

Abstract
The retina being a part of the central nervous system consumes large amounts of glucose and oxygen to generate ATP for its visual function. During ATP generation in the mitochondrial electron transport chain, mitochondrial Reactive Oxygen Species (mtROS) is generated as a byproduct. Although anti-oxidants are present in the mitochondrion to counter free radicals, excess mtROS causes damage to mitochondrial proteins, mtDNA, and membrane lipids. Furthermore, damaged mitochondria are inefficient in ATP production but continue to release ROS. Mitochondrial components, when released into the cytosol, are recognized as Danger-Associated Molecular Patterns (DAMPS) by pattern recognition NOD-like receptors including the NLRP3 inflammasome. NLRP3 inflammasomes process inactive pro-caspase-1 to an active caspase-1, which cleaves pro-inflammatory IL-1β to mature IL-1β causing inflammation and premature cell death. To counter the damaging action of mtROS and inflammasomes in fully differentiated retinal cells, the removal of dysfunctional mitochondria is needed by mitophagy, a specific form of lysosomal degradation via autophagy. Nonetheless, mitophagy deregulation, lysosome destabilization and NLRP3 inflammasome activations occur in Diabetic Retinopathy (DR) causing chronic inflammation and disease progression. Recently, the Thioredoxin-interacting protein, TXNIP, has been shown to be induced strongly by high glucose and diabetes inhibiting the anti-oxidant function of Thioredoxin. Subsequently, TXNIP causes mitochondrial dysfunction, oxidative stress, mitophagy deregulation, lysosome destabilization and inflammation in DR. Therefore, gene therapies targeting TXNIP, NLRP3 and/or the redox system have potentials to prevent/slow down retinal damages in DR.
Keywords: Diabetic Retinopathy, Gene Therapy, Lysosomal Destabilization, Mitophagic Flux, NLRP3 Inflammasome, TXNIP
Introduction
Diabetic Retinopathy (DR) is the number one cause of ocular complications and blindness both in Type 1 and Type 2 diabetes mellitus in developed countries. Additionally, due to increases in the number of obese people around the world including developing countries, individuals with diabetes and its complications are considered to increase significantly in coming years. Initially, DR was considered to be a microvascular complication involving capillary leakage due to breakdown of the inner blood-retinal barrier, pericyte dropout and proliferation of weak blood vessels (neovascularization) [1]. However, recent studies support the idea that retinal neurodegeneration occurs in early DR, which may influence microvascular abnormalities leading to blindness [2]. Nonetheless, a direct correlation between microvascular dysfunction and neurodegeneration in DR is yet to be established. It is also not fully understood which one of the two events (microvascular or neuronal dysfunction) occurs first. Therefore, so far, an effective therapeutic method has not been successfully devised to prevent or slow down the progression of DR, indicating finding new potential targets are necessary.
TXNIP and Diabetic Retinopathy
Thioredoxin-Interacting Protein (TXNIP) is strongly induced by diabetes and high glucose in all tissues examined including the retina [3-6]. TXNIP binds to and inhibits the antioxidant and thiol reducing capacity of Thioredoxin (Trx). Trx1 is found in the cytosol and nucleus while Trx2 is the mitochondrial isoform. Therefore, TXNIP has been defined as a pro-oxidative stress, pro-inflammatory and pro-apoptosis protein in diabetes and its complications. TXNIP is known to localize in all cellular compartments including the mitochondrion [7]. TXNIP is significantly induced in the diabetic rat retina and inhibition of TXNIP by intravitreal siRNA delivery reduces abnormalities of early DR including capillary basement membrane thickening, gliosis, and neuronal injury [3]. TXNIP also mediates high glucose-induced cellular oxidative stress, mitochondrial dysfunction and mitophagy deregulation in retinal Muller cells [6]. Furthermore, TXNIP has been shown to involve in the assembly of cytosolic NOD-like receptor, the NLRP3 Inflammasome under oxidative stress [7]. The NLRP3 Inflammasome activates pro-caspase-1 to caspase-1, which further processes pro-IL-1β to an active IL-1β. IL-1β is a cytokine that initiates innate immune responses and induces various cytokines including TNF-α IL-6 and CXC chemokines [8]. Therefore, TXNIP silencing may be an approach to reduce early retinal defects in diabetes and slow down or prevent the progression of ocular complications.
