

Abstract
Head and neck squamous cell carcinoma (HNSCC) is a debilitating and deadly disease with a high prevalence of recurrence and treatment failure. To develop better therapeutic strategies, understanding tumor microenvironmental factors that contribute to the treatment resistance is important. A major impediment to understanding disease mechanisms and improving therapy has been a lack of murine cell lines that resemble the aggressive and metastatic nature of human HNSCCs. Furthermore, a majority of murine models employ subcutaneous implantations of tumors which lack important physiological features of the head and neck region, including high vascular density, extensive lymphatic vasculature, and resident mucosal flora. The purpose of this study is to develop and characterize an orthotopic model of HNSCC. We employ two genetically distinct murine cell lines and established tumors in the buccal mucosa of mice. We optimize collagenase-based tumor digestion methods for the optimal recovery of single cells from established tumors. The data presented here show that mice develop highly vascularized tumors that metastasize to regional lymph nodes. Single-cell multiparametric mass cytometry analysis shows the presence of diverse immune populations with myeloid cells representing the majority of all immune cells. The model proposed in this study has applications in cancer biology, tumor immunology, and preclinical development of novel therapeutics. The resemblance of the orthotopic model to clinical features of human disease will provide a tool for enhanced translation and improved patient outcomes.
Keywords: Cancer Research, Issue 146, Head and neck cancer, orthotopic model, tumor microenvironment, immune evasion, flow cytometry, mass cytometry
Introduction
HNSCC is the fifth most common malignancy globally, with over 600,000 patients diagnosed annually1. Despite aggressive treatment involving chemotherapy and radiotherapy (RT), the overall survival (OS) rate for HNSCC patients without human papillomavirus (HPV) infection remains below 50% after 5 years2. This is largely attributed to a highly complex tumor microenvironment as tumors can originate from several distinct anatomical sites within the head and neck region, including the buccal mucosa, tongue, floor of the mouth, nasal cavity, oral cavity, pharynx, oropharynx, and hypopharynx. In addition, the head and neck region is highly vascularized and contains nearly half of all lymph nodes in the body3. A majority of studies investigating head and neck tumor biology rely on tumor models in the flank region. Such models can offer insight into tumor-intrinsic mechanisms, but the lack of the native head and neck microenvironment can significantly impact the translational potential of such findings. One method that has been used to induce oral tumors is through exposure to the carcinogen 9,10-dimethyl-1,2-benzanthracene (DMBA)4. However, this method is associated with a lengthy process, induces tumors in rats and hamsters but not in mice, and the resulting tumors do not possess many of the histological features of differentiated SCCs5,6. The introduction of the carcinogen4-nitroquinoline 1-oxide (4- NQO), a water-soluble quinolone derivative, resulted in mouse oral tumors when applied orally but also suffered from long exposure times (16 weeks) and a limited take rate within and between batches of mice7,8,9. In order to develop clinically relevant models, several groups utilized genetically engineered models involving the manipulation of driver oncogenes or tumor suppressor genes, including TP53, TGFB, KRAS, HRAS, and SMAD410. These models can offer insight into tumors with known driver genes but do not recapitulate the complex heterogeneity of human HNSCCs.
In this work, we demonstrate the feasibility of performing an intramucosal inoculation of squamous cell carcinoma cells in mice. Inoculated cells develop into aggressive tumors within 1 week of injection. Similar to human HNSCCs, the tumors metastasize to the regional lymph nodes. We characterize histological and clinical features of the disease and provide insight into the tumor immune microenvironment. We propose that this orthotopic model of HNSCC has applications in cancer biology, tumor immunology, and preclinical studies. Mechanisms of immune evasion, tumor progression, treatment resistance, and metastases represent areas of clinical significance that can be addressed using the proposed model.
Protocol
All animal procedures were performed in accordance with an approved institutional animal care and use committee (IACUC) protocol of the University of Colorado Denver (protocol # 00250).
