

Abstract
Background
Evidence from observational studies of telomere length (TL) has been conflicting regarding its direction of association with cancer risk. We investigated the causal relevance of TL for lung and head and neck cancers using Mendelian Randomization (MR) and mediation analyses.
Methods
We developed a novel genetic instrument for TL in chromosome 5p15.33, using variants identified through deep-sequencing, that were genotyped in 2051 cancer-free subjects. Next, we conducted an MR analysis of lung (16 396 cases, 13 013 controls) and head and neck cancer (4415 cases, 5013 controls) using eight genetic instruments for TL. Lastly, the 5p15.33 instrument and distinct 5p15.33 lung cancer risk loci were evaluated using two-sample mediation analysis, to quantify their direct and indirect, telomere-mediated, effects.
Results
The multi-allelic 5p15.33 instrument explained 1.49–2.00% of TL variation in our data (p = 2.6 × 10–9). The MR analysis estimated that a 1000 base-pair increase in TL increases risk of lung cancer [odds ratio (OR) = 1.41, 95% confidence interval (CI): 1.20–1.65] and lung adenocarcinoma (OR = 1.92, 95% CI: 1.51–2.22), but not squamous lung carcinoma (OR = 1.04, 95% CI: 0.83–1.29) or head and neck cancers (OR = 0.90, 95% CI: 0.70–1.05). Mediation analysis of the 5p15.33 instrument indicated an absence of direct effects on lung cancer risk (OR = 1.00, 95% CI: 0.95–1.04). Analysis of distinct 5p15.33 susceptibility variants estimated that TL mediates up to 40% of the observed associations with lung cancer risk.
Conclusions
Our findings support a causal role for long telomeres in lung cancer aetiology, particularly for adenocarcinoma, and demonstrate that telomere maintenance partially mediates the lung cancer susceptibility conferred by 5p15.33 loci.
Keywords: lung cancer, telomere length, chromosome 5p15.33, Mendelian Randomization, mediation analysis, TERT
Key Messages
Genetic predisposition to long telomeres increases the risk of lung cancer, predominately lung adenocarcinoma.
Genetic determinants of long telomeres are not associated with squamous carcinomas of the lung or head and neck.
Using two-sample mediation analysis, we determined that the novel 5p15.33 instrument for telomere length (TL) does not have direct effects on the outcome, and demonstrated that the association between 5p15.33 lung cancer susceptibility variants is partially mediated by TL, suggesting the presence of other relevant mechanisms.
Introduction
Telomeres are highly conserved stretches of tandem repeats of the TTAGGG sequence, which protect chromosome ends from degradation and maintain genome stability.1,2 Due to the incomplete replication of chromosomes during cell division, human telomeres lose between 50 and 200 base pairs with each replication.1–3 In checkpoint proficient cells, critically short telomeres trigger senescence, followed by apoptosis, which represents a barrier against cancer initiation by limiting cellular proliferation.4,5 As telomeres shorten, their ability to maintain chromosomal stability also diminishes, which may increase cancer susceptibility.6,7 However, long telomeres may also promote cancer development through an accumulation of mutations due to prolonged cell survival and proliferation. In fact, cancer cells are characterized by such a proliferative advantage, often through reactivation of telomerase, which is normally silent in somatic cells.4,5,8
Telomere length (TL) has been studied extensively in relation to cancer risk. However, findings of epidemiologic studies have been conflicting.6,9–11 Observational studies investigating TL measured after cancer diagnosis are particularly vulnerable to reverse causation and residual confounding, so shorter TL observed in cancer cases is likely to reflect underlying disease or the impact of cancer treatment.12,13 It is also difficult to isolate the influence of TL on cancer risk from that of other risk factors that influence both TL and cancer susceptibility, including biological or replicative age.10,14,15
Mendelian Randomization (MR) is an approach for evaluating causality by using single-nucleotide polymorphisms (SNPs) in relevant genes as instrumental variables (IVs).16 Genome-wide association studies (GWAS) identified a number of genetic regions involved in TL regulation, including genes encoding the catalytic subunit of telomerase reverse transcriptase (TERT) in chromosome 5p15.33 and its RNA template (TERC) in 3q26.2.17–21 By leveraging these associations, MR can provide a valid test of the causal hypothesis assuming the genetic IVs only affect cancer risk through TL regulation.
Previous studies using genetic proxies for TL suggest that longer telomeres confer an increased risk of lung cancer, especially adenocarcinoma,22–24 which is consistent with the findings of prospective observational studies.25–27 Lung cancer case–control studies report both increased28 and inverse6,29 associations for long TL, and some implicate high TL variability in lung cancer susceptibility.30 For head and neck cancers (HNC), which are predominantly squamous carcinomas, short TL is consistently associated with increased risk in case–control studies,6,31,32 whereas a recent MR analysis24 did find evidence supporting a causal relationship.
The overarching aim of this study is to investigate the causal relationship between TL and risk of lung and upper aero-digestive tract cancers. First, we developed a novel genetic instrument for TL in chromosome 5p15.33, given the extensive pleiotropy in this region and potential for violating MR assumptions.22,33 Next, we conducted the largest two-sample MR analysis of lung and HNC risk to date. Lastly, we quantified the direct and telomere-mediated effects of 5p15.33 genetic variants on cancer risk using a two-sample mediation analysis approach (Figure 1).
Figure 1.
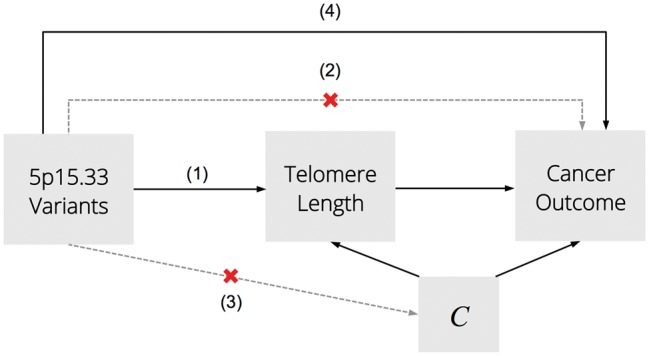
Conceptual diagram of Mendelian Randomization and mediation analyses. Mendelian Randomization is based the following assumptions (1–3): the genetic variant is strongly associated with telomere length; there is no direct association between the instrument and cancer outcome, except through telomere length; the genetic instrument is independent of any confounders (C). Mediation analyses of the 5p15.33 instrument for telomere length and 5p15.33 susceptibility variants test for the presence of direct effects (4) and quantify how much of the total genetic effect on lung cancer risk is mediated by telomere length.
Methods
Study populations
We used individual-level data from 23 pooled studies of lung cancer, with 16 396 cases (5690 adenocarcinoma, 4045 squamous carcinoma) and 13 013 controls; and 11 HNC studies with 4415 cases and 5013 controls, all part of the OncoArray collaboration34 (Supplementary Tables 1 and 2, available as Supplementary data at IJE online). Descriptions of studies and genotyping methods have been previously published34,35 (details in Supplementary File 1, available as Supplementary data at IJE online). Analyses were restricted to individuals of predominantly European ancestry (≥80% lung, >70% HNC).34,36 Studies received approval from institutional research ethics review boards and informed consent was obtained from the participants.
The novel 5p15.33 instrument was developed using data from two studies: the cancer-free controls from the Mount Sinai and Princess Margaret Hospital (MSH-PMH) case–control study in Toronto37 and cancer-free individuals from the Copenhagen General Population Study (CGPS),38 a population-based prospective cohort (Table 1). TL was measured in DNA from peripheral blood leukocytes using previously described quantitative polymerase chain reaction assays performed in MSH-PMH37 and CGPS23,38 (details in Supplementary File 2, available as Supplementary data at IJE online).
Table 1.
