

On-surface synthesis of π-extended triangulene enables the characterization of its spin-polarized edge states.
Abstract
The zigzag-edged triangular graphene molecules (ZTGMs) have been predicted to host ferromagnetically coupled edge states with the net spin scaling with the molecular size, which affords large spin tunability crucial for next-generation molecular spintronics. However, the scalable synthesis of large ZTGMs and the direct observation of their edge states have been long-standing challenges because of the molecules’ high chemical instability. Here, we report the bottom-up synthesis of π-extended [5]triangulene with atomic precision via surface-assisted cyclodehydrogenation of a rationally designed molecular precursor on metallic surfaces. Atomic force microscopy measurements unambiguously resolve its ZTGM-like skeleton consisting of 15 fused benzene rings, while scanning tunneling spectroscopy measurements reveal edge-localized electronic states. Bolstered by density functional theory calculations, our results show that [5]triangulenes synthesized on Au(111) retain the open-shell π-conjugated character with magnetic ground states.
INTRODUCTION
Clipping triangular motifs along the zigzag orientation of graphene creates an entire family of zigzag-edged triangular graphene molecules (ZTGMs) (also known as non-Kekulé polynuclear benzonoid compounds), which are predicted to have multiple unpaired π-electrons and high-spin ground states with large net spin scaling linearly with the number of carbon atoms of zigzag edges (N, N > 2) (Fig. 1A) (1–8). Therefore, the ZTGMs are regarded as promising candidates for molecular spintronic devices (3, 9, 10). However, the direct chemical synthesis of unsubstituted ZTGMs has been a long-standing challenge because of their high chemical instability (1). Recently, a tip-assisted approach has been used to synthesize unsubstituted [3]triangulene with detailed structural and electronic property characterization. However, because only one target molecule can be manipulated at one time, this method is useful only for particular applications but may not be suitable for other technological applications because of a lack of scalability (11). On the other hand, a bottom-up on-surface synthetic approach has revealed itself to have a great potential for the fabrication of atomically precise graphene-based nanostructures (12–17). This method often involves the cyclodehydrogenation of precursor monomers or polymerized monomers via intramolecular or intermolecular aryl-aryl coupling that occurs predominantly along the armchair direction rather than the zigzag direction (13, 15). It hence remains a great challenge to design appropriate molecular precursors for the synthesis of large homologs of zigzag-edged triangulenes with predicted large net spin.
Fig. 1. Illustration of open-shell ZTGMs and the synthetic strategy to π-extended [5]triangulene.
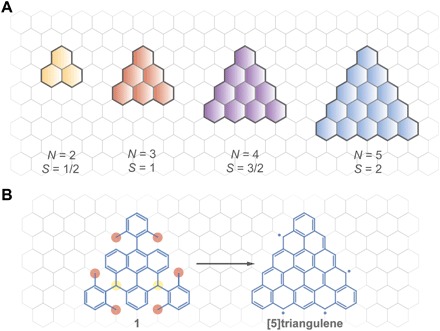
(A) Open-shell ZTGMs with different numbers of zigzag carbon atom (N) and predicted spin multiplicity (S). Yellow, monoradical phenalenyl (N = 2); red, biradical triangulene (N = 3); violet, π-extended triradical [4]triangulene (N = 4); blue, tetraradical [5]triangulene (N = 5). (B) Schematic illustration of the surface-assisted transformation of rationally designed precursor (compound 1) to [5]triangulene. The two yellow spots indicate the sites where the on-surface dehydrogenation initiated, and the six red spots represent the methyl groups that undergo the cyclodehydrogenation process.