1. Tumor Cell Culture
NOTE: B4B8 and LY2 cell lines were used to generate orthotopic HNSCC tumors: B4B8 tumor cells were derived from carcinogen-transformed mucosal keratinocytes (from BALB/C mice)11. LY2 tumor cells were derived from lymph node metastases of a spontaneously transformed BALB/ C keratinocyte line (Pam 212)12,13. Both cell lines were kindly provided by Dr. Nadarajah Vigneswaran (UTHealth, Houston, TX, USA).
Use Dulbecco’s modified Eagle’s medium (DMEM) F/12 supplemented with 10% fetal bovine serum (FBS) and 1% antimicrobial reagent for the cell line maintenance. Maintain these cell lines in a sterile incubator at 37 °C and 5% CO2. Cells should be used for inoculation before exceeding 15 passages.
-
Plate 2 × 106 to 4 × 106 cells in 175 cm2 cell culture flasks.
NOTE: Ensure the cells are less than 90% confluent to avoid inducing a stress response.
When the cells are 70% confluent (~48 h), remove the flask from the incubator and wash the cells 3× with cold phosphate-buffered saline (PBS).
- Detach the cells from the flask using 0.25% trypsin, enough to cover the surface of the plate.
- To do this, dispense 4 mL of trypsin in a 175 cm2 flask containing 70% confluent cells. Incubate the cells with trypsin for 3–4 min in the cell culture incubator at 37 °C and 5% co2.
- Visualize the cells under a microscope to ensure cell detachment.
- Add 12 mL of DMEM F/12 media containing FBS to neutralize trypsin activity.
Aspirate the cell suspension and place it into a 50 mL conical tube.
Shake the tube containing the cells by inverting it 3x–4x.
Optionally, mix a 10 μL aliquot of cell suspension with 10 μL of Trypan blue in a microcentrifuge tube and count the cells using a hemocytometer. Determine cell viability by subtracting the number of Trypan-blue-positive cells from the total number of cells and divide by the total number of cells.
Centrifuge the cell suspension at 300 × g for 5 min at 4 °C.
-
Resuspend the cells in serum-free and antibiotic-free DMEM at an appropriate volume so that 1 × 106 cells are present in 50 μL of media.
NOTE: Based on the in vivo cell line aggressiveness (determined empirically), 1 × 106 cells per injection site per mouse was deemed appropriate for B4B8 and LY2 cells.
Place the vial containing the cell suspension on ice.
-
Place the pre-thawed basement membrane matrix on ice.
NOTE: The basement membrane matrix was thawed overnight at 4 °C.
2. Cell Injection into Mice
Prepare a 1:1 mixture of cells:basement membrane matrix (50 μL each).
Add the cells first; then, gradually pipette the basement membrane matrix. Avoid introducing air bubbles. Ensure that the mixture is made immediately before the animal injection. Adding cells to basement membrane matrix for an extended period of time can result in cell settling in the matrix mixture, which makes the mixture difficult to shake rigorously. This will cause a considerable variability in tumor size between mice.
Mix gently. Ensure all steps involving matrix are performed on ice. Basement membrane matrix will polymerize at room temperature.
Prepare syringes for inoculation.
Load 0.5 mL insulin syringes (23 G) with 100 μL of the cell/basement membrane matrix solution.
Keep the syringes on ice to avoid basement membrane matrix polymerization.
Anesthetize mice by placing them in a chamber with isoflurane and oxygen (2.5%).
Ensure the mice are deeply anesthetized before performing the injection (by ensuring a lack of response to a toe pinch).
Insert the needle into the right or left buccal region. This is performed through the available open space on either side of the mouth.
-
Ensure that the mouse’s tongue is not in the way.
NOTE: It is easy to poke the tongue, which will result in tongue tumors. Move the tongue to the opposite side if necessary.
Keep the syringe parallel to the buccal region while inside the oral cavity.
When ready to inject, pull the syringe back and slowly insert the syringe at a 10° angle.
Inject 100 μL of the cell/basement membrane matrix suspension over a period of 5 s.
