

Abstract
Phenotypic heterogeneity in cancer cells is widely observed and is often linked to drug resistance. In several cases, such heterogeneity in drug sensitivity of tumors is driven by stochastic and reversible acquisition of a drug tolerant phenotype by individual cells even in an isogenic population. Accumulating evidence further suggests that cell-fate transitions such as the epithelial to mesenchymal transition (EMT) are associated with drug resistance. In this study, we analyze stochastic models of phenotypic switching to provide a framework for analyzing cell-fate transitions such as EMT as a source of phenotypic variability in drug sensitivity. Motivated by our cell-culture based experimental observations connecting phenotypic switching in EMT and drug resistance, we analyze a coarse-grained model of phenotypic switching between two states in the presence of cytotoxic stress from chemotherapy. We derive analytical results for time-dependent probability distributions that provide insights into the rates of phenotypic switching and characterize initial phenotypic heterogeneity of cancer cells. The results obtained can also shed light on fundamental questions relating to adaptation and selection scenarios in tumor response to cytotoxic therapy.
Subject terms: Cancer, Biophysics
Introduction
Acquisition of drug resistance constitutes a major challenge in cancer therapy1–13. Therapeutic agents (with widely varying biochemical mechanisms) often exhibit a common pattern of providing an initial reduction in tumor burden followed by recurrence of therapeutically resistant disease with more aggressive progression6,11,12,14. Tumor recurrence, which is a major obstacle for cancer cure, is primarily associated with the survival and growth of cell phenotypes that are resistant to chemotherapy15–17. Therefore, in order to develop new strategies for the effective treatment of human cancers, a quantitative understanding of the underlying processes leading to drug resistance is essential.
Cellular phenotypic heterogeneity is widely observed in many cancers3,5,8,18–20 as a tumor is often composed of multiple subpopulations21,22 that show different responses to chemotherapy9. In particular, cellular phenotypes that are not sensitive to drugs survive the treatment and can drive drug resistance. As the underlying processes that can lead to the emergence of resistant cells are often stochastic, tumors may locally contain varying numbers of resistant cells. Therefore, quantifying the statistics of drug resistant cells in a tumor is important for effective therapy. Specifically, we are interested in studying population heterogeneity at the start of therapy and aim to address an important issue of therapeutic importance, namely, how to quantify randomness in the numbers of resistant cells prior to drug treatment.
In analyzing population heterogeneity in tumors, a basic question that arises is: How are cell phenotypes that confer survival advantage in the presence of chemotherapeutic drugs generated? A common explanation for the emergence of such phenotypes revolves around Darwinian selection of pre-existing cellular heterogeneity that arises due to random genetic mutations23–25. However, the fact that resistant cells switch reversibly to sensitive cells, and that resistant cells often appear on short time intervals (hours to few days), starting from clonal populations, suggests that non-genetic factors play a major role in the generation of phenotypic heterogeneity5,7,13,26–31. Such non-genetic phenotypic heterogeneity can arise due to multistability in the underlying gene expression dynamics32,33 and noise in gene expression34–36. It is interesting to note that non-genetic factors are known to generate phenotypic variation and provide fitness benefits in diverse systems, e.g. in the evolution of microbial colonies under stress36. Similar advantages have been reported in the context of Saccharomyces cerevisiae wherein phenotypic transitions due to non-genetic factors are known to enhance fitness37. Furthermore, theoretical studies have shown how factors such as fluctuations in gene expression can lead to the long-term survival of drug-resistant populations38 and how stochastic switching between distinct cellular phenotypes under fluctuating environments can lead to optimal population growth39,40.
These observations suggest that there are two distinct, though not mutually exclusive, mechanisms for the onset of drug resistance in cancer cells: (1) cell phenotypes that are resistant to chemotherapy pre-exist in the tumor prior to treatment and are selected for during the treatment, and (2) cells are induced to develop or acquire resistance due to treatment. Thus, it is important to distinguish between selection of pre-existing populations which are inherently less chemosensitive versus adaptive changes in cancer cells that are activated by exposure to chemotherapy. In the cancer biology literature, the latter option is often referred to as adaptation41. This adaptation-selection scenario was first explored in the famous Luria-Delbrück experiments42 to understand the mechanism of bacterial resistance to bacteriophage infections. The corresponding analysis gave rise to the celebrated fluctuation test which is also used to estimate mutation rates in bacteria. It is important to note that, while in the Luria-Delbrück case phenotypic changes are driven by genetic mutations and thus an irreversible process, in our study, we are considering phenotypic changes that are reversible. Besides reversible phenotypic switching, it is important to consider intrinsic stochasticity in the underlying processes and to characterize cellular heterogeneity as highlighted by previous studies focusing on modeling drug resistance in cancer43,44.
