoma 2, Ikkα- Ikappa B kinase (IKK)-associated protein 1α, FOXO1/3a- Forkhead box 1/3a,Tpl2- tumor progression locus 2, mTOR- mammalian target of rapamycin, eNOS- endothelial nitric oxide synthase, HIF1α- hypoxia-inducible factor 1α, p27Kip1- Cyclin-dependent kinase inhibitor 1B, Myt1- Myelin transcription factor 1, PAK- p21 activated kinase, GSK3- Glycogen synthase kinase 3, AS160- Akt substrate of 160 kDa, PIP5K- Phosphatidylinositol-5-Phosphate kinase, PFKB2- Phosphofructokinase-2, TSC2-Tuberous sclerosis proteins (tuberin).
Due to its integral role in cell cycle and cellular functions, Akt has long been considered an important oncogene essential for tumor initiation and growth (10). In an early study published in 1987, Akt was found to be amplified 20-fold in some of the human gastric carcinoma tissue samples, however, authors thought that was a sporadic event (25). Later, another study conducted on Asian population showed a significantly enhanced level of Ser473 phosphorylated Akt in tumor compared to the normal tissues (26). The phosphorylated Akt was predominantly localized in the cell membrane and cytoplasm, and occasionally in the nuclei of the cancer cells; while it was restricted to the cytoplasm of the normal cells. Following this, Sun M et al reported a predominant activation of Akt1 in many other types of human cancers such as the prostate, breast and ovary carcinomas (27). Interestingly, they showed that phosphorylated Akt1 was restricted to the primary tumor cells and absent in the stromal tissues. Later, we demonstrated the importance of Akt1 and its cooperation with the MAP Kinase pathway on oncogenic transformation (21) and cancer growth in the prostate (28). Our studies have also indicated that pharmacological (29–32) and genetic suppression of Akt activity (28, 33) could inhibit prostate cancer cell function in vitro and tumor growth in vivo. On the same line, Akt1 upregulation has also been shown in mammary adenocarcinoma developed in Neu and PyMT transgenic mice and its ablation significantly aborted cancer cell survival, thus emphasizing on the major role of Akt1 in tumor initiation and growth in the mammary glands (34). The role of Akt1 has also been demonstrated in lung cancer mouse models. One study showed that Akt1 deletion in tobacco-induced lung cancer and K-Ras mutant mouse models prevented tumor initiation in the lungs (35). In another study, It was shown that mice overexpressing IGF-IR developed lung cancer and Akt1 ablation significantly suppressed it (36). Moreover, this study showed that selective inhibition of Akt1 using A-674563 enhanced cancer cell apoptosis compared to pan-Akt inhibitor MK-2206 suggesting that targeting Akt1 isoform specifically could be more effective than inhibiting all Akt isoforms in lung cancer. Among the breast cancer studies, Wu Y et al showed that transgenic mice expressing a constitutively active Akt1 (MMTVmyr-Akt1) as such did not develop any neoplasms, however, treatment with 7,12 dimethyl-1,2-benzanthracene (DMBA)-induced mammary tumorigenesis in glands of virgin and post-lactating mice was significantly higher in the transgenic compared to wild-type mice (42.9% vs. 7.1%, respectively), indicating that Akt1 activation is a major risk factor in the development of mammary carcinoma secondary to carcinogens exposure (37). Additional details regarding the role of Akt in tumorigenesis have been extensively reviewed elsewhere (38). A schematic representation of the role of Akt in early cancers is shown in Figure 1.
Akt1 in angiogenesis and vascular permeability
Angiogenesis and vascular permeability are essential for the tumor perfusion, growth and trans-endothelial migration of cancer cells (39). Researchers have identified Akt1 as the predominant Akt isoform in endothelial cells (ECs) that is responsible for their growth and survival (40, 41). The role of Akt1 in the regulation of angiogenesis and vascular tone is also well established (41, 42). Studies on conditional Akt1–/– and Akt2–/– mice revealed the non-redundant function of Akt1 isoform in angiogenesis, where Akt1–/–, but not Akt2–/– mice had significant inhibition of retinal angiogenesis (43). Moreover, since nitric oxide (NO) is a major modulator of angiogenesis and blood flow and its release is promoted by phosphorylation of eNOS, many researchers reported that inhibition of Akt1 was accompanied by a significant reduction in the levels of phosphorylated eNOS and NO, thus blocking angiogenesis (44, 45). In a hind limb ischemia model elucidating the role of Akt1 in adaptive angiogenesis, a study indicated a significant impairment in vascular regeneration and >50% of the reduction in eNOS phosphorylation was observed in the Akt1–/– mouse lungs after Vascular Endothelial Growth Factor-A (VEGF) administration compared to the wild type animals (46). In support of this, another study in a cutaneous wound healing model also revealed impaired angiogenesis and extracellular matrix remodeling in Akt1–/– mice (41). More recently, Akt1 has been shown to promote angiogenesis and cardiac remodeling following myocardial infarction (47). These studies indeed demonstrate the ability of Akt1 in the regulation of adaptive angiogenesis, tissue remodeling, and blood flow. Therefore, targeting Akt1 or pharmacological inhibition its activity would impair the adaptive angiogenesis.
Tumor angiogenesis is a unique process that occurs as a result of the interaction between tumor cells and the endothelial cells. Vascular endothelial growth factor (VEGF), as mentioned previously, is an important cytokine required for initiating and regulating physiological angiogenesis (48). However, the elevated level of VEGF in many solid tumors besides the hyper-activation of Akt1 confirms the crosstalk between these molecules and their importance in tumor angiogenesis (49–51). One of the underlying mechanisms through which VEGF and Akt1 interplay in regulating angiogenesis are through activation of integrin that is necssary for migration of endothelial cells (52). In order to recapitulate the tumor-like effect in non-tumor endothelial cells, researchers adopted Akt1 over-expression and knockout in non-tumor endothelial cells. An initial study using this approach reported that the overexpression of active form of Akt1, myristoylated-Akt1 (MyrAkt1), in the endothelial cells of mice resulted in the formation of enlarged blood vessels compared to the wild type mice (53). These vessels had features similar to those observed in tumor vessels with a significant increase in neovascularization. In another study, endothelial myrAkt1 overexpression in mice was lethal with the formation of abnormally tortuous blood vessels in the embryos, similar to the tumor blood vessels (54) once again suggesting that endothelial Akt1 is not only promoting tumor vascularization but also enhancing vascular permeability.