TXNIP, Mitochondrial Dysfunction and Mitophagy
Mitochondria are the major source of cellular energy (generation of ATP) by oxidative phosphorylation in the electron transport chain. During this process, elections leak which are captured by molecular oxygen generating Reactive Oxygen Radicals or Species (ROS). These mtROS damages mitochondrial components such as the mtDNA, proteins, and membrane lipids. In order to circumvent such damages, the mitochondrion contains several anti-oxidant proteins including GSH, SOD2, Trx2, and others. However, under chronic diseases and stress, the anti-oxidant capacity is over whelmed and mitochondrial membrane depolarization occurs. Damaged mitochondria release ROS but are ineffective in ATP production.
We recently showed that TXNIP is found in mitochondria and causes mitochondrial dysfunction in retinal cells under high glucose enviromnent [6]. Removal or segregation of the damaged mitochondria by mitophagy, a process of lysosomal degradation via autophagy, is critical for cell viability (Figure 1). Therefore, damaged mitochondria undergo fission/fragmentation, which involves proteins such as Drp1 and Fis1. These fragmented mitochondria are flagged by Pink1 and Parkin via ubiquitination of mitochondrial membrane proteins including VDAC1 and Mfn2. Then, the fragmented and tagged mitochondria are engulfed by an autophagophore containing LC3BII and ubiquitin adaptors such as optineurin and p62/Sequestosome 1 (SQSTM1). The initial formation of LC3BII phagosomes require multiple factors or proteins called Autophagy-Related Genes (ATGs) [9]. Among these, ATG4B has been shown to be involved in autophagophore generation, which involves TXNIP and REDD1 complex formation and redox regulation of ATG4B [10].
Figure 1:
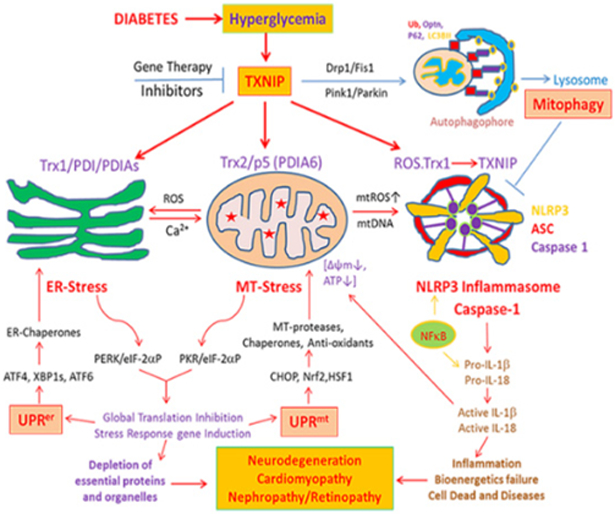
A potential role for TXNIP in hyperglycemia-induced oxidative stress, ER-stress, mitochondrial dysfunction, mitophagic flux and NLRP3 inflammasome assembly in diabetes and its complications including DR.
Once damaged mitochondria are completely engulfed by a double-membrane autophagophore, lysosomes fuse with them to form autolysosome. Lysosomal proteolytic enzymes such as cathepsins degrade mitochondrial components and cytosolic aggregates to their molecular components for recycling as nutrients. Autophagy and mitophagy processes are highly complexed cellular stress response and defense mechanism to maintain cellular homeostasis and survival. However, both under acute and chronic diseases, the process is derailed and cellular damage and disease progression occur.
Mitophagic Flux Deregulation, Lysosome Destabilization and Inflammasome Activation
As mentioned before TXNIP is highly induced by diabetes and high glucose and inhibits Trx1 and Trx2 causing cellular oxidative stress. Stressed mitochondria release ROS into the Endoplasmic Reticulum (ER) via the Mitochondria-Associated ER Membrane (MAM) and causes ER-stress [11,12]. In addition, TXNIP also interacts with protein disulfide isomerases (PDIs) in ER lumens and induces protein misfolding thereby evoking an ER-stress response (UPRer) [13]. In turn, ER-stress and UPRer increase calcium release into mitochondria via MAM further enhancing mitochondrial oxidative stress and evokes a mitochondrial stress response (UPRmt) (Figure 1). While the UPRer mechanism is well studied, the UPRmt is less known. Whether it begins with the mitochondrial stress or ER-stress, a vicious cycle of ER-mitochondrial injury is generated. Furthermore, uncontrolled UPRer itself induces an interaction between TXNIP and NLPR3 inflammasome leading to inflammation and cell death. This area of UPRer has been studied more extensively in pancreatic beta cell death in diabetes and innate immunity [13] and will not be a part of the present article.