In consideration of cellular mechanisms likely to be associated with drug resistance, the epithelial-mesenchymal transition (EMT) emerges as a logical candidate. EMT is a conserved cellular program that enables cells of epithelial lineage to transiently acquire traits of mesenchymal cells, including reversible loss of adherens junctions and gain of proteins associated with enhanced motility, adhesion to extracellular substrates and remodeling of the extracellular matrix45,46. In cancer cells, this ability to reversibly adopt a more motile phenotype has been linked to tumor invasion and metastasis47–49 but, more importantly for this study, EMT is also directly linked to chemotherapy resistance and cancer stem cell (CSC) properties50,51. The mechanisms through which cancer cells having undergone EMT become resistant to cancer therapeutics have been investigated in a number of studies and recently reviewed by Shibue and Weinberg52. EMT populations have been shown to be resistant to classical chemotherapy drugs via a combination of decreased apoptotic signal transduction, increased drug efflux (increased ATP binding cassette transporter expression), and decreased cell proliferation. Although other therapeutics are not explored in the present study it is also worth noting that EMT populations also exhibit resistance to molecular targeted and immunotherapy agents through other mechanisms which have also been studied. In the context of this background, experimental studies described herein focus on established markers of epithelial and mesenchymal phenotype in relation to chemotherapy response, which in this report involves pancreatic ductal adenocarcinoma (PDAC) cells. While recognizing that EMT is more likely a spectrum of intermediate states53–57, the strong correlation in phenotype and drug response reported here motivates the adoption of two coarse-grained states to be used in the model development. Specifically, a relatively drug-sensitive state with more pronounced epithelial characteristics (E); and a drug-resistant state with increased mesenchymal characteristics (M). In the following sections, we will consider these phenotypes to form the basis of a two-state model of the dynamics of phenotypic switching and associated survival of cancer cells under cytotoxic stress22,26.
The paper is organized as follows. We begin with a brief description and results from a set of motivating experiments, in which drug resistance is evaluated as a determinant of phenotype in pancreatic cancer cells in vitro, and conversely, phenotype as a determinant of drug response in the same cells. The two phenotype switching model inspired by the experimental data is then described. We then develop an analytic approach to quantify population heterogeneity at the start of therapy followed by a protocol for estimating phenotypic switching parameters. We then construct an approximate approach for characterizing the probability distribution of the fraction of resistant cells in a population. We conclude with remarks on possible extensions and future directions.
Results
Evaluation of drug resistance and phenotype in cell culture studies
We first sought to compare phenotypic traits in naive and drug-resistant pancreatic ductal adenocarcinoma (PDAC) cells. PANC1 cells (a quasimesnchymal PDAC cell line58) were exposed to increasing doses of oxaliplatin chemotherapy over successive passages until resistant cells were stable through multiple passages and cryopreservation. As shown in Fig. 1 (upper panels), acquisition of chemoresistance leads to a marked change in phenotype from naive cells displaying characteristically epithelial adherens junctions, to drug-resistant cells with highly branched morphology, no evident E-cadherin, and marked increase in cytoskeletal vimentin (IF quantification, upper right). This pattern of changes in E-cadherin and vimentin expression are classic and well-established markers of EMT. We further examined the reciprocal scenario, in which the same parental cells were directly induced45 to undergo EMT via administration of exogenous TGF-β (Fig. 1, lower panels). The resultant phenotype is strikingly similar to that of our drug-resistant cells and importantly, exhibits resistance to chemotherapy similar to when resistance was acquired directly through drug exposure. Collectively these results display a symmetry in that acquisition of drug resistance in epithelial cancer cells leads to increase in mesenchymal characteristics, while direct transition from epithelial to mesenchymal phenotype leads to drug resistance.
Figure 1.

Equivalence in the acquisition of chemotherapy resistance and epithelial-mesenchymal transition in pancreatic cancer cells.
Coarse-grained Model
Motivated by the preceding observations and by previous work22,26, we now consider a simple coarse-grained model (Fig. 2) for phenotypic heterogeneity in tumor cells. We consider that the population of cancer cells consists of two distinct subpopulations; drug-sensitive or drug-tolerant. Based on our experimental results, we denote the drug-sensitive population by E (for epithelial phenotype) and the drug-tolerant population by M (for mesenchymal phenotype). The processes that control the evolution of tumor heterogeneity are as follows: (1) birth: each E-type or M-type cell gives rise to birth of new cells of the same type with rates kE and kM, respectively; (2) death: each E-type (M-type) cell degrades with rates μE (μM); (3) phenotypic switching: an E cell can switch to a M cell with rate kEM, and M cell can switch back to E cell with rate kME. We assume that intrinsic rates of phenotypic switching and decay of each type of cancer cell are the same throughout the sample.