RESULTS
Synthesis of π-extended [5]triangulene
For the synthesis of π-extended [5]triangulene, we designed a unique molecular precursor (1), as shown in Fig. 1B. This precursor molecule consists of a central triangular core formed by six hexagonal rings and three 2,6-dimethylphenyl substituents attached at meso- positions of the core (Fig. 1B). This precursor design is expected to undergo cyclodehydrogenation and ring-closure reactions on the catalytic metal surface at elevated temperatures. The two hydrogen atoms linked to the two sp3-carbons of the core (shaded in yellow) undergo dehydrogenation first because of the higher stability of the as-formed triphenylmethyl radicals compared to benzylic radicals, which would be formed if the six methyl groups (shaded in red) were dehydrogenated instead. Subsequently, the dehydrogenation of the six methyl groups will take place to generate benzylic radicals, which can attack the neighboring carbon atoms of the triangular core. Last, an aromaticity-driven ring-closure reaction leads to the formation of the target π-extended [5]triangulene. We developed a new strategy for the synthesis of precursor 1. The synthetic route starts from 9-bromo-10-(2,6-dimethylphenyl)-anthracene (compound 2), which undergoes sequential Suzuki coupling and transformation to give a key dialdehyde intermediate (compound 8) (scheme S1). Subsequent addition of the aldehyde groups with 2,6-dimethylphenylmagnesiumbromide followed by BF3•Et2O-mediated Friedel-Crafts cyclization affords target precursor 1 with a 40% yield. After purification, precursor 1 was deposited onto atomically clean Cu(111) and Au(111) surfaces via thermal sublimation under ultrahigh vacuum conditions (see Materials and Methods for more details). Both substrates exhibit threefold lattice symmetry that closely matches the expected D3h symmetry of [5]triangulene. First, we chose the Cu(111) surface as the growth substrate because of its higher catalytic activity compared to the Au(111) substrate. To produce well-separated target molecules, we deposited a low amount of precursor 1 on the substrates and subsequently attempted to image these precursors using low-temperature scanning tunneling microscopy (LT-STM) at 4.5 K. However, it is challenging to resolve starting compound 1, presumably because of its nonplanar molecular skeleton giving a high imaging instability. Nevertheless, we found that annealing the precursor-decorated Cu(111) substrate at ~500 K induced a cyclodehydrogenation reaction and resulted in the formation of flat triangle-shaped molecules that can be matched to the expected [5]triangulene molecules, as will be discussed further in detail. In contrast to Cu(111), the synthesis of [5]triangulene on the relatively inert Au(111) substrate had to be carried out at a higher temperature (~600 K) and gave a much lower yield (~5%) of the product (~60% yield on the Cu substrate).
Structural characterization of [5]triangulene
A large-scale STM image (Fig. 2A) reveals several well-separated triangle-shaped molecules upon annealing of the precursor-decorated Cu(111) and Au(111) surfaces (Fig. 2, A and D). Magnified STM images recorded with a metallic tip (Fig. 2, B and E) show that individual molecules adopt a triangular and planar configuration on both substrates. The edges of as-obtained molecules on both substrates exhibit characteristic nodal features, resembling the zigzag termini or zigzag edges of graphene nanoribbons (GNRs) (13, 18). These observations indicate that the dehydrogenated methyl groups are bonded to the neighboring carbon atoms, which leads to the formation of a fully π-conjugated system with atomically precise zigzag edges.
Fig. 2. Structural characterization of single π-extended [5]triangulene synthesized on Cu(111) and Au(111) surfaces.
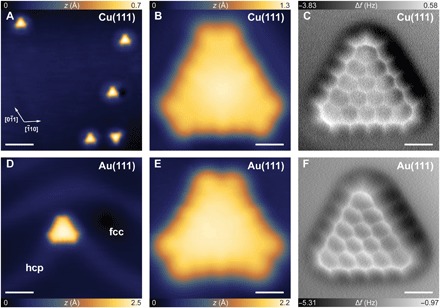
(A and D) Large-scale STM images of [5]triangulene molecules (A) on Cu(111) and (D) on Au(111) [(A) Vs = −1 V and I = 1 nA; scale bar, 5 nm; (D) Vs = 1 V and I = 0.2 nA; scale bar, 1.5 nm]. (B and E) Zoom-in STM images of a single [5]triangulene (B) on Cu(111) and (E) on Au(111) [(B) Vs = −0.8 V and I = 1 nA; (E) Vs = −0.8 V and I = 1 nA; scale bar, 4 Å]. (C and F) nc-AFM images of a single [5]triangulene (C) on Cu(111) and (F) on Au(111) acquired using a CO-functionalized tip [(C) ∆z = 0.15 Å, Vs = 30 mV, I = 0.3 nA; (F) ∆z = 0.15 Å, Vs = 10 mV, I = 0.5 nA; scale bar, 4 Å]. fcc, face-centered cubic; hcp, hexagonal close-packed.
