

Abstract
Activating mutations of calcium‐sensing receptor (CaSR) cause autosomal dominant hypocalcemia type 1 (ADH1). Patients with ADH1 exhibit similar features to patients with hypoparathyroidism, including reduced serum parathyroid hormone (PTH) and Ca with low bone turnover. Although persistent suppression of bone turnover may increase bone fragility, bone strength in ADH1 patients has been unclear. We created knock‐in mice harboring the A843E activating mutation of CaSR, mimicking severe features of ADH1 patients. The severe form of ADH1 model mice showed smaller body and bone size with lower bone mineral density (BMD) and cortical area of the femur compared with age‐matched wild‐type (WT) mice. Bone strength in the femur was lower in ADH1 mice even after correction by bone geometry and/or BMD. Microcracks were markedly increased in ADH1 mice, but were rarely detected in WT mice. There was a negative correlation between bone strength corrected by bone geometry and/or BMD and microcrack number or density in ADH1 and WT mice. Among ADH1 mice, negative correlation was still observed between bone strength and microcrack number or density. Microcracks increased with age in ADH1 mice, and were negatively correlated with bone strength. Treatment with PTH(1‐34) or a calcilytic, JTT‐305, increased bone turnover, reduced microcracks, and increased bone strength to similar levels to those in WT mice. The increase in microcracks was associated with a reduction in bone strength in ADH1 mice, and aging aggravates these changes. These results demonstrate that activating mutation of CaSR causes reduction in PTH secretion with suppressed bone turnover, that reduced bone turnover is associated with an age‐dependent increase in microcracks with a reduction in bone strength, and that both PTH(1‐34) and calcilytic ameliorate all these changes in bone turnover and strength. It is suggested that fracture susceptibility may be increased in severe types of ADH1 patients especially in the elderly. © 2019 The Authors. JBMR Plus published by Wiley Periodicals, Inc. on behalf of American Society for Bone and Mineral Research.
Keywords: CALCIUM‐SENSING RECEPTOR, MICROCRACK, ADH1
Introduction
Activating mutation of calcium‐sensing receptor (CaSR) causes autosomal dominant hypocalcemia 1 (ADH1).1, 2 ADH1 patients show reduced serum parathyroid hormone (PTH) level, and develop hypocalcemia and hyperphosphatemia, similar to the clinical features of hypoparathyroidism.3 In addition to these features, especially severe types of ADH1 patients exhibit remarkable hypercalciuria with nephrocalcinosis.4 Patients with ADH1 also exhibit low bone turnover associated with decreased PTH secretion.1, 2 In a large nationwide case‐finding study in Denmark, fractures of upper extremities were increased in hypoparathyroid patients involving ADH1 patients in 7% of the total cases.3 However, the reason for the increase in the prevalence of fractures is unclear, and there has been no study about bone strength of ADH1 patients.
It has been demonstrated that microcracks accumulate in low turnover bone,5, 6 and that the number of microcracks substantially increases with aging.7 Because bone turnover is substantially suppressed in ADH1 patients, there is a possibility that microcrack number may be increased, and the increase in microcracks may affect the strength of bone in ADH1 patients.
In order to answer these questions, we generated knock‐in mice harboring a human activating mutation of CaSR (A843E) as a model of ADH1. Patients with A843E CaSR mutation exhibit most severe phenotypes of ADH1,7 and A843E CaSR knock‐in mice mimicked almost all the severe features of ADH1 patients.8 In the present study, we examined bone strength, number and density of microcracks, and the effect of PTH(1‐34) and a calcilytic, JTT‐305, on bone turnover and strength as well as microcrack number and density, using A843E CaSR knock‐in ADH1 model mice.
Materials and Methods
A flowchart of this study is shown in Fig. 1.
Figure 1.
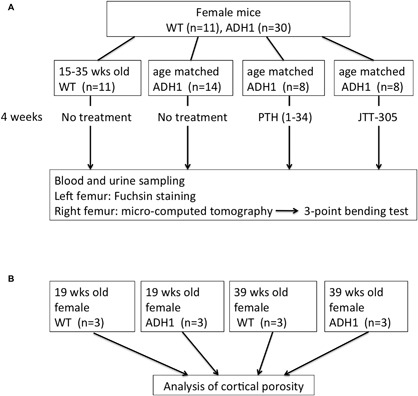
Flowchart of the present study. (A) Biochemical parameter, bone size, BMD, bone strength and microcracks were analyzed on WT and age matched ADH1 mice with or without treatment. (B) Three midshaft femurs of 19‐week and 39‐week‐old ADH1 mice and age matched WT mice were analyzed for cortical porosity.