Figure 2.
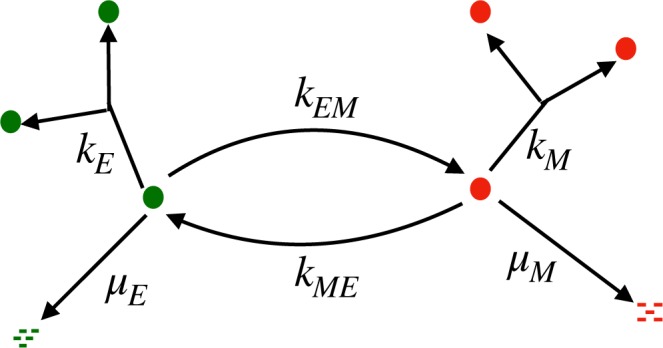
Schematic representation of the two phenotype EMT model of tumor growth: Sensitive and resistant phenotypes are shown as green and red circles respectively. The phenotypic switching rates are represented by KEM and KME, birth rates by kE and kM, and death rates by μE and μM.
At any time t, the state of the system is defined by the number of E and M cells. The temporal evolution of the corresponding probability distribution is given by the master equation:
| 1 |
where P(E, M, t) denotes the probability that there are E and M numbers of epithelial and messenchymal cells present at time t.
Within the framework of this model (Fig. 2), we now address a key issue: How to characterize the initial heterogeneity (i.e. prior to the start of drug exposure) in tumor cells. Consider a snapshot of the cell population in a tumor composed of distinct spatial regions. Each local neighborhood of the tumor can, in general, have widely different fractions of M cells in the local population; this is the heterogeneity we wish to quantify using our model. The proposed protocol for quantifying such heterogeneity involves drawing samples from different spatially distinct regions of the tumor, with each sample corresponding to a fixed number (N0) of cells, see Fig. 3. Let p0 = M/N0 denote the local fraction of M-cells in the sample population. Thus, the probability (p0) that a randomly chosen cell in the sample is a M cell can itself be considered to be a random variable. We denote the corresponding probability density function by ρ(p0) and characterize it by its mean 〈p0〉 and variance . Thus, initial tumor heterogeneity is characterized not just by the presence of drug-tolerant M cells in the sample but also by variations in the number of M cells from sample to sample, characterized by the distribution ρ(p0). Note that the sample size N0 should be large enough such that the sampling noise can be taken to be negligible. Thus the heterogeneity in the local fraction of M cells is reflective of the underlying biological heterogeneity rather than sampling noise.
Figure 3.

Schematic representation of tumor containing drug-sensitive (green circles) and drug-resistant cells (red circles) is shown at the center. For the sake of conceptual visualization, we have shown different samples taken from the tumor, each characterized by the same number of total cells (here N0 = 10) but different number of M-cells, and thus different values for p0 = M/N0.
Analytical results for parameter estimation
We now consider the stochastic process governing evolution of the tumor population upon treatment with drugs. Upon exposure to drugs, it is a reasonable assumption that growth is inhibited, so accordingly we set kE = kM = 0. As discussed in the preceding section, we consider the evolution of different sample populations, each of which has a fixed initial size N0 such that the fraction of M cells is drawn from a distribution ρ(p0). In this limit, the key parameters of the model are: . In what follows, we derive analytical results that can be used to estimate model parameters by analyzing the distribution of surviving cells upon drug exposure.
We note that, within our model, the evolution of each cell in the population is independent of the state of the remaining cells. Correspondingly, we first focus on the time evolution of a single tumor cell, which is initially either E-type or M-type with corresponding probabilities as 1 − p0 and p0. Let us denote by PE(PM) the probability that the cell is E(M)-type at time t, conditional on the initial probability p0 for it to be M-type. The corresponding probability generating function for the single cell, conditional on the value of p0 (g(z1, z2, t|p0) ), can be expressed as
| 2 |
It is straightforward to derive analytic expressions for PE(PM) and to thereby obtain an expression for g(z1, z2, t|p0) (Supplementary Material A). Now, let G(z1, z2, t) denote the probability generating function corresponding to P(E, M, t), the probability that we have E and M number of sensitive (E-type) and resistant (M-type) cells in the entire population at time t. Since each cell in the population (initial size N0) evolves independently, the probability generating function for the joint distribution at time t (averaging over the initial choice of p0) is given by
| 3 |
where ρ(p0) is the probability distribution function for the initial fraction of M-type cells (p0).