The genuine effects of Akt1 in tumor vasculature were elegantly revealed by a study published by Dr. Byzova’s group using an Akt1 deficient mouse model (40). Although VEGF-induced cell migration was hampered in Akt1–/– endothelial cells, the vasculature supporting melanoma xenografts implanted in Akt1–/– mice were significantly leaky compared to vessels surrounding xenografts implanted in the wild type group. Because of that, these vessels were considered young and immature. Strikingly and unlike previous studies, subcutaneous xenografts in these mice were more vascularized compared to the ones in the wild type group. Mechanistically, the increased vascular permeability and enhanced angiogenesis seen in Akt1 null mice were attributed to the decreased levels of anti-angiogenic factors, Thrombospondin 1 and 2 (TSP1 and TSP2, respectively) in both tumor and endothelial cells of these mice compared to the wild-type mice indicating that Akt1 can directly regulate TSP1 and 2 expressions. Further analysis of these mice revealed that constitutive generation of VEGF through Adenovirus in Akt1–/– mouse skin also resulted in increased vascular permeability. In support of these observations, a recent study from our group showed that shRNA-mediated Akt1 knockdown in endothelial cells led to a significant reduction in the expression of 20 genes encoding tight-junction proteins, claudins, and mice with tamoxifen-induced VE-CreAkt1 had enhanced VEGF-induced vascular permeability compared to the wild type group (55). Moreover, Akt1 knock out mice had lower expression of Angiopoietin-1 that is known for mediating vascular protection, thus confirming the indispensable role of Akt1 in vascular maturation. Recently, we showed that although endothelial Akt1 loss in mice with tamoxifen-induced VE-CreAkt1 had not affected xenograft tumor growth, loss of endothelial Akt1 enhanced lung metastasis in these mice, suggesting that targeting Akt1 in endothelial cells could enhance transmigration of cancer cells through the vascular/endothelial barrier, thus cancer metastasis (56).
Apart from the direct effects of Akt1 on the tumor endothelial cells and cancer cells, a recent study from our group also identified the reciprocal cross-talk between Akt1 and Src, a non-receptor tyrosine kinase, in regulating and maintaining vascular homeostasis (57), cancer growth and metastasis (58). In addition, we recently demonstrated that candesartan, an angiotensin receptor blocker, inhibited prostate tumor growth via a tumor endothelium-dependent and tumor cell-independent manner indicating that modulation of Akt1 in tumor endothelial cells via angiotensin-renin system can have therapeutic benefits in cancer (32). Interestingly, although candesartan had no significant effect on various signaling pathways in tumor cells, it specifically activated endothelial Akt1, promoted vascular normalization and reduced permeability thus inhibiting the growth of prostate tumor xenografts in mice. Similarly, simvastatin with its dual role of activation and inhibition of Akt1 in the endothelial (59) and cancer cells (29, 30) respectively had demonstrated prevention of prostate cancer metastasis via vascular normalization (31).
Other laboratories have also reported the endothelial-barrier protection offered by activated Akt1. Following the initial findings, where the Akt1 suppression led to increased vascular permeability in mouse tumor xenografts (40), studies from Liao group showed the presence of vascular lesions in Akt1 deficient mice as a result of impaired mTOR signaling (60). Another study reported the endothelial-barrier protective role of Akt1 as a result of VE-cadherin overexpression and clustering (61). Furthermore, the protective effect of Akt1 on lung edema was reported as a result of stimulation by sphingosine-1-phosphate (62). Interestingly, Pestell group demonstrated that although mammary tumor xenografts in ErbB2/Akt1+/+mice were larger than ErbB2/Akt1–/– mice, the vascular density surrounding tumors in the latter was significantly higher compared to the former (63). Overall, these studies strongly indicate that Akt1 plays a dual and context-specific role in modulating tumor angiogenesis and vascular permeability.
Akt1 in the advanced cancers and metastasis
Apart from its pro-cell survival and proliferation roles, Akt1 has also been involved in the migration and invasion of cancerous cells (64), highlighting its importance in cancer progression and metastasis. Cancer progression was significantly ameliorated in tumors formed by Akt1 deficient lung cancer cells compared to Akt2 deficient cells (65), thus revealing that silencing Akt1, but not Akt2, can abrogate cancer cell migration in vitro and lung invasion in vivo. In bladder cancer cells, whereas Akt1 suppression resulted in the inhibition of cell migration, suppression of Akt2 had no significant effect (23). In soft tissue sarcomas, tumor cell migration and invasion were mainly controlled by Akt1 isoform, and its silencing not only abolished these properties but also reduced the expression of specific epithelial to mesenchymal transition (EMT) markers such as vimentin, which is linked to invasive cancers (66). In prostate cancer, cell motility, invasion, and trans-endothelial migration were promoted through Akt1-mediated integrin activation (64). Expression of the constitutively active Akt1 (CA-Akt1) in prostate cancer cells promoted tumor cell interaction with the endothelial cells and ECM proteins via enhanced cancer cell integrin β3 affinity, an effect that was reversed with the overexpression of inactive, a dominant negative mutant of Akt1 (DN-Akt1) (64). The role of TGFβ1-Akt1 pathway has also been indicated in melanoma progression and metastasis, where TGFβ1-mediated Akt1 activation was necessary to induce SKP2 expression, enhance N-cadherin and reduce epithelial marker E-cadherin expression thereby exhibiting mesenchymal cell morphology (67). In a pancreatic ductal adenocarcinoma study, KRasG12D mutant mice expressing myristoylated Akt1 developed early liver and abdominal metastases compared to the control KRasG12D group (68). In a murine model of thyroid cancer, Akt1–/– mice had less invasive thyroid tumors with the absence of lung metastasis compared to the control group (69). In a breast cancer study, ErbB2/Akt1+/+ mice that developed mammary tumors also developed lung metastasis; however, this was blunted in ErbB2/Akt1–/– mice (63). This was also supported by a significant reduction of ErbB2/Akt1–/– cells migration and invasion in vitro compared to ErbB2/Akt1+/+ cells. However, as previously described, in spite of the larger mammary tumors size in ErbB2/Akt1+/+mice, ErbB2/Akt1–/– mice tumors were significantly highly vascularized despite their smaller size. In a gastric cancer study, Han Z et al reported that activated Akt1 was significantly higher in the advanced, poorly differentiated gastric tumors compared to the early stages indicating the importance of Akt1 activation in gastric cancer progression (26).
