

Abstract
Purpose
We develop an accessible and reliable RNA sequencing (RNA-seq) transcriptome database of healthy human eye tissues and a matching reactive web application to query gene expression in eye and body tissues.
Methods
We downloaded the raw sequence data for 1375 RNA-seq samples across 54 tissues in the Genotype-Tissue Expression (GTEx) project as a noneye reference set. We then queried several public repositories to find all healthy, nonperturbed, human eye-related tissue RNA-seq samples. The 916 eye and 1375 GTEx samples were sent into a Snakemake-based reproducible pipeline we wrote to quantify all known transcripts and genes, removes samples with poor sequence quality and mislabels, normalizes expression values across each tissue, perform 882 differential expression tests, calculate GO term enrichment, and output all as a single SQLite database file: the Eye in a Disk (EiaD) dataset. Furthermore, we rewrote the web application eyeIntegration (available in the public domain at https://eyeIntegration.nei.nih.gov) to display EiaD.
Results
The new eyeIntegration portal provides quick visualization of human eye-related transcriptomes published to date by database version, gene/transcript, 19 eye tissues, and 54 body tissues. As a test of the value of this unified pan-eye dataset, we showed that fetal and organoid retina are highly similar at a pan-transcriptome level, but display distinct differences in certain pathways and gene families, such as protocadherin and HOXB family members.
Conclusions
The eyeIntegration v1.0 web app serves the pan-human eye and body transcriptome dataset, EiaD. This offers the eye community a powerful and quick means to test hypotheses on human gene and transcript expression across 54 body and 19 eye tissues.
Keywords: app, geneexpression, database, transcriptome
From anterior to posterior along the light trajectory, the human eye is composed of the cornea, lens, retina, RPE, and choroid. The differentiation, maturation, and function of these tissues is mediated through spatial- and temporal-specific transcript and gene expression patterns, also known as the transcriptome. Today, RNA-sequencing (RNA-seq) is the predominant technology for quantifying the transcriptome. Analysis of the transcripts' expression across tissue, time, and perturbation allows researchers to decipher the genetic controls of eye development and function. To this end, a wide variety of human tissue sources have been used to assess gene function, including primary tissue (fetal and postmortem), differentiated stem cells, immortalized cell lines, and most recently, organoids. These tissue types have been deeply sequenced across the cornea,1–7 lens,8 retina,9–17 and RPE (choroid).14,17–34
The Genotype-Tissue Expression (GTEx) Gene Expression Web App Lacks Eye-Specific Tissues
The GTEx Project has generated RNA-seq data across dozens of postmortem human tissues from hundreds of unique donors, and presents the gene and transcript level data in a comprehensive and user-friendly web app (available in the public domain at https://gtexportal.org/); however eye tissues have not been included.35,36 Recently Ratnapriya et al.37 reported on a huge set of postmortem retina, normal and with varying degrees of AMD and the GTEx project is providing the data as a download link. These data, as of June 2019, are not available in the interactive GTEx visualizations.37 The Sequence Read Archive (SRA) and European Nucleotide Archive (ENA) are the primary repositories for all raw sequence data and two groups have quantified large portions of the RNA-seq data, including some human eye tissues, from the SRA: recount2 and ARCHS4.38,39 To date, no curation of the sample level metadata has been done; therefore, it is challenging to parse out which eye tissues are present and even more difficult to determine whether any samples were chemically or genetically perturbed. More targeted web resources that allow researchers to quickly assess gene expression in eye tissues include iSYTE, EXPRESS, and retina.Tigem.it.16,40,41 However iSYTE only includes lens samples, EXPRESS is limited to a subset of mouse lens and retina samples, and retina.Tigem.it is retina only. Thus, we aimed our efforts at developing an easily accessible and reliable RNA-seq based transcriptome database of healthy human eye tissues and a matching reactive web application to query gene expression in eye and body tissues.
The eyeIntegration App Interactively Serves Huge GTEx and Human Eye Tissue Datasets (Eye in a Disk [EiaD])
The eyeIntegration web resource (available in the public domain at https://eyeIntegration.nei.nih.gov), originally released in 2017 at version 0.6, provides the largest set of transcriptomes from hand-curated human eye tissues along with hundreds of GTEx tissue samples.42 This interactive web app allows for quick transcript and gene comparisons across many eye tissues and dozens of other body tissues. The dataset that the original eyeIntegration web app served was created with a series of scripts, several of which were run interactively to manually assess quality control for the samples. The interactive nature of some of the steps precluded efficient and regular data updates for the data.
To better meet the needs of the eye research community we have rewritten the bioinformatic pipeline that creates the eye and body RNA-seq dataset to allow for regular, versioned updates for eyeIntegration. We call this reproducible and versioned transcriptome dataset “Eye in a Disk” (EiaD). The pipeline automates the EiaD creation, ensures full reproducibility of the results, allow for external data comparison, provides consistent sample quality control, and improves efficiency for future sample updates. The 2019 EiaD dataset contains several new tissue types, full gene product quantification, along with hundreds of new samples and improved sample labeling. The eyeIntegration web app also has been rewritten to provide many new features, including versioned EiaD datasets, custom URL shortcut creation, new visualizations, improved data table searching, easy download of core datasets, and local install of the entire interactive resource with three commands. Additionally, we are prototyping new tools to display single cell RNA-seq (scRNA-seq) data to provide researchers access to cell type–specific information about gene expression across murine retinal development.
The EiaD Dataset Can Be Used to Identify Potential Avenues to Improve Retina Organoid Maturation
Retina organoids are an increasingly popular means to model human retina development. We used our pan-study EiaD dataset to show that, at a pan-transcriptome level, organoids are highly similar to early fetal retina tissue. We also showed that important temporal gene expression patterns in the fetal retina tissue are recapitulated in the organoids. As the organoid differentiation methods do not yet produce fully mature retina, we focused on identifying differentially expressed processes between organoid retina and embryonic retina, and detected, for example, identifying protocadherin and HOXB family gene expression differences that suggest targetable pathways to improve and benchmark organoid differentiation methods.