The expression derived for the generating function, Eq. (3), can be used to derive analytic expressions for all the moments of the marginal distributions corresponding to E-type and M-type cells at time t. For example, expressions for mean number of E and M cells can be obtained using , respectively (see Supplementary Material A). This leads to the following expression for mean number of surviving cells at time t, 〈N〉 = 〈E + M〉:
| 4 |
where
| 5 |
It is clear from the above expression that by fitting the curve corresponding to the mean number of surviving cells as a function of time, the three parameter combinations: α0, γ0, and μE − μEp0 + μMp0 can be determined.
To extract the remaining model parameters based on time-course data, we have to turn to analytic results for the higher moments. For example, we can use expressions for the Fano factor (F) associated with total number of surviving cells, which is given by with denoting the variance in the number of surviving cells. The expression for can be obtained using
| 6 |
where and are variances associated with the marginal distributions for the E and M cells respectively, and CEM = 〈EM〉 − 〈E〉〈M〉 is the correlation between numbers of E and M cells. We obtain an explicit expression for the Fano factor given by (see Supplementary Material A):
| 7 |
The Fano factor F is a measure of deviations from the Poisson distribution, for which F = 1. If F < 1 or F > 1, the distribution is sub-Poissonian or super-Poissonian, respectively. Before turning our attention to approaches for parameter estimation, let us first examine the expression derived for the Fano factor. We note that in the absence of initial variability in the fraction of M cells (i.e. ), the Fano factor of surviving population is simply given by F = 1 − 〈N〉/N0. As the cells are not dividing due to the exposure to drugs, the mean number of surviving cells (〈N〉) is less than the initial population of cells (N0) for t > 0. Thus, in this case, the Fano factor is always less than one and the distribution of cell population follows a sub-Poissonian distribution. However, given variability in the fraction of M-cells in the initial population, the Fano factor can potentially exceed one making the distribution super-Poissonian. This result implies that the observation of a Fano factor in excess of 1 in the distribution of surviving cells is an indicator of variance in the fraction of M-cells in the initial population. Thus the measurements of the moments of surviving cell populations can provide evidence for phenotypic heterogeneity in tumor populations prior to drug treatment.
To gain more quantitative insight into the initial heterogeneity, we need to estimate the parameters characterizing the mean and variance of ρ(p0). Let us rewrite Eq. (7) in a more compact form by regrouping terms in the expression to yield the following form
| 8 |
where the quantity can now be expressed entirely in terms of experimentally measurable quantities such as F and 〈N〉,
| 9 |
Using the expressions for the mean and Fano factor of the surviving population as functions of time, Eqs (4) and (8), we can estimate four of the parameter combinations, namely, α0, γ0, μE − μEp0 + μMp0, and . Correspondingly, we need additional experiments to determine the entire set of 6 model parameters. As we now show, a set of measurements that accomplish this can be obtained by starting from different initial conditions.
The proposed protocol is motivated by that fact that experimental techniques such as fluorescence-activated cell sorting (FACS) can be used to prepare the samples in specified initial states. With this in mind, we begin from an initial condition where all cells are E-type i.e. . Using the derived results, we can determine the parameters μE, α0 and γ0. We next consider the initial condition to be all M-type cells i.e. . Analysis of the corresponding time-course measurements of the number of surviving cells can now be used to estimate the parameter μM. Having estimated values of α0, γ0, μE, and μM, we can now find the switching rates, kEM and kME, from Eq. (5). Finally, using the expressions for the mean number of surviving cells and corresponding Fano factors for arbitrary p0, we can get explicit expressions for the probability p0 and variance (Supplementary Material A):
| 10 |
The above results are expressed in terms of experimentally measurable quantities, involving mean values 〈N〉0 (for p0 = 0) and 〈N〉1 (for p0 = 1) and the Fano-factor of the total surviving population, and thus can be used to estimate the population heterogeneity at the start of drug exposure based on time-course measurements of the surviving population size.