Although a plethora of such reports has been published on the promoting effect of Akt1 activation on cancer metastasis, more recent studies, particularly started on breast cancer, have challenged this concept (See Table 1). The first report on the observation that Akt1 activation inhibiting cancer cell migration and invasion came from Muller’s group (70). Although ErbB2/activated Akt1 mice (bi-transgenic mice) had accelerated mammary tumorigenesis, fewer invasions to the surrounding tissues and a significant reduction in lung metastatic lesions were observed in these mice compared to the control group indicating that tumors developed with activated Akt1 had less metastatic propensity compared to the tumors with a reduced level of active Akt1. Another study supporting this observation was published by Alex Toker’s group (71), where investigators showed that overexpression with activated Akt1 attenuated breast cancer cell migratory and invasive properties in vitro, and reduced the formation of the actin cytoskeletal stress fiber. On the other hand, siRNA-mediated Akt1 deletion rescued the migratory and invasive phenotype of cancer cells. Mechanistically, ubiquitination and proteasomal degradation of nuclear factor activated T cells (NFAT) mediated by HDM2 (the human homolog of the oncoprotein and E3 ubiquitin ligase MDM2) was observed with the activation of Akt1 and totally reversed with Akt1 gene silencing. Another study on the distinct role of Akt1 and Akt2 in breast cancer cell lines came from Virginia Novaro’s group (72). In spite of the reduction in IBH-6 cells proliferation in vitro with genetic deletion of Akt1, cells invasion was significantly enhanced compared to their controls. On the other hand, Akt2 expression was crucial for promoting cells migration and invasion with less evident effect on proliferation shown after its deletion. Mechanistically, while Akt1 deletion enhanced β1 integrin and focal adhesion kinase (FAK) expression around the edges of IBH-6 cells supporting their attachment during the invasion, Akt2 deletion reduced vimentin and F-actin expression. Therefore, Akt1 deletion and Akt2 overexpression are essential for peritumoral invasion and lung metastasis.
Table 1.
Key studies showing metastasis of different types of cancer after genetic or pharmacological inhibition of Akt1 expression or activity, respectively.
| Reference | Cancer type | Method of Akt1 activity modulation | Observations | Mechanisms |
|---|---|---|---|---|
| Hutchinson JN. Et al. Cancer Res. 2004;64(9):3171–8. | Breast cancer | • In vivo: Bitransgenic mice overexpressing ErbB-2 and activated Myr-Akt1. | Myr-Akt1 overexpression inhibited lung metastasis despite accelerating mammary tumorigenesis. | Unknown |
| Yoeli-Lerner M. et al. Mol Cell. 2005;20(4):539–50. | Breast cancer | • In vitro: Breast cancer cell lines overexpressing activated Myr-Akt1 or transfected with Akt1-siRNA. | Cells migration and invasion were attenuated with Myr-Akt1 overexpression but enhanced with Akt1-siRNA. | Myr-Akt1 phosphorylated HDM2 that enhanced degradation of migration/invasion promoting factor-NFAT. |
| Chen L. et al. Oncology reports. 2014;31(2):737–44. | Colorectal cancer | • In vitro: Colorectal cancer cell line overexpressing Akt1 with/out Wortmannin treatment (Akt1 activity inhibitor). | Akt1 overexpression inhibited cells migration and invasion, which was reversed by Wortmannin treatment. | Akt1 overexpression inhibited the expression of MMP2, MMP9, HIF1α and VEGF that have a critical role in cancer progression. However, Wortmannin rescued their levels. |
| Li CW. et al. Cancer Res. 2016;76(6):1451–62. | Breast cancer | • In vitro: Mouse embryonic fibroblasts (MEFs) overexpressing activated Myr-Akt1 • In vivo: Tumor bearing mice treated with MK-2206 (Akt1 activity inhibitor). |
Myr-Akt1 overexpression inhibited EMT in MEFs whereas MK-2206 enhanced lung metastasis of cancer cells. | Myr-Akt1 phosphorylated the migration/invasion promoting factor-Twist-1 and enhanced its degradation β-TrCP, whereas all reversed by MK-2206. |
| Rao G. et al. Sci Rep.2017;7(1):7066. | Non small cell lung cancer (NSCLC) | • In vitro: NSCLC cell line overexpressing activated Myr-Akt1 • In vivo: Tumor bearing mice treated with MK-2206 (Akt1 activity inhibitor). |
Myr-Akt1 overexpression inhibited EMT in NSCLC cell line whereas MK-2206 induced their migration, invasion and bone and brain metastasis | Akt1 inhibition by MK-2206 promoted MARCKS phosphorylation that is required for LAMC2 overexpression and the enhanced migration, invasion and metastasis resulted from Akt1 inhibition, however, this was reversed by Myr-Akt1 overexpression |
| Gao F. et al. Cancer Lett. 2017;402:177–189. | Prostate cancer (PCa) | • In vitro: PCa cell lines with Akt1 downregulation using shRNA • In vivo: • Mice administered Akt1 silenced cancer cells • Tumor bearing TRAMP mice treated with TCBN (Akt1 activity inhibitor). |
Silencing of Akt1 in PCa cell lines and inhibition of its activity in tumor bearing TRAMP mice induced EMT and lung and liver metastasis, respectively. | Downregulation of Akt1 expression or inhibition of its activity using TCBN suppressed β-catenin and enhanced TGFβ signaling that promoted EMT and cancer metastasis. |
| Riggio M. et. al. Sci Rep. 2017 Mar 13;7:44244. | Breast cancer | • In vitro: Breast cancer cell lines overexpressing Myr-Akt1 or Akt2, or transfected with Akt1 or Akt2 shRNA • In vivo: Mice administered Akt1 or Akt2 silenced or overexpressing breast cancer cells. |
Silencing of Akt1 enhanced lung metastasis despite the reduction in cells proliferation., while overexpression of Akt2 increased vimentin and Factin levels with less evident effect on cellular growth. | Inhibition of Akt1 increased β1 integrin and FAK expression that enhanced cells invasions, while overexpression of Akt2 increased vimentin and Factin levels that enhanced cells migration and invasion. |
| Brolih S. et al. BMC cancer. 2018;18(1):249. | Head and neck squamous cell carcinoma (HNSCC) | • In vitro: HNSCC cell line with Akt1 downregulation using shRNA or treated by MK-2206 (Akt1 activity inhibitor). | Silencing of Akt1 expression or inhibition of its activity by MK-2206 in HNSCC cell line promoted loss of epithelial morphology, induced EMTlike phenotype, and increased their invasive capacity. | Unknown |
A link between Akt1 activity and palladin, an actin-binding protein that anchors other proteins to actin fibers, was demonstrated in support of the suppressive effects of Akt1 activity on breast cancer cell invasion (73). Palladin, an Akt1 specific substrate phosphorylated at Ser507, is required to maintain spheroid cells structure, prevent invadopodia formation and strengthen cells adhesion. However, upon Akt1 depletion, reduction in palladin phosphorylation destabilized actin filaments and enhanced cell branching and migration as shown in Figure 2. Another study has shown that over-expression of myrAkt1 inhibited RhoA activity and led to inhibition of breast cancer cell motility and invasion (74). In addition, an alternative mechanism by which Akt1 suppression has a pro-migratory and invasion effect was demonstrated in the human mammary epithelial cells (MCF-10A cells), where Akt1 silencing (but not Akt2) enhanced ERK activation (75). This was accompanied by significant changes in MCF-10A cell morphology shown by losing cuboidal-epithelial shape and acquiring spindle-shaped EMT characteristics, as depicted in Figure 2, along with increased expression of vimentin and N-cadherin.