-
Hold the syringe in place for an additional 5 s to ensure all material is injected.
NOTE: For control nontumor-bearing mice, inject a mixture of serum-free media and matrix (as described above) without the tumor cells.
Withdraw the syringe gently.
Continue the above procedure with the remaining mice.
Allow for 1 week until tumors begin to appear grossly (50–200 mm3 for B4B8 and LY2 cells).
3. Mouse Monitoring
Continue to perform regular caliper measurements for tumor volume (1x–2x per week at regular intervals).
Measure animal weight to assess the effects of tumor growth on feeding.
4. Harvesting Tumors
-
When the experimental endpoint is reached, euthanize the animals using appropriate measures (e.g., CO2 asphyxiation, decapitation, or cervical dislocation).
NOTE: In this study, the endpoint was reached if the mice became moribund (with a weight loss of >15% of their initial weight, a lack of grooming, cachexia) and/or if the tumor size reached 1,000 mm3.
- Begin dissecting the animal by creating a long incision through the midline in the neck region.
- Use blunt forceps to grab the skin and sharp scissors to cut the skin.
Insert scissors gently under the skin covering the tumor and create air pockets by pushing the scissors across and into the skin.
-
Once the skin is sufficiently detached from the tumor, identify the draining lymph nodes (DLNs) and excise them to avoid having the tumor tissue confounded by the presence of lymph nodes.
NOTE: Large tumors may reach and/or cover the DLN. Depending on the type of assay being performed, the isolation of intact lymph nodes is essential to avoid spillage of immune cells into the tumor, which will result in skewing the assay results.
Cut through the borders of the tumor until the entire volume is detached.
5. Tumor Processing for Downstream Applications
-
For the downstream histologic examination, place tumors in 10% formalin at room temperature. To avoid overfixation, replace the formalin with 70% ethanol. For flow cytometry analysis, process the tumors as described below.
NOTE: The tissue can be kept in formalin for 72 h before overfixation can become problematic for certain types of staining.
Cut the tumor into 1–2 mm-sized pieces using a sterile razor blade or sharp scissors.
Place the cut tumor pieces in a 50 mL conical tube with collagenase III (4,250 units per sample), DNase I (0.1 mg per sample), and trypsin inhibitor (1 mg per sample).
-
Incubate at 37 °C for 30 min with shaking every 10 min.
NOTE: The samples can be placed in a resealable bag, sterilized with 90% ethanol, and placed in a cell culture incubator.
-
After 30 min, add 20 mL of Hank’s balanced salt solution (HBSS) and spin at 300 × g for 5 min.
NOTE: HBSS is comprised of potassium chloride, sodium chloride, sodium bicarbonate, sodium phosphate dibasic, sodium phosphate monobasic and glucose.
-
Discard the supernatant and resuspend the pellet in 2–3 mL of red blood cell (RBC) lysis buffer. Pipette rigorously.
NOTE:RBC lysis buffer is comprised of ammonium chloride, sodium bicarbonate, and disodium.
-
Incubate for 2 min at room temperature.
NOTE: Longer incubation can be toxic to other cell populations.
Add 20 mL of HBSS to neutralize the effect of lysis buffer.
Centrifuge at 300 × g for 5 min and discard the supernatant.
Resuspend the pellet in 10 mL of HBSS.
Pass the solution through a 70 μm nylon restrainer and centrifuge at 300 × g for 5 min. Discard the supernatant.
Repeat steps 5.8–5.10 and pass the cell suspension through a 40 μm nylon restrainer to ensure any additional debris is removed from the suspension.
6. Cell Staining and Data Acquisition
Resuspend the cell pellet in 1 mL of HBSS.
Count the cells using a hemocytometer or an automated cell counter, as described in step 1.7
-
Determine the total cell number and appropriate concentration for staining.
NOTE: The ideal cell concentration for flow cytometry staining is 1–2 million cells. For example, if staining is performed in a 96-well plate and there are 10 million cells, resuspend the sample in 1 mL and plate 100 μL for 1 million cells per well.
