

Abstract
Muscle Lim Protein (MLP) has emerged as a key regulator of striated muscle physiology and pathophysiology. Mutations in cysteine and glycine-rich protein 3 (CSRP3), the gene encoding MLP, are causative of human cardiomyopathies, whereas altered expression patterns are observed in human failing heart and skeletal myopathies. In vitro and in vivo evidence reveals a complex and diverse functional role of MLP in striated muscle, which is determined by its multiple interacting partners and subcellular distribution. Experimental evidence suggests that MLP is implicated in both myogenic differentiation and myocyte cytoarchitecture, although the full spectrum of its intracellular roles still unfolds.
Keywords: MLP, sarcomere, muscle structure, differentiation, heart failure, cardiomyopathies, skeletal myopathies
1. Introduction
The cysteine and glycine-rich protein 3 (CSRP3 or CRP3) gene is a member of cysteine-rich protein (CRP) family that consists of CRP1, CRP2 and CSRP3 (Weiskirchen et al., 1995). CRP1 and CRP2 are prominent in smooth muscle, whereas CSRP3 is expressed in striated muscle (Weiskirchen et al., 1995; Louis et al., 1997). MLP, the protein encoded by CSRP3, belongs to the LIM-only domain family, a large protein family with diverse functional roles, including transcriptional regulation, cell fate determination, cell adhesion and motility, cytoskeleton organization and signal transduction (Schmeichel and Beckerle, 1997; Kadrmas and Beckerle, 2004; Zheng and Zhao, 2007). Its diverse functional roles have a significant impact on cardiac and skeletal muscle physiology and pathology.
Since the identification of MLP, 20 years ago (Arber et al., 1994), a multitude of studies have focused on delineating its functional significance in striated muscle, with exciting findings to date. These investigations were intensified once MLP was implicated in muscle pathogenesis. Mutations in the CSRP3 gene have been directly associated with dilated (DCM) and hypertrophic (HCM) cardiomyopathies while the MLP protein levels appear to be significantly altered in human failing hearts and various skeletal myopathies (Knoll et al., 2002a; Gehmlich et al., 2008). Several in vitro and in vivo approaches have been used to decipher the MLP’s multifaceted role in pathological settings (Knoll et al., 2002a; Gehmlich et al., 2008). Herein, we discuss the intricate involvement of MLP in striated muscle function and disease, the emerging hypotheses, and open questions.
2. CSRP3 gene structure and transcription
CSRP3 was first identified in 1994 during a rat cDNA library screen for genes regulating muscle gene expression and neuromuscular synapses formation (Arber et al., 1994). Northern blot analysis demonstrated its predominant expression in striated muscles (Arber et al., 1994). In 1995, the human CSRP3 gene was mapped to chromosome 11p15.1 and subsequently isolated from a human cardiac cDNA library (Fung et al., 1995; Fung et al., 1996). The human gene spans a 20kb genomic region, producing a 0.8kb transcript that is organized in 6 exons (Fung et al., 1996; Knoll et al., 2002b). We recently reported the discovery of the first splice variant of CSRP3, originating from alternative splicing of exons 3 and 4 (Vafiadaki et al., 2014). This novel MLP isoform, designated MLP-b, was demonstrated to exhibit distinct expression and functional roles to the full length MLP, revealing the complexity of MLP expression and intracellular roles.
CSRP3 expression was found to be largely determined by an E-box sequence at the position −186 to −180 within the CSRP3 promoter region (Ji et al., 2009). Synergistic binding of the transcription factors myogenin and myocyte enhancer factor 2C (MEF2C) occurs at the E-box promoter region, and leads to transcriptional activation of CSRP3. Additional cis-acting elements such as the binding sites for AP1, Jun/Fos are suggested to be present in the promoter (Ji et al., 2009), however, their role in regulating the CSRP3 promoter is unclear.
3. MLP structure and physicochemical properties
MLP is a relatively small protein of 194 amino acids that is specifically expressed in skeletal and cardiac muscles. Structurally, it belongs to a family of proteins that harbor one or more LIM domains. The name of this protein domain was derived from the initial letters of Lin-11, Isl1 and Mec-3 that represent the first three members of this protein family to be identified (Schmeichel and Beckerle, 1997; Kadrmas and Beckerle, 2004). In general, LIM domains comprise of approximately 50–60 amino acids and share two characteristic zinc finger domains. These zinc fingers contain 8 highly conserved cysteine and histidine residues at specific positions, which coordinately bind two zinc ions (Zheng and Zhao, 2007). The overall consensus sequence of a LIM domain is CX2CX16_23HX2CX2CX2CX16_21CX2(C/H/D), where X denotes any amino acid (Schmeichel and Beckerle, 1997; Kadrmas and Beckerle, 2004). LIM domains may be found throughout the length of a protein. They are believed to act as a modular protein-binding interface that mediates diverse protein interactions and facilitates macromolecular complex formation, thus enabling LIM domain proteins to participate in a broad range of biological functions (Schmeichel and Beckerle, 1994; Arber and Caroni, 1996; Weiskirchen and Gunther, 2003; Kadrmas and Beckerle, 2004).
MLP contains two LIM domains (LIM1 and LIM2), each surrounded by glycine-rich repeat regions, and the two separated by more than 50 residues (Weiskirchen et al., 1995). The presence of these glycine-rich repeats is characteristic of all CRP family members and distinguishes them from other LIM-only containing proteins (Weiskirchen and Gunther, 2003). While the structural analysis of CRP1 and CRP2 proteins was completed several years ago (Perez-Alvarado et al., 1994; Konrat et al., 1997; Kontaxis et al., 1998; Yao et al., 1999), it was only recently that this information became available for MLP (Schallus et al., 2007; Schallus et al., 2009). Using nuclear magnetic resonance spectroscopy, it was deduced that the two MLP LIM domains act as independent units and that the adjacent linker region is fully flexible (Schallus et al., 2007; Schallus et al., 2009). These structural characteristics suggest that LIM domains could act as adaptors facilitating the formation of macromolecular complexes. Indeed, the two MLP LIM domains mediate interactions with a vast number of different proteins at different subcellular locations, including the cytoplasm and the nucleus (see section “MLP protein interactions and localization”) (Buyandelger et al., 2011b).
An emerging concept is this of MLP oligomerization. Using biochemical subcellular fractionation and immunocytochemistry techniques it was shown that MLP forms dimers, trimers and tetramers in myocytes (Boateng et al., 2007; Boateng et al., 2009). The oligomerization potential of MLP is believed to be critical for its cellular localization and function, with direct implications in muscle physiology.