Modeling generation of tumor heterogeneity
The analysis in the preceding section holds regardless of the source of initial heterogeneity in tumor populations. In this section, we explore how the model introduced for tumor cell dynamics can also be used to analyze a potential mechanism for generation of tumor heterogeneity. We note that the proposed model in Fig. 2 can be seen as a generalized version of the celebrated Luria-Delbrück (LD) model with the important addition that, in the present case, the transition between the two phenotypes is reversible (as opposed to the Luria-Delbrück case). However, while the LD model can be solved exactly59, the exact analytical solution of the reversible model in Fig. 2 is not known, to the best of our knowledge. Nevertheless, as we show below, exact expressions for the mean and variance of the number of E-type and M-type cells can be obtained and used to characterize heterogeneity in tumor cell populations. We can use the master equation, Eq. (1), to derive expressions for the mean number of E and M cells at any time t (Supplementary Material B). Using these expressions, the mean number of surviving cells 〈N〉 = 〈E〉 + 〈M〉 is given by:
| 11 |
where
| 12 |
with and representing the effective birth rates for E-type and M-type cells respectively, while E0 and M0 are the initial numbers of E and M cells at t = 0.
The results show that the mean number of surviving cells at any time t is characterized by six parameters: initial number of E and M cells (E0, M0), two effective birth rates (, ) and two switching rates (kEM, kME). Given that the initial population can be chosen in a controlled manner, we can use the results for the mean population size to determine some of the model parameters. Specifically, we can set M0 = 0 as the initial condition and fitting the data to obtain the coefficient of exponential terms in Eq. (11) will yield . Next, we can set E0 = 0 and Eq. (11) will allow us to extract . Once we estimate and , we can extract the switching rates using the estimated values of γ and α, using Eq. (12). That is, the proposed procedure allows us to estimate the parameter combinations, and as well as the parameters kEM and kME.
In order to estimate the remaining model parameters, we need to consider the higher moments. While obtaining analytical expressions for the full probability distribution is still an open problem, higher moments can be calculated in a straightforward manner. For example, using Eq. (1) the evolution equation for , and is given by
| 13 |
The above set of equations can be solved to get explicit expressions for 〈E2〉, 〈M2〉 and 〈EM〉 at any time t (Supplementary Material B), which can be used to get the variance () in the total number of surviving cells by using Eq. (6). The analytic expression for the variance, in combination with the expression for mean number of surviving cells, can be used to extract all model parameters. Furthermore, the extracted parameters can then be compared with the parameters derived based on tumor cell dynamics after exposure to drugs. The comparisons can provide insight into the relative roles of adaptation and selection in driving tumor heterogeneity. The scenario wherein the switching parameters kEM and kME are effectively unchanged upon exposure to drugs, whereas μE and μM increase favors selection as the dominant driver of tumor heterogeneity. However, significant changes in the switching rates kEM and kME are indicative of a role for adaptation as well in the generation of tumor heterogeneity.
Characterizing the distribution of the fraction of resistant cells
The results derived for the moments can also be used to characterize the probability distribution ρ(p0) for the fraction of M-type cells. Recall that we must have 0 ≤ p0 ≤ 1 and furthermore the first two moments of ρ(p0) can be obtained using the procedure outlined in the previous section. Thus a natural choice to characterize the distribution ρ(p0) is to take it to be the Beta distribution with the mean and variance as determined by measurements. The Beta distribution is a natural choice because the domain of a Beta distribution can be viewed as a probability. Another advantage of using a Beta distribution in describing the distribution (or likelihood) of a probability value is that it is a conjugate prior to binomial distribution. This means that, in carrying out Bayesian inference, if the likelihood function is binomial, then a Beta distribution prior will lead to a posterior that is also a Beta distribution (with renormalized parameters). Such a distribution is expressed in terms of two exponents (α and β) as
| 14 |
with mean The two parameters of the Beta distribution can be estimated using the experimentally determined mean and variance. Explicitly, these are given by
| 15 |
To test this approach for characterizing the initial heterogeneity, we compare the Beta distribution with the results obtained from stochastic simulations of the model. Specifically, we carried out stochastic simulations using the Gillespie algorithm60 for the model in Fig. 2 starting with 200 sensitive E-type cells and no resistant M-type cells. The empirically determined distribution for the fraction of M-type cells (ρ(p0)) is then compared to the Beta distribution with the same mean and variance as the empirical distribution. The results obtained are shown in Fig. 4, which indicate that the Beta distribution is an excellent approximation for the range of parameters explored.
Figure 4.

Simulation results for the distributions of fraction of M-cells ρ(p0) are shown as histograms and the continuous solid lines represent fits by Beta distributions. Top (a), middle (b) and lower (c) panels corresponds to kEM/kME = 0.1, 1 and 10 respectively. In each panel, distributions are shown at various time points, t = 0.1, 1, 10, 20 from left to right. Other parameters are: kE = 0.2, kM = 0.1, μE = 0.3, μM = 0.15.