- Add Fc block (CD16/CD32) at a concentration of 1:100 in order to prevent nonantigen-specific binding of immunoglobulins to the FCΥIII and FCΥII receptors. Incubate for 5 min at room temperature.
- Centrifuge and resuspend the cell pellet in 100 μL of flow cytometry cell staining buffer (comprised of saline solution containing 5% FBS, 2% EDTA, and 1% HEPES).
- Perform cell surface staining according to supplier-provided instructions (adding each antibody at the appropriate dilution). Incubate for 60 min at room temperature.
- Centrifuge and resuspend the cell pellet in 100 μL of HBSS. Repeat 2× to get rid of excess antibody.
- Run samples on an appropriate flow cytometer.
- If analyzing intracellular markers (e.g., foxp3), perform cell permeabilization and staining according to the supplier’s instructions.
Representative Results
The in vitro assessment of LY2 and B4B8 cell proliferation showed that both cell lines have similar doubling times (21 h and 23 h, respectively). In vivo, both cell lines formed a single, visible, and palpable mass within 1 week of inoculation (Figure 1A). In mice bearing LY2 tumors, the jaw was displaced by 3 weeks due to tumor burden (Figure 1A). Control mice that did not receive tumor cells did not develop tumors as anticipated. LY2 tumors grew at a higher rate compared to B4B8 tumors. The average tumor volume on day 21 in LY2 tumor-bearing mice was 632 ± 10 mm3 compared to 162 ± 4 mm3 in B4B8 tumors (Figure 1B). Mice exhibiting jaw displacement rapidly developed weight loss due to dysphagia (Figure 1C). Mice exhibiting significant weight loss defined as >15% of their initial weight and/or having a tumor volume of >1 cm3 were euthanized within 1 day of the noted observation. The median survival in LY2 mice was 22.5 days, compared to 52.0 days in B4B8 tumor-bearing mice (Figure 1D).
Figure 1: Monitoring mice bearing orthotopic HNSCC tumors.
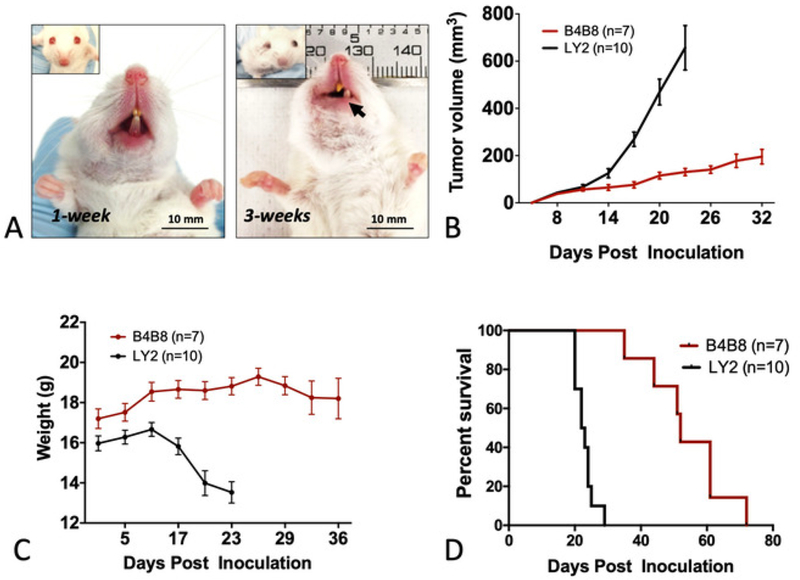
(A) Representative images of tumor-bearing mice. The inoculated buccal develops a tumor within 1 week of inoculation. At 3 weeks, displacement of the jaw is observed. (B) Tumor growth rate in B4B8 and LY2 tumor- bearing mice as determined by caliper measurements. (C) Changes in the body weight of mice bearing B4B8 or LY2 tumors. (D) Survival of B4B8 and LY2 tumor-bearing mice. The bars represent the standard error of the mean (SEM) of 7–10 mice per group.