Mitochondria are also main sites for cellular heme biosynthesis and several mitochondrial TCA cycle enzymes and electron transport chain complexes require Fe-S complexes for their holoenzyme assembly [14]. Therefore, when there is excess mitophagic flux to lysosomes, the accumulation of damaged mitochondria with heme complexes occur resulting in lysosomal enlargement and deficiencies in lysosomal hydrolytic enzyme activities (Figure 2). Furthermore, an accmnulation of free iron in lysosomes will result in increases in the Fenton reaction [15]. which releases reactive hydroxyl ions damaging the lysosomal membrane. We have also observed mitophagic flux and lysosomal enlargement in human retinal pigment epithelial cells under high glucose conditions and TXNIP silencing prevents these events (unpublished data). Under these conditions, lysosomal enzymes such as cathepsins B and L may leak out into the cytosol and act on mitochondrial outer membrane proteins [16]. Thus, another vicious cycle of mitochondria-lysosome injury and oxidative stress is generated. Also mentioned earlier, defective or depolarized mitochondria are inefficient in ATP production but release mtROS and mtDNA, which facilitate an assembly of the NLRP3 inflammasome containing ASC and pro-caspase-1 [17]. Assembled NLRP3 inflammasomes activate pro-caspase-1 and caspase 1 further processes pro-IL-1β to an active IL-1β. IL-1β is considered as an important initiator of innate immune responses and inflammation via activation of downstream cytokines and chemokines [18,19].
Figure 2:
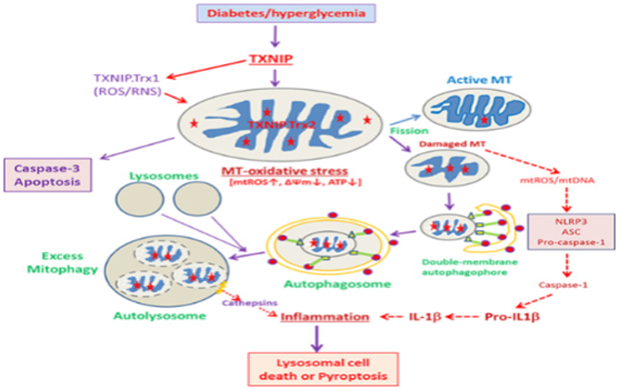
An illustration as to how excess mitophagic flux leads to lysosome enlargement, destabilization and inflammation under high glucose and TXNIP overexpression in diabetes and its complications including DR. Either a slow or excess mitophagic flux may lead to accumulation of damaged mitochondria or enlarged lysosomes leading to apoptosis, pyroptosis or autophagic/lysosomal cell death.
Lysosomal membrane permeabilization further leads to increased lysosomal pH and inactivation of acidic lysosomal hydrolytic enzymes, which subsequently may result in an incomplete digestion of mtDNA and create fragments of double-stranded CpG DNA [20]. These unmethylated mitochondrial CpG DNAs are recognized by lysosomal membrane Toll-like receptors such as TLR9 [20] and induce pro-inflammatory cytokine expression such as pro-IL-1β and Pro-IL-18. In addition, lysosomal cathepsins may also be able to process pro-IL-1β and Pro-IL-18 to their active forms and evoke inflammatory responses. Thus, several cell mechanisms under stress are integrated with lysosomal stress responses [21]. It is known that a mild lysosomal stress results in a lysosome-nuclear communication via translocation of lysosome-associated transcription factors such as TFEB and TFE3 to the nucleus and induce gene expressions for lysosomal proteins and ATGs [22]. Nonetheless, excess mitophagy flux will lead to lysosomal enlargement, destabilization and membrane permeability and therefore these defective lysosomes can’t properly fuse with autophagosomes. Eventually, damaged mitochondria, autophagosomes and endosomes accumulate in the cytosol. Such events lead to cytosolic crowding, loss of sanctity and premature cell death.