Experimental animals
As an ADH1 model, A843E mutant CaSR knock‐in mice (Accession No. CDB1054K; http://www2.clst.riken.jp/arg/mutant%20mice%20list.html) was generated as described (http://www2.clst.riken.jp/arg/methods.html). Mice were housed in a 25°C room under a 12‐hour daylight cycle from 7:00 a.m. to 7:00 p.m., with free access to water and diet during the night cycle, and fed with diet containing 1.2% calcium, 0.8% phosphorus, and 400 IU/100 g vitamin D9 after weaning. Genotyping analysis of mice was done by PCR to detect of both human CaSR and mouse CaSR using genomic DNA extracted from tail as described.8 All animal experimental procedures were performed in accordance with the guidelines of the Animal Research Committee of the University of Tokushima Graduate School.
Calcilytic JTT‐305 and PTH(1‐34) preparation
Calcilytic JTT‐305 (kindly provided by Japan Tobacco Inc., Tokyo, Japan) was prepared as a suspension in 0.5 wt/vol% methylcellulose (WAKO, Osaka, Japan), and was administered to mice by gastric gavage. Human PTH(1‐34) (obtained from Asahi Kasei Pharma, Tokyo, Japan) was injected subcutaneously. Doses of calcilytic and PTH(1‐34) were selected by their ability to increase serum Ca to a similar level to that in WT mice. As a result, 40 μg/g/day JTT‐305 or 100 μg/kg/day PTH (1‐34) were given to ADH1 model mice for 4 weeks.
Serum and urinary biochemistry
All mice were housed in metabolic cages to collect urine samples. Six‐hour urine was collected under a layer of mineral oil to prevent evaporation. Baseline blood and urine samples were taken at least 6 hours after the night cycle ended.
Urinary Ca, inorganic phosphate (Pi), cyclic AMP, and creatinine were measured at Special Reference Laboratory (SRL, Tokyo, Japan). Cardiac puncture was performed to collect blood samples. Serum creatinine, Ca, magnesium (Mg), Pi, and 1,25(OH)2D were measured at SRL. Serum osteocalcin and tartrate‐resistant acid phosphatase (TRAP) were measured using the Mouse Osteocalcin EIA kit (Biomedical Technologies Inc., Stroughton, MA, USA), and Mouse TRAP Assay (Immunodiagnostic Systems Ltd, Boldon, UK), respectively, following the manufacturers’ protocols.
Micro–CT
We used a LaTheta LCT‐200 (Hitachi‐Aloka, Tokyo, Japan) CT scanner. Nineteen‐week‐old to 38‐week‐old female ADH1 mice and both age‐ and sex‐matched WT mice were euthanized. The right femur was resected and then muscles and connective tissues were removed. Resected femurs were placed and scanned in a 24‐mm‐wide specimen holder with resolution of 48 μm pixel size. Resected bone slices were scanned at the distal metaphysis and diaphysis of the right femur. The cortical area and perimeter were calculated by the average of 30 slices in the mid‐shaft of femurs. BMD (mg/cm3) was calculated by LaTheta mCT software according to ASBMR guidelines.10
Fuchsin staining
Left femurs of ADH1 and WT mice were resected, and muscles and connective tissues were removed for staining. The procedure of fuchsin staining was described by Burr and Hooser.11 In order to eliminate artifacts at the time of bone section preparation,12 femurs were removed en‐block, and fuchsin staining was performed under negative pressure.11 Briefly, following preservation in 70% ethanol, total femurs were placed under 20‐psi vacuum in 1% basic fuchsin in 80% ethanol followed by new 1% fuchsin in 80% ethanol, and these steps were repeated by placing the femur in 1% basic fuchsin in 90% ethanol. After femurs were placed in 1% basic fuchsin in 100% ethanol twice, they were rinsed in 100% ethanol for 1 hour to remove excess stain. Specimen was embedded in methylmethacrylate and sliced to 20 μm thickness. Microcracks were evaluated by a microscope at ×20 magnification.
Micropores in cortical bone (cortical porosity)
Three midshaft femurs of 19‐week‐old and 39‐week‐old ADH1 mice and age‐matched WT mice were embedded in methylmethacrylate and sliced to 10 μm thickness. Micropores of cortex bone were evaluated in sequential 10 slices by a microscope at ×20 magnification.
Three‐point bending test
After evaluation of micro–CT (μCT), the right femur of ADH1 and WT mice were wrapped with saline‐soaked gauze to prevent dryness and stored at −80°C until use. The three‐point bending test was performed by KUREHA Special Laboratory (KSL, Co., Ltd, Tokyo, Japan) to analyze the biomechanical properties of femurs as reported.13 Briefly, load cells were perpendicularly positioned at the just middle point of the bone, which was assessed by the point of bone perimeter and cortical area. A 5‐N preload was applied in order to avoid specimen sliding. Then the bending force was applied at a constant deformation rate of 2 mm/min until fracture occurred. From the load‐deformation curve, the maximum breaking load (N), stiffness (N/mm), and energy to breaking max load (N.mm) were obtained.