Magnetic resonance imaging (MRI) of tumor-bearing mice showed well-demarcated tumors extending into the inner layer of the buccal mucosa (Figure 2A). Tumors did not invade into the tongue or other nearby organs (esophagus, bronchus, thymus) as shown with histological assessment. Signal heterogeneity on MR images was representative of the presence of vascularized hyperintense regions (denoted with V) and necrotic hypointense regions (denoted with N) (Figure 2A). A gross pathological examination showed enlarged DLNs (Figure 2B). We further assessed tumor volume with computed tomography (CT) to determine the reliability of the caliper measurements. Tumors were delineated using a digital imaging and communications in medicine (DICOM) image analysis software16. Assessment of LY2 tumor volume by calipers and CT imaging showed an excellent correlation between the two methods (R2 = 0.8493; Figure 2C). This indicates that tumor growth is primarily exophytic and caliper measurement is a reliable method for the assessment of tumor growth. Histologic examination showed that all LY2 tumor-bearing mice developed poorly differentiated squamous cell carcinoma (Figure 2D). Nine out of nine LY2 tumor-bearing mice developed metastases to the first and second echelon lymph nodes. Nodal metastases were primarily subcapsular (with seven out of nine mice) and within sinuses (with five out of nine mice), with two out of nine mice demonstrating intracapsular invasion. In contrast, mice bearing B4B8 tumors developed moderately differentiated squamous cell carcinoma (Figure 2E) and did not metastasize to regional or distant sites, including DLNs. Necrosis was assessed based on a previously established three-grade scale: 0 = no visible necrosis, 1 = scant, 2 = moderate, and 3 = severe17. All LY2 tumor-bearing mice had histologically confirmed necrosis with the majority (seven out of ten) demonstrating moderate to severe necrosis (Figure 2F). No evidence of necrosis was observed in B4B8 tumors.
Figure 2: Imaging and histologic features of murine HNSCC tumors.
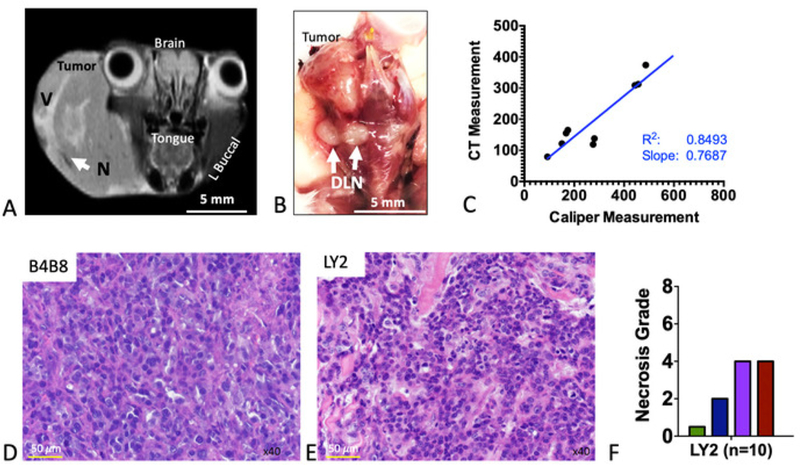
(A) Representative coronal MR image of an LY2 tumor-bearing mouse. Anatomic sites are labeled. Areas of signal hyperintensity represent vascularized regions (denoted V). Regions of signal hypointensity represent necrotic areas (denote N). (B) Representative gross image of a dissected tumor-containing region. White arrows point to enlarged draining lymph nodes (DLNs). (C) Correlation of tumor volume as assessed by caliper measurement versus computed tomography (CT)-based assessment. Slope, correlation coefficient (R2), and line of best fit are shown. (D) Representative hematoxylin and eosin (H&E) image of LY2 tumor. (E) Representative H&E image of B4B8 tumor. (F) Quantification of necrosis in LY2 tumors, based on an H&E four-grade scoring system.