For accurate chemical determination of reaction products, we conducted noncontact atomic force microscopy (nc-AFM) measurements of individual triangular molecules in constant height mode with a CO-functionalized tip (19–22). In this mode, the contrast of the images collected can be correlated to the magnitude of short-range repulsive chemical forces (21, 23, 24). Hence, the bright areas exhibiting high-frequency shift represent surface regions with higher electron density. As shown in Fig. 2 (C and F), individual triangle-shaped reaction products consisting of 15 fused benzene rings with zigzag-edged topology can be clearly resolved. Upon closer examination, noticeable “line features” attached to individual zigzag carbon atoms can be observed, suggesting the possibility of a monohydrogenated zigzag edge. CH2 termination can be excluded because of the absence of distinct maxima at the edges (11, 13, 21, 25). Moreover, the absence of bonding signature of [5]triangulene to the supporting substrates can be ascribed to the π radical nature of its zigzag edges, which is in stark contrast to σ radicals (26). Note that experimental nc-AFM images have excellent agreement with simulated ones for various tip-molecule distances using the numerical model developed by Hapala et al. (24). The slight elongation of corner hexagonal rings can be rationalized by nonhomogeneous force field potential affecting the lateral bending of probe particle (CO) at the corners of [5]triangulene (11, 24). Hence, the observed molecular morphology corresponds to the expected [5]triangulene. Moreover, the possibility to acquire the high-resolution images resolving all benzene rings with the same brightness over all molecules in the constant height mode indicates that the molecule adopted the flat structure. This excludes a strong nonlocal interaction with an elbow of the Au(111) surface.
Probing electronic properties of [5]triangulene
The freestanding [5]triangulene contains four unpaired π-electrons, as predicted by different theoretical models (1, 3, 7, 27). A thorough investigation of its magnetic ground states and excited states is of great interest, but this is outside the scope of this paper. The primary focus of this work lies in probing the frontier molecular orbitals of [5]triangulene, which will be further compared with theory. To unveil its peculiar electronic properties, we performed scanning tunneling spectroscopy (STS) measurements of single [5]triangulene grown on the weakly interacting Au(111) substrates using a metallic tip (14, 17, 28, 29). The characteristic differential conductance (dI/dV) point spectra recorded at specific positions over the center (black curve) and edge (blue curve) of [5]triangulene along with spectrum collected on the bare Au(111) surface (red curve) are illustrated in Fig. 3A. The dI/dV spectrum recorded at the molecular edge (blue curve) shows two prominent peaks at Vs = −0.62 ± 0.04 V (P1) and Vs = 1.07 ± 0.04 V (P2). By contrast, dI/dV spectra acquired at the center of the molecule do not show these two features (black curve). Moreover, the dI/dV spectrum collected on the clean Au surface is rather featureless, apart from the surface state–induced peak at −0.48 ± 0.05 V. All these observations suggest that P1 and P2 states are associated with molecular states. Furthermore, color-coded dI/dV spectra (spaced by 0.11 nm) recorded along the molecular edge (Fig. 3B) and across the molecular center (Fig. 3C) reveal that both P1 and P2 states are observed exclusively at the zigzag edges of [5]triangulene.
Fig. 3. Characterization of electronic properties of individual [5]triangulene.
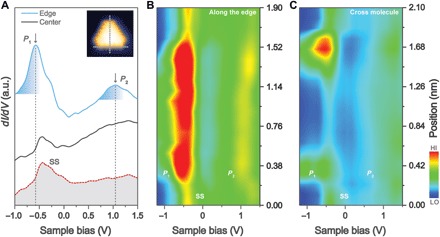
(A) Point dI/dV spectra acquired over different sites of the [5]triangulene molecule and the Au(111) substrate. dI/dV curves taken at the edge (solid blue line) and at the center (solid black line) of [5]triangulene and taken on the clean Au(111) surface (red dotted line). a.u., arbitrary units. (B and C) Color-coded dI/dV spectra (spaced by 0.11 nm) taken along the zigzag edge (B) and across the center of [5]triangulene [(C), starting from the apex]. The actual positions where the dI/dV spectra were taken are indicated by gray dots in the inset STM image in (A). SS, surface-state.
To capture the spatial distribution of the above-discussed electronic states, we conducted dI/dV mapping over a single [5]triangulene molecule at different sample biases using a metallic tip. Figure 4, B and C, shows dI/dV maps recorded at the biases corresponding to the energetic positions of the aforementioned P1 and P2 states, respectively. Differential conductance map collected at the energy of the P1 state (−0.62 eV) reveals five bright lobes located at the edge of [5]triangulene represented by a characteristic nodal pattern (Fig. 4B). The dI/dV map acquired at 1.07 V (Fig. 4C) resolves that the P2 state exhibits an edge-localized pattern, resembling that of the P1 state, albeit with a slightly blurred nodal structure. This characteristic feature is similar to the nodal pattern of spin-polarized electronic states observed at the zigzag termini and zigzag edge of GNRs (13). As for now, we ascribe P1 and P2 states to spin-polarized edge states (6, 8).