Methods
Identification of Potential Eye Samples
We exhaustively searched the SRA with the SRAdb R package for eye related tissues using the query ‘cornea|retina|RPE|macula|fovea|choroid|sclera|iris|lens|eye' across all columns and rows in the ‘SRA' table.43,44 As the SRAdb is being deprecated, we also ran searches on the SRA and Gene Expression Omnibus (GEO) web pages with as follows: ((“Homo sapiens”[orgn:__txid9606]) AND (transcriptomic[Source]) AND (“2019/01/01”[Publication Date] : “3000”[Publication Date]) AND (retina[Text Word] OR RPE[Text Word] OR macula[Text Word] OR fovea[Text Word] OR choroid[Text Word] OR sclera[Text Word] OR iris[Text Word] OR lens[Text Word] OR cornea[Text Word] OR ‘trabecular meshwork'[Text Word] OR ‘canals of schlemm'[Text Word] OR ‘cillary body'[Text Word] OR ‘optic nerve'[Text Word] OR ‘laminar cribosa'[Text Word] OR retina[Title] OR RPE[Title] OR macula[Title] OR fovea[Title] OR choroid[Title] OR sclera[Title] OR iris[Title] OR lens[Title] OR cornea[Title] OR ‘trabecular meshwork'[Title] OR ‘canals of schlemm'[Title] OR ‘cillary body'[Title] OR ‘optic nerve'[Title] OR ‘laminar cribosa'[Title]))”. We hand selected relevant studies and selected healthy, control or unmodified samples spanning primary adult tissue, primary fetal tissue, induced pluripotent stem cell (iPSC)–derived tissue, stem cell–derived organoids, and immortalized cell lines. To compare gene expression in the eye against expression in other body tissues, we obtained samples from 54 different body tissues from the GTEx project. Using SRA metadata from each study, we extracted sample and run accessions, library type, tissue of origin, and subtissue of origin. Any of the preceding information missing from the SRA metadata was added by hand, when available. Stem cell-derived tissues and cell lines are marked as subtissues of the tissue they model.
Raw Data Download and Quantification
We downloaded the relevant SRA files for each sample directly from the NCBI ftp server using the file transfer software Aspera. SRA files were converted to FASTQ format using the tool fastq-dump from the SRAtoolkit software package.43 Samples only available in the BAM format were converted to FASTQ format using SAMTools.45 Sample transcriptomes were quantified using the alignment free quantification software Salmon, using transcriptomic index built from gencode v28 protein coding transcript sequences using the transcriptomic aligner Salmon.46,47 Using the resulting expression quantification, we identified lowly or unused transcripts within the gencode annotation, and removed transcripts that accounted for 5% or less of the total expression for its parent gene as per Soneson et al.48 Samples were requantified against a transcriptomic index built on the filtered transcript sequences. The Salmon count values were quantified as (transcript) length scaled Transcripts Per Million (TPM) to the transcript and gene level using tximport.49
Quality Control
We first removed samples with a Salmon calculated mapping rate less than 40%. This value was selected as being the far left tail of the distribution of mapping rates across samples (Supplementary Fig. S2). We removed lowly expressed genes by calculating the median expression across all samples for each gene and kept genes that had a median count >200 across all samples. To reduce the noise from experimental variability between each study, we normalized samples by sequence library size using the calcNormFactors function from the edgeR R package, and then quantile smoothed expression data using the R package qsmooth at the tissue level.50,51 In a change from our previous eyeIntegration work,42 we now correct our counts for mapping rate and tissue type with the limma batchEffects function.52 The transformed values are used for the box plot and t-SNE visualizations.
To identify outliers we followed an approach similar to a method reported by Wright et al.53 Briefly, we first selected the 3000 genes with the highest variance across all samples and then for each subtissue type 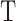 and each sample
and each sample 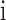 in
in 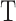 , we first calculated
, we first calculated 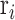 , the average correlation between
, the average correlation between 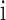 and all other samples in
and all other samples in 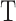 . Next, we calculated
. Next, we calculated 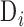 , where
, where 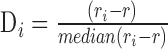 and
and 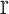 is the grand mean of all
is the grand mean of all 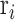 for
for 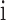 in
in 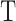 . We removed samples with
. We removed samples with 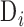 −17.5; we determined this threshold by generating a tSNE plot of our samples, and visually identifying outliers in adult retina tissue. The (
−17.5; we determined this threshold by generating a tSNE plot of our samples, and visually identifying outliers in adult retina tissue. The (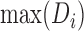 ) among these outliers was −17.58 and from this we chose −17.5 as our outlier threshold.
) among these outliers was −17.58 and from this we chose −17.5 as our outlier threshold.
To calculate Pearson correlation (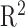 ) between GTEx-calculated TPM gene values and our GTEx TPM gene values, we downloaded “GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_tpm.gct.gz” and matched against our GTEx TPM values, running the Pearson correlation with
) between GTEx-calculated TPM gene values and our GTEx TPM gene values, we downloaded “GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_tpm.gct.gz” and matched against our GTEx TPM values, running the Pearson correlation with 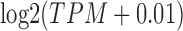 values as per Zhang et al. with the cor function in R.54
values as per Zhang et al. with the cor function in R.54
Differential Gene Expression Analysis and GO Term Enrichment
We used the nontransformed length scaled TPM values to determine differential gene expression between different subtissue types. First, we generated a synthetic body set to serve as single representative subtissue type for pan-body gene expression by randomly sampling GTEx tissues. We used the voom function from the limma R package to convert gene expression to precision weights, and then performed pairwise differential expression tests for all combinations of eye subtissues (using mapping rate as a covariate), the synthetic body tissue, and human body tissues using an empirical Bayes test.52,55 We extracted significant genes (FDR P < 0.01) for all 882 comparisons and used these to calculate GO enrichment. The significant gene list for each eye subtissue was split into upregulated and down regulated sets and each set was tested for enrichment using the enrichGO function from the clusterProfiler R package (q-value < 0.01).56
eyeIntegration Web App and R Package
The data generated in the above steps is consolidated into a SQLite database, with the original dataset for eyeIntegration and the new 2019 EiaD dataset each getting a separate database file. The code that creates the eyeIntegration web app is written in Shiny and R and has been wrapped into an R package (available in the public domain at https://github.com/davemcg/eyeIntegration_app/) that can be deployed on a local computer or a web server (available in the public domain at https://eyeIntegration.nei.nih.gov). The app can be deployed on a local computer with 50 GB of free disk space by running three commands in R: “devtools::install_github(‘davemcg/eyeIntegration_app')”, “eyeIntegrationApp::get_eyeIntegration_datasets()”, and “eyeIntegrationApp::run_eyeIntegration()”.