4. MLP post-translational modifications
Post-translational modifications represent key determinants of protein function. The precise effect of post-translational modifications on MLP is to date largely unknown. The only available experimental evidence shows the acetylation/deacetylation of MLP at a lysine residue at position 69 (K69), by acetyltransferase (PCAF) and histone deacetylase 4 (HDAC4), respectively (Gupta et al., 2008). Since the K69 residue is within the predicted nuclear localization signal of MLP (Fung et al., 1996), it was proposed that acetylation of K69 may influence MLP nucleocytoplasmic shuttling. K69 acetylation was further associated with calcium sensitivity and myofilament contractility (Gupta et al., 2008). These findings reveal the emerging role of reversible acetylation in the regulation of muscle contraction.
In addition to acetylation, it has been postulated that MLP may undergo phosphorylation or sumoylation, which could in turn modulate its localization and/or function (Buyandelger et al., 2011b). Bioinformatical analysis predicts the existence of several putative phosphorylation sites on MLP (Fig. 1), however, no evidence on the occurrence of MLP phosphorylation has been published to date so experimental confirmation and functional characterization is pending.
Fig.1.
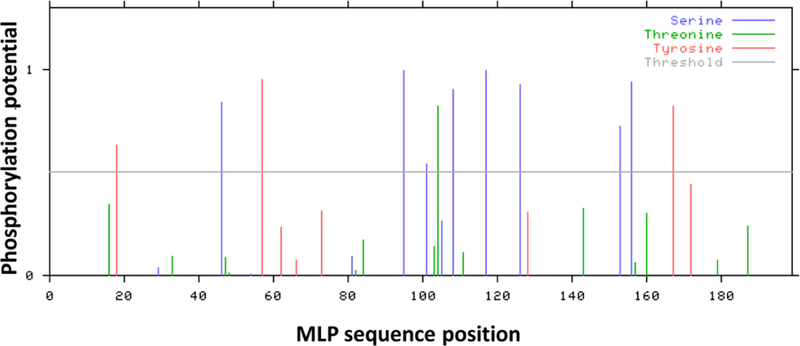
Bioinformatical predictions of phosphorylation sites in MLP protein sequence. Graphical output showing multiple serine, threonine or tyrosine residues with predicted phosphorylation potential above the threshold. Bioinformatical analysis was performed using the publicly available NetPhos 2.0 Server (http://www.cbs.dtu.dk/services/NetPhos/) (Blom et al., 1999).
5. MLP protein interactions and localization
Determining the localization and binding partners of MLP has been an integral part of the quest for its spectrum of intracellular roles. This has led to an increasingly large list of interacting proteins with diverse functional properties, as well as variable subcellular localizations within muscle cells. In particular, in the cytoplasm MLP has been detected at the level of the sarcomeres (the basic muscle contractile unit, delimited by Z-discs and an M-band in the center), intercalated discs (located at the bipolar ends of cardiomyocytes, inter-connecting cardiomyocytes, enabling the synchronized contraction of cardiac tissue), costameres (a critical component of striated muscle morphology connecting the sarcomeres to the sarcolemma), and even the cell membrane. At the sarcomeres MLP interacts with the Z-disc proteins telethonin (T-cap), α-actinin, cofilin-2 (CFL2), calcineurin, HDAC4, as well as MLP and MLP-b (Louis et al., 1997; Zolk et al., 2000; Knoll et al., 2002b; Heineke et al., 2005; Gupta et al., 2008; Papalouka et al., 2009; Vafiadaki et al., 2014). Although LIM domains are major protein-binding interfaces, we have demonstrated that the MLP inter-LIM region, containing a glycine-rich repeat, can itself serve as the minimal binding region, with proteins such as CFL2 and the MLP-b isoform (Papalouka et al., 2009; Vafiadaki et al., 2014). The functional implications of these interactions are discussed in the next section. M-band distribution of MLP has also been described, although the binding partners mediating this are currently unknown (Boateng et al., 2007; Knoll et al., 2010). Additionally to the sarcomere, MLP localizes to the costameres, where it binds to zyxin, integrin linked kinase (ILK) and β1-spectrin (Louis et al., 1997; Flick and Konieczny, 2000; Postel et al., 2008), as well as to the intercalated discs, where it associates with the nebulin-related anchoring protein (N-RAP) (Ehler et al., 2001).
Beyond its cytoplasmic roles, MLP can also localize to the nucleus, where it binds to the nuclear transcription factors MyoD, myogenin and myogenic regulatory factor 4 (MRF4) (Kong et al., 1997). Furthermore, it can shuttle between these two cellular compartments. This nucleocytoplasmic shuttling appears to be driven by its nuclear localization signal, as shown by cell permeable synthetic peptides containing the predicted nuclear localization signal of MLP (Boateng et al., 2009).
The oligomeric/monomeric state of MLP appears to be tightly correlated with its subcellular localization. Specifically, the oligomeric form of MLP is encountered in the cytoplasm, whereas nuclear MLP is monomeric. Based on findings from the analysis of a C-terminal flag containing construct that prevented MLP oligomer formation, it was proposed that the carboxyl-terminal of MLP is required for oligomerization (Boateng et al., 2007). This, however, was not confirmed by a recent study which demonstrated the involvement of the N-terminal LIM domain of MLP in its self-association using bimolecular fluorescence complementation assays (Hoffmann et al., 2014).
Although the variable MLP subcellular distribution documented by different studies appears to be consistent with its multiple, diverse protein binding partners, some inconsistencies in its localization patterns have led to concerns about the specificity of the available MLP antibodies (Gehmlich et al., 2008). The use of different polyclonal antibodies raised against MLP may be associated with potential non-specific reactivity problems. Moreover, the generation of a mouse monoclonal antibody with high specificity to MLP and no cross-reactivity to CRP1 or CRP2, was reported to primarily detect a diffuse cytoplasmic localization for MLP (Geier et al., 2008). However, a different mouse monoclonal antibody raised against the C-terminus of MLP, detected both cytoplasmic and nuclear distribution of the protein (Boateng et al., 2007). Although the full details of its subcellular localization remain to be uncovered, there is a consensus that MLP can be found in, interact with, and consequently participate in, a variety of locations, proteins, and functions, respectively.
6. MLP functions
MLP has been found to interact with different protein partners in each of the subcellular locations where it is detected. Through these interactions it is implicated in a series of physiological processes.