Figure 2. Akt1 in angiogenesis and endothelial-barrier function with respect to tumor cell trans-vascular migration and metastasis.
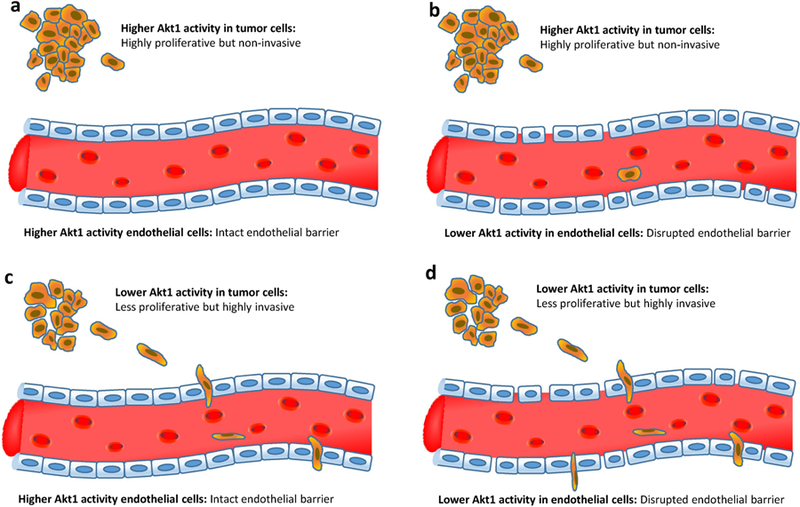
Four different scenarios are represented in the presence or absence of Akt1 activity in tumor and endothelial cells. (a) The tumor cells with higher Akt1 activity proliferate at a higher rate but are less invasive while endothelial barrier becomes impermeable due to the higher Akt1 activity thus blocking intravasation. (b) Despite less invasive, Akt1 active tumor cells may cross the Akt1 suppressed endothelial barrier due to the disrupted endothelial barrier. (c) Akt1 suppressed tumor cells proliferate at a lower rate but their highly invasive ability helps them to cross the Akt1 active endothelial barrier. (d) The highly invasive Akt1 suppressed tumor cells easily penetrate the highly permeable Akt1 suppressed endothelial barrier thereby causing intravasation and extravasation at a higher degree.
Another study on MCF-10A cells demonstrated that Akt1 down-regulation reduced miR-200 abundance, resulting in reduced E-cadherin expression and enhanced TGFβ1-mediated EMT (76). Interestingly, a paradoxical role of Akt1 and Akt2 isoforms in mammary tumorigenesis and metastasis was observed in transgenic models upon co-expression of these isoforms with activated ErbB2 or polyomavirus middle T antigen (PyVmT Y315/322F) (77). Whereas Akt1 promoted mammary gland tumorigenic activity, it did not impact the metastatic phenotype observed in these mice. In contrast, co-expression of Akt2 promoted invasion and lung metastasis in these mice. A more recent study conducted in breast cancer cells highlighted the role of Akt1 as a negative regulator of EMT and metastasis (78). Mechanistically, Akt1 was shown to regulate the function of Twist-1, a transcription factor involved in EMT and promotion of breast cancer metastasis. Akt1 was responsible for the direct phosphorylation of Twist-1 leading to its ubiquitination by β-TrCP and proteolytic degradation, however, inhibition of Akt1 by MK-2206 led to Twist-1 stabilization and enhanced breast cancer migration and invasion in vitro, associated with increased N-cadherin and vimentin, and decreased E-cadherin expression. Moreover, stabilization of Twist-1 was associated with enhanced lung metastasis in vivo.
Interestingly, context-specific effects of Akt1 on cancer progression and metastasis in a mouse model were reported in a recent study published by Nissim Hay’s group. Their study demonstrated that inhibition of hepatic Akt1 in systemic Akt2–/– mice led to hepatic carcinogenesis however that was not observed with inhibition of either hepatic Akt1 in Akt2+/– mice or with inhibition of one allele of hepatic Akt1 in Akt2–/– mice (79). Of note, Akt2–/– mice did not develop hepatic cancer although Akt2 was the main isoform expressed in the liver. Akt1–/– mice treated with diethylnitrosamine (DEN) developed macroscopic tumors via activation of FoxO1 and resultant liver inflammation. Overall, this study not only demonstrated the mutual role of Akt1 and Akt2 in maintaining liver homeostasis but also challenged the dogma that Akt1 is a tumor promoter in all contexts. A reinvestigation on the role of Akt1 in hepatocellular carcinoma (HepG2) and colorectal cancer (HCT-116) cell lines revealed the paradoxical effects of Akt1 activation on cell motility and invasion (80). Whereas Akt1 over-expressing HepG2 cells exhibited enhanced cell migration and invasion, these were impaired in HCT-116 cells. Of note, using PI3 kinase inhibitor wortmannin in both cell lines significantly reversed these results. Surprisingly, while the expression of matrix metalloproteinases MMP2 and MMP9 was elevated in HepG2 with Akt1 activation, the same was decreased in HCT-116 cells upon Akt1 activation, which led to the conclusion that the effect of Akt1 inhibition on motility could be cell-type specific.
In prostate cancer, Akt1 silencing in androgen-sensitive and androgen-resistant prostate cancer cell lines as well as prostate epithelial cells or treatment with pan-Akt inhibitor triciribine in androgen-resistant prostate cancer cell line (PC3) increased integrin β1 localization at the periphery and induced its activity leading to enhanced focal adhesions to the extracellular matrix, which augmented spreading and invasion ability of these cells (81). Intriguingly, silencing of Akt2 also exhibited similar effects. However, Akt1 loss promoted migration via enhanced expression and activity of receptor tyrosine kinases such as EGFR and hepatocyte growth factor receptor (cMET), whereas Akt2 loss was associated with induction of miR-200a/b that has been implicated in EMT and cells invasion, suggesting that Akt1 and Akt2 may modulate different pathways in the regulation of cell motility and invasion. Recently, we have identified a new role of Akt1 in prostate cancer. By using the transgenic adenocarcinoma of the mouse prostate (TRAMP) mice, which develop neuroendocrine prostate cancer spontaneously by the age of 24 weeks, we reported that although Akt1 silencing abrogated oncogenic transformation in these mice, inhibition of Akt1 during the advanced stages promoted lung metastasis (33). Moreover, silencing of Akt1 in androgen-resistant prostate cancer cell lines (PC3 and DU145) enhanced EMT, shown by increased N-cadherin, Snail, and reduced E-cadherin. Mechanistically, we demonstrated that suppression of the Akt1-βcatenin pathway during the advanced prostate cancer enhanced TGFβ1-mediated EMT and cancer metastasis. The most recent findings from our laboratory have also identified the microRNA signatures responsible for the early tumorigenic effects of Akt activation and late metastasis promoting effects of Akt suppression in prostate cancer (82), once again supporting the dual, stage-specific effects of Akt1 activity on cancer.