Fig. 4. Electronic structure of [5]triangulene.
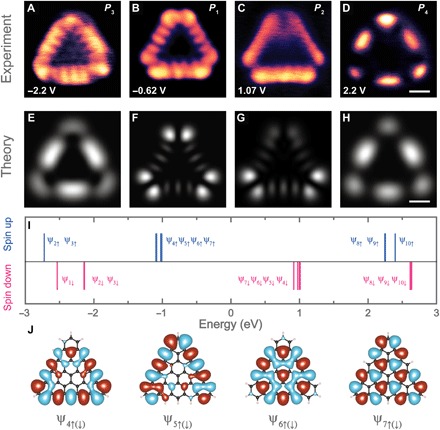
(A to D) Experimental dI/dV maps recorded at different energy positions [−2.2 V for (A), −0.62 V for (B), 1.07 V for (C), and 2.2 V for (D); scale bar, 4 Å]. (E to H) Simulated dI/dV maps of [5]triangulene acquired at different energy positions corresponding to different sets of orbitals: (E) ψ2↓ and ψ3↓, (F) ψ4↑ to ψ7↑, (G) ψ4↓ to ψ7↓ (note: the weight of ψ5↓ is set to 0.7; refer to fig. S8 for more details), and (H) ψ8↑ and ψ9↑. Scale bar, 4 Å. (I) Calculated spin-polarized molecular orbital energies of an isolated [5]triangulene. Blue and red refers to spin-up and spin-down states, respectively. (J) DFT-calculated wave functions of four pairs of spin-polarized orbitals [ψ4 ↑ ( ↓ ), ψ5 ↑ ( ↓ ), ψ6 ↑ ( ↓ ), and ψ7 ↑ ( ↓ )]. Red and blue colors indicate the wave functions with positive or negative values, respectively.
To gain deeper insights into the electronic structure of [5]triangulene, we performed spin-polarized density functional theory (DFT) calculations. The calculations reveal that the quintuple ground state of Au-supported [5]triangulene is lower in energy by 0.13, 0.27, and 0.38 eV than its triplet, singlet open-shell, and singlet closed-shell states, respectively. The energy ordering of these electronic states is consistent with the previous calculation of similar graphene molecule systems (5, 11, 13, 30). The calculation also reveals a total magnetic moment of 3.58 μb for [5]triangulene on the Au substrate, suggesting that its magnetic ground state could be retained on the Au(111) surface (Supplementary Materials). Hereinafter, we will assume that Au-supported [5]triangulene has a ferromagnetic alignment of the unpaired electrons.
Modeling the electronic structure of [5]triangulene
We note that DFT can provide reliable information about ground-state energy ordering and the spatial shape of molecular orbitals, although it tends to underestimate the size of the electronic gap, which is offset by the metallic substrate screening in our case (13, 14). Figure 4I depicts the calculated single-particle DFT spin-polarized energies of a free [5]triangulene in the ferromagnetic quintuple state. The frontier molecular orbitals are four pairs of orbitals [refer to ψ4 ↑ ( ↓ ), ψ5 ↑ ( ↓ ), ψ6 ↑ ( ↓ ),and ψ7 ↑ ( ↓ ) in Fig. 4I] with the corresponding wave function plots, as shown in Fig. 4J. Each spin-down orbital is accompanied by the counterpart spin-up orbital arising from the spin-polarized orbital splitting (section S4). From the energy-level alignment, it is expected that the neutral state of [5]triangulene should produce frontier orbital-associated resonances below and above the Fermi level (Fig. 4I and fig. S4). As illustrated in Fig. 4I, the nearly degenerate spin-up states (ψ4↑, ψ5↑, ψ6↑, and ψ7↑) and spin-down states (ψ4↓, ψ5↓, ψ6↓, and ψ7↓) are energetically close to P1 (−0.62 V) and P2 (1.07 V) states, respectively. However, we find that none of the wave function patterns (Fig. 4J) of these spin-polarized orbitals matches well with the spatial resolution of the dI/dV maps acquired at −0.62 V (P1) and 1.07 V (P2). This can be explained by the fact that dI/dV maps are determined by selection rules ruling the tunneling matrix elements between the tip orbital and linear combination of wave functions of nearly degenerated frontier orbitals (31, 32). We calculated the dI/dV images (Fig. 4, F and G) of [5]triangulene using a tip with s-like orbital, which mimics well the electronic structure of metallic tips (32). The calculated dI/dV images reveal three characteristic lobes located at the center of the edge with a pair of brighter lobes at the apex of the triangle. Such an edge-localized nodal pattern shows a good agreement with our experimental dI/dV map recorded at the energy position of P1. We note that the P2 state is also exclusively localized at the edge, albeit with a slightly blurred nodal structure. The slightly different contrast of dI/dV maps between P1 and P2 states may arise from the different degrees of degeneracy of spin-up and spin-down edge states, as predicted in our theoretical results (Fig. 4I and fig. S6). It means that in particular energy where the dI/dV map was acquired, the weight of certain molecular orbitals is lower because of different degrees of degeneracy of the frontier orbitals, giving rise to a slightly asymmetric contrast.