1) At the nucleus, MLP serves as a positive regulator of myogenesis, promoting myogenic differentiation (Arber et al., 1994). Accordingly, various studies have demonstrated that MLP overexpression results in enhanced myotube differentiation (Arber et al., 1994; Kong et al., 1997; Vafiadaki et al., 2014). This effect is attributed to the direct association of MLP with muscle differentiation transcription factors such as MyoD, myogenin and MRF4. While MLP does not bind to DNA directly, it was proposed to serve as a cofactor for these myogenic transcription factors, by increasing their interaction with specific DNA regulatory elements, as demonstrated by electrophoretic mobility shift assays, enhancing their activity and thus leading to increase myotube differentiation (Kong et al., 1997). In agreement with this concept, we recently demonstrated that MLP-b, which does not bind to the above transcription factors, cannot promote myotube differentiation (Vafiadaki et al., 2014). While the precise role of MLP in the nucleus is being investigated, valuable insights can come from CRP1 and CRP2 that also localize to the nucleus and promote (smooth) muscle differentiation (Arber and Caroni, 1996; Louis et al., 1997). In particular, they interact with the serum response factor (SRF) and GATA transcription factors and are believed to act as cofactors, activating smooth muscle cell gene expression (Chang et al., 2003). CRP2 associates with its N-terminal LIM domain to SRF and its C-terminal LIM domain to GATA4, and has therefore been proposed to act as a bridging molecule, resulting in the strong activation of smooth muscle specific gene promoters, such as smooth muscle K-actin and h1calponin (Chang et al., 2003). Analysis of a CRP2 transgenic animal model demonstrated that CRP2 acts as a potent transcriptional co-adaptor that remodels silent myocyte chromatin and activates of smooth muscle gene expression (Chang et al., 2007). A similar mode of action could be envisioned for MLP, although the details remain to be uncovered.
2) At the cytoplasm, MLP has a prominent structural role at multiple levels. Evidence for its critical function in the establishment and maintenance of the (cardio)myocyte cytoskeleton arose from studies on patient biological material and animal models, carrying human mutations or lacking MLP. These studies demonstrate severe disruption of cardiac cytoarchitecture and myofibrillar disarray (Arber et al., 1997; Knoll et al., 2002b; Knoll et al., 2010). This cytoskeletal effect is primarily mediated through its localization at the Z-disc and its interactions with different Z-disc components (Louis et al., 1997; Zolk et al., 2000; Knoll et al., 2002b; Heineke et al., 2005; Clark et al., 2007; Gupta et al., 2008; Papalouka et al., 2009; Clark and Kadrmas, 2013; Vafiadaki et al., 2014). MLP has therefore been suggested to act as a scaffold protein that promotes the assembly of macromolecular complexes along sarcomeres and the actin-based cytoskeleton (Arber and Caroni, 1996; Arber et al., 1997; Flick and Konieczny, 2000; Ehler et al., 2001; Knoll et al., 2002b).
The Z-disc represents a dynamic macromolecular structure, anchoring a variety of different proteins that contribute to muscle contraction (Frank et al., 2006; Frank and Frey, 2011; Sequeira et al., 2014). In addition to its structural role, it is a nodal point for signaling networks, acting as a stretch sensor and being implicated in mechano-signaling and mechano-trasduction (Luther, 2009; Buyandelger et al., 2011a; Frank and Frey, 2011; Gautel, 2011). The Z-disc anchors the ends of actin filaments and in this way mediates force transmission from one sarcomere to the next, ultimately reaching the ends of the cardiomyocyte (Buyandelger et al., 2014). Examples of molecular components acting as mechanosensors include the giant proteins titin, nebulin and obscurin (Maillet et al., 2013). MLP is believed to be involved in these mechano-signaling process, with cardiomyocytes from MLP transgenic or knock-out mice exhibiting defective intrinsic stretch responses, due to selective loss of passive stretch sensing (Knoll et al., 2002b; Knoll et al., 2010). It was initially suggested that loss of MLP destabilizes anchoring of the Z disc to the proximal end of the T-cap/titin complex, leading to a conformational alteration of the intrinsic titin molecular spring elements and consequently to defects in the cardiac muscle stretch sensor machinery. Based on the complex subcellular distribution of MLP, the role of MLP as mechanical stretch sensor has been questioned and has instead been proposed to represent a mediator of stretch signaling, acting downstream in mechano-signaling cascades (Gehmlich et al., 2008; Gehmlich et al., 2010). The mechanism through which MLP mediates signal transduction remains unclear, however, the possible role of post-translational modifications is being considered.
The MLP’s mechano-signaling role may also be mediated through its interaction with the costameric proteins β1 spectrin and zyxin. The costameres have a critical role in lateral force transmission. Specifically, they transmit force bidirectionally from the sarcomere to the sarcolemma and the extracellular matrix, maintaining mechanical integrity of the sarcolemma and orchestrating mechanically related signaling in order to modulate myofibril growth and contraction (Bershadsky et al., 2003; Bloch and Gonzalez-Serratos, 2003; Peter et al., 2011). However, the precise role of MLP at the costameres has not been extensively investigated yet.
Another function of MLP relates to actin dynamics. We previously reported the interaction of MLP with cofilin-2 (CFL2), a muscle-specific protein that belongs to the actin depolymerization factor (ADF)/cofilin family (Papalouka et al., 2009). Using biochemical studies, we determined that the interaction of MLP with CFL2 enhances the CFL2-dependent F-actin depolymerization, a finding with direct implications on actin cytoskeleton dynamics (Papalouka et al., 2009). Stoichiometry of MLP/CFL2 was shown to be of importance, with changes in the MLP/CFL2 ratio resulting in either enhancement or suppression of the MLP effect on CFL2-dependent F-actin depolymerization, supporting the dynamic nature of this reaction. In agreement to this, a recent study has also demonstrated the impact of MLP on the actin cytoskeleton dynamics, suggesting that it binds, stabilizes and crosslinks actin filaments into bundles (Hoffmann et al., 2014). Similarly, CRP1 and CRP2 have been described to influence actin filament bundling (Grubinger and Gimona, 2004; Tran et al., 2005; Jang and Greenwood, 2009), thus suggesting a common molecular mechanism of this protein family in regulating the actin cytoskeleton.
7. CSRP3 in human disease
The critical role of MLP in striated muscle function is evident by the major implications of its absence or aberrant function. A range of CSRP3 mutations (10 missense and 1 frameshift) have been published and associated with the development of DCM or HCM (Fig. 2)(Geier et al., 2003; Mohapatra et al., 2003; Bos et al., 2006; Geier et al., 2008; Hershberger et al., 2008). Interestingly, all known mutations are located within the first 100 amino acids of the protein, but none within the C-terminus. The publicly available single nucleotide polymorphisms (SNPs) database (NCBI dbSNP Build 142, http://www.ncbi.nlm.nih.gov/snp/) includes 13 rare non-synonymous and 5 synonymous SNPs, with a frequency less than 1:1000 in Caucasians. These SNPs correspond to the MLP protein region between amino acid residue 115 and the C-terminus, however, their functional significance is currently unknown and no correlation with human disease has been reported. In other databases, such as the Exome Variant Server (http://evs.gs.washington.edu/EVS/) and ExAc Browser (http://exac.broadinstitute.org/about), many different but rare CSRP3 variants of currently unknown function are reported. The increasing use of high throughput technologies, such as next generation sequencing, for the study of large, well-characterized population and patient cohorts, will shed light to these open questions. Overall, the data published to date suggest a key role of the MLP N-terminus, with alterations in its sequence leading to significant aberrations in the corresponding molecular functions and ultimately the development of a severe cardiac pathology.