Statistical analysis
All the data are presented as means ± SD. Statistical differences were analyzed by ANOVA test followed by appropriate post hoc test for multiple comparisons, using statistical software SPSS version 21.0 (IBM Corp., Armonk, NY, USA). Bonferroni‐corrected Mann‐Whitney U test or Dunn test was used for multiple comparisons. A p value of <0.05 was considered to be statistically significant.
Results
Improvement of biochemical features of ADH1 mice by PTH(1‐34) and calcilytic
In ADH1 mice, serum Ca and Mg levels were decreased, while serum Pi was increased compared to those in WT mice (Table 1). Serum 1,25(OH)2D tended to be lower in ADH1 mice than WT mice, but the difference was not significant. Urinary Ca to creatinine ratio (urinary Ca/Cr) was markedly increased. Serum creatinine tended to be higher in ADH1 compared to WT mice. Urinary cyclic AMP (cAMP), as an index of PTH action, was reduced in ADH1 mice. Serum osteocalcin as a bone formation marker and serum TRAP as a bone resorption marker were both reduced in ADH1 mice, indicating that bone turnover was reduced in ADH1 mice (Table 1). These phenotypes of the mutant mice mimic almost all the features of human ADH1. Oral administration of 40 μg/g/day JTT‐305 or subcutaneous injection of 100 μg/kg/day PTH (1‐34) for 4 weeks reversed all the phenotypes of ADH1 except for urinary Ca/Cr in PTH(1‐34)‐treated group, in which no reduction in urinary Ca/Cr was observed (Table 1). Severe adverse events including death were not observed by these doses of PTH(1‐34) or JTT‐305. Serum Ca reached the highest plateau 3 to 4 hours after treatment, and the plateau was kept around 12 hours. Highest serum Ca levels at the plateau were 10.6 and 10.4 mg/dL after treatment with PTH(1‐34) or JTT‐305, respectively.
Table 1.
Serum and Urinary Ca and P, and Bone Turnover Markers in ADH1 Model Mice at Baseline and After Treatment With PTH or Calcilytic JTT‐305
| WT | ADH1 | ADH1 + PTH | ADH1 + JTT | |
|---|---|---|---|---|
| Serum Ca (mg/dL) | 10.02 ± 0.36 | 5.60 ± 0.56 * | 9.84 ± 0.86 # | 8.93 ± 1.38 # |
| Serum Mg (mg/dL) | 3.70 ± 0.10 | 3.08 ± 0.30 * | 3.76 ± 0.15 # | 3.46 ± 0.35 # |
| Serum Pi (mg/dL) | 10.60 ± 1.33 | 16.17 ± 1.07 * | 11.10 ± 1.03 # | 11.79 ± 1.89 # |
| Serum 1,25(OH)2D (pg/mL) | 87.8 ± 7.0 | 76.2 ± 15.0 | 157.5 ± 47.3 # | 198.0 ± 36.7 # |
| Serum Cr (mg/dL) | 0.083 ± 0.029 | 0.117 ± 0.029 | 0.121 ± 0.084 | 0.093 ± 0.035 |
| Urinary Ca/Cr | 0.267 ± 0.002 | 0.747 ± 0.190 * | 0.674 ± 0.151 | 0.344 ± 0.810 # |
| Urinary cAMP/Cr | 617.5 ± 39.2 | 445.7 ± 28.7 * | 757.2 ± 67.3 # | 681.9 ± 75.2 # |
| Serum OCN (ng/mL) | 196.3 ± 21.6 | 122.3 ± 37.3 * | 189.6 ± 45.3 # | 187.1 ± 39.3 # |
| Serum TRAP (U/L) | 2.56 ± 0.05 | 1.95 ± 0.04 ** | 3.25 ± 0.28 # | 3.16 ± 0.07 # |
Values are mean ± SD. The ADH1 model mouse mimicked almost all ADH1 phenotypes. Ca/P parameter and bone turnover markers showed control and A843E knock‐in mice including those treated with JTT‐305 or PTH (1‐34) at 15 to 35 weeks of age. Four weeks of treatment with 20 μg/g calcilytic JTT‐305 or 100 μg/kg PTH reversed all ADH1 phenotypes. The number of WT mice, ADH1 mice, ADH1 mice treated with PTH (1‐34), and ADH1 mice treated with JTT‐305 were 11, 14, 8, and 8, respectively.
OCN = osteocalcin; TRAP = tartrate‐resistant acid phosphatase.
p < 0.05 versus WT littermate.
p < 0.01 versus WT littermate.
p < 0.05 versus untreated ADH1.