In addition, the dI/dV spectrum taken at the edge of [5]triangulene resolves two sharp rises near the sample biases of −2.2 and 2.2 V (section S3). The experimental dI/dV maps acquired at these energies are shown in Fig. 4 (A and D). The nodal patterns of these two states are in agreement with the calculated dI/dV maps at the corresponding energetic positions. The spatial dI/dV maps of P3 and P4 states are notably different from those of P1 and P2 states, which further confirm that P3 and P4 states are originated from different molecular orbitals.
Last, we used GW approximation of many-body perturbation (section S4) to calculate the quasiparticle energies of a free [5]triangulene. The quasiparticle gap of a free [5]triangulene is predicted to be 2.81 eV based on the GW calculations. To directly compare the GW gap with the experimental value, we have to include the dynamic screening from electrons residing in the molecule and the underlying Au(111) substrate, which is anticipated to substantially reduce the quasiparticle gap of [5]triangulene. The energy gap of Au-supported [5]triangulene is determined to be ~1.7 eV (Fig. 3A). A direct comparison between the experimentally determined gap and the GW-predicted value yields a gap reduction of around 1 eV attributed to the metallic substrate screening, consistent with a previous study of the GNRs (28) and other molecular systems with comparable size (33–37). All the observations discussed above point toward a magnetic ground state of [5]triangulene on Au(111), which is further supported by our DFT calculations (section S4).
DISCUSSION
In conclusion, we demonstrate a feasible bottom-up approach for the atomically precise synthesis of unsubstituted π-extended [5]triangulene on metallic surfaces. Our nc-AFM imaging ambiguously confirms the zigzag edge topology of [5]triangulene, and STM measurements resolve the edge-localized electronic states. The successful synthesis of π-extended triangulenes not only enables the magnetism and spin transport properties to be explored at the single-molecule level but also opens a new avenue for fabricating larger triangular zigzag-edged graphene quantum dots with atomic precision for spin and quantum transport applications. Therefore, it is of great interest to generate similar systems with different sizes and spin numbers and uncover their properties on various substrates by spin-polarized STM studies.
MATERIALS AND METHODS
Synthesis and characterization of organic precursor
Compound 1 was synthesized using the procedures illustrated in scheme S1. Details of the synthesis and spectroscopic characterizations of the precursor are presented in the Supplementary Materials.
Sample preparation
Au(111) and Cu(111) single crystal (MaTeck GmbH) was cleaned by multiple cycles of Ar+ sputtering and annealing. We used Knudsen cell (MBE-Komponenten GmbH) for the deposition of precursor molecules (compound 1) onto clean Cu(111) and Au(111) surfaces under ultrahigh vacuum conditions (base pressure, <2 × 10−10 mbar). After deposition of precursors, the sample was annealed at 500 K (600 K for Au) for 20 min for the synthesis of [5]triangulene. Subsequently, the sample was transferred into the STM/AFM head for imaging and characterization.
STM/AFM characterization
The experiments were conducted in an Omicron LT-STM/AFM system operated under ultrahigh vacuum (base pressure, P < 2 × 10−11 mbar) at a temperature of T = 4.5 K. The microscope was equipped with qPlus sensors with a resonance frequency of f0 = 22839.5 Hz, a stiffness of k = 1800 N m−1, and a quality factor of 23,000. The qPlus sensor allows simultaneous access to It and df channels. nc-AFM images were collected at a constant-height frequency modulation mode using an oscillation amplitude of A = 100 pm. The tip-sample distance with respect to an STM set point is indicated in the figure caption for each AFM image. The dI/dV spectra were collected using an external lock-in amplifier (SR830, Stanford Research System) with a modulation frequency of 779 Hz and an amplitude of 10 mV. The color-coded dI/dV spectra were taken at the constant-current mode with the same initial set points (1.5 V and 1 nA) over the molecule and the bare Au(111) substrate. dI/dV maps were collected using the same parameter with spectra in a constant-current mode (fig. S3) (17, 29). We used chemically etched tungsten tips treated by multiple voltage pulsing in combination with a controlled poking into clean metal surfaces. The CO molecule was picked up by a clean and sharp metal tip following a routine procedure (38).