Snakemake Reproducible Pipeline
While the sample search and metadata parsing in a semicurated process, the processing from the raw data to the creation of the SQLite EiaD database underlying eyeIntegration is wrapped in a Snakemake pipeline, which ensures full reproducibility of the results.57 We make the code for the pipeline available at https://github.com/davemcg/EiaD_build.
scRNA-seq Processing
The eyeIntegration site, as of June 2019, hosts two large scRNA-seq datasets from Macosko et al.58 and Clark et al.59 We use the processed gene count data directly from each group, as well as their cluster assignments, which specify what cell type each individual cell is. The count data are mean averaged to the cell type, age, and gene level for the single cell expression section of eyeIntegration. We also displayed t-SNE and UMAP-based two-dimensional visualizations of the Macosko and Clark data, respectively, in the web app. For detail so the t-SNE processing we did on the Macosko dataset, see the methods of Bryan et al.42
Power Calculation
We used the ssizeRNA R package to calculate power (p) across samples (n) at an FDR of 0.05.60 Important parameters for ssize RNA include the variability (dispersions for the samples and genes), which were calculated directly from our EiaD length scaled TPM values by the edgeR packages estimateCommonDisp and estimateTagwiseDisp. The code to calculate the power is given as ‘power_calc.R.'
Manuscript as Code and Reproducibility
The figures, tables, and most numbers, are all created and laid out in an R markdown document that interweaves code and text. The knitr and pandoc program is used to lay out the figures and tables and output a docx file. The code that generates this study can be found at https://github.com/davemcg/eyeIntegration_v1_app_manuscript.
The relevant code-bases (https://github.com/davemcg/eyeIntegration_v1_app_manuscript, https://github.com/davemcg/EiaD_build) and the EiaD dataset itself have been deposited into Zenodo with accession 10.5281/zenodo.3238677 to ensure the data can be accessed in the future, even should eyeIntegration and GitHub become inaccessible in the future.
Results
EiaD 2019 Contains 24 New Human Eye RNA-seq Studies, 448 New Retina AMD Samples, 207 New Eye Samples, and 16 Total Eye Subtissue Types
Our query on May 8, 2019 to the SRA found 107 potentially relevant studies. We removed nonpertinent studies and selected healthy or unmodified tissue from each relevant study for a total, including 46 studies, 30 of which are new to the 2019 EiaD dataset. The 2019 EiaD dataset contains 835 human eye tissue samples and also includes 1314 GTEx samples across 54 tissues for easy comparison (Table; Supplementary Table S1). The 2019 EiaD contains six undifferentiated iPSC, 56 cornea, four lens, 648 retina, and 121 RPE (choroid) samples; in total we have added 655 new samples to the 2019 EiaD (Fig. 1). We refer to native-tissue extracted RPE as RPE (choroid) because it is not possible to remove the choroid from the RPE without culturing.
Table.
EiaD Contains a Large Set of Diverse Eye Tissues, Including Embryonic Stem Cells (ESC)
|
Tissue |
Pre-QC Count |
Count |
Subtissue Types (Count) |
| Cornea | 62 | 56 | Adult tissue (25), cell line endothelium (9), endothelium (16), fetal endothelium (2), stem cell endothelium (4) |
| ESC | 12 | 6 | Stem cell line (6) |
| Eye lid | 4 | 0 | |
| Lens | 9 | 4 | Stem cell line (4) |
| Retina | 681 | 648 | 3D organoid stem cell (52), adult tissue (107), adult tissue AMD mgs 2 (172), adult tissue AMD mgs 3 (112), adult tissue AMD mgs 4 (61), adult tissue mgs 1 (103), fetal eye (3), fetal tissue (35), RGC stem cell (3) |
| Retinal endothelium | 4 | 0 | |
| RPE | 144 | 121 | Adult tissue (48), cell line (50), fetal tissue (7), stem cell line (16) |
Eyelid and retina endothelium samples were included, but all failed to pass our QC filters.
Figure 1.

Substantial increase in eye tissue count and type from 2017 (180, lighter color) to 2019 (835, darker color) EiaD. We also improved the metadata labeling, the cornea samples (green) now delineates endothelial and epithelial tissues and the retina samples (orange) distinguish retina organoid and RGC from stem cells. Counts for each bar plot given in the boxes. The y-axis is a log2 transformed count of samples passing our QC filters.
Stem cell-derived cornea, stem cell-derived lens, and fetal retina are three new types of subtissues that are now available in EiaD. We also have substantially improved the granularity of the cornea tissue metadata, now delineating whether the tissue is from the endothelium or epithelium (Fig. 1); previously these had been grouped together as adult tissue. Another substantial addition to the 2019 EiaD are nonprotein coding genes; while protein-coding is the most common gene and transcript typse, there are dozens of different noncoding classes. The 2017 version of eyeIntegration only quantified protein coding genes and transcripts. We now quantify expression across 41 gene and 45 transcript types, including protein coding, retained intron, lincRNA, antisense, and pseuodogenes (Supplementary Table S2).
We also have added the large retina AMD postmortem Ratnapriya et al.37 cohort to EiaD 2019. This cohort contains hundreds of samples ranging from non-AMD (Minnesota Grading System [MGS] 1) to severe AMD (MGS 4). While eyeIntegration is intended to be a source for normal tissues, we have made an exception for this study, as this is a large cohort and AMD is a common disease. We found our corrections methods did not group the non-AMD Ratnapriya et al.37 samples with our other collected retina samples (see Retina MGS in Supplementary Fig. S3). This may be related to the lower mapping rate of the Ratnapriya et al.37 data (see Retina MGS in Supplementary Fig. S2).
467 More GTEx Samples and Nine New GTEx to 2019 EiaD
Our previous dataset for eyeIntegration version 0.6 held approximately 20 samples per GTEx tissue type. We ran power calculations to assess our ability to detected ≥1 log2(Fold Change) in gene expression between two conditions to determine whether this is a sufficient number of samples (Supplementary Fig. S4). Our calculations suggested, for example, that we had 83% power to detect a 1 log2(Fold Change) difference in gene expression with two groups of 20 samples. To increase our power to make significant eye-to-body comparisons, we added approximately 10 more samples per GTEx tissue types (which at 30 samples, would give approximately 90% power). We also took this opportunity to add bladder, bone marrow, cervix uteri, fallopian tube, ovary, prostate, testis, uterus, and vagina GTEx tissue samples (Supplementary Table S1).