Characteristics of the Toronto (MSH-PMH) and Copenhagen (CGPS) OncoArray studies that comprise the dataset for the development of genetic instruments for telomere length in chromosome 5p15.33
| Characteristic and description | Toronto (MSH-PMH) |
Copenhagen (CGPS) |
Total |
||||
|---|---|---|---|---|---|---|---|
| N | (%) | N | (%) | N | (%) | ||
| Age (years) | <50 | 135 | (17.4) | 287 | (24.5) | 422 | (20.6) |
| 50–59 | 241 | (28.6) | 259 | (22.1) | 500 | (24.4) | |
| 60–69 | 313 | (35.0) | 264 | (22.5) | 577 | (28.1) | |
| 70–79 | 143 | (14.7) | 237 | (20.2) | 380 | (18.5) | |
| ≥80 | 47 | (4.3) | 125 | (10.7) | 172 | (8.4) | |
| Mean (SD) | 61.0 | (11.7) | 61.3 | (12.8) | 61.2 | (12.3) | |
| Sex | Males | 436 | (49.6) | 470 | (40.1) | 906 | (44.2) |
| Females | 443 | (50.4) | 702 | (59.9) | 1145 | (55.8) | |
| Smoking status | Never smokers | 438 | (50.1) | 410 | (36.4) | 848 | (41.3) |
| Ever smokers | 436 | (49.6) | 717 | (61.2) | 1153 | (56.2) | |
| Former smokers | 366 | (41.7) | 717 | (61.2) | 1083 | (52.8) | |
| Current smokers | 59 | (6.7) | 0 | (0) | 59 | (2.9) | |
| Unknown | 5 | (0.6) | 45 | (3.8) | 50 | (2.4) | |
| Mean cigarette pack-years (SD) | 8.7 | (17.2) | 14.4 | (20.2) | 12.0 | (19.2) | |
| Total | 879 | 1172 | 2051 | (100.0) | |||
CGPS, Copenhagen General Population Study; MSH-PMH, Mount Sinai Hospital-Princess Margaret Hospital study; SD, standard deviation.
Statistical analysis
MR analysis
The genetic instruments for TL included independent SNPs showing strong prior evidence of association with TL, such as p < 5 × 10–8 in the discovery stage of at least one GWAS and replication in a separate GWAS or meta-analysis.17–21 In addition to the new 5p15.33 instrument described below, we selected seven additional loci involved in telomere maintenance: rs10165485 (proxy for rs11125529, r2 = 1.0) in ACYP2 (2p16.2), rs6772228 in PXK (3p14.3), rs10936599 in TERC (3q26.2), rs11100479 (proxy for rs7675998, r2 = 0.99) in NAF1 (4q32.2), rs9420907 in OBFC1 (10q24.3), rs10419926 in ZNF676 (19p12) and rs755017 near RTEL1 and ZBTB46 (20q13). Only genotyped, non-imputed variants were used.
For the purpose of developing a new instrument in the 5p15.33 region, TL values were converted to Z-scores in MSH-PMH (n = 879) and CGPS (n = 1172) studies separately, and pooled to increase statistical power. Linear regression was used to estimate the association between 899 variants in 5p15.33 and TL, adjusting for age, sex, study and the top five genetic ancestry principal components (PCs).
Selection of variants for the 5p15.33 instrument was based on statistical significance, consistency across the two studies and instrument strength, measured by the F statistic, which depends on the variance in TL explained by the genetic predictors (R2), sample size (n) and number of instruments (k): . Variants were considered for inclusion in the 5p15.33 instrument if they met the following criteria:
F ≥ 5 and p < 0.05 in the Toronto and Copenhagen combined dataset (n = 2051);
F < 5 and p < 0.05 overall (n = 2051) and F > 5 among never smokers (n = 848);
consistent direction of allelic effects in MSH-PMH and CGPS;
minor allele detected in at least two individuals.
Independent genetic variants (r2 < 0.2) that met the selection criteria were combined into an allele score representing the 5p15.33 region to increase the power of the resulting instrument.39,40
The MR analysis combined summary statistics across the genetic IVs to estimate the causal parameter βIV, which is the log odds ratio (OR) describing the causal effect of increasing TL on cancer risk (Supplementary Figure 1, available as Supplementary data at IJE online). Parameters for the MR analysis included βTL and βY, where βTL is a vector of SNP-TL associations and βY is a vector of per-allele cancer log ORs for each instrument. For genetic instruments outside of 5p15.33, βTL and corresponding standard errors (SEs) were obtained from the literature and scaled to represent a 1000 base-pair (kbp) increase in leukocyte TL, a proxy for TL in relevant tissues.19–21 For all instruments, βY and corresponding SE were estimated directly using individual-level OncoArray lung and HNC data. Logistic regression models were adjusted for age, sex, study and 10 PCs.
The causal parameter βIV was estimated using the maximum likelihood-based (ML) approach and the inverse-variance weighted (IVW) method.41,42 This was complemented by sensitivity analyses using the weighted median estimator (WME), which provides valid estimates of the causal parameter even when up to 50% of the statistical weights are contributed by genetic instruments violate MR assumptions.43
Mediation analysis
The aim of the mediation analysis was to quantify how much of the lung cancer association in the 5p15.33 region is mediated by TL. First, we validated the 5p15.33 instrument by decomposing its total effect on lung cancer into direct and indirect effects, mediated by TL. Next, we extended this analysis to independent (r2 < 0.20) variants that capture the lung cancer association signal in 5p15.33 (details in Supplementary File 3, available as Supplementary data at IJE online).
Our mediation approach is based on the counterfactual framework44,45 and extends the sensitivity analysis using two randomized–controlled trials proposed by Vanderweele, which allows the mediator–outcome (θ2) and exposure–mediator (β1) relationships to be estimated in separate studies.46 Application of this approach in the present context assumes that a valid estimate for the mediator–outcome relationship can be obtained from independent MR or cohort studies. Based on previously published formulae for mediation analysis,44,45 the total effect (TE) of increasing the exposure from reference level a* to level a on lung cancer () conditional on covariates c can be decomposed into natural direct effects (NDE) and natural indirect effects (NIE):
| (1) |
Assuming a rare outcome and absence of exposure–mediator interaction, mediated effects are given by:
| (2) |
where θ2 is log-OR per one-unit increment in TL and β1 is the effect of the 5p15.33 instrument on TL. Based on Equation 1, NDE can be obtained by subtracting the NIE from the TE:
| (3) |
In the presence of interaction between the exposure and mediator, the NIE is given by:
| (4) |
where θ2 now represents the main effect of the mediator, TL, and θ3 is the exposure–mediator interaction parameter, with NDE having a more complicated form given by Valeri and VanderWeele.45 Formulae for a dichotomized mediator are provided in Supplementary File 4, available as Supplementary data at IJE online.
The β1 parameter for the 5p15.33 instrument is equivalent to βTL estimated in the cancer-free subset of the MSH-PMH and CGPS studies, adjusting for appropriate covariates. For 5p15.33 cancer susceptibility variants, β1 estimates were selected from Bojesen et al.47—the largest fine-mapping analysis of common 5p15.33 loci and TL with 15 567 cancer-free controls. Per-allele associations were reported as percent increase in TL and base-pair change. ORTE for all variants was estimated in 23 lung cancer OncoArray studies, and is equivalent to βY for the 5p15.33 instrument.
External estimates of the mediator–outcome relationship (θ2) were substituted into Equation (2) to avoid estimating the effect of TL on lung cancer risk directly using MSH-PMH case–control data, which are likely to be biased due to the post-diagnostic timing of TL measurement. The effect of TL on lung cancer risk was obtained from two studies: an MR analysis TL by Zhang et al.22 and a meta-analysis of prospective studies by Zhu et al.11 (Supplementary Figure 2, available as Supplementary data at IJE online).