Fig.2.
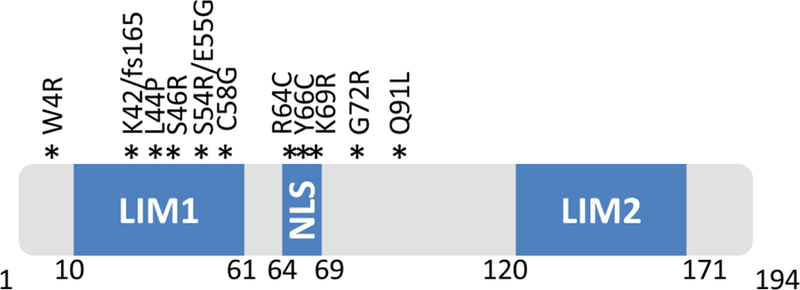
Diagrammatic representation of MLP illustrating the location of published human mutations. The positions of the two LIM domains (LIM1 and LIM2) as well as the nuclear localization signal (NLS) are indicated.
In agreement with this notion, nearly half of the reported mutations in patients are located within the N-terminus. The most common MLP mutation in cardiomyopathy patients is the W4R. It was originally reported in 2002 by Knoll et al. (Knoll et al., 2002b), and has since been detected in patients of different ethnic origins (Mohapatra et al., 2003; Newman et al., 2005; Bos et al., 2006; Geier et al., 2008; Knoll et al., 2010). Biochemical analysis determined that this mutation leads to complete loss of binding to T-cap and causes mislocalization of T-cap from Z-disc, supporting the significance of MLP and T-cap interactions in Z-disc stability (Knoll et al., 2002b). It is of interest that the W4R mutation has been found in both DCM and HCM patients (Knoll et al., 2002b; Mohapatra et al., 2003; Newman et al., 2005; Bos et al., 2006; Geier et al., 2008). While the underlying mechanisms mediating the variable clinical outcome is unclear, it has been proposed to implicate modifier gene variants and/or epigenetic factors (Knoll et al., 2010). For example, in some cases MLP mutations have been detected in conjunction with genetic variants in other myofilament-encoding genes (e.g. MLP-W4R and beta myosin heavy chain or myosin binding protein C variants) (Bos et al., 2006).
Although a large number of MLP mutations have been associated with DCM and HCM, few have been functionally characterized. In addition to W4R, examples include the C58G and the K69L mutations. Functional characterization of the C58G mutation initially determined that the mutant MLP protein shows reduced binding to N-RAP and α-actinin in vitro (Geier et al., 2003; Gehmlich et al., 2004). However, in vivo observations in myocardial biopsy and in transfected mammalian cells suggest that C58G-MLP exhibits reduced stability due to enhanced ubiquitin-dependent proteasomal degradation (Geier et al., 2008). These findings led to the hypothesis that the relative lack of functional MLP protein rather than the presence of the C58G mutation is causative of heart disease (Geier et al., 2008). The K69L mutation on the other hand, is within the predicted nuclear localization signal of MLP. Analysis in cell culture systems demonstrated that MLP-K69L exhibits altered subcellular distribution, with predominant perinuclear localization (Mohapatra et al., 2003). MLP LIM1 domain is also involved in α-actinin binding and K69L-MLP is not co-precipitated with wild-type α-actinin (Mohapatra et al., 2003). Additionally, the lysine residue at position 69 is predicted to be acetylated. Consequently some of the molecular and cellular aberrations observed in these patients could be attributed to the altered acetylation of this mutant MLP.
In addition to gene mutations, alterations in MLP levels or oligomerization have also been associated with human cardiac disease. Specifically, MLP is significantly down regulated at the protein level in chronic heart failure patients with dilated or ischemic cardiomyopathy (Zolk et al., 2000). Reduced levels of MLP may play an essential role in myofibril derangement or impaired myofibril rearrangement in the human failing myocardium, thus promoting deterioration of contractile function (Zolk et al., 2000). Interestingly, MLP mRNA levels were not altered in these patients, suggesting that the changes occurred at the translational level and were proposed to reflect decreased MLP synthesis or enhanced MLP protein turnover (Zolk et al., 2000). Noteworthy, mechanical support of the failing heart with left ventricular assist device could not restore MLP protein levels in the myocardium of DCM patients (Gehmlich et al., 2008).
Aberrations in MLP oligomerization have also been associated with cardiac disease. Specifically, MLP oligomers are reduced in human failing hearts with a concomitant increase in monomeric MLP levels (Boateng et al., 2007). This decline in oligomeric, cytoplasmic, MLP is hypothesized to impair the cardiomyocyte mechanical sensor mechanism, a defect known to disrupt cardiac function to give rise to heart disease (Boateng et al., 2007).
Beyond heart disease, deregulation of MLP levels has also been reported in different skeletal muscle diseases, including facioscapulohumeral muscular dystrophy, nemaline myopathy and limb girdle muscular dystrophy type 2B (Winokur et al., 2003; von der Hagen et al., 2005; Sanoudou et al., 2006). The reason and implications of these expression changes remain largely unknown; they could be an unsuccessful compensatory mechanism against the widespread structural disarray encountered in the affected skeletal muscles, or a reflection of the impaired myogenic process that is sometimes observed. In parallel to MLP, we have demonstrated that the MLP-b isoform may also be abnormally expressed, and/or the physiologically important MLP:MLP-b ratio disturbed in different neuromuscular diseases (e.g. limb girdle muscular dystrophy type 2A, Duchenne muscular dystrophy and dermatomyositis) (Vafiadaki et al., 2014).
Delineating the precise role of MLP in all these different settings would allow a deeper understanding of the different pathogenetic mechanisms and potentially the utilization of MLP as a therapeutic target.