Rigorous Quality Control and Reproducible Workflow System Ensures High Quality Transcriptomes That Consistently Cluster Together by Tissue Type
We built an automated pipeline for processing and analyzing all data for the web app using the program Snakemake, a python-based workflow management system that allows for efficient parallel execution of the analysis, facilitates reproduction by others, and simplifies long-term maintenance of the EiaD data (Fig. 2; Supplementary Fig. S5).57 To create a high quality final dataset across the 2291 initial samples (Supplementary Table S3) and 67,315,523,736 reads, we developed a rigorous quality control procedure as part of our analysis, considering a sample's read mapping rate and median count level as well as behavior relative to samples of the same subtissue type (see Methods). To identify tube mislabeling or sample extraction issues, we used sample-level gene correlation metrics (see Methods) to identify variability within samples of the same subtissue and ensure overall consistency in data processing (Fig. 3). After these steps 81 eye samples and 61 GTEx samples were removed.
Figure 2.
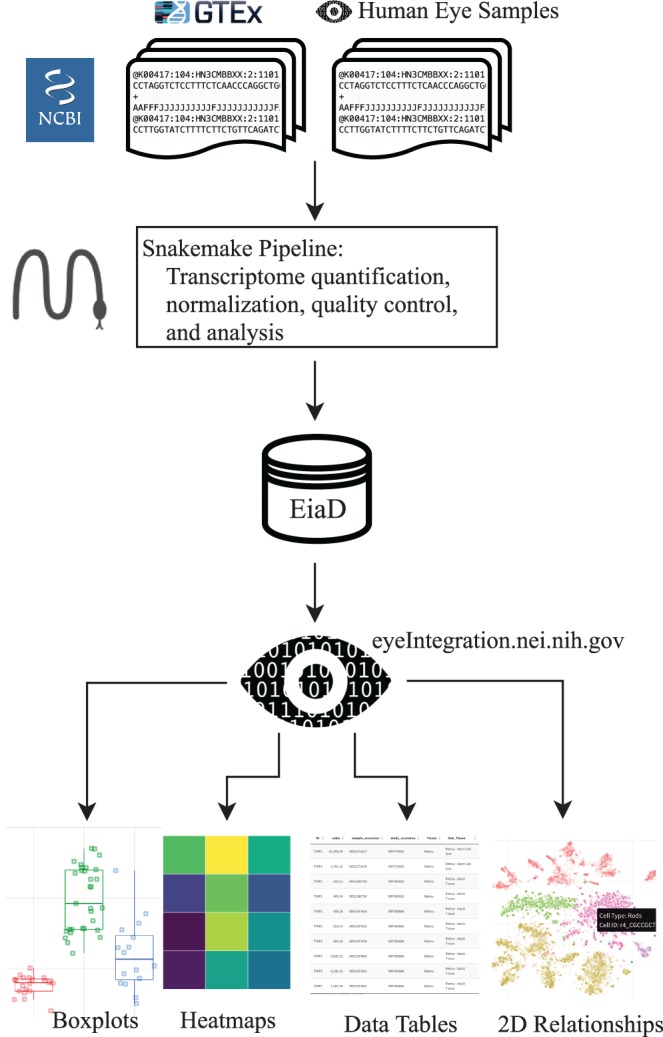
Raw RNA-seq data from the SRA is run through our pipeline to create the EiaD, which is used by eyeIntegration app to serve interactive gene expression visualizations across 73 tissues.
Figure 3.

t-SNE two-dimensional transcriptome profiles by sample demonstrate effective quality control and transcriptome processing. Colors match different tissue types and shapes of points define the origin of the tissues.
To ensure there are no substantial differences in quantification of gene TPM values, we calculated the 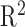 between GTEX and EiaD generated TPM values for our shared GTEx samples (see Methods); we computed an
between GTEX and EiaD generated TPM values for our shared GTEx samples (see Methods); we computed an 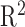 of 0.89. Zhang et al.54 reported that RNA-seq quantifications done between alignment-free methods (used in EiaD) and alignment-based methods (used by GTEx) get a
of 0.89. Zhang et al.54 reported that RNA-seq quantifications done between alignment-free methods (used in EiaD) and alignment-based methods (used by GTEx) get a 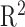 ranging from 0.89 to 0.93. As Zhang et al.54 compared quantification methods with identical gene references (we use Gencode GRCh38 gene models and GTEx uses hg19) and did not scale TPM score differently, our result falls in line with expectations.
ranging from 0.89 to 0.93. As Zhang et al.54 compared quantification methods with identical gene references (we use Gencode GRCh38 gene models and GTEx uses hg19) and did not scale TPM score differently, our result falls in line with expectations.
After our quality control and processing workflow, we found that samples of the same tissue type and origin cluster well together (Fig. 3; Supplementary Table S4). For example, in the retina group, primary adult tissue clusters tightly and distinctly from other cell types, and retinal organoids and fetal retina samples cluster together. Our ability to uniformly cluster data by known biological source independent of study origin demonstrates that our workflow can effectively account for technical variation between studies.
While t-SNE is a powerful algorithm for grouping samples, it is not consistent for determining the relationships between clusters.61 PCA is more useful in this regard. We ran a PCA dimensionality reduction (Supplementary Fig. S6) on all samples, finding that the eye tissues still generally group together and apart from all other human body tissues. Adult retina is most similar to the brain tissue. RPE and cornea are most similar to blood, bone marrow, and skin.
The eyeIntegration Web App Provides Interactive Visual Portal to All Data
The EiaD 2019 dataset is used directly by the eyeIntegration web app (available in the public domain at https://eyeIntegration.nei.nih.gov). The web app was designed to provide a simple interface that has the same general concept – select specific genes and tissue and view relevant information. The web-app is divided into four general categories: expression, two-dimension sample relationships, gene networks, and data tables.
Custom Gene and Tissue Expression Boxplots
The ‘Expression' tab of the webpage provides a wealth of information about gene- and transcript-level expression for eye and body tissues, giving the user the ability to compare the expression of different genes within a single tissue, as well as the expression of genes across multiple tissues (Fig. 4A). The user first selects either the 2017 or 2019 gene or transcript EiaD dataset, then Hugo Gene Nomeclature Committee (HGNC) genes names (or ENSEMBL transcripts), then tissues. A boxplot then is generated after hitting the “Re(Draw) Plot” button with overlaid individual data points. On mouse-over, the metadata for the individual sample is displayed. A tabular report is generated based on selected genes and tissues: a table with links to Ensembl, GeneCard, and OMIM for each gene for quick referencing, and a table containing expression levels for each selected gene in each selected tissue. The tables can be arranged or sorted to the user's preference and can be easily downloaded for local use.
Figure 4.