Potentials of Gene Therapy for DR Using Nucleic Acid Constructs Bearing A TXNIP Promoter
TXNIP is strongly induced by diabetes and inhibited by insulin and IGF1 [3-6,23,24]. In diabetes, Type 1 (insulin deficiency) or Type2 (insulin resistance), hyperglycemia prevails and induces sustained TXNIP up-regulation [3]. TXNIP silencing or knocking out prevents various cellular dysfunctions and increases cell viability under high glucose environment in the retina [24]. The transcription factors responsible for the induction of TXNIP mRNA include Mondo A and ChREBP as well as transcriptional co-factor p300 [5, 25-27]. Therefore, TXNIP silencing by Post- Transcriptional Gene Silencing siRNAs (PTGS) targeting the mRNA or promoter-targeted transcriptional gene silencing by double-stranded siRNAs to gene promoters (TGS) prevents early abnormalities of DR [5]. Furthermore, CRISPR/Cas9 and TXNIP gRNA reduces mitochondrial damage and mitophagic flux in rat retinal Muller cells [6]. We have further shown that the TXNIP promoter exists as an opened and poised configuration that high glucose and histone deacetylase inhibitors activate TXNIP transcription strongly and immediately [3,5].
We, therefore, propose that nucleic acid constructs containing the proximal promoter sequences from the transcriptional start site linked to a gene of interest and/or siRNAs could be constructed for gene therapies using various vectors such as pcDNA3.1, Adeno-Associated Vectors (AAVs) or lentivirus (Figure 3) [28]. AAV serotypes, such as AAV2, AAV9 and AAV2/8, exist in nuclei as episomes upon transduction as compared to lentivirus that incorporates into the genome therefore, AAV vectors have been used to deliver genes efficiently in the retina via an intravitreal or sub retinal injection method [29,30]. Such TXNIP promoter gene constructs can sense elevated glucose environments and induce the linked gene expression while maintaining a basal expression under normal glucose levels.
Figure 3:
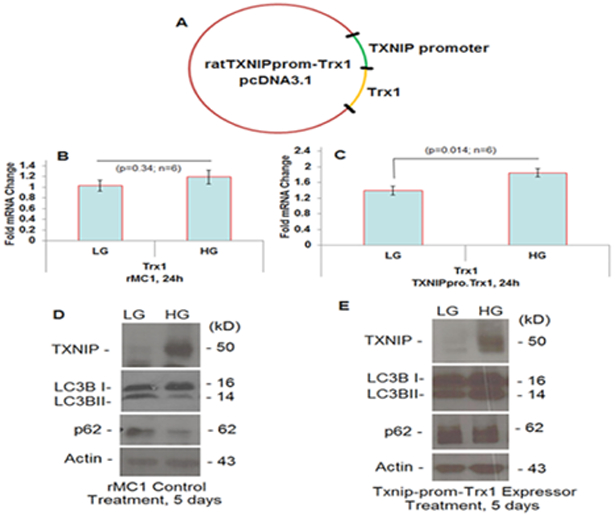
Nucleic acid construct containing a rat TXNIP promoter linked to a rat Trx1 cDNA(in a pcDNA3.1 vector) and transfected in rMC1 cells and its effects on TXNIP and autophagic receptor levels were examined. [28]
For initial experiments, we prepared a rat TXNIP promoter (−1 to −1597 bp; Gene ID: 117514) linked with a rat Trx1 cDNA (NM_053800.3) and examined its expression in a rat Muller cell line, rMC1 (Figure 3) [28]. These transfected cells when treated with high glucose have higher Trx1 mRNA levels than low glucose conditions while Trx1 message was unchanged in control rMC1 cells. On the other hand, TXNIP is strongly induced by high glucose in control rMC1 cells than in low glucose, which also correlates with decreases in the level of autophagy markers - LC3BII and p62/Sequestosome1. However, in the TXNIP-prom-Trx1 expressing rMC1 cells, the TXNIP level is marginally down under high glucose (than that observed in control rMC1) while both LC3BII and p62 levels are increased. These observations provide an initial proof of concept that the TXNIP promoter can be linked with a gene of interest or siRNA that will respond under diabetic conditions. In addition, an anti-sense long noncoding RNA targeted against the sense TXNIP promoter may also be employed to regulate TXNIP and the redox system (Figure 4) [28]. Further studies will be conducted to test effectiveness of these and other TXNIP-promoter constructs both in vitro and in vivo systems.