Improvement of BMD, body, and bone size in ADH1 mice by PTH(1‐34) and calcilytic
Body weight (BW), bone length, and cortical area were lower by 20%, 24%, and 19%, respectively, in ADH1 mice than in WT mice (Fig. 2 A–C) as reported.8 Total, cortical and cancellous BMD of the femur were lower in ADH1 mice than in age‐matched WT littermates (Fig. 2 D–F). Subcutaneous injection of 100 μg/kg PTH(1‐34) or oral administration of 40 μg/g JTT‐305 for 4 weeks partially reversed the reduction in body weight, bone length, cortical area, and total and cancellous BMD of ADH1 mice (Fig. 2 A–D, F).
Figure 2.
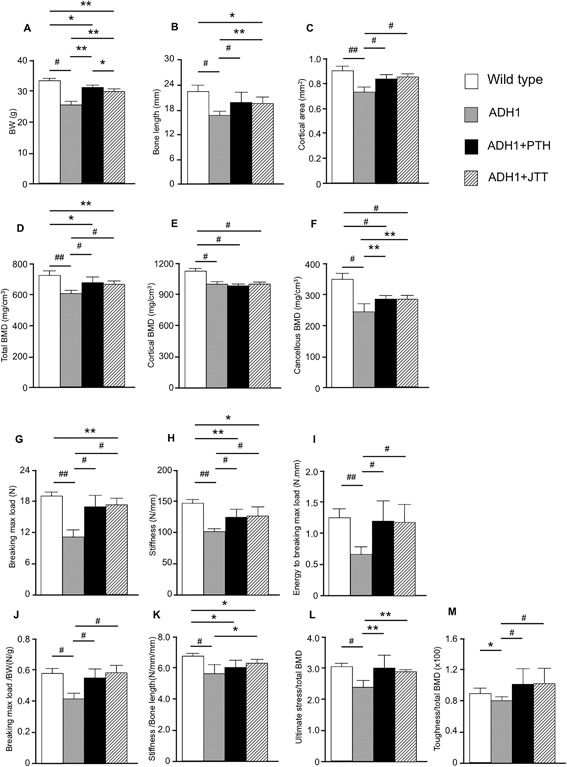
Body and bone size, BMD, and bone strength parameters of ADH1 mice with or without treatment with PTH(1‐34) or calcilytic, JTT‐305. Parameter of bone strength of 19‐week‐old to 39‐week‐old WT littermates (white bars), ADH1 (gray bars), PTH(1‐34)‐treated ADH1 (black bars), and JTT‐305–treated ADH1 mice (diagonal bars). (A) Body weight (BW), (B) bone length of femur, (C) cortical area, (D) total BMD, (E) cortical BMD, (F) cancellous BMD, (G) breaking maximum load, (H) stiffness, (I) energy to breaking maximum load, (J) breaking maximum load/BW, (K) stiffness/bone length, (L) ultimate stress/total BMD, and (M) toughness/total BMD. Results are presented as means ± SD of 8 or more measurements. *p < 0.05, **p < 0.01, #p < 0.005, ##p < 0.001, compared with the group at the other end of bars.
Reduced bone strength of ADH1 mice after correction by geometry of bone and/or BMD
Breaking maximum load, stiffness, and energy to breaking maximum load of ADH1 mice were decreased by 43%, 32%, and 50%, respectively, compared to WT mice (Fig. 2 G–I). PTH(1‐34) and calcilytic partially ameliorated the decrease in breaking maximum load and stiffness (Fig. 2 G, H) and completely reversed the reduction in energy to breaking maximum load of ADH1 mice (Fig. 2 I). Breaking maximum load is influenced by BMD, and strong correlation was observed between breaking maximum load and total, cortical, and cancellous BMD (Supporting Fig. 1A). Because the strength of long bone also depends on the size and geometry of bone,14 and breaking maximum load was significantly correlated with cortical area and perimeter (Supporting Fig. 1B), there was a possibility that the reduction in the parameters of bone strength may at least in part be due to the smaller size of bone. However, although the reduction in total BMD of ADH1 was 17% (Fig. 2 D), the reduction in breaking maximum load, stiffness, and energy to breaking maximum load were much larger in ADH1 (43%, 32%, and 50%, respectively) (Fig. 2 G–I). In addition, the increase in the breaking maximum load, stiffness, and energy to breaking maximum load after treatment of ADH1 mice with PTH(1‐34) or calcilytic was much larger (25% to 98%) than the changes in the BW, bone length, cortical area, and total BMD (13% to 22%) (Fig. 2 A–D, G–I). Therefore, we corrected the parameters of bone strength by BW or bone size and/or BMD of each mouse. Because breaking maximum load is reported to be influenced by BW, and stiffness is influenced by bone length, we corrected these parameters by BW and bone length, respectively.15 After correction by BW, there was still a significant reduction in breaking maximum load of ADH1 mice but the difference became smaller (43% to 28%), and breaking maximum load/BW reached the similar level to that in WT mice after PTH(1‐34) or calcilytic treatment (Fig. 2 G, J). The decrease in stiffness corrected by bone length became also smaller (32% to 15%) compared to that without correction, and was partially recovered by calcilytic treatment (Fig. 2 H, K). When breaking maximum load is divided by cortical area, the parameter is known as ultimate stress, and when energy to breaking maximum load is divided by both cortical area and bone length, the parameter is called toughness.16 When ultimate stress and toughness were divided by total BMD, the corrected bone strength parameters (breaking maximum load/BW, stiffness/bone length, ultimate stress/total BMD, and toughness/total BMD) were still lower in ADH1 mice than WT littermates (–28%, p < 0.005; −15%, p < 0.005; −24%, p < 0.005; −11%, p < 0.05; respectively, versus WT) (Fig. 2 J–M). Treatment of ADH1 mice with PTH(1‐34) or calcilytic almost completely reversed the reduction in those bone strength parameters, except for the partial recovery in stiffness/bone length (Fig. 2 J–M).