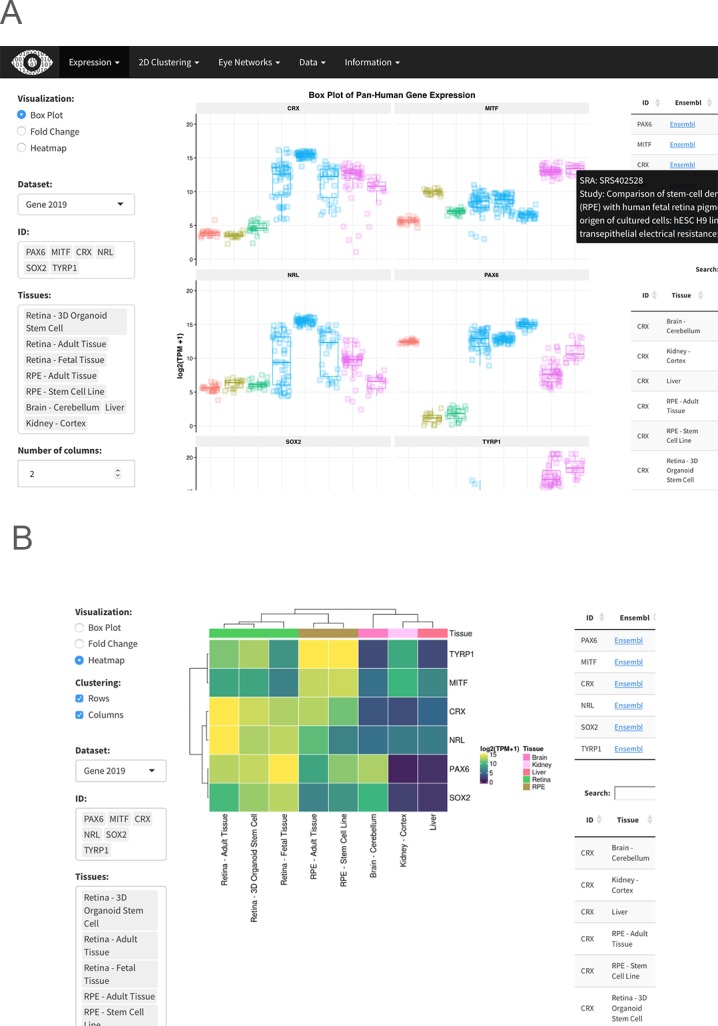
Screenshots from eyeIntegration web app. (A) Pan-tissue gene expression box plots with accompanying data tables. The data tables display the rank (lower is more highly expressed) of each gene in each sub tissue, decile of the rank (10 is the highest decile of expression), and gene's mean log2(TPM + 1) score for each sub tissue. (B) Heatmap visualization.
Heatmap built by the R package ComplexHeatmaps based on expression can be drawn for selected genes and tissues and gene expression can be compared across many genes and tissues (Fig. 4B).62 Finally, a session can be saved or shared by building a custom link for the session with the “Build URL Shortcut” button.
Differential Expression and Gene Ontology Enrichment Tests Allow Quick Comparison of Gene Differences Between Groups
We performed multiple differential comparisons at the subtissue level within all eye tissues and against a pan-body synthetic set comprised of a stratified sample of all tissues present in our subset of the GTEx dataset, allowing quick identification of eye-specific genes across 882 different comparisons. We expanded the differential tests in the 2019 EiaD by adding the GTEx tissues as direct comparisons to our eye subtissues. The user can view the results selecting ‘Differential' under the ‘Expression' tab (Supplementary Fig. S7D). As with ‘Expression,' the user can select which version of the web app to draw data from as well as select for gene- or transcript-level comparisons. The user additionally has the option to select different gene classes to examine, for example, protein coding, lincRNA.
The results of differential expression are presented in a tabular format showing log2 fold change, average expression, and P values. Depending on the comparison, there are 1 to 33,380 differentially expressed genes (Supplementary Files). The table can be easily searched for any given gene, viewed and ordered to the user's preference, and downloaded in CSV format. Differential expression can be visualized through fold change bar graphs with the ‘Pan-tissue plots' selection under ‘Expression.' Additionally, we performed GO enrichment for all differential comparisons. Enriched GO terms are presented first as a word cloud, for quick comparison of GO enrichment. We provide tables, with similar viewing options as the differential expression table, for enriched GO terms in each class of a given differential comparison.
Murine scRNA-seq Enables Testing of Retina Cell Type Specific Expression
We incorporated scRNA-seq data from murine retina across two studies.58,59 This allows researchers to quickly examine gene expression across individual cell types in the retina. Single cell gene expression data are visualized through a heatmap showing the expression of a gene across multiple retinal cell types and different developmental time points, from embryonic day (E)11 to postnatal day (P)14 (when available), and a table of expression values is generated containing the expression data used to draw the heatmap (Fig. 4C). We also provide t-SNE/UMAP–based clustering using cell type–specific labeling created by the publishing authors (Fig. 4D, see Methods). The plots show all cell types present at a given developmental stage, and highlights cells expressing a gene above a user-selected given level.
EiaD 2019 Suggests That iPSC-Derived Organoids and Fetal Retina Have Closely-Related Transcriptomes
There are currently two major approaches to studying developing human retina: postmortem fetal tissue and stem-cell derived organoids. We looked at how well these approaches to studying developing retina compare at a transcriptomic level, for tissue–organoid relationships and how well they correlate across early development.
To evaluate how the tissues and organoids compare at a transcriptome level, we looked at the same t-SNE plot from Figure 3 and focused in on the three types of retina tissue (adult, fetal, and organoid; Fig. 5A). Here, we saw three distinct groupings: adult retina (1), developing fetal retina and stem cell-derived organoid (2), and undifferentiated and early differentiating stem cells (3). We identified several organoid samples in cluster 3, but these share one important difference from the rest of the organoid samples in cluster 3: they have been differentiating for less than 30 days (shape ‘X'). All of the organoid retina samples in cluster 2 were older than 50 days.
Figure 5.

Organoid retina, stem cell retina, and fetal retina tissue have highly similar transcriptomes. The zoom inset (A) shows the retina samples. The “Subtissue Cluster” shading shows the cluster membership of the three major groups. The shapes of the points show the different origin types – notable types include the square for adult, the ‘X' for organoid under 30 days of differentiation, and the diamond for organoid over 30 days of differentiation. (B) Major markers of retina progenitor, photoreceptors (cone and rod), and RGCs have similar gene expression patterns across development in retina fetal tissue and organoids.