Since interaction between the 5p15.33 instrument and TL is plausible, we conducted sensitivity analyses under different magnitudes of θ3 (details in Supplementary File 4, available as Supplementary data at IJE online). Confidence intervals (CIs) for the NIE and NDE were approximated as Bayesian credible intervals. Analyses were conducted using R version 3.3.3.
Results
Characteristics of the combined Toronto and Copenhagen dataset (n = 2051), used to develop the 5p15.33 instrument, are summarized in Table 1. The cancer-free participants in the MSH-PMH and CGPS studies were of similar mean age—61.0 and 61.30 years, respectively. Age was the strongest predictor of TL (p = 2.6 × 10–30), whereas sex, smoking status and cigarette pack-years among smokers were not associated with relative TL (Supplementary Table 3, available as Supplementary data at IJE online).
Novel 5p15.33 instrument for TL
The 5p15.33 variants comprising this instrument were not used in any previous MR studies of TL. After excluding 17 singletons and other SNPs that did not meet our criteria, 14 variants were included in the multi-allelic instrument for 5p15.33 (Table 2; regional plot and linkage disequilibrium (LD) illustrated in Supplementary Figure 3, available as Supplementary data at IJE online). Most variants were located in non-coding intronic regions of several genes, including SLC6A3, TERT, LPCAT1 and a long-non-coding RNA (LINC01511) except for rs35033501, a synonymous TERT variant. The resulting multi-allelic 5p15.33 IV accounted for 1.49% of variation in the telomere Z-score in all subjects (F = 35.83; βTL = 0.14, SE = 0.02) and 2.00% in never smokers (F = 20.81), but was not predictive of smoking status (p = 0.19) or cigarette pack-years among smokers (p = 0.59) (Table 3). The 5p15.33 instrument was positively associated with lung cancer (OR = 1.04, 95% CI: 1.01–1.07) and lung adenocarcinoma (OR = 1.06, 95% CI: 1.03–1.10), but not squamous lung carcinomas (OR = 1.03, 95% CI: 0.98–1.07). An inverse association was observed for HNC (OR = 0.95, 95% CI: 0.90–1.00) and oral cavity cancer (OR = 0.93, 95% CI: 0.87–0.98).
Table 2.
Genetic variants included in the novel 5p15.33 instrumental variable and their associations with the telomere length Z-score in the combined dataset (n = 2051)
| Variant | Gene | Alleles |
EAF | Per-allele estimate |
P-value | ||
|---|---|---|---|---|---|---|---|
| Long TL | Other | βa,b | (SE) | ||||
| rs956942 | LINC01511 | A | G | 2.4 × 10–3 | 1.11 | (0.29) | 1.7 × 10–4 |
| Chr5: 1383486 | CLPTM1L-SLC6A3 | A | G | 4.9 × 10–4 | 2.09 | (0.65) | 1.4 × 10–3 |
| Chr5: 1404329 | SLC6A3 | T | C | 9.8 × 10–4 | 1.28 | (0.46) | 5.8 × 10–3 |
| Chr5: 1501109 | LPCAT1 | A | G | 7.4 × 10–4 | 1.46 | (0.53) | 6.1 × 10–3 |
| Chr5: 1297379 | TERT | A | C/G | 1.5 × 10–3 | 0.68 | (0.27) | 0.01 |
| rs80022192 | LINC01511 | G | A | 4.9 × 10–4 | 1.60 | (0.65) | 0.01 |
| rs35033501 | TERT | A | G | 0.03 | 0.22 | (0.09) | 0.01 |
| rs28363089 | SLC6A3 | A | G | 0.03 | 0.23 | (0.02) | 0.01 |
| Chr5: 1434327 | SLC6A3 | A | T | 0.99 | 0.89 | (0.38) | 0.02 |
| Chr5: 1402812 | SLC6A3 | T | C | 4.9 × 10–4 | 1.49 | (0.65) | 0.02 |
| rs79717857 | CLPTM1L | A | C | 0.02 | 0.21 | (0.09) | 0.02 |
| rs35334674 | TERT | G | A | 0.97 | 0.19 | (0.08) | 0.02 |
| rs7733853 | LPCAT1 | A | G | 0.24 | 0.08 | (0.03) | 0.02 |
| rs72715516 | SLC6A3 | G | A | 0.96 | 0.21 | (0.10) | 0.04 |
EAF, effect allele frequency, where the effect allele is the long telomere allele; SE, standard error; LINC01511, long intergenic non-protein coding RNA 151; CLPTM1L, cleft lip and palate associated transmembrane protein 1-like; SLC6A3, solute carrier family 6 member 3; LPCAT1, lysophosphatidylcholine acyltransferase 1; TERT, telomerase reverse transcriptase. aLinear regression models adjusted for age, sex, study and ethnicity principal components. bRegression coefficients are standardized and correspond to a 1 standard deviation (1 unit) change in the telomere length Z-score, approximately 1000 base pairs.
Table 3.
Per-allele associations for the 5p15.33 genetic instrument and relevant telomere and cancer endpoints
| Outcome | Sample size (cases, controls) | βa/ORb | (SE)/95% CI | P-value | F statistic | R 2 (%) | |
|---|---|---|---|---|---|---|---|
| Telomere length | 2051 | 0.14 | (0.02) | 2.6 × 10–9 | 35.83 | 1.49 | |
| Telomere length in never smokers | 848 | 0.18 | (0.04) | 7.0 × 10–6 | 20.81 | 2.02 | |
| Smoking status (ever/never) | 2051 | –0.08 | (0.06) | 0.19 | – | – | |
| Cigarette pack-years | 1101 | 0.40 | (0.73) | 0.59 | 0.29 | 0.00 | |
| Lung cancer | 16 396 | 13 013 | 1.04 | 1.01, 1.07 | 4.89 × 10–3 | – | – |
| Adenocarcinoma | 5690 | 13 013 | 1.06 | 1.03, 1.10 | 1.4 × 10–3 | – | – |
| Squamous cell carcinoma | 4045 | 13 013 | 1.03 | 0.98, 1.07 | 0.23 | – | – |
| Head and neck cancer | 4415 | 5013 | 0.95 | 0.90, 1.00 | 0.04 | – | – |
| Oral cavity | 2284 | 5013 | 0.93 | 0.87, 0.98 | 0.01 | – | – |
| Oropharynx | 1849 | 5013 | 0.96 | 0.90, 1.03 | 0.26 | – | – |
| Never smokers | |||||||
| Lung cancer | 1619 | 3923 | 1.06 | 0.99, 1.14 | 0.08 | – | – |
| Adenocarcinoma | 836 | 3923 | 1.12 | 1.02, 1.22 | 0.02 | – | – |
| Head and neck cancer | 773 | 1827 | 0.85 | 0.77, 0.95 | 3.8 × 10–3 | – | – |
| Alcohol non-drinkers | |||||||
| Head and neck cancer | 614 | 795 | 0.86 | 0.74, 0.99 | 0.04 | – | – |
R 2, coefficient of determination estimating the proportion of the variance in the telomere length Z-score that is explained by the 5p15.33 genetic instrument; SE, standard error; TL, telomere length. aLinear regression models were adjusted for age, sex, study and top five ethnicity principal components; bLogistic regression models were adjusted for age, sex, study and top 10 ethnicity principal components.
TL and cancer risk
Results of the MR analysis based on eight genetic instruments are presented in Table 4 and Figure 2. The likelihood-based model estimated a 41% increase in lung cancer risk per kbp increase in TL (ORML = 1.41, 95% CI: 1.20–1.65). Estimates of the causal OR for lung cancer remained consistent across MR estimation methods. Genetic determinants of TL were predominantly associated with adenocarcinoma (ORML = 1.92, 95% CI: 1.51–2.45), and appeared unrelated to squamous carcinoma (ORML = 1.04, 95% CI: 0.83–1.29) and small cell carcinoma (ORML = 1.03, 95% CI: 0.76–1.39).