Increased microcracks with reduced bone strength in ADH1 mice
Because bone strength of ADH1 mice was reduced independent of BMD or bone geometry, bone quality may be deteriorated in ADH1 mice. Among indices of bone quality, bone mineralization was not different between ADH1 and WT mice.8 Microcracks may accumulate in low turnover bone, and bone turnover is reduced in ADH1 mice (Table 1). Therefore, we evaluated microcracks in ADH1 and WT mice. Microcracks were rarely detectable in the femur of WT mice, whereas both the number and density of microcracks were markedly increased in ADH1 mice (p < 0.005 and p < 0.001, respectively) (Fig. 3 A–D). Furthermore, treatment with PTH(1‐34) or calcilytic reduced microcrack number and density to similar levels to those in WT mice (Fig. 3 C, D). When treated or untreated ADH1 and WT mice were analyzed together, breaking maximum load/BW was negatively correlated with microcrack number and density (p < 0.001, p < 0.001, respectively) (Fig. 3 E, F, solid line). When only untreated ADH1 mice were analyzed, strong correlation was still observed between breaking maximum load/BW and microcrack number or density (p < 0.001, p < 0.001, respectively) (Fig 3 E, F, broken line). There was also a significant negative correlation between stiffness/bone length and microcrack number or density regardless of whether treated and untreated ADH1 and WT mice were analyzed together (p < 0.001, p < 0.001, respectively) (Fig. 3 G, H, solid line) or only untreated ADH1 mice were analyzed (p < 0.01, p < 0.01) (Fig. 3 G, H, broken line). Ultimate stress/total BMD was significantly correlated with microcrack number or density when treated and untreated ADH1 and WT mice were analyzed together (p < 0.001, p < 0.001, respectively) (Fig. 3 I, J, solid line). In untreated ADH1 mice, negative correlation was observed between ultimate stress/total BMD and microcrack density (p < 0.05) (Fig. 3 J, broken line). Toughness/total BMD was also significantly correlated with microcrack number and density in treated and untreated ADH1 and WT mice together (p < 0.01, p < 0.01, respectively) (Fig. 3 K, L, solid line), and was negatively correlated with microcrack density in untreated ADH1 mice (p < 0.05) (Fig. 3 L, broken line).
Figure 3.