Discussion
To summarize, we have studied a coarse-grained stochastic model to quantify phenotypic heterogeneity in a population of cancer cells. Motivated by the experimental observation that both chemoresistance and TGF-β induced EMT lead to similar outcomes, the model assumes that a cell has two phenotypes corresponding to whether it is drug-sensitive or drug-resistant. Importantly, the model is also consistent with epigenetic mechanisms for generating phenotypic heterogeneity in cancer, given that it allows reversible phenotypic switching between sensitive and resistant cells.
For the model considered, we have derived analytic results, both in the presence and absence of chemotherapeutic agents, which provide insights into the role of phenotypic switching in generating population heterogeneity. One of the issues that we address through these results focuses on quantifying initial heterogeneity in the local fraction of resistant cells. This heterogeneity is characterized by mean fraction of resistant cells 〈p0〉 in a sample and its variance . We propose a protocol that can be used to estimate the model parameters based on measurements of mean and variance of the surviving population of tumor cells. Furthermore, our analysis also leads to a condition, in terms of experimentally measurable quantities, whose value serves as an indicator for the presence of initial heterogeneity in the fraction of resistant cells.
While the proposed method allows us to estimate the mean and variance of the fraction of resistant cells prior to therapy, obtaining an exact analytical form for the entire distribution appears to be challenging. However, our simulation results suggest that this distribution is well approximated by the Beta distribution, which can be characterized by using the mean and variance of the surviving population. Besides characterizing initial heterogeneity in the cancer cell population, the estimated model parameters can also be useful in analyzing the complex roles of adaptation and selection in the acquisition of chemoresistance. Furthermore, the results obtained provide exact analytical expressions characterizing the distribution of of tumor cell population under treatment by drugs. The simple model considered in this work can serve as a building block for studying models with more explicit spatial dependence. Going forward we envision further model development in dialog with experiments that longitudinally monitor phenotypic changes in time lapse microscopy studies, either by quantitative analysis of morphometric parameters or implementing fluorescent reporters of EMT which have been recently developed61. These results can serve as important inputs to future work focusing on evaluation of the hypothesis that model-informed design of treatment schedule and dose parameters may reduce the emergence of chemoresistance.
Methods
Cell culture and reagents
PANC1 cells were obtained from the American Type Culture Collection (Manassas, VA), and grown in T-75 cell culture flasks according to ATCC guidelines. DMEM medium (HyClone; Logan, UT) was supplemented with 10% FBS (HyClone; Logan, UT), 100 IU/mL penicillin and 1% streptomycin (HyClone; Logan, UT), and 0.5 ug/mL Amphotericin B (Corning; Corning, NY). The drug-resistant subline, PANC1-OR was generated as described previously62. Briefly, increasing concentrations of oxaliplatin were added to each cell type in regular media over successive passages until a stable proliferative phenotype without chemotherapy was observed and maintained following cryopreservation and confirmed by comparative dose response and measurement of a statistically significant increase in IC50.
Immunofluorescence sample preparation and imaging
Formaldehyde-fixed cells in optical-bottom multiwell plates were incubated overnight at 4 °C with primary antibodies against e-cadherin and vimentin (Cell Signaling EMT Duplex; Danvers, MA). After washing with PBS, cells were incubated for 1 hour with mouse or rabbit Alexa Fluor secondary antibodies (Cell Signaling; Danvers, MA). Cells were mounted with ProLong Gold Antifade reagent containing DAPI (ThermoFisher Scientific Molecular Probes; Waltham, MA) and imaged after 24 hours using a Zeiss LSM 880 confocal microscope with the same detector settings and excitation laser power settings across groups. Images were analyzed using custom Matlab scripts where fluorescent signal for each protein was normalized to the number of cells based on DAPI-stained nuclei.
Therapeutic response assessment
In sample wells receiving chemotherapy treatment, oxaliplatin (Selleck Chemical; Houston, TX) was added to the media at doses ranging from 0.1 to 500 μM for 48 hours. In experiments where EMT was induced via TGF-beta, 10 ng/mL human recombinant TGF-beta (Gibco, Thermo Fisher Scientific) in 1% FBS DMEM was added to designated wells for 48 hours and respective comparison groups were also grown in 1% FBS for the same duration. In all therapeutic studies treatment conditions were prepared in at least triplicate within each batch including internal controls with sham manipulations. Therapeutic response was assessed via the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega; Madison, WI) at 490 nm absorbance in a BioTek® Epoch Microplate Spectrophotometer.