Table 4.
Mendelian Randomization estimates of the causal odds ratios for lung and head and neck cancers per 1000 base-pair increase in telomere length
| Outcome | Cases | Controls | Estimation method |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximum likelihood |
Inverse-variance weighted |
Weighted median estimator |
|||||||||
| ORa | 95% CI | P-value | ORa | 95% CI | P-value | ORa | 95% CI | P-value | |||
| Lung cancer | 16 396 | 13 013 | 1.41 | 1.20, 1.65 | 2.0 × 10–5 | 1.39 | 1.21, 1.60 | 3.7 × 10–6 | 1.37 | 1.12, 1.67 | 2.0 × 10–3 |
| Adenocarcinoma | 5690 | 13 013 | 1.92 | 1.51, 2.45 | 1.3 × 10–7 | 1.83 | 1.51, 2.22 | 5.5 × 10–10 | 1.63 | 1.23, 2.16 | 6.5 × 10–4 |
| Squamous | 4045 | 13 013 | 1.04 | 0.83, 1.29 | 0.74 | 1.04 | 0.83, 1.29 | 0.74 | 1.09 | 0.82, 1.46 | 0.57 |
| Small cell | 1846 | 13 013 | 1.03 | 0.76, 1.39 | 0.86 | 1.03 | 0.76, 1.38 | 0.86 | 0.96 | 0.66, 1.38 | 0.82 |
| Head and neck cancer | 4415 | 5013 | 0.90 | 0.70, 1.15 | 0.39 | 0.90 | 0.70, 1.15 | 0.41 | 0.71 | 0.51, 0.98 | 0.04 |
| Oral cavity | 2284 | 5013 | 0.88 | 0.65, 1.19 | 0.40 | 0.88 | 0.65, 1.19 | 0.40 | 0.67 | 0.44, 1.03 | 0.07 |
| Oropharynx | 1849 | 5013 | 0.83 | 0.59, 1.16 | 0.28 | 0.83 | 0.60, 1.16 | 0.28 | 0.72 | 0.46, 1.12 | 0.14 |
| Ever smokers | |||||||||||
| Lung cancer | 14 498 | 8815 | 1.36 | 1.14, 1.63 | 5.3 × 10–4 | 1.36 | 1.15, 1.60 | 2.6 × 10–4 | 1.31 | 1.05, 1.63 | 0.02 |
| Adenocarcinoma | 4754 | 8815 | 1.72 | 1.33, 2.24 | 4.2 × 10–5 | 1.66 | 1.33, 2.07 | 5.2 × 10–6 | 1.71 | 1.26, 2.32 | 6.1 × 10–4 |
| Squamous | 3835 | 8815 | 1.06 | 0.84, 1.35 | 0.60 | 1.06 | 0.84, 1.35 | 0.61 | 1.08 | 0.80, 1.47 | 0.63 |
| Head and neck | 3108 | 2865 | 1.12 | 0.79, 1.58 | 0.54 | 1.11 | 0.79, 1.56 | 0.54 | 0.91 | 0.60, 1.39 | 0.69 |
| Never smokers | |||||||||||
| Lung cancer | 1619 | 3923 | 1.78 | 1.22, 2.61 | 3.1 × 10–3 | 1.76 | 1.23, 2.52 | 2.0 × 10–3 | 1.55 | 0.98, 2.46 | 0.06 |
| Adenocarcinoma | 836 | 3923 | 2.68 | 1.70, 4.24 | 2.4 × 10–5 | 2.68 | 1.70, 4.24 | 2.4 × 10–5 | 2.24 | 1.18, 4.27 | 0.01 |
| Squamous | 149 | 3923 | 0.72 | 0.26, 1.97 | 0.52 | 0.72 | 0.26, 1.95 | 0.51 | 0.80 | 0.22, 2.90 | 0.75 |
| Head and neck | 773 | 1827 | 0.72 | 0.42, 1.22 | 0.22 | 0.72 | 0.42, 1.22 | 0.22 | 0.71 | 0.32, 1.55 | 0.39 |
| Early-onset (≤50 years) | |||||||||||
| Lung cancer | 1868 | 1557 | 1.68 | 1.07, 2.62 | 0.02 | 1.67 | 1.08, 2.59 | 0.02 | 1.76 | 0.98, 3.22 | 0.06 |
| Alcohol non-drinkers | |||||||||||
| Head and neck | 614 | 795 | 0.76 | 0.37, 1.56 | 0.45 | 0.76 | 0.37, 1.57 | 0.46 | 0.45 | 0.17, 1.16 | 0.10 |
CI, confidence intervals; OR, odds ratio. aRegression models for each genetic instrument were adjusted for age, sex, study and the top 10 ethnicity principal components.
Figure 2.

Scatter plots showing the association estimates for telomere length (βTL) and cancer risk (βY) for each instrumental variable (IV), overlaid on the causal log odds ratio for the effect of increasing telomere length on cancer risk (solid red line) and corresponding 95% confidence intervals (dotted red lines), estimated using the likelihood-based method.
The effect of long TL on lung cancer risk was larger in magnitude among never smokers (ORML = 1.78, 95% CI: 1.22–2.61) compared with smokers (ORML = 1.36, 95% CI: 1.14–1.63), although the former was attenuated in sensitivity analyses (ORWME = 1.55, 95% CI: 0.98–2.46). Effects on adenocarcinoma risk were also substantial in never smokers (ORML = 2.68, 95% CI: 1.70–4.24). Genetic determinants of long telomeres conferred a 68% increase in lung cancer risk (ORML = 1.68, 95% CI: 1.07–2.62) in subjects aged 50 years or younger. In contrast to lung cancer, genetic predisposition for longer TL did not seem related to risk of HNC overall (ORML = 0.90, 95% CI: 0.70–1.05), oral cavity (ORML = 0.88, 95% CI: 0.65–1.19) and oropharynx cancers (ORML = 0.83, 95% CI: 0.59–1.16).
Several additional sensitivity analyses were undertaken to further interrogate the MR results. Since smoking is an established risk factor for both HNC and lung cancer, MR analyses were repeated with adjustment for cigarette pack-years and smoking status. No appreciable changes were observed in the causal effect estimates for lung cancer overall (ORML = 1.50, 95% CI: 1.27–1.78), lung adenocarcinoma (ORML = 1.95, 95% CI: 1.53–2.49), HNC (ORML = 0.91, 95% CI: 0.67–1.23), oral cavity (ORML = 0.82, 95% CI: 0.57–1.18) or oropharynx cancers (ORML = 0.86, 95% CI: 0.57–1.31).
The potential for directional pleiotropy was evaluated by checking for asymmetry in the plots depicting ratio estimates for each instrument, βY/βTL, plotted against instrument strength, βTL/SE(βY) (Supplementary Figure 4, available as Supplementary data at IJE online). These results were not suggestive of pleiotropy and none of the genetic instruments was associated with cigarette smoking status or pack-years (Supplementary Table 4, available as Supplementary data at IJE online). Lastly, selected causal effects were re-estimated using the weighted mode-based estimator (MBE), which is robust to horizontal pleiotropy when the largest number of similar causal effect estimates are based on valid instruments, even if the majority of instruments are invalid.48 Estimates for lung cancer overall (ORMBE = 1.34, 95% CI: 1.08–1.66), lung adenocarcinoma (ORMBE = 1.55, 95% CI: 1.14–2.12) and adenocarcinoma in never smokers (ORMBE = 2.04, 95% CI: 1.04–4.04) were consistent with the primary results in Table 4.