The latest findings in breast, prostate and liver cancers on the reciprocal regulation of cancer growth and metastasis by Akt1 isoform have now been extended to the non-small lung cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC). A study conducted by the Giaccone group showed that Akt1 inactivation in NSCLC cell lines with K-RAS and EGFR mutant background enhanced cancer cell migration and metastasis (83). More importantly, oral administration of MK-2206 enhanced brain metastasis in mice administered A549 cells via the intracardial route. Despite the absence of such results with Akt2 or Akt3 inhibition, their finding highlights the contribution of genetic background on determining the effect of Akt1 inactivation on cancer metastasis. In HNSCC, Picco group reported that Akt1 activation in cancer cell lines is not only essential for tumorigenesis but also for maintaining epithelial phenotype (84). However, inhibition of Akt1 expression by shRNA or suppression of its activity using MK-2206 decreased cell-cell contact and enhanced their invasive capacity, as shown in Figure 2 (84). Altogether these studies demonstrated the inhibitory effect of Akt1 on tumor cell migration and invasion in multiple cancers, thereby challenging the concept that Akt1 activation is necessary for promoting cancer metastasis. A schematic representation of the role of Akt1 in advanced cancer cells and tumor vasculature in a metastatic stage is shown in Figure 2.
In addition to the abundance of studies on the differential role of Akt1 in the early and advanced cancers, few studies on Akt2 have been debating the same concept. As discussed previously (75) although Akt1 silencing induced EMT-like phenotype in MCF-10A cells through activation of the ERK pathway, this was reversed by inhibition of Akt2 in these cells. Moreover, as mentioned early (72), despite the enhanced lung metastasis of the breast cancer cell lines after Akt1 deletion, overexpression of Akt2 promoted EMT in these cells and resulted in a similar effect. Another study showed that upregulation of Akt2 by Twist led to enhanced invasion and migration in the breast cancer cell lines (85). In the same study, this association was also observed in human breast cancer samples in which Akt2 and Twist expression were significantly elevated in the late stage compared to the early stage specimens. Cumulatively, these studies suggest the positive role of Akt2 in promoting breast cancer progression and metastasis. Interestingly, a study published by Sarah Wootton group to dissect the role of Akt isoforms in NSCLC showed that in spite of the necessity of Akt1 for NSCLC initiation and progression in AJEJJenv infected mice, Akt2 and Akt3, to some extent, appeared to have a protective role against tumorigenesis (86) conferring that the role of Akt2 and Akt3 can be a cancer type-specific. The role of Akt3 has also been debated in the literature. Clark and Toker group showed that silencing of Akt3 in triple negative breast cancer (TNBC), MCF10DCIS, MDA-MB468, and BT-549 cells, enhanced their migration in vitro, with no effect on invasion, and inhibited MDA-MB231 cells spheroid growth (87). On the other hand, another study from Manfred Jücker group showed that Akt3 downregulation in Balb-neuT mice, a transgenic model for ErbB2-induced breast cancer, reduced cancer progression and enhanced its sensitivity to tamoxifen, by reducing expression and activity of ERBb2 and ERBb3 and enhancing ERα expression (88). Overall, this conflict in Ak2 and Akt3 functions urges for more research to understand their role in different types and stages of cancers.
Akt pathway in exacerbating the effects of diabetes on cancer
The effect of diabetes, obesity and other metabolic diseases on exacerbating the cancer burden has been extensively reviewed elsewhere (89). However, the specific role of the Akt pathway in diabetes-exacerbated cancer growth or metastasis is not clear. Akt1 has been shown to be expressed in insulin-sensitive tissues such as liver, skeletal muscles and adipose tissue (90). Akt is crucial for initiating intracellular responses post-insulin receptor substrate-1 (IRS-1) phosphorylation (91) that eventually regulates glucose as well as lipid metabolism (92–94). Akt plays a major role in enhancing glucose uptake by inducing downstream molecules such as translocating glucose transporters (GLUTs) (91). Furthermore, overexpression of Akt or enhancing its activity in type 2 diabetes (T2DM) was found to increase glucose uptake in skeletal muscles thereby maintaining euglycemia (90, 91). On the other end, attenuated Akt signaling was found to be associated with insulin resistance in the metabolic tissues thereby leading to T2DM (92, 95). In addition, Akt plays a crucial role in insulin-mediated glucose uptake in the liver (96, 97) as well as suppression of glucagon secretion from the pancreatic α-cells to reduce hepatic glucose production (90). Beta cell survival is enhanced by the activation of Akt whereas free fatty acids inactivate it by inhibiting its translocation to the plasma membrane thereby preventing PDK1-mediated phosphorylation at Threonine 308 (98). Furthermore, transgenic expression of Akt in β cells caused an increase in their mass as a result of prolonged survival and enlargement in their size with no significant effect on neogenesis or cell replication.
Although the relationship between diabetes and cancer is not mechanistically clear, several studies have demonstrated that these diseases share pathways, such as PI3K/Akt and ERK and p38 MAP kinase pathways, which regulate proliferation and cell survival hence supporting tumorigenesis. It is also believed that hyperinsulinemia, hyper-glycemia, and inflammation observed in diabetes may enhance the initiation and progression of neoplastic lesions (99). Indeed, the growth of tumor orthografts in the hyperinsulinemic mice of IGF-IR-lysine-arginine (MKR) model was enhanced due to the elevated PI3K/Akt/mTOR signaling (100). On the other end, inhibition of the PI3K pathway in these mice led to a reduction in tumor growth. High glucose has been demonstrated to promote tumor cell invasion and expression of metastasis promoting molecules in human lung epithelial cells through the PI3K/Akt signaling pathway (101). In support of this, the anti-diabetic drug Metformin, which inhibits the PI3K/Akt/mTOR pathway has been implicated in the suppression of ovarian cancer cells function in vitro (102). In another study, activation of glucagon-like peptide-1 receptor inhibited tumor growth and metastasis of human pancreatic cancer cells via targeting the PI3K/Akt pathway (103). Overall, in spite of involving different pathways in diabetes, the role of Akt could potentially explain why diabetic patients are at a higher risk of cancer than the general population and suggest that diabetes and obesity may further exacerbate the burden in patients diagnosed with cancer. (104).