Calculation methods
For all calculation details, please refer to section S4.
Supplementary Material
Acknowledgments
Funding: J.L. acknowledges the support from NUS start-up grant (R-143-000-621-133) and MOE Tier 2 grants (R-143-000-682-112 and R-143-000-A06-112). The authors acknowledge NRF-CRP grant NRF-CRP16-2015-02: “Two dimensional covalent organic framework: Synthesis and applications.” J.W. acknowledges financial support from the MOE Tier 3 program (MOE2014-T3-1-004). F.-C.C. acknowledges support from the National Center for Theoretical Sciences and the Ministry of Science and Technology of Taiwan under grant no. MOST-107-2628-M-110-002-MY3. P.J. acknowledges support from Praemium Academiae of the Czech Academy of Sciences, the Ministry of Education of the Czech Republic grant LM2015087, and GACR project no. 18-09914S. Author contributions: J.L. supervised the project. J.S. and M.T. performed STM, nc-AFM measurements, and data analysis. P.H. and H.Z. synthesized and characterized the molecular precursor under the supervision of J.W. G.M. and Z.-Q.H. performed the theoretical calculations under the supervision of H.L. and F.-C.C. P.M. and P.J. performed simulation of dI/dV maps and their analysis. All authors contributed to the scientific discussion and helped in writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaav7717/DC1
Section S1. Synthesis and characterization of the precursor (compound 1)
Section S2. Additional nc-AFM images and simulated AFM images of [5]triangulene
Section S3. dI/dV spectra and maps of [5]triangulene synthesized on the Au(111) surface
Section S4. Theoretical calculation of electronic properties of [5]triangulene
Scheme S1. The synthetic route of the precursor (compound 1).
Fig. S1. Experimental and simulated AFM images of [5]triangulene.
Fig. S2. STS spectra collected over the [5]triangulene molecule on the Au(111) surface.
Fig. S3. Constant current dI/dV maps of P1 to P4 with the corresponding STM images.
Fig. S4. Calculated energy diagram and spin density distribution of [5]triangulene with different magnetic configurations.
Fig. S5. Wave function patterns and orbital densities of ψ4 ↑ ( ↓ ) to ψ7 ↑ ( ↓ ).
Fig. S6. Wave functions and charge densities of molecular orbitals in the singlet closed-shell, singlet open-shell, and quintuplet states.
Fig. S7. The calculated energy diagrams of a free [5]triangulene via DFT, HSE06, and GW methods.
Fig. S8. Simulated dI/dV maps of [5]triangulene taken at an energetic position of the P2 state contributed by different weights of orbitals ψ4↓ to ψ7↓.
Table S1. Total system energies of quintuplet (Q), triplet (T), singlet open-shell (SO), and singlet closed-shell (SC) states with respect to that of the SC state.
Table S2. The calculated energy gap of [3]triangulene and [5]triangulene.