We focused on the LY2 tumor model for further characterization of the tumor-immune microenvironment. Tumors were harvested at 3 weeks after the tumor inoculation and digested using collagenase III, followed by the staining of the cell surface and intracellular markers for flow/ mass cytometry (Figure 3A). Samples were processed, using a mass cytometer, at the University of Colorado Denver Flow Cytometry Shared Resource. Gating and data analysis were performed using flow cytometry commercial software. The data obtained showed the presence of numerous immune cell populations to varying degrees. Total immune cells (CD45+) represented 7.3% of the total tumor (Figure 3B). In absolute numbers, we observed, on average, 26 CD45+ cells (SD = 0.81) per milligram of tumor tissue (Figure 3B). Myeloid cells (CD11b+) represented 37.8% of all CD45+ cells (Figure 3C). Myeloid cells were largely comprised of macrophages (F4/80+, 63.0%), followed by myeloid-derived suppressor cells (MDSCs) (Gr1 +, 8.51%) and neutrophils (Ly6G+, 5.87%). Total T cells comprised 15.9% of CD45+ cells with CD4+ T cells comprising the majority of all T cells (79.4%), of which 53.4% were regulatory T cells (Tregs) (CD4+FoxP3+) (Figure 3D). Natural killer (NK) cells comprised 1.75% of all CD45+ cells. These data highlight the presence of various immune infiltrates in LY2 orthotopic HNSCC tumors which likely play a role in mediating tumor progression and immune evasion.
Figure 3: Analysis of baseline intratumoral immune populations using cytometry by time-of-flight (CyTOF).
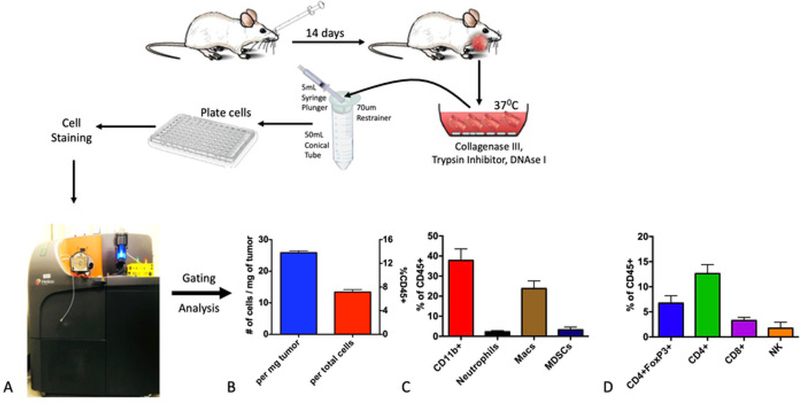
Tumors were processed into a single-cell suspension using collagenase-based enzymatic digestion and, then, stained with cell surface and intracellular markers. (A) Mass cytometry was performed using the CyTOF platform. (B) Absolute and relative quantitative assessment of total immune cells (CD45+). (C) Quantification of myeloid-immune subpopulations. (D) Quantification of lymphoid immune subpopulations.
Discussion
Rigorous analysis and characterization of the tumor microenvironment represent an important strategy for understanding mechanisms of tumor development, progression, and metastases and for the development of effective therapies. Head and neck cancer is a complex disease that can originate from multiple anatomical sites within the head and neck region. A major impediment to understanding disease mechanisms and improving therapy has been a lack of murine mouse cell lines that resemble the aggressive and metastatic nature of human HNSCCs. Furthermore, numerous murine models employ a subcutaneous implantation of tumors which lack important physiological features of the head and neck region, including a high vascular density, an extensive lymphatic vasculature, and resident mucosal flora.
In this study, we characterized an orthotopic model of murine HNSCC. We chose the buccal mucosa as the site of tumor inoculation for a number of reasons, including easy access to the region without the need for image guidance, the ability to assess tumor growth using calipers, the presence of the tumor within the mucosal flora, the presence of surrounding lymphatics, and the ease of reproducibility. Although the injection of cells into the buccal mucosa is straightforward, the positioning of the needle and the depth of penetration are critical to prevent puncture through the skin and ensure cells are not injected subcutaneously. We recommend inserting the needle intra-orally while it is perfectly parallel to the buccal region and tilting at an angle no greater than 10° when ready to inject.