Angiogenic abnormalities have also been associated with diabetes (105). While diabetes-induced excessive angiogenesis leads to retinopathy, nephropathy, neuropathy, and atherosclerotic plaques formation, impaired wound healing and myocardial perfusion with diabetes exhibit lack of blood vessels (105). Akt plays a crucial role in the angiogenesis observed in diabetes complications. Knowing that Ang1-induced prevention of endothelial cells apoptosis is dependent on Akt phosphorylation (106), and vascular impairment in patients suffering from T2DM occurs as a result of imbalance between Ang1 and Ang2 that are crucial angiogenic growth factors regulating vascular formation and maintaining homeostasis(107), interfering with Akt function can be of immense help to treat vascular complication in diabetes mellitus. Advanced glycation end-products (AGEs), which are elevated under diabetic conditions and associated with insulin resistance, endothelial dysfunction and vascular inflammation in humans is a known inhibitor of Akt-eNOS pathway thereby compromising endothelial-barrier and promoting vasoconstriction (108), either of which could promote cancer cell metastasis. Overall, in spite of the various signaling pathways in diabetes, the versatile role of Akt in endothelial cells and tumor cells could potentially explain why diabetic patients are at a higher risk of cancer than the general population and suggest that diabetes and obesity may further exacerbate the burden in patients diagnosed with cancer.
Summary and Conclusion
Akt1 has a critical role in modulating tumor angiogenesis and cancer metastasis. Despite the dogma that targeting Akt1 could be a beneficial approach to treat cancer, the paradoxical effect of Akt1 in the advanced cancer stages must be considered in cancer therapy. Although inhibition of Akt1 or targeting its activity can suppress tumorigenesis during the early cancer stages, this could be detrimental on the advanced cancers as it compromises the endothelial-barrier function, enhances EMT in tumor cells, and induces tumor cells-transendothelial migration, thus promoting cancer metastasis. Apart from these, due to the integral role of Akt in insulin receptor signaling and metabolic homeostasis, more caution should be employed while targeting the Akt pathway in cancer patients with comorbidities such as diabetes and obesity. Overall, this review highlights the importance of the role of Akt1 in tumor progression and invasion and accentuates its cell-type and cancer-stage specific effects.
ACKNOWLEDGMENTS
Funds were provided by the NHLBI grant R01HL103952, NCATS grant UL1TR002378, Wilson Pharmacy Foundation (intramural) and Translational Research Initiative grant (intramural). This work has been accomplished using the resources and facilities at the VA Medical Center in Augusta, GA. The funders had no role in the study design, data collection, analysis, and decision to publish the data. The contents of the manuscript do not represent the views of the Department of Veteran Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Annals of medicine 2014;46(6):372–83. [DOI] [PubMed] [Google Scholar]
- 2.Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. British journal of clinical pharmacology 2016. [DOI] [PMC free article] [PubMed]
- 3.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery 2014;13(2):140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gkretsi V, Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front Oncol 2018;8:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell cycle (Georgetown, Tex) 2009;8(16):2502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hida K, Maishi N, Torii C, Hida Y. Tumor angiogenesis--characteristics of tumor endothelial cells. Int J Clin Oncol 2016;21(2):206–12. [DOI] [PubMed] [Google Scholar]
- 7.Naito Y, Yoshioka Y, Yamamoto Y, Ochiya T. How cancer cells dictate their microenvironment: present roles of extracellular vesicles. Cell Mol Life Sci 2017;74(4):697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan W, Shen X, Lei J, Xu Q, Yu Y, Li R, et al. Hyperglycemia, a neglected factor during cancer progression. Biomed Res Int 2014;2014:461917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel A, Sabbineni H, Clarke A, Somanath PR. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci 2016;157:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in endothelial cell and angiogenesis. Cell cycle (Georgetown, Tex) 2006;5(5):512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin YR, Toker A. Akt isoform-specific signaling in breast cancer. Cell Adhesion & Migration 2014;5(3):211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imache MR, Pawlotsky JM, Lerat H. Isoform-specific activation of Akt involvement in hepatocarcinogenesis. Hepatic Oncology 2015;2(3):213–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal 2009;21(4):470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin Y, Xie Y, Ostriker AC, Zhang X, Liu R, Lee MY, et al. Opposing Actions of AKT (Protein Kinase B) Isoforms in Vascular Smooth Muscle Injury and Therapeutic Response. Arterioscler Thromb Vasc Biol 2017;37(12):2311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 2004;30(2):193–204. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Zhang S, Yamane H, Wahl R, Ali A, Lofgren JA, et al. Kinetic mechanism of AKT/PKB enzyme family. J Biol Chem 2006;281(20):13949–56. [DOI] [PubMed] [Google Scholar]
- 17.Lee RS, House CM, Cristiano BE, Hannan RD, Pearson RB, Hannan KM. Relative Expression Levels Rather Than Specific Activity Plays the Major Role in Determining In Vivo AKT Isoform Substrate Specificity. Enzyme Res 2011;2011:720985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol 2010;298(3):C580–91. [DOI] [PubMed] [Google Scholar]
- 19.Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal 2011;23(10):1515–27. [DOI] [PubMed] [Google Scholar]
- 20.Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene 2005;24(50):7391–3. [DOI] [PubMed] [Google Scholar]
- 21.Somanath PR, Vijai J, Kichina JV, Byzova T, Kandel ES. The role of PAK-1 in activation of MAP kinase cascade and oncogenic transformation by Akt. Oncogene 2009;28(25):2365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441(7092):424–30. [DOI] [PubMed] [Google Scholar]
- 23.Sabbineni H, Alwhaibi A, Goc A, Gao F, Pruitt A, Somanath PR. Genetic deletion and pharmacological inhibition of Akt1 isoform attenuates bladder cancer cell proliferation, motility and invasion. Eur J Pharmacol 2015;764:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fortier AM, Asselin E, Cadrin M. Functional specificity of Akt isoforms in cancer progression. Biomolecular concepts 2011;2(1–2):1–11. [DOI] [PubMed] [Google Scholar]
- 25.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 1987;84(14):5034–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han Z, Wu K, Shen H, Li C, Han S, Hong L, et al. Akt1/protein kinase B alpha is involved in gastric cancer progression and cell proliferation. Digestive diseases and sciences 2008;53(7):1801–10. [DOI] [PubMed] [Google Scholar]
- 27.Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. The American journal of pathology 2001;159(2):431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goc A, Al-Husein B, Kochuparambil ST, Liu J, Heston WW, Somanath PR. PI3 kinase integrates Akt and MAP kinase signaling pathways in the regulation of prostate cancer. International Journal of Oncology 2010;38(1). [PubMed] [Google Scholar]