Supplementary information
Supplementary Material for: Stochastic modeling of phenotypic switching and chemoresistance in cancer cell populations
Acknowledgements
The authors gratefully acknowledge funding support from the NIH through grants 3U54CA156734-05S3 (as part of the UMass Boston/Dana Farber-Harvard Cancer Center U54 partnership) and from the National Cancer Institute R00CA155045 (PI: JPC), and a Sanofi Genzyme doctoral fellowship which supported Gwendolyn Cramer. We would also like to acknowledge funding support from the UMass Boston Healey award.
Author Contributions
N.K., B.S., J.C. and R.V.K. designed research; N.K., S.Z.D. and R.K. carried out theoretical/computational aspects of research, G.C. carried out experimental aspects of research; N.K., B.S., J.C. and R.V.K. wrote the paper and all authors reviewed the paper.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-46926-x.
References
- 1.Housman G, et al. Drug resistance in cancer: an overview. Cancers. 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottesman MM. Mechanisms of cancer drug resistance. Annu. review medicine. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 3.Chisholm RH, Lorenzi T, Clairambault J. Cell population heterogeneity and evolution towards drug resistance in cancer: Biological and mathematical assessment, theoretical treatment optimisation. Biochimica et Biophys. Acta (BBA)-General Subj. 2016;1860:2627–2645. doi: 10.1016/j.bbagen.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Bozic I, Nowak MA. Resisting resistance. Annu. Rev. Cancer Biol. 2017;1:203–221. doi: 10.1146/annurev-cancerbio-042716-094839. [DOI] [Google Scholar]
- 5.Pogrebniak, K. L. & Curtis, C. N. Harnessing tumor evolution to circumvent resistance. Trends Genet. (2018). [DOI] [PMC free article] [PubMed]
- 6.Nikolaou, M., Pavlopoulou, A., Georgakilas, A. G. & Kyrodimos, E. The challenge of drug resistance in cancer treatment: a current overview. Clin. & Exp. Metastasis 1–10 (2018). [DOI] [PubMed]
- 7.Salgia, R. & Kulkarni, P. The genetic/non-genetic duality of drug ‘resistance’ in cancer. Trends cancer (2018). [DOI] [PMC free article] [PubMed]
- 8.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat. reviews Clin. oncology. 2018;15:81. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 9.Zhou B-BS, et al. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat. reviews Drug discovery. 2009;8:806. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 10.Zahreddine H, Borden K. Mechanisms and insights into drug resistance in cancer. Front. pharmacology. 2013;4:28. doi: 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013;13:714. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 12.Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer discovery. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 13.Shaffer SM, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nat. 2017;546:431. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chabner BA, Roberts TG. Chemotherapy and the war on cancer. Nat. Rev. Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 15.Gatenby R, Brown J. The evolution and ecology of resistance in cancer therapy. Cold Spring Harb. perspectives medicine. 2018;8:a033415. doi: 10.1101/cshperspect.a033415. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Gallaher, J., Enriquez-Navas, P. M., Luddy, K. A., Gatenby, R. A. & Anderson, A. R. Spatial heterogeneity and evolutionary dynamics modulate time to recurrence in continuous and adaptive cancer therapies. Cancer research canres–2649 (2018). [DOI] [PMC free article] [PubMed]
- 17.Castorina P, Carcò D, Guiot C, Deisboeck TS. Tumor growth instability and its implications for chemotherapy. Cancer research. 2009;69:8507–8515. doi: 10.1158/0008-5472.CAN-09-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer. 2003;3:895. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 19.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nat. 2013;501:328. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer. 2012;12:323. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 21.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat. medicine. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 22.Zhou JX, Pisco AO, Qian H, Huang S. Nonequilibrium population dynamics of phenotype conversion of cancer cells. PloS one. 2014;9:e110714. doi: 10.1371/journal.pone.0110714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowell PC. The clonal evolution of tumor cell populations. Sci. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 24.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nat. 2013;501:338. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 26.Pisco AO, et al. Non-darwinian dynamics in therapy-induced cancer drug resistance. Nat. communications. 2013;4:2467. doi: 10.1038/ncomms3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘what does not kill me strengthens me’. Br. journal cancer. 2015;112:1725. doi: 10.1038/bjc.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer. 2014;14:747. doi: 10.1038/nrc3819. [DOI] [PubMed] [Google Scholar]
- 29.Su, Y. et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. 201712064 (2017). [DOI] [PMC free article] [PubMed]