8. Deciphering the role of MLP in cardiac muscle pathophysiology through animal model studies
The utilization of animal models has provided valuable evidence on MLP’s functional significance in striated muscle pathophysiology. The generation of a mouse model lacking Mlp (MLP−/−) demonstrated the essential role of this protein in mammalian striated muscle (Arber et al., 1997). Although both skeletal and cardiac muscles were affected by MLP deficiency, defects were most dramatic in the latter. MLP−/− mice initially exhibit defects in passive muscle mechanics, followed by severe cardiac dysfunction including alterations in cardiac pressure and volume, leading to the development of dilated cardiomyopathy with hypertrophy and progressive heart failure (Arber et al., 1997; Esposito et al., 2000; Omens et al., 2002; Lorenzen-Schmidt et al., 2005). At the histological level, MLP deficiency leads to a dramatic disruption of cardiomyocyte cytoarchitecture with a pronounced myofibril disorganization, abnormal alignment of Z-discs and significant fibrosis that justify the described abnormalities in the intrinsic contractility of these hearts (Arber et al., 1997; Su et al., 2001). Further aberrations relate to the intercalated disc ultrastructure and the alignment of costameres, which suggest defects in cell–matrix and cell–cell contact structures in cardiomyocytes lacking MLP (Ehler et al., 2001; Wilson et al., 2014). This was associated with an up-regulation of intercalated disk proteins including N-RAP, β-catenin, vinculin and plakoglobin, while levels of the gap-junctional protein connexin-43 were reduced (Ehler et al., 2001). Overall, the structural defects triggered by MLP deficiency were proposed to indicate that deficits in cytoarchitectural organization lead to impaired cell and tissue tension. Consequently, upon hypertrophic stimuli, MLP deficient cardiomyocytes expand but fail to generate sufficient tension to control hypertrophic responses, leading to a chronic hypertrophic state and dilated cardiomyopathy (Arber et al., 1997).
Intriguingly, the absence of MLP led to local loss of mitochondria and energy deficiency, indicating a direct link between cytoskeletal defects and energy metabolism (van den Bosch et al., 2005). Alterations in intracellular calcium handling and defects in excitation-contraction coupling were also described (Esposito et al., 2000; Su et al., 2001; Kemecsei et al., 2010). The role of calcium appears to be of utmost importance in the process of pathogenesis: the absence of the SR calcium uptake regulator phospholamban (PLN) in a double knockout model of MLP and PLN, rescued the morphological and ultrastructural defects associated with MLP ablation including myofibrillar disarray and fibrosis, and enhanced cardiac function (Minamisawa et al., 1999). This phenotypic rescue was lifelong and prevented the onset of DCM (Minamisawa et al., 1999). Following this pioneering study, a number of different mouse models have been crossed with the MLP−/− in order to assess the effect of different molecular players and mechanisms in reversing the MLP−/−phenotype (Rockman et al., 1998; Heineke et al., 2005; Yamamoto et al., 2007; Fajardo et al., 2013). Ablation of β2-adrenergic receptor (β2-AR) was effective in rescuing MLP−/− mice from cardiomyopathy, by enhancing myocyte shortening, improving Ca2+ availability, restoring cardiac function and survival (Fajardo et al., 2013). The vulnerability of MLP−/− hearts to β2-adrenergic stimulation points up the importance of MLP in regulating myofilament contractility. The double knockout mouse model of MLP and calcineurin, a calcium/calmodulin-dependent protein phosphatase, resulted in increased apoptosis, decreased left ventricular function, enhanced fibrosis and cardiomyopathy. Conversely, modest calcineurin overexpression in MLP−/− mice improved cardiac function and reduced fibrosis (Heineke et al., 2010). Since calcineurin is involved in transcriptional control of myocyte hypertrophy through MEF2 (Olson and Williams, 2000) and MLP cooperates with the same factor (Ji et al., 2009), it could be postulated that calcineurin overexpression restores transcriptional regulatory functions of MLP.
Besides ablation of MLP, a transgenic mouse model with cardiac-specific overexpression of MLP has also been described (Kuhn et al., 2012). These transgenic mice exhibit normal contractile function and show no pathological phenotype, under baseline conditions. MLP overexpression did not modulate cardiac hypertrophy induced by transverse aortic constriction or chronic infusion of angiotensin-II (Kuhn et al., 2012), suggesting that cardiac overexpression of MLP in vivo does not influence response to different forms of pathological stress.
Apart from the levels of MLP, its structural integrity is also of paramount importance in normal cardiac function. In order to understand how human CSRP3 mutations lead to heart disease, knock-in mouse models are being used. For example, studies on the knock-in mouse harboring the human W4R mutation proved that it suffices to induce HCM and heart failure (Knoll et al., 2010). This is at least partly mediated by increased nuclear and reduced Z-disc localization of MLP, myocardial disarray and significant fibrosis (Knoll et al., 2010).
Alterations in nucleocytoplasmic shuttling have also been implicated in hypertrophy and heart failure, independently of mutations (Boateng et al., 2009). It has been hypothesized that MLP becomes mislocalized after prolonged mechanical overload, resulting in impaired mechanosensing in cardiomyocytes and promotion of hyperthrophy. Indeed, in the ventricles from aortic-banded and myocardially infarcted rat hearts, nuclear MLP increased by twofold (Boateng et al., 2007). The reduced levels of cytoplasmic MLP may be part of a “declining mechanosensor” phenomenon. Importantly, the translocation of MLP to the nucleus was shown to lead to stimulation of ribosomal protein synthesis and hypertrophic marker brain natriuretic peptide (BNP), processes associated with hypertrophy. Conversely, inhibition of the MLP translocation to the nucleus, by synthetic peptides, led to sarcomeric disarray and decreased α-actinin expression (Boateng et al., 2009). MLP localization as well as its nucleocytoplasmic shuttling potential appears to be associated with the MLP oligomeric state, as shown in rats either after myocardial infarction or aortic banding (Boateng et al., 2007).
While a large number of studies have focused on mouse as animal model, additional animals have also been generated for MLP. No report on zebrafish being utilized as animal models for MLP has been described so far but studies using Drosophila have resulted in highly concordant data to mouse. Genetic ablation of Mlp84B, the Drosophila homolog of MLP, was associated with pupal lethality and impaired muscle function (Clark et al., 2007), with significantly reduced muscle stiffness and decreased power generation (Clark et al., 2011). The cardiac-specific ablation of Mlp84B resulted in decreased lifespan, impaired diastolic function and disturbances in cardiac rhythm. Surprisingly, deficiency of Mlp84B did not result in major structural abnormalities, a finding that was proposed to reflect either that loss of Mlp84B does not compromise muscle structure, or that subtle structural defects may exist and suffice to induce muscle failure (Clark et al., 2007; Mery et al., 2008).
Collectively, findings from numerous animal model studies have provided valuable insights into the functional significance of MLP in striated muscle physiology and pathophysiology.