- 29.Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther 2011;336(2):496–505. [DOI] [PubMed] [Google Scholar]
- 30.Goc A, Kochuparambil ST, Al-Husein B, Al-Azayzih A, Mohammad S, Somanath PR. Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis. BMC Cancer 2012;12:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al-Husein B, Goc A, Somanath PR. Suppression of interactions between prostate tumor cell-surface integrin and endothelial ICAM-1 by simvastatin inhibits micrometastasis. J Cell Physiol 2013;228(11):2139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alhusban A, Al-Azayzih A, Goc A, Gao F, Fagan SC, Somanath PR. Clinically relevant doses of candesartan inhibit growth of prostate tumor xenografts in vivo through modulation of tumor angiogenesis. J Pharmacol Exp Ther 2014;350(3):635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao F, Alwhaibi A, Sabbineni H, Verma A, Eldahshan W, Somanath PR. Suppression of Akt1-beta-catenin pathway in advanced prostate cancer promotes TGFbeta1-mediated epithelial to mesenchymal transition and metastasis. Cancer Lett 2017;402:177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer research 2007;67(1):167–77. [DOI] [PubMed] [Google Scholar]
- 35.Hollander MC, Maier CR, Hobbs EA, Ashmore AR, Linnoila RI, Dennis PA. Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene 2011;30(15):1812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franks SE, Briah R, Jones RA, Moorehead RA. Unique roles of Akt1 and Akt2 in IGF-IR mediated lung tumorigenesis. Oncotarget 2016;7(3):3297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y, Kim J, Elshimali Y, Sarkissyan M, Vadgama JV. Activation of Akt1 accelerates carcinogen-induced tumorigenesis in mammary gland of virgin and post-lactating transgenic mice. BMC Cancer 2014;14:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer IA, Arteaga CL. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med 2016;67:11–28. [DOI] [PubMed] [Google Scholar]
- 39.Ye W The Complexity of Translating Anti-angiogenesis Therapy from Basic Science to the Clinic. Dev Cell 2016;37(2):114–25. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nature medicine 2005;11(11):1188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Somanath PR, Chen J, Byzova TV. Akt1 is necessary for the vascular maturation and angiogenesis during cutaneous wound healing. Angiogenesis 2008;11(3):277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu H, Littlewood T, Bennett M. Akt isoforms in vascular disease. Vascular pharmacology 2015;71:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proceedings of the National Academy of Sciences of the United States of America 2014;111(35):12865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999;399(6736):601–5. [DOI] [PubMed] [Google Scholar]
- 45.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999;399(6736):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schleicher M, Yu J, Murata T, Derakhshan B, Atochin D, Qian L, et al. The Akt1-eNOS axis illustrates the specificity of kinase-substrate relationships in vivo. Science signaling 2009;2(82):ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du J, Zhang L, Wang Z, Yano N, Zhao YT, Wei L, et al. Exendin-4 induces myocardial protection through MKK3 and Akt-1 in infarcted hearts. Am J Physiol Cell Physiol 2016;310(4):C270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008;11(2):109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 1998;273(46):30336–43. [DOI] [PubMed] [Google Scholar]
- 50.Aoki T, Nagakawa Y, Tsuchida A, Kasuya K, Kitamura K, Inoue K, et al. Expression of cyclooxygenase-2 and vascular endothelial growth factor in pancreatic tumors. Oncology reports 2002;9(4):761–5. [PubMed] [Google Scholar]
- 51.Kirkpatrick K, Ogunkolade W, Elkak A, Bustin S, Jenkins P, Ghilchik M, et al. The mRNA expression of cyclo-oxygenase-2 (COX-2) and vascular endothelial growth factor (VEGF) in human breast cancer. Current medical research and opinion 2002;18(4):237–41. [DOI] [PubMed] [Google Scholar]
- 52.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, et al. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Molecular cell 2000;6(4):851–60. [PubMed] [Google Scholar]
- 53.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer cell 2006;10(2):159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, et al. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci U S A 2005;102(1):128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao F, Artham S, Sabbineni H, Al-Azayzih A, Peng XD, Hay N, et al. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol Life Sci 2016;73(20):3917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao F, Alwhaibi A, Artham S, Verma A, Somanath PR. Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover. Br J Cancer 2018;118(11):1464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao F, Sabbineni H, Artham S, Somanath PR. Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling. J Cell Physiol 2017;232(10):2599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goc A, Al-Husein B, Katsanevas K, Steinbach A, Lou U, Sabbineni H, et al. Targeting Src-mediated Tyr216 phosphorylation and activation of GSK-3 in prostate cancer cells inhibit prostate cancer progression in vitro and in vivo. Oncotarget 2014;5(3):775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature medicine 2000;6(9):1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukai Y, Rikitake Y, Shiojima I, Wolfrum S, Satoh M, Takeshita K, et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J Clin Invest 2006;116(2):334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 2008;10(8):923–34. [DOI] [PubMed] [Google Scholar]
- 62.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, et al. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res 2009;104(8):978–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, et al. Akt1 governs breast cancer progression in vivo. Proceedings of the National Academy of Sciences of the United States of America 2007;104(18):7438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goc A, Liu J, Byzova TV, Somanath PR. Akt1 mediates prostate cancer cell microinvasion and chemotaxis to metastatic stimuli via integrin beta(3) affinity modulation. Br J Cancer 2012;107(4):713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim EK, Yun SJ, Ha JM, Kim YW, Jin IH, Yun J, et al. Selective activation of Akt1 by mammalian target of rapamycin complex 2 regulates cancer cell migration, invasion, and metastasis. Oncogene 2011;30(26):2954–63. [DOI] [PubMed] [Google Scholar]
- 66.Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011;30(4):457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qu X, Shen L, Zheng Y, Cui Y, Feng Z, Liu F, et al. A signal transduction pathway from TGF-beta1 to SKP2 via Akt1 and c-Myc and its correlation with progression in human melanoma. The Journal of investigative dermatology 2014;134(1):159–67. [DOI] [PubMed] [Google Scholar]