To assess how similarly the fetal and organoid retina develop through time, we plotted expression of retinal progenitors, photoreceptors, and retinal ganglion markers by time in days (Fig. 5B). Each row is a gene marker of either retinal progenitor, photoreceptor, or RGC. The rows are hierarchically clustered to put more similar expression patterns closer together, as denoted by the height of the dendrogram. We split the organoid tissues into three groups: Kaewkhaw et al.12 GFP+ and GFP- samples, and Eldred et al.63 samples. The Kaewkhaw samples are flow sorted for a GFP marker (GFP+) under the control of the CRX promoter, an important regulator of photoreceptor development. GFP+ cells would be enriched in photoreceptor populations. We saw that the retinal progenitor, photoreceptor, and RGC groups are largely clustered together, with patterns of expression consistent across the fetal retinal and organoid groups.
Differential Gene Expression of Organoid Retina Versus Fetal Tissue Identifies Sets of Genes Relating to Patterning (HOXB Family), Cell Adhesion (Protocadherin Family), and RGC Identity (BRN3/POU4F, NEFL, GAP43, SNCG)
To identify specific changes between retinal organoid and fetal retina tissue, we performed differential gene expression and GO term enrichment analyses. The GO term enrichment identified cell adhesion (protocadherins) and patterning (HOXB family) as enriched gene sets in retinal organoids. As there is some evidence suggesting that protocadherins influence RGC viability and we noticed that several RGC markers appeared to have lower expression in the organoids compared to the fetal tissue Figure 5B, we looked more closely into RGC marker expression.64
We plotted HOXB family, protocadherin family, and RGC genes in a heatmap visualization, with columns as age in days of fetal or organoid retina. Rows are genes, split by the three different groups of genes and are internally clustered by how similar the expression patterns are. We observed that there are strong, consistent gene expression differences in these three groups of genes between fetal retina and the organoid samples (Supplementary Fig. S8). We also plotted the differential expression values between all organoids and all fetal retina samples; all genes across all three groups are significantly differentially expressed with an FDR corrected P < 0.01.
Limitations of the RNA-seq Quantification in EyeIntegration
Salmon quantification, while highly performant and accurate, has a higher variance for lower read depth samples and shorter transcripts.54 Extra care should be taken with comparisons with lower counts of samples (cornea, RGC) as smaller sample numbers decrease the confidence in differential expression. We do not recommend you directly compare our TPM values with your counts data as there are many important variables that will differ. Instead run our Snakemake pipeline (available in the public domain at https://www.github.com/davemcg/EiaD_build), adding your samples. Finally, we would like to remind any users that RNA-seq methods measure mRNA levels, but the functional unit is the protein; Westerns are still the gold standard with which to evaluate expression and localization.
Data Accessibility
Individual data files for gene expression and sample metadata can be downloaded from the ‘Data' tab on the web app. All data and code used to generate the web app can be installed from the R command line by running devtools::install_github(‘davidmcg/eyeIntegration_app'). The code for the EiaD data processing pipeline can be found at https://github.com/davemcg/EiaD_build.
Discussion
EiaD 2019 contains a large set of carefully curated, reproducibly processed human eye RNA-seq datasets alongside a human body tissue comparison set from the GTEx project. It is available for local install as an R package at https://www.github.com/davemcg/eyeIntegration_app and it is served via a web app, eyeIntegration at https://eyeIntegration.nei.nih.gov. The web app offers a wide range of user-driven visualizations to compare expression of genes across dozens of human body and eye tissues. Furthermore, murine scRNA-seq datasets have been incorporated, allowing for examination of retina cell type–specific gene expression. Several human and nonhuman primate studies have been posted in the past year on the preprint server bioRxiv and as the raw data becomes publicly available, we will be updating this section of eyeIntegration.65–67
If you wish to have your data added to EiaD in the future, we suggest you: (1) deposit data into GEO/SRA; (2) use clear, descriptive, consistent, and detailed metadata for each sample; and (3) (optional) contact the corresponding author. Contacting the corresponding author is only necessary if you feel your data should be included in EiaD and were deposited into the SRA before May 8, 2019.
As human fetal tissue is difficult to obtain and, thus, not very amenable for chemical or genetic modification, it is crucial for organoid-based models to be developed. Our merging of these datasets and analysis at the transcriptome level (compared to cross-analyzing using a limited number of known marker genes) indicated that these two approaches successfully recapitulate fetal retina tissue, to a first approximation, at the whole transcriptome level. However as organoids do not develop to full function, it is important to look at how gene expression differs between retinal organoid and fetal tissue so as to suggest areas for improvement.
We used our large dataset to narrow in on three core processes that differ significantly and substantially between retinal organoids and fetal retina. First we showed that the HOXB family is overexpressed in the organoids. The homeobox family is well known to initiate polarity of the embryo during early development.68 Retinoic acid is applied at approximately day 20 in culture to help differentiate stem cell to organoids and also is known to activate gene members of the HOXB family. The lack of HOXB expression at any age in fetal retina and the broad chromatin and gene expression changes HOXB family members can mediate suggests that HOXB activity may be unwanted for organoid maturation.
Next, we detected several protocadherins more highly expressed in the fetal tissue, relative to the organoids. Protocadherins mediate cell-to-cell connections and, in the developing mice, are shown to be important for spinal internneurons and RGC survival.64,69 We would predict that decreased protocadherin expression reduces the number and maturation of RGC. Indeed, we observed that many canonical RGC markers, while present in detectable levels in the organoids, are significantly underexpressed relative to fetal tissue. This result suggested that modifying culture conditions to promote protocadherin expression may result in higher RGC yield and survival.
We built the EiaD dataset and the accompanying web app, eyeIntegration in the hopes that easily accessible gene expression across tissue space and time will be a useful tool for hypothesis generation and refinement in eye research. Wrapping all of the data processing steps in a Snakemake pipeline has several important advantages for the community: our code is publicly available for review, our analyses are reproducible, future sample updates can be streamlined in with less effort, and because all the processing is in modular pieces it is easier to add new analysis steps. In the future, we plan on regularly adding new samples to EiaD, offering de novo eye tissue transcriptomes, expanding the single cell RNA-seq expression tooling, adding nonhuman eye samples, and epigenetic datasets.