Figure 4:
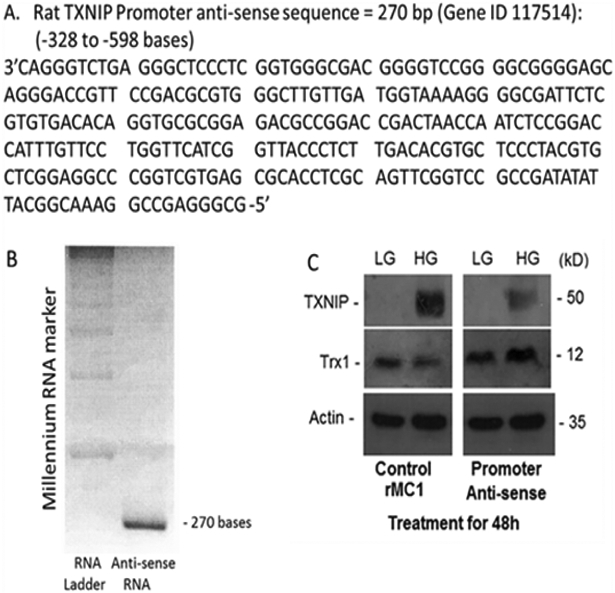
A long noncoding anti-sense RNA (270 bases) targeted to the sense DNA of the proximal TXNIP promoter, based on the anti-sense DNA sequence, was generated and transfected in rMC1 cells and its effects on TXNIP and Trx1 were examined. [28]
Conclusion
Diabetic retinopathy is a complex disease developed after a prolonged exposure to hyperglycemia in diabetes. Therefore, its etiology and disease mechanisms are multifaceted. Currently, it is recognized that both the microvasculature and neuroretina are affected in DR. Therefore, identifying disease causing and/or stage specific disease-associated abnormalities will be of critical important. While there are a few treatment methods available for DR patients, there is no satisfactory outcome. Hence, a search for and development of new gene targets and improved treatment methods continues to remain open. TXNIP is strongly up-regulated by diabetes and high glucose in all tissues tested including the retina as long as hyperglycemia prevails in the system. Furthermore, TXNIP targets multiple cellular metabolic pathways including the redox system, glucose transporters, and membrane receptors at multiple organelles [3-6]. These actions of TXNIP cause cellular oxidative stress, inflammation, neurovascular damage and potentially the progression of DR. Therefore, we advocate that TXNIP itself, its redox partners, and/or the NLRP3 inflammasome constitute effective gene therapy targets. In this regard, nucleic acid constructs containing the TXNIP promoter linked with a target gene or an inhibitory RNA packaged in AAV vectors may be practical for gene therapies in DR.
Acknowledgement:
Funding from NIH/NEI: R01 EY023992 (LPS); NIH Core Grant: P30EY004068 and Research to Prevent Blindness (RPB) unrestricted grant to the Department of Ophthalmology, Visual and Anatomical Sciences (OVAS) are acknowledged.