Mediation analysis of the 5p15.33 instrument
We conducted mediation analyses to quantify direct (ORNDE) and indirect effects (ORNIE) of the 5p15.33 instrument on lung cancer. The ORNIE we report is the proportional change in the odds of lung cancer for a change in TL that occurs when the 5p15.33 allele score increases by one from the reference level, corresponding to the mean of the allele score distribution. The estimate of the TL effect on lung cancer (θ2) was selected from the strict model reported by Zhang et al.22 (OR per kbp increase: 1.37, 95% CI: 1.12–1.68), which excluded rs2736100 (TERT). ORTE for the 5p15.33 IV was re-estimated after removing overlapping subjects (n = 3498) between the OncoArray and Zhang et al.22 Assuming no interaction between the 5p15.33 IV and TL, the lung cancer effect appeared to be almost entirely mediated by TL (ORNIE = 1.05, 95% CI: 1.01–1.08), whereas the direct effects of the 5p15.33 IV appeared null (ORNDE = 1.00, 95% CI: 0.95–1.04) (Figure 3; Supplementary Table 5, available as Supplementary data at IJE online). For lung adenocarcinoma, the 5p15.33 effects mediated by TL were larger in magnitude (ORNIE = 1.11, 95% CI: 1.05–1.18) than direct effects, which were close to unity (ORNDE = 0.97, 95% CI: 0.90–1.03).
Figure 3.

Odds ratio (OR) plot summarizing the direct effects (triangle, dotted line) and indirect effects (circle, solid line) of the 5p15.33 genetic instrument on lung cancer risk. Estimates of the direct and indirect effects are presented across different levels of interaction and for different versions of the mediator (dichotomous and continuous), indicated by different colours.
Interaction sensitivity analyses for the NIE and NDE were carried out across three levels of θ3: 0.10, 0.20 and 0.30. As the magnitude of the interaction parameter increased, so did the NIE, whereas TL-independent effects were not observed (Figure 3). Indirect effects on lung cancer risk mediated by TL ranged from ORNIE = 1.06 (95% CI: 1.03–1.10) for θ3 = 0.10, to ORNIE = 1.09 (95% CI: 1.05–1.15) for θ3 = 0.30. For adenocarcinoma, increasing the magnitude of interaction between the 5p15.33 IV and TL was also associated with increasing NIE and diminishing direct effects.
The prospective meta-analysis estimate of θ2 from Zhu et al.11 reported an OR of 1.28 (95% CI: 1.09–1.50) for lung cancer comparing long vs short TL. Based on this binary mediator, the NIE mediated by TL was attenuated, but remained statistically significant (ORNIE = 1.01, 95% CI: 1.00–1.03). A positive direct effect on lung cancer risk was also observed (ORNDE = 1.03, 95% CI: 1.00–1.06). Assuming interaction between the 5p15.33 instrument and TL, the mediated effects ranged from ORNIE = 1.02 (95% CI: 1.01–1.03) when θ3 = 0.10, to ORNIE = 1.03 (95% CI: 1.01–1.05) when θ3 = 0.30, whereas the direct effects decreased (Figure 3; Supplementary Table 5, available as Supplementary data at IJE online).
Mediation analysis of 5p15.33 lung cancer susceptibility loci
Five common (MAF > 0.05), independent (r2 < 0.20) variants were selected to represent the lung cancer susceptibility signal in 5p15.33 (details in Supplementary File 3, available as Supplementary data at IJE online): rs7705526 (PAdeno = 4.6 × 10–13; PLung = 8.0 × 10–7), rs2736108 (PAdeno = 1.7 × 10–12; PLung = 1.8 × 10–11), rs421629 (PAdeno = 6.2 × 10–9; PLung = 1.2 × 10–16), rs13167280 (PAdeno = 1.4 × 10–8; PLung = 1.1 × 10–6) and rs56345976 (PAdeno = 2.2 × 10–7; PLung = 3.6 × 10–9). These variants have been associated with lung cancer and lung adenocarcinoma in previous studies,37,49–51 and are representative of the genetic susceptibility architecture in this region.
Estimates of β1 were obtained from Bojesen et al.47 and three TERT lung cancer risk variants were significantly associated with TL: rs7705526 (PTL = 2.3 × 10–14), rs2736108 (PTL = 5.8 × 10–7) and rs13167280 (PTL = 1.2 × 10–5). Estimates of θ2 were selected from the MR analysis22 and ORTE were re-estimated for each variant after removing the overlapping subjects. For all variants, the TL-increasing allele was positively associated with cancer risk, and both direct and indirect TL-mediated effects were significant (Supplementary Table 6, available as Supplementary data at IJE online).
For lung cancer, the proportion mediated (PM) by TL was the largest for rs13167280 (ORNIE = 1.05, 95% CI: 1.03–1.07; PM = 40.5%), followed by rs7705526 (ORNIE = 1.03, 95% CI: 1.01–1.05; PM = 28.7%) and rs2736108 (ORNIE = 1.02, 95% CI: 1.01–1.03; PM = 13.7%). The magnitude and proportion of the SNP effects that were mediated by TL were larger for adenocarcinoma compared with lung cancer overall: rs7705526 (ORNIE = 1.07, 95% CI: 1.04–1.10; PM = 36.5%), rs13167280 (ORNIE = 1.05, 95% CI: 1.03–1.07; PM = 24.8%) and rs2736108 (ORNIE = 1.04, 95% CI: 1.03–1.06; PM = 22.9%).
Discussion
We observed an association between genetic determinants of long telomeres and increased risk of lung cancer, but not HNC. Our findings lend support to a causal relationship between longer leukocyte TL and increased risk of lung adenocarcinoma, but not squamous or small cell carcinoma. The magnitude of the increased risk was larger in never smokers and participants aged 50 or younger, consistently with a stronger influence of genetic susceptibility in individuals with a lower burden of modifiable risk factors.52 Although histology and smoking status are closely linked, our results suggest that the associations were histology-specific for adenocarcinoma.53,54 Lastly, our mediation analysis demonstrated that mechanisms resulting in long telomeres mediate a proportion of the increase in lung cancer and lung adenocarcinoma risk conferred by 5p15.33 loci, and that the proportion of genetic susceptibility attributed to telomere maintenance differs between distinct 5p15.33 susceptibility loci.
Other analyses using multi-SNP telomere scores have also observed excess risks of lung cancer22–24 and lung adenocarcinoma,22,24 but did not observe an effect of TL on oral cancer risk.23,24 Opposite directions of effect for the 5p15.33 instrument on lung and HNC are consistent with earlier reports of opposing allelic effects for 5p15.33 SNPs on lung and oral cancer, respectively.35,55 Leukocyte TL and functional TERT variants were previously reported to be unrelated to squamous HNC risk,56 although one study linked short TL to increased HNC risk based on rs2736100, which may be an invalid instrument.22,57 With the exception of the 5p15.33 IV, the instruments used in this study overlap with those used in other MR analyses of TL.22–24
Our findings lend support to the hypothesis that a greater number of telomere-increasing alleles increase lung cancer susceptibility. Although the precise molecular mechanisms remain to be elucidated, telomere maintenance may promote carcinogenesis by enabling prolonged cell survival and accumulation of mutations. This is supported by the hallmark observation that telomerase is overexpressed in 85–90% of adult tumours,8,58 as well as recent data showing that long telomeres increase chromosomal instability59 and promote immortalization of cancer cells.60 Excessively long telomeres may also be more fragile and dysfunctional, which is supported by the observation that TERT not only replenishes telomeres, but also regulates a trimming process to maintain TL homeostasis.61–63
Differences in the effect of TL persisted after stratifying by smoking status, suggesting that underlying mechanisms differ across tissues and histological types. Longer TL does not appear to increase risk of small cell lung cancer or squamous lung carcinoma, the histology that also comprises 90% of HNC tumours, and for which the causal effect of tobacco smoking is the strongest.64 Since our genetic instruments are unrelated to smoking, confounding is unlikely to account for these differences. It is plausible that genetic predisposition for telomere maintenance offers some protection against genomic instability due to oxidative stress, declining regenerative capacity and immune function.7,65,66 Although human papillomavirus (HPV), a known cause of oropharynx cancer,67 has been reported to correlate with TL,31 the similarity of associations observed for oropharynx and oral cancers, only 2% of which are attributed to HPV,68 suggests that HPV infection is unlikely to modify the influence of TL.