Appendix S1. 1H-13C nuclear magnetic resonance spectra and mass spectra for all new compounds
REFERENCES AND NOTES
- 1.Clar E., Stewart D. G., Aromatic hydrocarbons. LXV. Triangulene derivatives. J. Am. Chem. Soc. 75, 2667–2672 (1953). [Google Scholar]
- 2.Melle-Franco M., When 1 + 1 is odd. Nat. Nanotechnol. 12, 292–293 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Morita Y., Suzuki S., Sato K., Takui T., Synthetic organic spin chemistry for structurally well-defined open-shell graphene fragments. Nat. Chem. 3, 197–204 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Lieb E., Two theorems on the Hubbard model. Phys. Rev. Lett. 62, 1201–1204 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Potasz P., Güçlü A. D., Hawrylak P., Zero-energy states in triangular and trapezoidal graphene structures. Phys. Rev. B. 81, 033403 (2010). [Google Scholar]
- 6.Fernández-Rossier J., Palacios J. J., Magnetism in graphene nanoislands. Phys. Rev. Lett. 99, 177204 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Wang W. L., Meng S., Kaxiras E., Graphene nanoflakes with large spin. Nano Lett. 8, 241–245 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Castro Neto A. H., Guinea F., Peres N. M. R., Novoselov K. S., Geim A. K., The electronic properties of graphene. Rev. Mod. Phys. 81, 109–162 (2009). [Google Scholar]
- 9.Wang W. L., Yazyev O. V., Meng S., Kaxiras E., Topological frustration in graphene nanoflakes: Magnetic order and spin logic devices. Phys. Rev. Lett. 102, 157201 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Han W., Kawakami R. K., Gmitra M., Fabian J., Graphene spintronics. Nat. Nanotechnol. 9, 794–807 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pavliček N., Mistry A., Majzik Z., Moll N., Meyer G., Fox D. J., Gross L., Synthesis and characterization of triangulene. Nat. Nanotechnol. 12, 308–311 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Cai J., Ruffieux P., Jaafar R., Bieri M., Braun T., Blankenburg S., Muoth M., Seitsonen A. P., Saleh M., Feng X., Müllen K., Fasel R., Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 466, 470–473 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Ruffieux P., Wang S., Yang B., Sánchez-Sánchez C., Liu J., Dienel T., Talirz L., Shinde P., Pignedoli C. A., Passerone D., Dumslaff T., Feng X., Müllen K., Fasel R., On-surface synthesis of graphene nanoribbons with zigzag edge topology. Nature 531, 489–492 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Chen Y.-C., Cao T., Chen C., Pedramrazi Z., Haberer D., de Oteyza D. G., Fischer F. R., Louie S. G., Crommie M. F., Molecular bandgap engineering of bottom-up synthesized graphene nanoribbon heterojunctions. Nat. Nanotechnol. 10, 156–160 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Treier M., Pignedoli C. A., Laino T., Rieger R., Müllen K., Passerone D., Fasel R., Surface-assisted cyclodehydrogenation provides a synthetic route towards easily processable and chemically tailored nanographenes. Nat. Chem. 3, 61–67 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Cai J., Pignedoli C. A., Talirz L., Ruffieux P., Söde H., Liang L., Meunier V., Berger R., Li R., Feng X., Müllen K., Fasel R., Graphene nanoribbon heterojunctions. Nat. Nanotechnol. 9, 896–900 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Nguyen G. D., Tsai H.-Z., Omrani A. A., Marangoni T., Wu M., Rizzo D. J., Rodgers G. F., Cloke R. R., Durr R. A., Sakai Y., Liou F., Aikawa A. S., Chelikowsky J. R., Louie S. G., Fischer F. R., Crommie M. F., Atomically precise graphene nanoribbon heterojunctions from a single molecular precursor. Nat. Nanotechnol. 12, 1077–1082 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Talirz L., Söde H., Cai J., Ruffieux P., Blankenburg S., Jafaar R., Berger R., Feng X., Müllen K., Passerone D., Fasel R., Pignedoli C. A., Termini of bottom-up fabricated graphene nanoribbons. J. Am. Chem. Soc. 135, 2060–2063 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Giessibl F. J., Advances in atomic force microscopy. Rev. Mod. Phys. 75, 949–983 (2003). [Google Scholar]
- 20.Mohn F., Schuler B., Gross L., Meyer G., Different tips for high-resolution atomic force microscopy and scanning tunneling microscopy of single molecules. Appl. Phys. Lett. 102, 073109 (2013). [Google Scholar]
- 21.Gross L., Mohn F., Moll N., Liljeroth P., Meyer G., The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Giessibl F. J., High-speed force sensor for force microscopy and profilometry utilizing a quartz tuning fork. Appl. Phys. Lett. 73, 3956–3958 (1998). [Google Scholar]