9. Conclusions
MLP is a fascinating molecule, specifically expressed in striated muscles, yet implicated in a broad range of functions therein. Its two LIM domains enable interactions with multiple other proteins, and together with the nuclear localization signal, place it at diverse subcellular locations. In vitro and in vivo experiments have demonstrated that beyond its somewhat anticipated complex structural and myogenic role, it is indirectly related to calcium homeostasis and mitochondrial energy metabolism. Importantly, MLP deficiency, and aberrant structure, localization or oligomerization, can give rise to pathological phenotypes across species. It has therefore been evolutionary conserved and possesses irreplaceable functional attributes. Despite the extensive investigations, a lot remains to be discovered on MLP, such as the potential existence of other binding partners or MLP isoforms and their roles, the full implications of its oligomerization, post-translational modification, and how different conditions regulate its nucleocytoplasmic shuttling. At the clinical level, MLP could serve not only as a diagnostic marker (disease specific mutations), but also as a potential prognostic marker. For instance, MLP is released in blood circulation of patients after myocardial infarction and could therefore serve as a biomarker for myocardial infarction (Cordwell et al., 2012). Taking into account the direct relation of MLP to disease and its multitude of functions, therapeutic strategies targeting any of these could potentially help alleviate muscle dysfunction.
Highlights.
MLP is a key regulator of striated muscle physiology
Multiple binding partners and localization dictate MLP’s structural and myogenic role
MLP mutations and aberrant expression are implicated in striated muscle pathology
Oligomerization regulates MLP nucleocytoplasmic shuttling, physio/pathological functions.
Acknowledgement
This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series--a series resulting from collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM083924 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors would like to thank the Hellenic Cardiological Society, Fondation Sante, General Secretariat for Research and Technology-ARISTEIA II and the European Community’s Seventh Framework Program FP7/2007-2013 under grant agreement no HEALTH-F2-2009-241526, EUTrigTreat.
Abbreviations:
- MLP
Muscle Lim Protein
- CSRP3
cysteine and glycine-rich protein 3
- CRP3
cysteine-rich protein
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- CFL2
cofilin-2
- MRF4
myogenic regulatory factor 4
- PLN
phospholamban
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Authors have no conflict of interest.
References
- Arber S and Caroni P Specificity of single LIM motifs in targeting and LIM/LIM interactions in situ. Genes Dev 10 (1996), pp. 289–300. [DOI] [PubMed] [Google Scholar]
- Arber S, Halder G and Caroni P Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell 79 (1994), pp. 221–31. [DOI] [PubMed] [Google Scholar]
- Arber S, Hunter JJ, Ross J Jr., Hongo M, Sansig G, Borg J, Perriard JC, Chien KR and Caroni P MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell (1997), pp. 393–403. [DOI] [PubMed] [Google Scholar]
- Bershadsky AD, Balaban NQ and Geiger B Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol 19 (2003), pp. 677–95. [DOI] [PubMed] [Google Scholar]
- Bloch RJ and Gonzalez-Serratos H Lateral force transmission across costameres in skeletal muscle. Exerc Sport Sci Rev 31 (2003), pp. 73–8. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S and Brunak S Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294 (1999), pp. 1351–62. [DOI] [PubMed] [Google Scholar]
- Boateng SY, Belin RJ, Geenen DL, Margulies KB, Martin JL, Hoshijima M, de Tombe PP and Russell B Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am J Physiol Heart Circ Physiol 292 (2007), pp. H259–69. [DOI] [PubMed] [Google Scholar]
- Boateng SY, Senyo SE, Qi L, Goldspink PH and Russell B Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol 47 (2009), pp. 426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JM, Poley RN, Ny M, Tester DJ, Xu X, Vatta M, Towbin JA, Gersh BJ, Ommen SR and Ackerman MJ Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab 88 (2006), pp. 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyandelger B, Mansfield C and Knoll R Mechano-signaling in heart failure. Pflugers Arch 466 (2014), pp. 1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyandelger B, Ng KE, Miocic S, Gunkel S, Piotrowska I, Ku C and Knoll R Genetics of mechanosensation in the heart. J Cardiovasc Transl Res 4 (2011a), pp. 238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyandelger B, Ng KE, Miocic S, Piotrowska I, Gunkel S, Ku CH and Knoll R MLP (muscle LIM protein) as a stress sensor in the heart. Pflugers Arch 462 (2011b), pp. 135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DF, Belaguli NS, Chang J and Schwartz RJ LIM-only protein, CRP2, switched on smooth muscle gene activity in adult cardiac myocytes. Proc Natl Acad Sci U S A 104 (2007), pp. 157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DF, Belaguli NS, Iyer D, Roberts WB, Wu SP, Dong XR, Marx JG, Moore MS, Beckerle MC, Majesky MW and Schwartz RJ Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev Cell 4 (2003), pp. 107–18. [DOI] [PubMed] [Google Scholar]
- Clark KA, Bland JM and Beckerle MC The Drosophila muscle LIM protein, Mlp84B, cooperates with D-titin to maintain muscle structural integrity. J Cell Sci 120 (2007), pp. 2066–77. [DOI] [PubMed] [Google Scholar]
- Clark KA and Kadrmas JL Drosophila melanogaster muscle LIM protein and alpha-actinin function together to stabilize muscle cytoarchitecture: a potential role for Mlp84B in actin-crosslinking. Cytoskeleton (Hoboken) 70 (2013), pp. 304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KA, Lesage-Horton H, Zhao C, Beckerle MC and Swank DM Deletion of Drosophila muscle LIM protein decreases flight muscle stiffness and power generation. Am J Physiol Cell Physiol 301 (2011), pp. C373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordwell SJ, Edwards AV, Liddy KA, Moshkanbaryans L, Solis N, Parker BL, Yong AS, Wong C, Kritharides L, Hambly BD and White MY Release of tissue-specific proteins into coronary perfusate as a model for biomarker discovery in myocardial ischemia/reperfusion injury. J Proteome Res 11 (2012), pp. 2114–26. [DOI] [PubMed] [Google Scholar]
- Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM and Perriard JC Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol 153 (2001), pp. 763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ and Rockman HA Cellular and functional defects in a mouse model of heart failure. Am J Physiol Heart Circ Physiol 279 (2000), pp. H3101–12. [DOI] [PubMed] [Google Scholar]