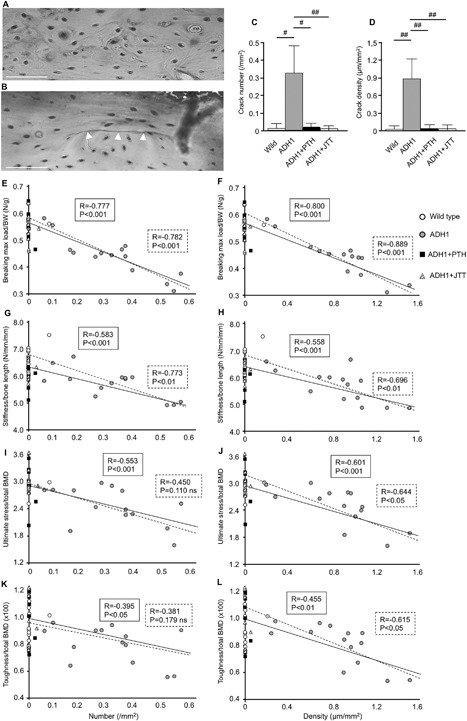
Microcrack number and density in the femur of ADH1 mice, and correlation between parameters of bone strength and microcrack number or density corrected by bone geometry and/or BMD. Fuchsin staining of the femoral midshaft of (A) WT mice and (B) ADH1 model mice. White arrowhead indicates microcrack. White scare bar = 50 μm. (C) Number and (D) density of microcracks in WT (white bars), untreated ADH1 mice (gray bars), PTH(1‐34)‐treated ADH1 mice (black bars), and JTT‐305‐treated ADH1 mice (diagonal bars). Results are presented as means ± SD of 8 or more measurements. #p < 0.005, ##p < 0.001 versus untreated ADH1 mice. (E) Correlation between breaking maximum load/BW and microcrack number or (F) microcrack density, using femurs of WT (white circle), untreated ADH1 (gray circle), PTH(1‐34)‐treated ADH1 (black square), and JTT‐305‐treated ADH1 mice (diagonal triangle). Solid line represents correlation between parameters of bone strength and number or density of all the mice including WT and ADH1 mice. Broken line represents analysis using untreated ADH1 mice alone. (G) Correlation between stiffness/bone length and microcrack number or (H) microcrack density, (I) correlation between ultimate stress/total BMD and microcrack number or (J) microcrack density, (K) correlation between toughness/total BMD and microcrack number or (L) microcrack density.
Because cortical porosity is also regarded as a part of cortical bone quality,17 we evaluated cortical porosity in ADH1 mice. Regardless of the presence or absence of CaSR mutation or aging of mice, very few micropores representing cortical porosity were observed, and %volume of micropores was very similar between WT and ADH1 or between younger and older mice (Supporting Fig. 2).
Increased microcracks and reduced bone strength with aging of ADH1 mice
Microcracks are known to increase with aging under low bone turnover.18 In ADH1 mice, both microcrack number and density increased with the age of these mice (p < 0.05, p < 0.01, respectively) (Fig. 4 A, B). In parallel with the increase in the number and density of microcracks, there was a reduction in breaking maximum load/BW, stiffness/bone length, and ultimate stress/total BMD with aging of ADH1 mice (p < 0.05, p < 0.005 and p < 0.05, respectively) (Fig. 4 C–E).
Figure 4.
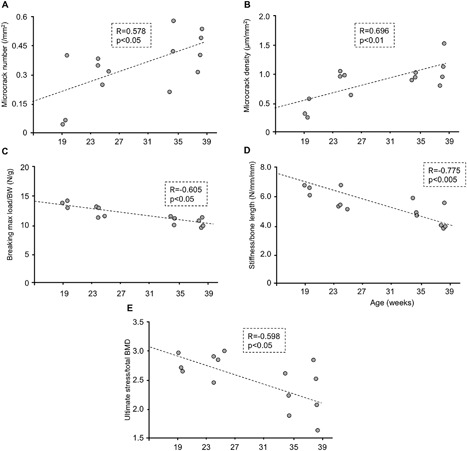
Correlation between the age of ADH1 mice and microcrack number, microcrack density, and parameters of bone strength corrected by bone geometry and/or BMD. (A) Correlation between mice age and microcrack number, (B) microcrack density, (C) corrected breaking maximum load/BW, (D) stiffness/bone length, and (E) ultimate stress/total BMD.
Discussion
The present observations show that femoral BMD and strength were reduced in severe type of ADH1 mice. In a previous study, we also created C129S CaSR knock‐in mice, which showed a less severe phenotype with milder hypercalciuria and hypocalcemia. In those mild ADH1 model mice, BMD was higher than WT mice with low bone turnover,8 similar to the changes seen in hypoparathyroid mice. Because hypercalciuria in the current ADH1 model of A843E CaSR knock‐in mice was much more severe than that in C129S CaSR knock‐in mice, we assume that the reduced BMD in A843E CaSR knock‐in mice in the present study was due to the negative Ca balance created by the marked hypercalciuria due to the strong activation of renal tubular CaSR.
Strength of long bones is mainly influenced by BMD,19 but geometric variables, including cortical area, cortical thickness, and bone perimeter14, 20 also affect long bone strength. In the present study, total, cortical, and cancellous BMD, as well as cortical area and cortical perimeter were strongly correlated with the breaking maximum load in ADH1 and WT mice. Furthermore, these parameters were markedly reduced in ADH1 mice. In order to compare bone strength without the influence of bone geometry and/or BMD, we corrected the parameters of bone strength by BW or bone size and/or BMD of each mouse. After correction by these factors, ADH1 mice still showed low bone strength, and both PTH(1‐34) and calcilytic treatment almost completely rescued the decreased bone strength parameters in ADH1 mice, and the improvement of low bone turnover and of microcrack number/density by PTH or calcilytic showed a mirror image with the corrected bone strength parameters, with close correlation to each other. In addition, cortical porosity was not increased in ADH1 mice, and there was no difference in the number, size, or the volume of micropores between WT and ADH1 mice in both younger and older mice. These results are consistent with the assumption that the reduction in bone strength of ADH1 mice was due to a reduction in bone quality, or more specifically an increase in microcracks.