The cell lines we used were murine cell lines allowing their implantation in immunocompetent mice of the strain they were derived from. There are over 39 HNSCC human cell lines that have been studied in the literature but cannot be used in the presence of a native microenvironment18. Recent developments allowed for the design of humanized mouse models which integrate human bone-marrow-derived chimeras in NOD scid gamma mice (also known as NSG mice), allowing the development of a human immune system that can interact with human tumors in an immunodeficient mouse model19. However, such models are costly, require laborious breeding methods, and do not recapitulate all the components of the tumor microenvironment, including endothelial cells, fibroblasts, and lymphatics. The murine mouse models we employed in this study recapitulate critical components of human HNSCCs, including the presence of various immune infiltrates. To ensure maximum retrieval of viable single cells from tumors, the use of digestion enzymes is necessary. Collagenase-based digestion enzymes can be harsh on cells and the optimization of the concentration and type of collagenase may be necessary for different tumor types. We compared five digestion enzymes before determining that collagenase III, which was used in this study, is the optimal method for digestion of squamous cell tumors.
The tumor models used in this study are particularly useful for studying mechanisms of HNSCC immune evasion and preclinical assessment of various therapies. We previously demonstrated that both B4B8 and LY2 tumors are radioresistant and refractory to the immune checkpoint inhibitor anti-PD-L120. This is consistent with the majority of human HNSCCs. Recent clinical trials showed that less than 15% of HNSCC patients are responsive to anti-PD-1/PD-L1 therapy 21,22,23,24,25 In addition, it is well established that HNSCCs are radioresistant26,27. Targeting pathways responsible for radioresistance in HNSCC is an important step for enhancing treatment response. The landmark Bonner et al. study showed a significant benefit in overall survival with the addition of cetuximab to RT compared to RT alone in locally advanced HNSCC patients28. Recent data demonstrate that the inhibition of EphB4-Ephrin-B2 signaling in orthotopic HNSCCs can further improve the response to RT or cetuximab-RT by promoting tumor apoptosis and inhibiting tumor proliferation29,30. In addition, combining RT with rational immune therapies can augment antitumor immune responses and enhance both local tumor control and control of distant metastases31. We have recently demonstrated that targeting Tregs in combination with immune checkpoint blockade and RT results in tumor eradication and immunologic memory32. Future studies employing orthotopic models of HNSCC will better inform the design of clinical trials and improve the understanding of mechanisms of tumor immune evasion and treatment resistance.
Footnotes
Disclosures
The authors have nothing to disclose.
Video Link
The video component of this article can be found at https://www.jove.com/video/59195/
References
- 1.Chaturvedi AK et al. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. Journal of Clinical Oncology. 31 (36), 4550–4559 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ang KK, Sturgis EM Human papillomavirus as a marker of the natural history and response to therapy of head and neck squamous cell carcinoma. Seminars in Radiation Oncology. 22 (2), 128–142 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Amin MB et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA: A Cancer Journal for Clinicians. 67 (2), 93–99 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Salley JJ Experimental carcinogenesis in the cheek pouch of the Syrian hamster. Journal of Dental Research. 33 (2), 253–262 (1954). [DOI] [PubMed] [Google Scholar]
- 5.Morris AL Factors influencing experimental carcinogensis in the hamster cheek pouch. Journal of Dental Research. 40, 3–15 (1961). [DOI] [PubMed] [Google Scholar]
- 6.MacDonald DG Comparison of epithelial dysplasia in hamster cheek pouch carcinogenesis and human oral mucosa. Journal of Oral Pathology & Medicine. 10 (3), 186–191 (1981). [DOI] [PubMed] [Google Scholar]