Supplementary Material
Acknowledgements
The authors thank the dozens of groups who provided the raw data required to create this project. We keep a running list of the projects and associated citations at https://github.com/davemcg/eyeIntegration_app/blob/master/inst/citations.md and strongly encourage anyone who uses EiaD and eyeIntegration to cite relevant projects. We also thank Kapil Bharti, Robert Hufnagel, and Brian Brooks for their continuous set of critiques and suggestions in the development of eyeIntegration app over the past 2 years, and Tiziana Cogliati for editing this manuscript. We also thank the two anonymous reviewers for their careful reading and constructive criticisms. Finally, this work used the computational resources of the National Institutes of Health (NIH; Bethesda, MD) HPC Biowulf cluster (available in the public domain at http://hpc.nih.gov).
Supported by the Intramural Research Program of the National Eye Institute.
Disclosure: V. Swamy, None; D. McGaughey, None
Acknowledgments
Amended August 6, 2019: In the third paragraph of the article, the URL https://eyeIntegration.nei.nih was corrected to be https://eyeIntegration.nei.nih.gov.
References
- 1.Chen Y, Huang K, Nakatsu MN, Xue Z, Deng SX, Fan G. Identification of novel molecular markers through transcriptomic analysis in human fetal and adult corneal endothelial cells. Hum Mol Genet. 2013;22:1271–1279. doi: 10.1093/hmg/dds527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chng Z, Peh GSL, Herath WB, et al. High throughput gene expression analysis identifies reliable expression markers of human corneal endothelial cells. PLoS One. 2013;8:e67546. doi: 10.1371/journal.pone.0067546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung DD, Frausto RF, Lin BR, Hanser EM, Cohen Z, Aldave AJ. Transcriptomic profiling of posterior polymorphous corneal dystrophy. Invest Ophthalmol Vis Sci. 2017;58:3202–3214. doi: 10.1167/iovs.17-21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frausto RF, Le DJ, Aldave AJ. Transcriptomic analysis of cultured corneal endothelial cells as a validation for their use in cell replacement therapy. Cell Transplant. 2016;25:1159–1176. doi: 10.3727/096368915X688948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabza M, Karolak JA, Rydzanicz M, et al. Collagen synthesis disruption and downregulation of core elements of TGF-β Hippo, and Wnt pathways in keratoconus corneas. Eur J Hum Genet. 2017;25:582–590. doi: 10.1038/ejhg.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouyang H, Xue Y, Lin Y, et al. WNT7A and PAX6 define corneal epithelium homeostasis and pathogenesis. Nature. 2014;511:358–361. doi: 10.1038/nature13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Q, Yuan S, An Q, et al. Directed differentiation of human embryonic stem cells to corneal endothelial cell-like cells: a transcriptomic analysis. Exp Eye Res. 2016;151:107–114. doi: 10.1016/j.exer.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Han C, Li J, Wang C, et al. Wnt5a Contributes to the differentiation of human embryonic stem cells into lentoid bodies through the noncanonical Wnt/JNK signaling pathway. Invest Ophthalmol Vis Sci. 2018;59:3449–3460. doi: 10.1167/iovs.18-23902. [DOI] [PubMed] [Google Scholar]
- 9.Aldiri I, Xu B, Wang L, et al. The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron. 2017;94:550–568.e10. doi: 10.1016/j.neuron.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farkas MH, Grant GR, White JA, Sousa ME, Consugar MB, Pierce EA. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics. 2013;14:486. doi: 10.1186/1471-2164-14-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoshino A, Ratnapriya R, Brooks MJ, et al. Molecular anatomy of the developing human retina. Develop Cell. 2017;43:763–779.e4. doi: 10.1016/j.devcel.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaewkhaw R, Kaya KD, Brooks M, et al. Transcriptome dynamics of developing photoreceptors in three-dimensional retina cultures recapitulates temporal sequence of human cone and rod differentiation revealing cell surface markers and gene networks. Stem Cells. 2015;33:3504–3518. doi: 10.1002/stem.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaewkhaw R, Swaroop M, Homma K, et al. Treatment paradigms for retinal and macular diseases using 3-D retina cultures derived from human reporter pluripotent stem cell lines. Invest Ophthalmol Vis Sci. 2016;57:ORSFl1–ORSFl11. doi: 10.1167/iovs.15-17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, Jia C, Kazmierkiewicz KL, et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum Mol Genet. 2014;23:4001–4014. doi: 10.1093/hmg/ddu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mustafi D, Kevany BM, Bai X, et al. Transcriptome analysis reveals rod/cone photoreceptor specific signatures across mammalian retinas. Hum Mol Genet. 2016;25:4376–4388. doi: 10.1093/hmg/ddw268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinelli M, Carissimo A, Cutillo L, et al. An atlas of gene expression and gene co-regulation in the human retina. Nucl Acids Res. 2016;44:5773–5784. doi: 10.1093/nar/gkw486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitmore SS, Wagner AH, DeLuca AP, et al. Transcriptomic analysis across nasal, temporal, and macular regions of human neural retina and RPE/choroid by RNA-Seq. Exp Eye Res. 2014;129:93–106. doi: 10.1016/j.exer.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darrow EM, Huntley MH, Dudchenko O, et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. PNAS. 2016;113:E4504–E4512. doi: 10.1073/pnas.1609643113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harenza JL, Diamond MA, Adams RN, et al. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Scientific Data. 2017;4:170033. doi: 10.1038/sdata.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu G, Huang K, Yu J, et al. Identification of miRNA signatures during the differentiation of hESCs into retinal pigment epithelial cells. PLoS One. 2012;7:e37224. doi: 10.1371/journal.pone.0037224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nozawa R-S, Boteva L, Soares DC, et al. SAF-A regulates interphase chromosome structure through oligomerization with chromatin-associated RNAs. Cell. 2017;169:1214–1227.e18. doi: 10.1016/j.cell.2017.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberstein A, Shenk T. Cellular responses to human cytomegalovirus infection: induction of a mesenchymal-to-epithelial transition (MET) phenotype. PNAS. 2017;114:E8244–E8253. doi: 10.1073/pnas.1710799114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng S, Gan G, Qiu C, et al. Engineering a blood-retinal barrier with human embryonic stem cell-derived retinal pigment epithelium: transcriptome and functional analysis. Stem Cells Transl Med. 2013;2:534–544. doi: 10.5966/sctm.2012-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radeke MJ, Radeke CM, Shih Y-H, et al. Restoration of mesenchymal retinal pigmented epithelial cells by TGFβ pathway inhibitors: implications for age-related macular degeneration. Genome Medicine. 2015;7:58. doi: 10.1186/s13073-015-0183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saini JS, Corneo B, Miller JD, et al. Nicotinamide ameliorates disease phenotypes in a human iPSC model of age-related macular degeneration. Cell Stem Cell. 2017;20:635–647.e7. doi: 10.1016/j.stem.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuel W, Jaworski C, Postnikova OA, et al. Appropriately differentiated ARPE-19 cells regain phenotype and gene expression profiles similar to those of native RPE cells. Mol Vis. 2017;23:60–89. [PMC free article] [PubMed] [Google Scholar]