Both PTH(1‐34) and calcilytic reversed the reduction in femoral bone strength of ADH1 model mice. Although both treatment increased bone turnover markers to similar levels to those in WT mice, only calcilytic corrected hypercalciuria to normal level, whereas PTH(1‐34) could not normalize hypercalciuria. This is because only calcilytic can counteract the inhibition of renal tubular Ca reabsorption by mutant CaSR. Nevertheless, both PTH(1‐34) and calcilytic corrected the negative Ca balance and increased BMD similarly in ADH1 mice.
Because CaSR is present in both osteoclasts and osteoblast lineage cells, there was a possibility that the severe activating mutation of CaSR might have a direct influence on the functions of these cells and cause alterations in bone turnover and quality. However, the present study demonstrates that PTH(1‐34) can ameliorate almost all the deteriorations in bone quality and strength similar to a calcilytic. These results suggest that the changes in bone quality and strength observed in ADH1 mice are not due to the direct influence of the CaSR mutation in bone cell functions but due mainly to the reduction in bone turnover. However, the possibility cannot be ruled out that there may be some direct effect of CaSR in bone cells that could not be detected under the treatment regimens of PTH(1‐34) and calcilytic in this study.
In our ADH1 model mice, microcracks increased with aging. It has been reported that long‐term suppression of bone remodeling increases the accumulation of microcracks in the bone.4, 5 Therefore, it is plausible to speculate that the increase in microcracks in ADH1 model mice is due to low bone turnover. These results suggest that fracture incidence may be increased with aging of ADH1 patients. However, there has been no such report showing higher increase in fracture incidence with aging of ADH1 patients compared to other population. Underbjerg and colleagues3 performed a nationwide survey in Denmark of patients with nonsurgical hypoparathyroidism including ADH1 in 7% of the patients. The incidence of forearm, proximal humerus, and any fractures in the upper extremities was significantly higher in these patients compared to the age‐matched controls. Fractures in the other sites including vertebra, proximal femur, ankle, and any fractures in the lower extremities were also higher in number in these patients, but the difference was not significant from controls. The incidence of fractures was significantly higher in female, and all fractures were those of fragility fractures. Therefore, there may be an increased risk with aging, but age distribution of fractures was not reported.
Our study has limitations. First, bone strength was assessed only in the femur of ADH1 model mice. Because fracture incidence was higher in the upper extremities of hypoparathyroid patients compared to control subjects, there might be larger difference in the strength of upper extremities of the ADH1 model mice. Second, only one strain of ADH1 model mice was examined. Because the phenotype of A843E CaSR mutant mice used in this study was more severe than C129S CaSR mutant mice,8 there might not be as much difference in bone strength of C129S mutant mice as that observed in this study. Third, trabecular structure, such as trabecular thickness and connectivity, also influences femoral bone strength. However, because femoral bone strength examined by three‐point bending test is influenced mostly by BMD and cortical structure, trabecular structure was not analyzed in this study. Finally, although PTH(1‐34) at an optimum dose to correct hypocalcemia ameliorated the deteriorations in bone quality and strength similar to a calcilytic, there might be a direct influence of CaSR mutation on bone quality and strength. Such possibility has to be clarified in future studies.
In conclusion, bone strength is reduced in ADH1 model mice even after correction by geometry and/or BMD of the femur. Microcrack number and density are markedly increased in ADH1 model mice in parallel with the reduction in bone strength. Microcracks are increased with aging, and the increase in microcracks is correlated with the reduction in bone strength. Both PTH(1‐34) and calcilytic reverse the suppression of bone turnover and reduce microcrack number/density of ADH1 model mice, with improvement in bone strength. These results demonstrate that activating mutation of CaSR causes reduction in PTH secretion resulting in a suppression of bone turnover, that the reduced bone turnover is associated with an age‐dependent increase in microcracks, which is associated with reduced bone strength, and that both PTH(1‐34) and calcilytic ameliorate all these changes in bone turnover and strength.
Disclosures
All authors state that they have no conflicts of interest.
Supporting information
Supporting Figure S1.
Supporting Figure S2.
Acknowledgments
This study was supported in part by Grants‐in‐Aid for Scientific Research to TM and MA from the Ministry of Education, Culture, Science and Sports of Japan. Calcilytic JTT‐305/MK‐5442 was kindly provided by Japan Tobacco Inc., Tokyo, Japan.