- Fajardo G, Zhao M, Urashima T, Farahani S, Hu DQ, Reddy S and Bernstein D Deletion of the beta2-adrenergic receptor prevents the development of cardiomyopathy in mice. J Mol Cell Cardiol 63 (2013), pp. 155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick MJ and Konieczny SF The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. J Cell Sci 113 (Pt 9) (2000), pp. 1553–64. [DOI] [PubMed] [Google Scholar]
- Frank D and Frey N Cardiac Z-disc signaling network. J Biol Chem 286 (2011), pp. 9897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D, Kuhn C, Katus HA and Frey N The sarcomeric Z-disc: a nodal point in signalling and disease. J Mol Med (Berl) 84 (2006), pp. 446–68. [DOI] [PubMed] [Google Scholar]
- Fung YW, Wang R and Liew CC Characterization of a human cardiac gene which encodes for a LIM domain protein and is developmentally expressed in myocardial development. J Mol Cell Cardiol 28 (1996), pp. 1203–10. [DOI] [PubMed] [Google Scholar]
- Fung YW, Wang RX, Heng HH and Liew CC Mapping of a human LIM protein (CLP) to human chromosome 11p15.1 by fluorescence in situ hybridization. Genomics 28 (1995), pp. 602–3. [DOI] [PubMed] [Google Scholar]
- Gautel M The sarcomeric cytoskeleton: who picks up the strain? Curr Opin Cell Biol 23 (2011), pp. 39–46. [DOI] [PubMed] [Google Scholar]
- Gehmlich K, Ehler E, Perrot A, Furst DO and Geier C “MLP: A Stress Sensor Goes Nuclear” by Gunkel Sylvia, Heineke Jorg, Hilfiker-Kleiner Denise, Knoll Ralph, J Mol Cell Cardiol 2009;47(4):423–5. J Mol Cell Cardiol 48 (2010), pp. 424–5; author reply 426–7. [DOI] [PubMed] [Google Scholar]
- Gehmlich K, Geier C, Milting H, Furst D and Ehler E Back to square one: what do we know about the functions of muscle LIM protein in the heart? J Muscle Res Cell Motil 29 (2008), pp. 155–8. [DOI] [PubMed] [Google Scholar]
- Gehmlich K, Geier C, Osterziel KJ, Van der Ven PF and Furst DO Decreased interactions of mutant muscle LIM protein (MLP) with N-RAP and alpha-actinin and their implication for hypertrophic cardiomyopathy. Cell Tissue Res 317 (2004), pp. 129–36. [DOI] [PubMed] [Google Scholar]
- Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, Cardim N, Wenzel K, Erdmann B, Krackhardt F, Posch MG, Osterziel KJ, Bublak A, Nagele H, Scheffold T, Dietz R, Chien KR, Spuler S, Furst DO, Nurnberg P and Ozcelik C Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet 17 (2008), pp. 2753–65. [DOI] [PubMed] [Google Scholar]
- Geier C, Perrot A, Ozcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PF, Furst DO, Vornwald A, von Hodenberg E, Nurnberg P, Scheffold T, Dietz R and Osterziel KJ Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 107 (2003), pp. 1390–5. [DOI] [PubMed] [Google Scholar]
- Grubinger M and Gimona M CRP2 is an autonomous actin-binding protein. FEBS Lett 557 (2004), pp. 88–92. [DOI] [PubMed] [Google Scholar]
- Gupta MP, Samant SA, Smith SH and Shroff SG HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 283 (2008), pp. 10135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H and Wollert KC Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci U S A 102 (2005), pp. 1655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Wollert KC, Osinska H, Sargent MA, York AJ, Robbins J and Molkentin JD Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J Mol Cell Cardiol 48 (2010), pp. 1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J and Litt M Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 1 (2008), pp. 21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Moreau F, Moes M, Luthold C, Dieterle M, Goretti E, Neumann K, Steinmetz A and Thomas C Human muscle LIM protein dimerizes along the actin cytoskeleton and cross-links actin filaments. Mol Cell Biol 34 (2014), pp. 3053–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HS and Greenwood JA Glycine-rich region regulates cysteine-rich protein 1 binding to actin cytoskeleton. Biochem Biophys Res Commun 380 (2009), pp. 484–8. [DOI] [PubMed] [Google Scholar]
- Ji ZX, Du C, Wu GS, Li SY, An GS, Yang YX, Jia R, Jia HT and Ni JH Synergistic up-regulation of muscle LIM protein expression in C2C12 and NIH3T3 cells by myogenin and MEF2C. Mol Genet Genomics 281 (2009), pp. 1–10. [DOI] [PubMed] [Google Scholar]
- Kadrmas JL and Beckerle MC The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5 (2004), pp. 920–31. [DOI] [PubMed] [Google Scholar]
- Kemecsei P, Miklos Z, Biro T, Marincsak R, Toth BI, Komlodi-Pasztor E, Barnucz E, Mirk E, Van der Vusse GJ, Ligeti L and Ivanics T Hearts of surviving MLP-KO mice show transient changes of intracellular calcium handling. Mol Cell Biochem 342 (2010), pp. 251–60. [DOI] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M and Chien KR Muscle LIM protein in heart failure. Exp Clin Cardiol 7 (2002a), pp. 104–5. [PMC free article] [PubMed] [Google Scholar]
- Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP and Chien KR The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111 (2002b), pp. 943–55. [DOI] [PubMed] [Google Scholar]
- Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA and Chien KR A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res 106 (2010), pp. 695–704. [DOI] [PubMed] [Google Scholar]
- Kong Y, Flick MJ, Kudla AJ and Konieczny SF Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol 17 (1997), pp. 4750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrat R, Weiskirchen R, Krautler B and Bister K Solution structure of the carboxyl-terminal LIM domain from quail cysteine-rich protein CRP2. J Biol Chem 272 (1997), pp. 12001–7. [DOI] [PubMed] [Google Scholar]
- Kontaxis G, Konrat R, Krautler B, Weiskirchen R and Bister K Structure and intramodular dynamics of the amino-terminal LIM domain from quail cysteine- and glycine-rich protein CRP2. Biochemistry 37 (1998), pp. 7127–34. [DOI] [PubMed] [Google Scholar]
- Kuhn C, Frank D, Dierck F, Oehl U, Krebs J, Will R, Lehmann LH, Backs J, Katus HA and Frey N Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res Cardiol 107 (2012), p. 262. [DOI] [PubMed] [Google Scholar]
- Lorenzen-Schmidt I, Stuyvers BD, ter Keurs HE, Date MO, Hoshijima M, Chien KR, McCulloch AD and Omens JH Young MLP deficient mice show diastolic dysfunction before the onset of dilated cardiomyopathy. J Mol Cell Cardiol 39 (2005), pp. 241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis HA, Pino JD, Schmeichel KL, Pomies P and Beckerle MC Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. J Biol Chem 272 (1997), pp. 27484–91. [DOI] [PubMed] [Google Scholar]