This analysis has several important strengths. Genetic instruments are unaffected by reverse causality and are more likely to reflect causality due to the independence of genotypes from confounding factors. In addition to the large sample size, our analysis leveraged rich genetic data in 5p15.33, including rare sequence variations, to develop a robust, novel instrument. Furthermore, the use of multiple genetic instruments from essential genes for telomere maintenance mitigates the possibility for weak instruments bias and genetic confounding due to pleiotropy. The association between genetic predisposition to long TL and increased lung cancer risk persisted in analyses using the weighted median and MBEs, which further supports the causal interpretation of these results.
Our mediation analysis offers insight not only by validating the new 5p15.33 instrument, by demonstrating an absence of direct effects, but also by formally quantifying the contribution of telomere-related mechanisms to the observed association between the established lung and adenocarcinoma susceptibility loci and lung cancer risk in this region. Although we confirmed that TL is an important molecular mechanism underlying the associations observed for 5p15.33 lung cancer risk loci, our results also indicated that only a fraction of these genetic effects operate through telomere maintenance. For instance, only 3–8% of the total effect of rs421629 (CLPTM1L) was mediated TL, and approximately half of the association between the TERT loci and lung cancer risk can be attributed to telomere mechanisms.
These findings are consistent with our knowledge that 5p15.33 is a complex susceptibility locus for multiple cancers33,55,69 and GWAS peaks in this region also encompass non-cancer traits, such as red blood cell counts, prostate-specific antigen levels and lung diseases.69–72 In addition, non-canonical functions of TERT, related to proliferation and differentiation via regulation of Wnt/β-catenin and Myc signalling, have been proposed.73 Therefore, although telomere maintenance is clearly an important 5p15.33 mechanism, cancer susceptibility loci in this region likely invoke additional pathways.
Several limitations of this work should be acknowledged. The time lag between genotype assignment at conception and the assessment of genetic effects on TL and cancer risk, as well as the time-varying nature of TL, pose challenges for interpreting MR estimates of the causal effect.74 However, whereas genetic instruments do not recapitulate all aspects of telomere function and dynamics, they can still provide a valid test of the causal hypothesis that inherited predisposition to telomere maintenance increases lung cancer susceptibility.75 Second, genetic instruments for leukocyte TL may not be accurate proxies for TL in target tissues, which would reduce the power of our genetic instruments. However, the validity of instruments based on leukocyte TL is supported by correlation between TL in leukocytes and other tissues, including lung, and comparable rates of telomere shortening across somatic tissues.76–78 Third, our MR analysis may be affected by winner’s curse, with the magnitude and strength of association with TL observed in the discovery dataset likely to be exaggerated, particularly the 5p15.33 instrument. However, since the instrument discovery and MR analysis populations are independent, any potential bias in the causal parameter due to winner’s curse or limited instrument strength will be towards the null.79 A related concern involves our ability to detect subtle effects of TL on cancer risk due to the modest proportion of variation in TL explained by our genetic instruments (approximately 5%), which is comparable to most genetic instruments for complex phenotypes.80–82 Based on our power calculations, this analysis was adequately powered (>80%) to detect effects with OR of 1.5 and above for all lung and HNC histological subtypes and smoking-stratified analyses.
Lastly, the validity of our mediation analysis depends in part on the validity of the published estimates of the mediator–outcome relationship. MR-based estimates of the mediator–outcome relationship are likely to satisfy the assumption of no unmeasured confounding, but must assume that all instruments used in Zhang et al.22 were valid. Whereas observational studies are more susceptible to confounding and bias due measurement error in the molecular mediator,83 a synthesis of prospective studies provides complementary evidence that does not depend on MR assumptions, and is less vulnerable to reverse causation than case–control designs.
In summary, we demonstrated that genetic determinants of long telomeres are associated with an increased risk of lung cancer, particularly adenocarcinoma. The associations observed for HNC were less consistent with a causal relationship, although we cannot preclude the possibility of very subtle telomere effects (OR < 1.5). Using mediation analysis that incorporates independent published data, we validated the novel 5p15.33 instrument and quantified the proportion of the lung cancer association signal in 5p15.33 that is mediated by TL. Whereas this work provides insight into the role of TL in cancer aetiology, further research is needed to identify appropriate ways of utilizing this complex biomarker in the context of disease prevention or clinical intervention.
Funding
L.K. is a fellow in the Canadian Institutes of Health Research (CIHR) Strategic Training in Advanced Genetic Epidemiology (STAGE) programme and is supported by the CIHR Doctoral Research Award from the Frederick Banting and Charles Best Canada Graduate Scholarships (GSD-137441). Transdisciplinary Research for Cancer in Lung (TRICL) of the International Lung Cancer Consortium (ILCCO) was supported by the National Institutes of Health (U19-CA148127, CA148127S1). Genotyping for the TRICL-ILCCO OncoArray was supported by in-kind genotyping at Centre for Inherited Disease Research (CIDR) (26820120008i-0–6800068-1). Genotyping for the Head and Neck Cancer OncoArray performed at CIDR was funded by the US National Institute of Dental and Craniofacial Research (NIDCR) grant 1X01HG007780–0. CAPUA study was supported by FIS-FEDER/Spain grant numbers FIS-01/310, FIS-PI03–0365 and FIS-07-BI060604, FICYT/Asturias grant numbers FICYT PB02–67 and FICYT IB09–133, and the University Institute of Oncology (IUOPA), of the University of Oviedo and the Ciber de Epidemiologia y Salud Pública. CIBERESP, SPAIN. The work performed in the CARET study was supported by the National Institute of Health (NIH)/National Cancer Institute (NCI): UM1 CA167462 (PI: Goodman), National Institute of Health UO1-CA6367307 (PIs Omen, Goodman); National Institute of Health R01 CA111703 (PI Chen), National Institute of Health 5R01 CA151989 (PI Doherty). The Liverpool Lung Project is supported by the Roy Castle Lung Cancer Foundation. The Harvard Lung Cancer Study was supported by the NIH (National Cancer Institute) grants CA092824, CA090578 and CA074386. The Multiethnic Cohort Study was partially supported by NIH Grants CA164973, CA033619, CA63464 and CA148127. The work performed in MSH-PMH study was supported by the Canadian Cancer Society Research Institute (020214), Ontario Institute of Cancer and Cancer Care Ontario Chair Award to R.J.H. and G.L. and the Alan Brown Chair and Lusi Wong Programs at the Princess Margaret Hospital Foundation. The Norway study was supported by Norwegian Cancer Society, Norwegian Research Council. The work in TLC study has been supported in part the James & Esther King Biomedical Research Program (09KN-15), National Institutes of Health Specialized Programs of Research Excellence (SPORE) Grant (P50 CA119997) and by a Cancer Center Support Grant (CCSG) at the H. Lee Moffitt Cancer Center and Research Institute, an NCI designated Comprehensive Cancer Center (grant number P30-CA76292). The dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by institutional funding and by the Vanderbilt CTSA grant UL1 TR000445 from NCATS/NIH. Dr Melinda Aldrich is supported by the by NIH/National Cancer Institute 5K07CA172294. The Copenhagen General Population Study (CGPS) was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council and Herlev Hospital. The NELCS study: Grant Number P20RR018787 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Kentucky Lung Cancer Research Initiative (KLCRI) was supported by the Department of Defense (Congressionally Directed Medical Research Program, U.S. Army Medical Research and Materiel Command Program) under award number: 10153006 (W81XWH-11–1-0781). Views and opinions of, and endorsements by the author(s) do not reflect those of the US Army or the Department of Defense. This research was also supported by unrestricted infrastructure funds from the UK Center for Clinical and Translational Science, NIH grant UL1TR000117 and Markey Cancer Center NCI Cancer Center Support Grant (P30 CA177558) Shared Resource Facilities: Cancer Research Informatics, Biospecimen and Tissue Procurement, and Biostatistics and Bioinformatics. The research undertaken by M.D.T., L.V.W. and M.S.A. was partly funded by the National Institute for Health Research (NIHR). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. M.D.T. holds a Medical Research Council Senior Clinical Fellowship (G0902313). The Tampa study was funded by Public Health Service grants P01-CA68384 and R01-DE13158 from the National Institutes of Health. The University of Pittsburgh head and neck cancer case–control study is supported by US National Institutes of Health grants P50 CA097190 and P30 CA047904. The Carolina Head and Neck Cancer Study (CHANCE) was supported by the National Cancer Institute (R01CA90731). The Head and Neck Genome Project (GENCAPO) was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grants 04/12054–9 and 10/51168–0). The authors thank all the members of the GENCAPO team. This publication presents data from the Head and Neck 5000 study. The study was a component of independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research scheme (RP-PG-0707–10034). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Human papillomavirus (HPV) serology was supported by a Cancer Research UK Programme Grant, the Integrative Cancer Epidemiology Programme (grant number: C18281/A19169). The Alcohol-Related Cancers and Genetic Susceptibility Study in Europe (ARCAGE) was funded by the European Commission’s fifth framework programme (QLK1– 2001-00182), the Italian Association for Cancer Research, Compagnia di San Paolo/FIRMS, Region Piemonte and Padova University (CPDA057222). The Rome Study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) awards IG 2011 10491 and IG 2013 14220 to S.B. and by Fondazione Veronesi to S.B. The IARC Latin American study was funded by the European Commission INCO-DC programme (IC18-CT97–0222), with additional funding from Fondo para la Investigación Científica y Tecnológica (Argentina) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (01/01768–2). The IARC Central Europe study was supported by the European Commission’s INCO-COPERNICUS Program (IC15-CT98–0332), US NIH/National Cancer Institute grant CA92039 and World Cancer Research Foundation grant WCRF 99A28. The IARC Oral Cancer Multicenter study was funded by grant S06 96 202489 05F02 from Europe against Cancer; grants FIS 97/0024, FIS 97/0662 and BAE 01/5013 from Fondo de Investigaciones Sanitarias, Spain; the UICC Yamagiwa-Yoshida Memorial International Cancer Study; the National Cancer Institute of Canada; Associazione Italiana per la Ricerca sul Cancro; and the Pan-American Health Organization. Coordination of the EPIC study is financially supported by the European Commission (DG SANCO) and the International Agency for Research on Cancer.
Supplementary Material
Acknowledgements
The authors would like to acknowledge all of the participants involved in this research and the funders and support.
Conflict of interest: None declared.
References
- 1. Blackburn EH. Structure and function of telomeres. Nature 1991;350:569–73. [DOI] [PubMed] [Google Scholar]
- 2. de Lange T. Protection of mammalian telomeres. Oncogene 2002;21:532–40. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Y, Sfeir AJ, Zou Y. et al. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell 2009;138:463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bodnar AG, Ouellette M, Frolkis M. et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349–52. [DOI] [PubMed] [Google Scholar]
- 5. Hanahan D, Weinberg RA.. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 6. Wu X, Amos CI, Zhu Y. et al. Telomere dysfunction: a potential cancer predisposition factor. J Natl Cancer Inst 2003;95:1211–18. [DOI] [PubMed] [Google Scholar]
- 7. Bernardes de Jesus B, Blasco MA.. Telomerase at the intersection of cancer and aging. Trends Genet 2013;29:513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newbold RF. The significance of telomerase activation and cellular immortalization in human cancer. Mutagenesis 2002;17:539–50. [DOI] [PubMed] [Google Scholar]
- 9. Wentzensen IM, Mirabello L, Pfeiffer RM, Savage SA.. The association of telomere length and cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:1238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prescott J, Wentzensen IM, Savage SA, De Vivo I.. Epidemiologic evidence for a role of telomere dysfunction in cancer etiology. Mutat Res 2012;730:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu X, Han W, Xue W. et al. The association between telomere length and cancer risk in population studies. Sci Rep 2016;6:22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benitez-Buelga C, Sanchez-Barroso L, Gallardo M. et al. Impact of chemotherapy on telomere length in sporadic and familial breast cancer patients. Breast Cancer Res Treat 2015;149:385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li P, Hou M, Lou F, Bjorkholm M, Xu D.. Telomere dysfunction induced by chemotherapeutic agents and radiation in normal human cells. Int J Biochem Cell Biol 2012;44:1531–40. [DOI] [PubMed] [Google Scholar]
- 14. Huzen J, Wong LS, van Veldhuisen DJ. et al. Telomere length loss due to smoking and metabolic traits. J Intern Med 2014;275:155–63. [DOI] [PubMed] [Google Scholar]
- 15. Bojesen SE. Telomeres and human health. J Intern Med 2013;274:399–413. [DOI] [PubMed] [Google Scholar]
- 16. Davey Smith G, Hemani G.. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Mol Genet 2014;23:R89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levy D, Neuhausen SL, Hunt SC. et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc Natl Acad Sci USA 2010;107:9293–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prescott J, Kraft P, Chasman DI. et al. Genome-wide association study of relative telomere length. PLoS One 2011;6:e19635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mangino M, Hwang SJ, Spector TD. et al. Genome-wide meta-analysis points to CTC1 and ZNF676 as genes regulating telomere homeostasis in humans. Human Mol Genet 2012;21:5385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pooley KA, Bojesen SE, Weischer M. et al. A genome-wide association scan (GWAS) for mean telomere length within the COGS project: identified loci show little association with hormone-related cancer risk. Human Mol Genet 2013;22:5056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Codd V, Nelson CP, Albrecht E. et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 2013;45:422–27, 7e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C, Doherty JA, Burgess S. et al. Genetic determinants of telomere length and risk of common cancers: a Mendelian randomization study. Human Mol Genet 2015;24:5356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rode L, Nordestgaard BG, Bojesen SE.. Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol 2016;45:1634–43. [DOI] [PubMed] [Google Scholar]
- 24.The Telomeres Mendelian Randomization Collaboration. Association between telomere length and risk of cancer and non-neoplastic diseases: a Mendelian randomization study. JAMA Oncol 2017;3:636–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seow WJ, Cawthon RM, Purdue MP. et al. Telomere length in white blood cell DNA and lung cancer: a pooled analysis of three prospective cohorts. Cancer Res 2014;74:4090–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lan Q, Cawthon R, Gao Y. et al. Longer telomere length in peripheral white blood cells is associated with risk of lung cancer and the rs2736100 (CLPTM1L-TERT) polymorphism in a prospective cohort study among women in China. PLoS One 2013;8:e59230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen M, Cawthon R, Rothman N. et al. A prospective study of telomere length measured by monochrome multiplex quantitative PCR and risk of lung cancer. Lung Cancer 2011;73:133–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanchez-Espiridion B, Chen M, Chang JY. et al. Telomere length in peripheral blood leukocytes and lung cancer risk: a large case-control study in Caucasians. Cancer Res 2014;74:2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jang JS, Choi YY, Lee WK. et al. Telomere length and the risk of lung cancer. Cancer Sci 2008;99:1385–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun B, Wang Y, Kota K. et al. Telomere length variation: a potential new telomere biomarker for lung cancer risk. Lung Cancer 2015;88:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y, Sturgis EM, Dahlstrom KR. et al. Telomere length in peripheral blood lymphocytes contributes to the development of HPV-associated oropharyngeal carcinoma. Cancer Res 2013;73:5996–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bau DT, Lippman SM, Xu E. et al. Short telomere lengths in peripheral blood leukocytes are associated with an increased risk of oral premalignant lesion and oral squamous cell carcinoma. Cancer 2013;119:4277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z, Zhu B, Zhang M. et al. Imputation and subset-based association analysis across different cancer types identifies multiple independent risk loci in the TERT-CLPTM1L region on chromosome 5p15.33. Human Mol Genet 2014;23:6616–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amos CI, Dennis J, Wang Z. et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev 2016;26:126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lesseur C, Diergaarde B, Olshan AF. et al. Genome-wide association analyses identify new susceptibility loci for oral cavity and pharyngeal cancer. Nat Genet 2016;48:1544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Byun J, Cai G. et al. FastPop: a rapid principal component derived method to infer intercontinental ancestry using genetic data. BMC Bioinformatics 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kachuri L, Amos CI, McKay JD. et al. Fine mapping of chromosome 5p15.33 based on a targeted deep sequencing and high density genotyping identifies novel lung cancer susceptibility loci. Carcinogenesis 2016;37:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weischer M, Bojesen SE, Cawthon RM, Freiberg JJ, Tybjaerg-Hansen A, Nordestgaard BG.. Short telomere length, myocardial infarction, ischemic heart disease, and early death. Arterioscler Thromb Vasc Biol 2012;32:822–29. [DOI] [PubMed] [Google Scholar]