References
- 1.Duh EJ, Sun JK, Stitt AW (2017) Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2: 93571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roy S, Kern TS, Song B, Stuebe C (2017) Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am J Pathol 187: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh LP, Perrone L (2013) Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J Clin Exp Ophthalmol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP (2009) Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol 221: 262–272. [DOI] [PubMed] [Google Scholar]
- 5.Perrone L, Devi TS, Hosoya Kl, Terasaki T, Singh LP (2010) Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 1: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devi TS, Somayajulu M, Kowluru RA, Singh LP (2017) TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis 8: e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxena G, Chen J, Shalev A (2010) Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. The Journal of biological chemistry 285: 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh LP, Devi TS, Yumnamcha T (2017) The Role of Txnip in Mitophagy Dysregulation and Inflammasome Activation in Diabetic Retinopathy: A New Perspective. JOJ Ophthalmol 4: 555643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Redmann M, Darley-Usmar V, Zhang J (2016) The Role of Autophagy, Mitophagy and Lysosomal Functions in Modulating Bioenergetics and Survival in the Context of Redox and Phototoxic Damage: Implications for Neurodegenerative Diseases. Aging Dis 7:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D et al. (2015) A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun 6:7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rieusset J (2017) Role of Endoplasmic Reticulum-Mitochondria Communication in Type 2 Diabetes. Adv Exp Med Biol 997:171–186. [DOI] [PubMed] [Google Scholar]
- 12.Park SJ, Lee SB, Suh Y, Kim SJ, Lee N et al. (2017) DISC1 Modulates Neuronal Stress Responses by Gate-Keeping ER-Mitochondria Ca2+ Transfer through the MAM. Cell Rep 21: 2748–2759. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Min Kim S, Dotimas J, Li L, Feener EP et al. (2014) Thioredox-in-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol Med 6: 732–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barupala DP, Dzul SP, Riggs-Gelasco PJ, Stemmier TL (2016) Synthesis, delivery and regulation of eukaryotic heme and Fe-S cluster cofactors. Arch Biochem Biophys 592: 60–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terman A, Kurz T (2013) Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 18: 888–898. [DOI] [PubMed] [Google Scholar]
- 16.Jaishy B, Abel ED (2016) Lipids, lysosomes, and autophagy. J Lipid Res 57: 1619–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rovira-Llopis S, Apostolova N, Bañuls C, Muntané J, Rocha M et al. (2018) Mitochondria, the NLRP3 Inflammasome, and Sit-ins in Type 2 Diabetes: New Therapeutic Targets. Antioxid Redox Signal. [DOI] [PubMed] [Google Scholar]
- 18.De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A et al. (2016) Autophag osome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol 18: 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KD et al. (2012) TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res 2012: 438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindqvist LM. (2016) Can the cargo control the car? Mitochondrial DNA as a stimulator of TLR9-mediated autophagosomes-lysosome fusion. Cell Death Differ 23: 1737–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aits S, Jäättelä M, Nylandsted J (2015) Methods for the quantification of lysosomal membrane permeabilization: a hallmark of lysosomal cell death. Methods Cell Biol 126: 261–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nabar NR, Kehrl JH (2017) The Transcription Factor EB Links Cellular Stress to the Immune Response. Yale J Biol Med 90: 301–315. [PMC free article] [PubMed] [Google Scholar]
- 23.Thielen L, Shalev A (2018) Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. CurrOpinEndocrinol Diabetes Obes 25: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan J, Du C, Shi Y, Liu D, Ma J (2018) Thioredoxin-interacting protein deficiency ameliorates diabetic retinal angiogenesis. Int J Biochem Cell Biol 94: 61–70. [DOI] [PubMed] [Google Scholar]
- 25.Stoltzman CA, Kaadige MR, Peterson CW, Ayer DE (2011) MondoA senses non-glucose sugars: regulation ofthioredoxin-interacting protein (TXNIP) and the hexose transport curb. J Biol Chem 286:38027–38034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cha-Molstad H, Saxena G, Chen J, Shalev A. (2009) Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem 284: 16898–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Marinis Y, Cai M, Bompada P, Atac D, Kotova O et al. (2016) Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 89: 342–353. [DOI] [PubMed] [Google Scholar]
- 28.Pukhrambam LS (2017) nucleic acid constructs including a Txnip promoter for the treatment of disease. US Patent Application 20170252376A1.
- 29.Khabou H, Garita-Hernandez M, Chaffiol A, Reichman S, Jaillard C, et al. (2018) Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight 3: 96029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seitz IP, Michalakis S, Wilhelm B, Reichel FF, Ochakovski GA et al. (2017) Superior Retinal Gene Transfer and Bio distribution Profile of Sub retinal Versus Intravitreal Delivery ofAAV8 in Nonhuman Primates. Invest Ophthalmol Vis Sci 58: 5792–5801 [DOI] [PubMed] [Google Scholar]