- 30.Inde Z, Dixon SJ. The impact of non-genetic heterogeneity on cancer cell death. Critical reviews biochemistry molecular biology. 2018;53:99–114. doi: 10.1080/10409238.2017.1412395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, et al. Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc. Natl. Acad. Sci. 2014;111:E364–E373. doi: 10.1073/pnas.1304840111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nat. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang S, Eichler G, Bar-Yam Y, Ingber DE. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. review letters. 2005;94:128701. doi: 10.1103/PhysRevLett.94.128701. [DOI] [PubMed] [Google Scholar]
- 34.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 35.Zhuravel D, et al. Phenotypic impact of regulatory noise in cellular stress-response pathways. Syst. synthetic biology. 2010;4:105–116. doi: 10.1007/s11693-010-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fraser D, Kaern M. A chance at survival: gene expression noise and phenotypic diversification strategies. Mol. microbiology. 2009;71:1333–1340. doi: 10.1111/j.1365-2958.2009.06605.x. [DOI] [PubMed] [Google Scholar]
- 37.Acar M, Mettetal JT, Van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nat. genetics. 2008;40:471. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 38.Charlebois DA, Abdennur N, Kaern M. Gene expression noise facilitates adaptation and drug resistance independently of mutation. Phys. review letters. 2011;107:218101. doi: 10.1103/PhysRevLett.107.218101. [DOI] [PubMed] [Google Scholar]
- 39.Belete MK, Balázsi G. Optimality and adaptation of phenotypically switching cells in fluctuating environments. Phys. Rev. E. 2015;92:062716. doi: 10.1103/PhysRevE.92.062716. [DOI] [PubMed] [Google Scholar]
- 40.Kussell E, Leibler S. Phenotypic diversity, population growth, and information in fluctuating environments. Sci. 2005;309:2075–2078. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- 41.Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: the origins of metastatic behavior. Cancer research. 2007;67:11476–11480. doi: 10.1158/0008-5472.CAN-07-1653. [DOI] [PubMed] [Google Scholar]
- 42.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genet. 1943;28:491. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kessler DA, Austin RH, Levine H. Resistance to chemotherapy: patient variability and cellular heterogeneity. Cancer research. 2014;74:4663–4670. doi: 10.1158/0008-5472.CAN-14-0118. [DOI] [PubMed] [Google Scholar]
- 44.Komarova N. Stochastic modeling of drug resistance in cancer. J. theoretical biology. 2006;239:351–366. doi: 10.1016/j.jtbi.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The J. clinical investigation. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. reviews Mol. cell biology. 2014;15:178. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heerboth S, et al. Emt and tumor metastasis. Clin. translational medicine. 2015;4:6. doi: 10.1186/s40169-015-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhang, Y. & Weinberg, R. A. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. medicine 1–13 (2018). [DOI] [PMC free article] [PubMed]
- 50.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Singh A, Settleman J. Emt, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibue T, Weinberg RA. Emt, cscs, and drug resistance: the mechanistic link and clinical implications. Nat. reviews Clin. oncology. 2017;14:611. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu, M., Jolly, M. K., Levine, H., Onuchic, J. N. & Ben-Jacob, E. Microrna-based regulation of epithelial–hybrid–mesenchymal fate determination. Proc. Natl. Acad. Sci. 201318192 (2013). [DOI] [PMC free article] [PubMed]
- 54.Jolly MK, et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. oncology. 2015;5:155. doi: 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jolly MK, et al. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016;7:27067. doi: 10.18632/oncotarget.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong T, et al. An ovol2-zeb1 mutual inhibitory circuit governs bidirectional and multi-step transition between epithelial and mesenchymal states. PLoS computational biology. 2015;11:e1004569. doi: 10.1371/journal.pcbi.1004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li C, Balazsi G. A landscape view on the interplay between emt and cancer metastasis. NPJ systems biology applications. 2018;4:34. doi: 10.1038/s41540-018-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collisson EA, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. medicine. 2011;17:500. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Antal T, Krapivsky P. Exact solution of a two-type branching process: models of tumor progression. J. Stat. Mech. Theory Exp. 2011;2011:P08018. doi: 10.1088/1742-5468/2011/08/P08018. [DOI] [Google Scholar]
- 60.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. The journal physical chemistry. 1977;81:2340–2361. doi: 10.1021/j100540a008. [DOI] [Google Scholar]
- 61.Toneff M, et al. The z-cad dual fluorescent sensor detects dynamic changes between the epithelial and mesenchymal cellular states. BMC biology. 2016;14:47. doi: 10.1186/s12915-016-0269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cramer GM, Jones DP, El-Hamidi H, Celli JP. Ecm composition and rheology regulate growth, motility, and response to photodynamic therapy in 3d models of pancreatic ductal adenocarcinoma. Mol. Cancer Res. 2017;15:15–25. doi: 10.1158/1541-7786.MCR-16-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material for: Stochastic modeling of phenotypic switching and chemoresistance in cancer cell populations