Authors’ roles: BD and IE performed most experiments. Data were analyzed by IE and YO. IE supervised the experiments and wrote the first draft of the manuscript. YO, YM, KK, and MK provided μCT data. MH, JT, HT, SF, and MA provided helpful discussion. TM designed the overall study, analyzed results, and wrote the manuscript.
References
- 1. Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G‐protein subunit α11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013; 368: 2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013; 45: 93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Underbjerg L, Sikjaer T, Mosekilde L, et al. The epidemiology of nonsurgical hypoparathyroidism in Denmark: a nationwide case finding study. J Bone Miner Res. 2015; 30: 1738–44 [DOI] [PubMed] [Google Scholar]
- 4. Sato K, Hasegawa Y, Nakae J, et al. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain‐of‐function mutations of the calcium‐sensing receptor gene. J Clin Endocrinol Metab. 2002; 87: 3068–73. [DOI] [PubMed] [Google Scholar]
- 5. Komatsubara S, Mori S, Mashiba T, et al. Suppressed bone turnover by long‐term bisphosphonate treatment accumulates microdamage but maintains intrinsic material properties in cortical bone of dog rib. J Bone Miner Res. 2004; 19: 999–1005. [DOI] [PubMed] [Google Scholar]
- 6. Mashiba T, Turner CH, Hirano T, et al. Effects of suppressed bone turnover by bisphosphonates on microdamage accumulation and biomechanical properties in clinically relevant skeletal sites in beagles. Bone. 2001; 28: 524–31. [DOI] [PubMed] [Google Scholar]
- 7. Sobelman OS, Gibeling JC, Stover SM, et al. Do microcracks decrease or increase fatigue resistance in cortical bone? J Biomech. 2004; 37: 1295–303. [DOI] [PubMed] [Google Scholar]
- 8. Dong B, Endo I, Ohnishi Y, et al. Calcilytic ameliorates abnormalities of mutant calcium‐sensing receptor (CaSR) knock‐in mice mimicking autosomal dominant hypocalcemia (ADH). J Bone Miner Res. 2015; 30: 1980–93. [DOI] [PubMed] [Google Scholar]
- 9. Hough TA, Bogani D, Cheeseman MT, et al. Activating calcium‐sensing receptor mutation in the mouse is associated with cataracts and ectopic calcification. Proc Natl Acad Sci U S.A. 2004; 101: 13566–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bouxsein ML, Boyd SK, Christiansen BA, et al. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res. 2010; 25: 1468–86. [DOI] [PubMed] [Google Scholar]
- 11. Burr DB, Hooser M. Alterations to the en bloc basic fuchsin staining protocol for the demonstration of microdamage produced in vivo. Bone. 1995; 17: 431–3. [DOI] [PubMed] [Google Scholar]
- 12. Lee TC, Mohsin S, Taylor D, et al. Detecting microdamage in bone. J Anat. 2003; 203: 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azboy İ, Özkaya M, Demir T, et al. Biomechanical properties of osteoporotic rat femurs after different hormonal treatments: genistein, estradiol, and estradiol/progesterone. SICOT.J. 2016; 2: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benhamou CL. Effects of osteoporosis medications on bone quality. Joint Bone Spine. 2007; 74: 39–47. [DOI] [PubMed] [Google Scholar]
- 15. Jämsä T, Jalovaara P, Peng Z, et al. Comparison of three‐point bending test and peripheral quantitative computed tomography analysis in the evaluation of the strength of mouse femur and tibia. Bone. 1998; 23: 155–61. [DOI] [PubMed] [Google Scholar]
- 16. Allen MR, Burr DB. Three years of alendronate treatment results in similar levels of vertebral microdamage as after one year of treatment. J Bone Miner Res. 2007; 22: 1759–65. [DOI] [PubMed] [Google Scholar]
- 17. Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab. 2008; 26: 1–8. [DOI] [PubMed] [Google Scholar]
- 18. Akkus O, Knott DF, Jepsen KJ, et al. Relationship between damage accumulation and mechanical property degradation in cortical bone: microcrack orientation is important. J Biomed Mater Res A. 2003; 65: 482–8. [DOI] [PubMed] [Google Scholar]
- 19. Stenström M, Olander B, Lehto‐Axtelius D, et al. Bone mineral density and bone structure parameters as predictors of bone strength: an analysis using computerized microtomography and gastrectomy‐induced osteopenia in the rat. J Biomech. 2000; 33: 289–97. [DOI] [PubMed] [Google Scholar]
- 20. Bonnet N, Benhamou C, Brunet‐Imbault B, et al. Severe bone alterations under b2 agonist treatments: bone mass, microarchitecture and strength analyses in female rats. Bone. 2005;37:622e33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure S1.
Supporting Figure S2.