- 7.Hawkins BL et al. 4NQO carcinogenesis: a mouse model of oral cavity squamous cell carcinoma. Head & Neck. 16 (5), 424–432 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clinical Cancer Research. 10 (1 Pt 1), 301–313 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Vered M, Yarom N, Dayan D 4NQO oral carcinogenesis: animal models, molecular markers and future expectations. Oral Oncology. 41 (4), 337–339 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Bornstein S et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. Journal of Clinical Investigation. 119 (11), 3408–3419 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas GR, Chen Z, Oechsli MN, Hendler FJ, Van Waes C Decreased expression of CD80 is a marker for increased tumorigenicity in a new murine model of oral squamous-cell carcinoma. International Journal of Cancer. 82 (3), 377–384 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Yuspa SH, Hawley-Nelson P, Koehler B, Stanley JR A survey of transformation markers in differentiating epidermal cell lines in culture. Cancer Research. 40 (12), 4694–4703 (1980). [PubMed] [Google Scholar]
- 13.Chen Z, Smith CW, Kiel D, Van Waes C Metastatic variants derived following in vivo tumor progression of an in vitro transformed squamous cell carcinoma line acquire a differential growth advantage requiring tumor-host interaction. Clinical & Experimental Metastasis. 15 (5), 527–537 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Euhus DM, Hudd C, LaRegina MC, Johnson FE Tumor measurement in the nude mouse. Journal of Surgical Oncology. 31 (4), 229–234 (1986). [DOI] [PubMed] [Google Scholar]
- 15.Tomayko MM, Reynolds CP Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemotherapy and Pharmacology. 24 (3), 148–154 (1989). [DOI] [PubMed] [Google Scholar]
- 16.Yushkevich PA et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. NeuroImage. 31 (3), 1116–1128 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Dutta S et al. The relationship between tumour necrosis, tumour proliferation, local and systemic inflammation, microvessel density and survival in patients undergoing potentially curative resection of oesophageal adenocarcinoma. Britisch Journal of Cancer. 106 (4), 702–710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Molecular Cancer Research. 12 (4), 571–582 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh NC et al. Humanized Mouse Models of Clinical Disease. Annual Review of Pathology. 12, 187–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oweida A et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. OncoImmunology. 6 (10), e1356153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferris R et al. Further evaluations of nivolumab (nivo) versus investigator’s choice (IC) chemotherapy for recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CheckMate 141. Journal of Clinical Oncology. 34 (suppl; abstr 6009) (2016). [Google Scholar]
- 22.Ferris RL et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. The New England Journal of Medicine. 375 (19), 1856–1867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrington KJ et al. Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health-related quality-of-life results from a randomised, phase 3 trial. The Lancet Oncology. 18 (8), 1104–1115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiyota N et al. A randomized, open-label, Phase III clinical trial of nivolumab vs. therapy of investigator’s choice in recurrent squamous cell carcinoma of the head and neck: A subanalysis of Asian patients versus the global population in checkmate 141. Oral Oncology. 73, 138–146 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Ling DC, Bakkenist CJ, Ferris RL, Clump DA Role of Immunotherapy in Head and Neck Cancer. Seminars in Radiation Oncology. 28 (1), 12–16 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Perri F et al. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head & Neck. 37 (5), 763–770 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto VN, Thylur DS, Bauschard M, Schmale I, Sinha UK Overcoming radioresistance in head and neck squamous cell carcinoma. Oral Oncology. 63, 44–51 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Bonner JA et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. The Lancet Oncology. 11 (1), 21–28 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Bhatia S et al. Inhibition of EphB4-Ephrin-B2 Signaling Enhances Response to Cetuximab-Radiation Therapy in Head and Neck Cancers. Clinical Cancer Research. 24 (18), 4539–4550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhatia S et al. Enhancing radiosensitization in EphB4 receptor-expressing Head and Neck Squamous Cell Carcinomas. Scientific Reports.6, 38792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang C et al. Combining radiation and immunotherapy: a new systemic therapy for solid tumors? Cancer Immunology Research. 2 (9), 831–838 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oweida A et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clinical Cancer Research. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]