- 68.Albury TM, Pandey V, Gitto SB, Dominguez L, Spinel LP, Talarchek J, et al. Constitutively active Akt1 cooperates with KRas(G12D) to accelerate in vivo pancreatic tumor onset and progression. Neoplasia (New York, NY) 2015;17(2):175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saji M, Narahara K, McCarty SK, Vasko VV, La Perle KM, Porter K, et al. Akt1 deficiency delays tumor progression, vascular invasion, and distant metastasis in a murine model of thyroid cancer. Oncogene 2011;30(42):4307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer research 2004;64(9):3171–8. [DOI] [PubMed] [Google Scholar]
- 71.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Molecular cell 2005;20(4):539–50. [DOI] [PubMed] [Google Scholar]
- 72.Riggio M, Perrone MC, Polo ML, Rodriguez MJ, May M, Abba M, et al. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci Rep 2017;7:44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chin YR, Toker A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Molecular cell 2010;38(3):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proceedings of the National Academy of Sciences of the United States of America 2006;103(11):4134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. The Journal of cell biology 2005;171(6):1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Science signaling 2009;2(92):ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer research 2009;69(12):5057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, et al. AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer research 2016;76(6):1451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, et al. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer cell 2016;29(4):523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen L, Kang QH, Chen Y, Zhang YH, Li Q, Xie SQ, et al. Distinct roles of Akt1 in regulating proliferation, migration and invasion in HepG2 and HCT 116 cells. Oncology reports 2014;31(2):737–44. [DOI] [PubMed] [Google Scholar]
- 81.Virtakoivu R, Pellinen T, Rantala JK, Perala M, Ivaska J. Distinct roles of AKT isoforms in regulating beta1-integrin activity, migration, and invasion in prostate cancer. Molecular biology of the cell 2012;23(17):3357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alwhaibi A, Gao F, Artham S, Hsia BM, Mondal A, Kolhe R, et al. Modulation in the microRNA repertoire is responsible for the stage-specific effects of Akt suppression on murine neuroendocrine prostate cancer. Heliyon 2018;4(9):e00796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rao G, Pierobon M, Kim IK, Hsu WH, Deng J, Moon YW, et al. Inhibition of AKT1 signaling promotes invasion and metastasis of non-small cell lung cancer cells with K-RAS or EGFR mutations. Sci Rep 2017;7(1):7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brolih S, Parks SK, Vial V, Durivault J, Mostosi L, Pouyssegur J, et al. AKT1 restricts the invasive capacity of head and neck carcinoma cells harboring a constitutively active PI3 kinase activity. BMC Cancer 2018;18(1):249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer research 2007;67(5):1979–87. [DOI] [PubMed] [Google Scholar]
- 86.Linnerth-Petrik NM, Santry LA, Petrik JJ, Wootton SK. Opposing functions of Akt isoforms in lung tumor initiation and progression. PloS one 2014;9(4):e94595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer research 2014;74(3):964–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grabinski N, Mollmann K, Milde-Langosch K, Muller V, Schumacher U, Brandt B, et al. AKT3 regulates ErbB2, ErbB3 and estrogen receptor alpha expression and contributes to endocrine therapy resistance of ErbB2(+) breast tumor cells from Balb-neuT mice. Cellular signalling 2014;26(5):1021–9. [DOI] [PubMed] [Google Scholar]
- 89.Suarez AL. Burden of cancer attributable to obesity, type 2 diabetes and associated risk factors. Metabolism 2018. [DOI] [PubMed]
- 90.Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci 2018;14(11):1483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Z, Liu H, Liu J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res Clin Pract 2017. [DOI] [PubMed]
- 92.Yu N, Fang X, Zhao D, Mu Q, Zuo J, Ma Y, et al. Anti-Diabetic Effects of Jiang Tang Xiao Ke Granule via PI3K/Akt Signalling Pathway in Type 2 Diabetes KKAy Mice. PLoS One 2017;12(1):e0168980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kang C, LeRoith D, Gallagher EJ. Diabetes, obesity and breast cancer. Endocrinology 2018. [DOI] [PMC free article] [PubMed]
- 94.Anuradha R, Saraswati M, Kumar KG, Rani SH. Apoptosis of beta cells in diabetes mellitus. DNA Cell Biol 2014;33(11):743–8. [DOI] [PubMed] [Google Scholar]
- 95.Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell 2017;169(3):381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Viollet B, Lantier L, Devin-Leclerc J, Hebrard S, Amouyal C, Mounier R, et al. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front Biosci 2009;14:3380–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao Y, Zhang M, Wu T, Xu M, Cai H, Zhang Z. Effects of D-Pinitol on Insulin Resistance through the PI3K/Akt Signaling Pathway in Type 2 Diabetes Mellitus Rats. J Agric Food Chem 2015;63(26):6019–26. [DOI] [PubMed] [Google Scholar]
- 98.Dickson LM, Rhodes CJ. Pancreatic -cell growth and survival in the onset of type 2 diabetes-a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab 2004;287(2):E192–8. [DOI] [PubMed] [Google Scholar]
- 99.Cignarelli A, Genchi VA, Caruso I, Natalicchio A, Perrini S, Laviola L, et al. Diabetes and cancer: Pathophysiological fundamentals of a ‘dangerous affair’. Diabetes Res Clin Pract 2018;143:378–88. [DOI] [PubMed] [Google Scholar]
- 100.Gallagher EJ, Fierz Y, Vijayakumar A, Haddad N, Yakar S, LeRoith D. Inhibiting PI3K reduces mammary tumor growth and induces hyperglycemia in a mouse model of insulin resistance and hyperinsulinemia. Oncogene 2012;31(27):3213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kang X, Kong F, Wu X, Ren Y, Wu S, Wu K, et al. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the TGF-beta1/PI3K/Akt signaling pathway. Cell Physiol Biochem 2015;35(3):1008–22. [DOI] [PubMed] [Google Scholar]
- 102.Zhang F, Chen H, Du J, Wang B, Yang L. Anticancer Activity of Metformin, an Antidiabetic Drug, Against Ovarian Cancer Cells Involves Inhibition of Cysteine-Rich 61 (Cyr61)/Akt/Mammalian Target of Rapamycin (mTOR) Signaling Pathway. Med Sci Monit 2018;24:6093–101. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Zhao H, Wang L, Wei R, Xiu D, Tao M, Ke J, et al. Activation of glucagon-like peptide-1 receptor inhibits tumourigenicity and metastasis of human pancreatic cancer cells via PI3K/Akt pathway. Diabetes Obes Metab 2014;16(9):850–60. [DOI] [PubMed] [Google Scholar]
- 104.Habib SL, Rojna M. Diabetes and risk of cancer. ISRN Oncol 2013;2013:583786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Costa PZ, Soares R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci 2013;92(22):1037–45. [DOI] [PubMed] [Google Scholar]
- 106.Papapetropoulos A, Fulton D, Mahboubii K, Kalb RG, O’Connori DS, Lii F, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt:survivin pathway. J Biol Chem 2000;275(13):9102–5. [DOI] [PubMed] [Google Scholar]
- 107.Isidori AM, Venneri MA, Fiore D. Angiopoietin-1 and Angiopoietin-2 in metabolic disorders: therapeutic strategies to restore the highs and lows of angiogenesis in diabetes. J Endocrinol Invest 2016;39(11):1235–46. [DOI] [PubMed] [Google Scholar]
- 108.Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc Diabetol 2017;16(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]