- Luther PK The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil 30 (2009), pp. 171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet M, van Berlo JH and Molkentin JD Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol 14 (2013), pp. 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mery A, Taghli-Lamallem O, Clark KA, Beckerle MC, Wu X, Ocorr K and Bodmer R The Drosophila muscle LIM protein, Mlp84B, is essential for cardiac function. J Exp Biol 211 (2008), pp. 15–23. [DOI] [PubMed] [Google Scholar]
- Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J Jr., Kranias EG, Giles WR and Chien KR Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99 (1999), pp. 313–22. [DOI] [PubMed] [Google Scholar]
- Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, Chrisco MA, Murphy RT, Lurie PR, Schwartz RJ, Elliott PM, Vatta M, McKenna W, Towbin JA and Bowles NE Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 80 (2003), pp. 207–15. [DOI] [PubMed] [Google Scholar]
- Newman B, Cescon D, Woo A, Rakowski H, Erikkson MJ, Sole M, Wigle ED and Siminovitch KA W4R variant in CSRP3 encoding muscle LIM protein in a patient with hypertrophic cardiomyopathy. Mol Genet Metab 84 (2005), pp. 374–5. [DOI] [PubMed] [Google Scholar]
- Olson EN and Williams RS Remodeling muscles with calcineurin. Bioessays 22 (2000), pp. 510–9. [DOI] [PubMed] [Google Scholar]
- Omens JH, Usyk TP, Li Z and McCulloch AD Muscle LIM protein deficiency leads to alterations in passive ventricular mechanics. Am J Physiol Heart Circ Physiol 282 (2002), pp. H680–7. [DOI] [PubMed] [Google Scholar]
- Papalouka V, Arvanitis DA, Vafiadaki E, Mavroidis M, Papadodima SA, Spiliopoulou CA, Kremastinos DT, Kranias EG and Sanoudou D Muscle LIM protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Mol Cell Biol 29 (2009), pp. 6046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Alvarado GC, Miles C, Michelsen JW, Louis HA, Winge DR, Beckerle MC and Summers MF Structure of the carboxy-terminal LIM domain from the cysteine rich protein CRP. Nat Struct Biol 1 (1994), pp. 388–98. [DOI] [PubMed] [Google Scholar]
- Peter AK, Cheng H, Ross RS, Knowlton KU and Chen J The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog Pediatr Cardiol 31 (2011), pp. 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel R, Vakeel P, Topczewski J, Knoll R and Bakkers J Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Dev Biol 318 (2008), pp. 92–101. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J Jr., Lefkowitz RJ and Koch WJ Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A 95 (1998), pp. 7000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoudou D, Corbett MA, Han M, Ghoddusi M, Nguyen MA, Vlahovich N, Hardeman EC and Beggs AH Skeletal muscle repair in a mouse model of nemaline myopathy. Hum Mol Genet 15 (2006), pp. 2603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallus T, Edlich C, Stier G and Muhle-Goll C 1H, 13C, and 15N assignment of the muscular LIM protein MLP/CRP3. Biomol NMR Assign 1 (2007), pp. 41–3. [DOI] [PubMed] [Google Scholar]
- Schallus T, Feher K, Ulrich AS, Stier G and Muhle-Goll C Structure and dynamics of the human muscle LIM protein. FEBS Lett 583 (2009), pp. 1017–22. [DOI] [PubMed] [Google Scholar]
- Schmeichel KL and Beckerle MC The LIM domain is a modular protein-binding interface. Cell 79 (1994), pp. 211–9. [DOI] [PubMed] [Google Scholar]
- Schmeichel KL and Beckerle MC Molecular dissection of a LIM domain. Mol Biol Cell 8 (1997), pp. 219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira V, Nijenkamp LL, Regan JA and van der Velden J The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta 1838 (2014), pp. 700–22. [DOI] [PubMed] [Google Scholar]
- Su Z, Yao A, Zubair I, Sugishita K, Ritter M, Li F, Hunter JJ, Chien KR and Barry WH Effects of deletion of muscle LIM protein on myocyte function. Am J Physiol Heart Circ Physiol 280 (2001), pp. H2665–73. [DOI] [PubMed] [Google Scholar]
- Tran TC, Singleton C, Fraley TS and Greenwood JA Cysteine-rich protein 1 (CRP1) regulates actin filament bundling. BMC Cell Biol 6 (2005), p. 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafiadaki E, Arvanitis DA, Papalouka V, Terzis G, Roumeliotis TI, Spengos K, Garbis SD, Manta P, Kranias EG and Sanoudou D Muscle lim protein isoform negatively regulates striated muscle actin dynamics and differentiation. FEBS J 281 (2014), pp. 3261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch BJ, van den Burg CM, Schoonderwoerd K, Lindsey PJ, Scholte HR, de Coo RF, van Rooij E, Rockman HA, Doevendans PA and Smeets HJ Regional absence of mitochondria causing energy depletion in the myocardium of muscle LIM protein knockout mice. Cardiovasc Res 65 (2005), pp. 411–8. [DOI] [PubMed] [Google Scholar]
- von der Hagen M, Laval SH, Cree LM, Haldane F, Pocock M, Wappler I, Peters H, Reitsamer HA, Hoger H, Wiedner M, Oberndorfer F, Anderson LV, Straub V, Bittner RE and Bushby KM The differential gene expression profiles of proximal and distal muscle groups are altered in pre-pathological dysferlin-deficient mice. Neuromuscul Disord 15 (2005), pp. 863–77. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R and Gunther K The CRP/MLP/TLP family of LIM domain proteins: acting by connecting. Bioessays 25 (2003), pp. 152–62. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, Pino JD, Macalma T, Bister K and Beckerle MC The cysteine-rich protein family of highly related LIM domain proteins. J Biol Chem 270 (1995), pp. 28946–54. [DOI] [PubMed] [Google Scholar]
- Wilson AJ, Schoenauer R, Ehler E, Agarkova I and Bennett PM Cardiomyocyte growth and sarcomerogenesis at the intercalated disc. Cell Mol Life Sci 71 (2014), pp. 165–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winokur ST, Chen YW, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y and Flanigan KM Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet 12 (2003), pp. 2895–907. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Akazawa H, Ito K, Toko H, Sano M, Yasuda N, Qin Y, Kudo Y, Sugaya T, Chien KR and Komuro I Angiotensin II type 1a receptor signals are involved in the progression of heart failure in MLP-deficient mice. Circ J 71 (2007), pp. 1958–64. [DOI] [PubMed] [Google Scholar]
- Yao X, Perez-Alvarado GC, Louis HA, Pomies P, Hatt C, Summers MF and Beckerle MC Solution structure of the chicken cysteine-rich protein, CRP1, a double-LIM protein implicated in muscle differentiation. Biochemistry 38 (1999), pp. 5701–13. [DOI] [PubMed] [Google Scholar]
- Zheng Q and Zhao Y The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell 99 (2007), pp. 489–502. [DOI] [PubMed] [Google Scholar]
- Zolk O, Caroni P and Bohm M Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation 101 (2000), pp. 2674–7. [DOI] [PubMed] [Google Scholar]