- 39. Burgess S, Thompson SG.. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013;42:1134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pierce BL, Ahsan H, Vanderweele TJ.. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol 2011;40:740–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burgess S, Butterworth A, Thompson SG.. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thompson JR, Minelli C, Abrams KR, Tobin MD, Riley RD.. Meta-analysis of genetic studies using Mendelian randomization—a multivariate approach. Stat Med 2005;24:2241–54. [DOI] [PubMed] [Google Scholar]
- 43. Bowden J, Davey Smith G, Haycock PC, Burgess S.. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. VanderWeele TJ. A three-way decomposition of a total effect into direct, indirect, and interactive effects. Epidemiology 2013;24:224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valeri L, Vanderweele TJ.. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods 2013;18:137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. VanderWeele TJ. Explanation in Causal Inference: Methods for Mediation and Interaction. New York, NY: Oxford University Press, 2015. [Google Scholar]
- 47. Bojesen SE, Pooley KA, Johnatty SE. et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet 2013;45:371–84, 84e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hartwig FP, Davey Smith G, Bowden J.. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017;46:1985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McKay JD, Hung RJ, Gaborieau V. et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet 2008;40:1404–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pande M, Spitz MR, Wu X, Gorlov IP, Chen WV, Amos CI.. Novel genetic variants in the chromosome 5p15.33 region associate with lung cancer risk. Carcinogenesis 2011;32:1493–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McKay JD, Hung RJ, Han Y. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet 2017;49:1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brennan P, Hainaut P, Boffetta P.. Genetics of lung-cancer susceptibility. Lancet Oncol 2011;12:399–408. [DOI] [PubMed] [Google Scholar]
- 53. Samet JM, Avila-Tang E, Boffetta P. et al. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res 2009;15:5626–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ.. Lung cancer in never smokers—a review. Eur J Cancer 2012;48:1299–311. [DOI] [PubMed] [Google Scholar]
- 55. Rafnar T, Sulem P, Stacey SN. et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu Z, Ma H, Wei S, Li G, Sturgis EM, Wei Q.. Telomere length and TERT functional polymorphisms are not associated with risk of squamous cell carcinoma of the head and neck. Cancer Epidemiol Biomarkers Prev 2011;20:2642–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gu Y, Yu C, Miao L. et al. Telomere length, genetic variants and risk of squamous cell carcinoma of the head and neck in Southeast Chinese. Sci Rep 2016;6:20675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shay JW, Bacchetti S.. A survey of telomerase activity in human cancer. Eur J Cancer 1997;33:787–91. [DOI] [PubMed] [Google Scholar]
- 59. Bull CF, Mayrhofer G, O'Callaghan NJ. et al. Folate deficiency induces dysfunctional long and short telomeres; both states are associated with hypomethylation and DNA damage in human WIL2-NS cells. Cancer Prev Res 2014;7:128–38. [DOI] [PubMed] [Google Scholar]
- 60. Borah S, Xi L, Zaug AJ. et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015;347:1006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng YL, Zhang F, Sun B. et al. Telomerase enzymatic component hTERT shortens long telomeres in human cells. Cell Cycle 2014;13:1765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martinez P, Thanasoula M, Munoz P. et al. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 2009;23:2060–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rivera T, Haggblom C, Cosconati S, Karlseder J.. A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol 2017;24:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pai SI, Westra WH.. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu Rev Pathol 2009;4:49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci 2000;908:99–110. [DOI] [PubMed] [Google Scholar]
- 66. Hohensinner PJ, Goronzy JJ, Weyand CM.. Telomere dysfunction, autoimmunity and aging. Aging Dis 2011;2:524–37. [PMC free article] [PubMed] [Google Scholar]
- 67. Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C.. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J Clin Oncol 2015;33:3235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. de Martel C, Plummer M, Vignat J, Franceschi S.. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer 2017;141:664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu YH, Graff RE, Passarelli MN. et al. Identification of pleiotropic cancer susceptibility variants from genome-wide association studies reveals functional characteristics. Cancer Epidemiol Biomarkers Prev 2018;27:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gudmundsson J, Besenbacher S, Sulem P. et al. Genetic correction of PSA values using sequence variants associated with PSA levels. Sci Transl Med 2010;2:62ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kamatani Y, Matsuda K, Okada Y. et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 2010;42:210–15. [DOI] [PubMed] [Google Scholar]
- 72. Fingerlin TE, Murphy E, Zhang W. et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013;45:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Low KC, Tergaonkar V.. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem Sci 2013;38:426–34. [DOI] [PubMed] [Google Scholar]
- 74. Swanson SA, Tiemeier H, Ikram MA, Hernan MA.. Nature as a trialist?: deconstructing the analogy between Mendelian randomization and randomized trials. Epidemiology 2017;28:653–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P.. Methodological challenges in Mendelian randomization. Epidemiology 2014;25:427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Friedrich U, Griese E, Schwab M, Fritz P, Thon K, Klotz U.. Telomere length in different tissues of elderly patients. Mech Ageing Dev 2000;119:89–99. [DOI] [PubMed] [Google Scholar]
- 77. Saferali A, Lee J, Sin DD, Rouhani FN, Brantly ML, Sandford AJ.. Longer telomere length in COPD patients with alpha1-antitrypsin deficiency independent of lung function. PLoS One 2014;9:e95600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Daniali L, Benetos A, Susser E. et al. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat Commun 2013;4:1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G.. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr 2016;103:965–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hartwig FP, Borges MC, Horta BL, Bowden J, Davey Smith G.. Inflammatory biomarkers and risk of schizophrenia: a 2-sample mendelian randomization study. JAMA Psychiatry 2017;74:1226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carreras-Torres R, Johansson M, Haycock PC. et al. Obesity, metabolic factors and risk of different histological types of lung cancer: a Mendelian randomization study. PLoS One 2017;12:e0177875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dimitrakopoulou VI, Tsilidis KK, Haycock PC. et al. Circulating vitamin D concentration and risk of seven cancers: Mendelian randomisation study. BMJ. 2017;359:j4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Richmond RC, Hemani G, Tilling K, Davey Smith G, Relton CL.. Challenges and novel approaches for investigating molecular mediation. Human Mol Genet 2016;25:R149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.