- 27.Santaguida S, Vasile E, White E, Amon A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015;29:2010–2021. doi: 10.1101/gad.269118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao Z, Wang H, Zhou X, et al. Spontaneous generation of a novel foetal human retinal pigment epithelium (RPE) cell line available for investigation on phagocytosis and morphogenesis. Cell Proliferation. 2017;50:e12386. doi: 10.1111/cpr.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih Y-H, Radeke MJ, Radeke CM, Coffey PJ. Restoration of mesenchymal RPE by transcription factor-mediated reprogramming. Invest Ophthalmol Vis Sci. 2017;58:430–441. doi: 10.1167/iovs.16-20018. [DOI] [PubMed] [Google Scholar]
- 30.Smith JR, Todd S, Ashander LM, et al. Retinal pigment epithelial cells are a potential reservoir for ebola virus in the human eye. Trans Vis Sci Tech. 2017;6(4):12. doi: 10.1167/tvst.6.4.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevenson NL, Bergen DJM, Skinner REH, et al. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltransferase expression. J Cell Sci. 2017;130:4132–4143. doi: 10.1242/jcs.212308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tresini M, Warmerdam DO, Kolovos P, et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature. 2015;523:53–58. doi: 10.1038/nature14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheway G, Schmidts M, Mans DA, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature Cell Biol. 2015;17:1074–1087. doi: 10.1038/ncb3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Au ED, Fernandez-Godino R, Kaczynksi TJ, Sousa ME, Farkas MH. Characterization of lincRNA expression in the human retinal pigment epithelium and differentiated induced pluripotent stem cells. PLoS One. 2017;12:e0183939. doi: 10.1371/journal.pone.0183939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carithers LJ, Ardlie K, Barcus M, et al. A novel approach to high-quality postmortem tissue procurement: the GTEx Project. Biopreserv Biobank. 2015;13:311–319. doi: 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratnapriya R, Sosina OA, Starostik MR, et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet. 2019;51:606–610. doi: 10.1038/s41588-019-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collado-Torres L, Nellore A, Kammers K, et al. Reproducible RNA-seq analysis using Recount2. Nat Biotechnoy. 2017;35:319–321. doi: 10.1038/nbt.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lachmann A, Torre D, Keenan AB, et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun. 2018;9:1366. doi: 10.1038/s41467-018-03751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budak G, Dash S, Srivastava R, Lachke SA, Janga SC. Express: a database of transcriptome profiles encompassing known and novel transcripts across multiple development stages in eye tissues. Exp Eye Res. 2018;168:57–68. doi: 10.1016/j.exer.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kakrana A, Yang A, Anand D, et al. iSyTE 2.0: a database for expression-based gene discovery in the eye. Nucleic Acids Res. 2018;46:D875–D885. doi: 10.1093/nar/gkx837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryan JM, Fufa TD, Bharti K, Brooks BP, Hufnagel RB, McGaughey DM. Identifying core biological processes distinguishing human eye tissues with precise systems-level gene expression analyses and weighted correlation networks. Hum Mol Genet. 2018;27:3325–3339. doi: 10.1093/hmg/ddy239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leinonen R, Sugawara H, Shumway M. The sequence read archive. Nucleic Acids Res. 2011;39:D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu Y, Stephens RM, Meltzer PS, Davis SR. SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics. 2013;14:19. doi: 10.1186/1471-2105-14-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soneson C, Matthes KL, Nowicka M, Law CW, Robinson MD. Isoform prefiltering improves performance of count-based methods for analysis of differential transcript usage. Genome Biol. 2016;17:12. doi: 10.1186/s13059-015-0862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2016;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hicks SC, Okrah K, Paulson JN, Quackenbush J, Irizarry RA, Bravo HC. Smooth quantile normalization. Biostatistics. 2018;19:185–198. doi: 10.1093/biostatistics/kxx028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright FA, Sullivan PF, Brooks AI, et al. Heritability and genomics of gene expression in peripheral blood. Nat Genet. 2014;46:430–437. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C, Zhang B, Lin L-L, Zhao S. Evaluation and comparison of computational tools for RNA-seq isoform quantification. BMC Genomics. 2017;18:583. doi: 10.1186/s12864-017-4002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 58.Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark BS, Stein-O'Brien GL, Shiau F, et al. Single-cell RNA-Seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron. 2019;102:1111–1126. doi: 10.1016/j.neuron.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bi R, Liu P. Sample size calculation while controlling false discovery rate for differential expression analysis with RNA-sequencing experiments. BMC Bioinformatics. 2016;17:146. doi: 10.1186/s12859-016-0994-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wattenberg M, Viégas F, Johnson I. How to use t-SNE effectively. Distill. 2016;1:e2. [Google Scholar]
- 62.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 63.Eldred KC, Hadyniak SE, Hussey KA, et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science. 2018;362:eaau6348. doi: 10.1126/science.aau6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lefebvre JL, Zhang Y, Meister M, Wang X, Sanes JR. Γ-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development. 2008;135:4141–4151. doi: 10.1242/dev.027912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lukowski S, Lo C, Sharov A, et al. Generation of human neural retina transcriptome atlas by single cell RNA sequencing. bioRxiv. 2018. p. 425223.
- 66.Hu Y, Wang X, Hu B, et al. Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis. PLoS Biol. 2019;17:e3000365. doi: 10.1371/journal.pbio.3000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peng Y-R, Shekhar K, Yan W, et al. Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell. 2019;176:1222–1237. doi: 10.1016/j.cell.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Philippidou P, Dasen JS. Hox Genes: choreographers in neural development, architects of circuit organization. Neuron. 2013;80:12–34. doi: 10.1016/j.neuron.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayashi S, Takeichi M. Emerging roles of protocadherins: from self-avoidance to enhancement of motility. J Cell Sci. 2015;128:1455–1464. doi: 10.1242/jcs.166306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual data files for gene expression and sample metadata can be downloaded from the ‘Data' tab on the web app. All data and code used to generate the web app can be installed from the R command line by running devtools::install_github(‘davidmcg/eyeIntegration_app'). The code for the EiaD data processing pipeline can be found at https://github.com/davemcg/EiaD_build.