- 23.Gross L., Recent advances in submolecular resolution with scanning probe microscopy. Nat. Chem. 3, 273–278 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Hapala P., Kichin G., Wagner C., Tautz F. S., Temirov R., Jelínek P., Mechanism of high-resolution STM/AFM imaging with functionalized tips. Phys. Rev. B. 90, 085421 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Pavlicěk N., Majzik Z., Collazos S., Meyer G., Peŕez D., Guitiań E., Peña D., Gross L., Generation and characterization of a meta-aryne on Cu and NaCl Surfaces. ACS Nano 11, 10768–10773 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Schule B., Fatayer S., Mohn F., Moll N., Pavliček N., Meyer G., Peña D., Gross L., Reversible Bergman cyclization by atomic manipulation. Nat. Chem. 8, 220–224 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Randić M., Aromaticity of polycyclic conjugated hydrocarbons. Chem. Rev. 103, 3449–3605 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Kimouche A., Ervasti M. M., Drost R., Halonen S., Harju A., Joensuu P. M., Sainio J., Liljeroth P., Ultra-narrow metallic armchair graphene nanoribbons. Nat. Commun. 6, 10177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizzo D. J., Veber G., Cao T., Bronner C., Chen T., Zhao F., Rodriguez H., Louie S. G., Crommie M. F., Fischer F. R., Topological band engineering of graphene nanoribbons. Nature 560, 204–208 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Ezawa M., Metallic graphene nanodisks: Electronic and magnetic properties. Phys. Rev. B 76, 245415 (2007). [Google Scholar]
- 31.Chen C. J., Tunneling matrix elements in three-dimensional space: The derivative rule and the sum rule. Phys. Rev. B 42, 8841–8857 (1990). [DOI] [PubMed] [Google Scholar]
- 32.Krejčí O., Hapala P., Ondráček M., Jelínek P., Principles and simulations of high-resolution STM imaging with a flexible tip apex. Phys. Rev. B 95, 045407 (2017). [Google Scholar]
- 33.Ruffieux P., Cai J., Plumb N. C., Patthey L., Prezzi D., Ferretti A., Molinari E., Feng X., Müllen K., Pignedoli C. A., Fasel R., Electronic structure of atomically precise graphene nanoribbons. ACS Nano 6, 6930–6935 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Zheng Y. J., Huang Y. L., Chen Y., Zhao W., Eda G., Spataru C. D., Zhang W., Chang Y.-H., Li L.-J., Chi D., Quek S. Y., Wee A. T. S., Heterointerface screening effects between organic monolayers and monolayer transition metal dichalcogenides. ACS Nano 10, 2476–2484 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Neaton J. B., Hybertsen M. S., Louie S. G., Renormalization of molecular electronic levels at metal-molecule interfaces. Phys. Rev. Lett. 97, 216405 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Chen Y.-C., de Oteyza D. G., Pedramrazi Z., Chen C., Fischer F. R., Crommie M. F., Tuning the band gap of graphene nanoribbons synthesized from molecular precursors. ACS Nano 7, 6123–6128 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Liu Z.-F., Egger D. A., Refaely-Abramson S., Kronik L., Neaton J. B., Energy level alignment at molecule-metal interfaces from an optimally tuned range-separated hybrid functional. J. Chem. Phys. 146, 092326 (2017). [Google Scholar]
- 38.Bartels L., Meyer G., Rieder K.-H., Controlled vertical manipulation of single CO molecules with the scanning tunneling microscope: A route to chemical contrast. Appl. Phys. Lett. 71, 213–215 (1997). [Google Scholar]
- 39.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Kohn W., Sham L. J., Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). [Google Scholar]
- 41.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 42.Kresse G., Hafner J., Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993). [DOI] [PubMed] [Google Scholar]
- 43.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Hohenberg P., Kohn W., Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964). [Google Scholar]
- 45.Krukau A. V., Vydrov O. A., Izmaylov A. F., Scuseria G. E., Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 125, 224106 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Shishkin M., Kresse G., Implementation and performance of the frequency-dependent GW method within the PAW framework. Phys. Rev. B 74, 035101 (2006). [Google Scholar]
- 47.Shishkin M., Kresse G., Self-consistent GW calculations for semiconductors and insulators. Phys. Rev. B 75, 235102 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Shishkin M., Marsman M., Kresse G., Accurate quasiparticle spectra from self-consistent GW calculations with vertex corrections. Phys. Rev. Lett. 99, 246403 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Fuchs F., Furthmüller J., Bechstedt F., Shishkin M., Kresse G., Quasiparticle band structure based on a generalized Kohn-Sham scheme. Phys. Rev. B 76, 115109 (2007). [Google Scholar]
- 50.Klimeš J., Bowler D. R., Michaelides A., Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 22, 022201 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaav7717/DC1
Section S1. Synthesis and characterization of the precursor (compound 1)
Section S2. Additional nc-AFM images and simulated AFM images of [5]triangulene
Section S3. dI/dV spectra and maps of [5]triangulene synthesized on the Au(111) surface
Section S4. Theoretical calculation of electronic properties of [5]triangulene
Scheme S1. The synthetic route of the precursor (compound 1).
Fig. S1. Experimental and simulated AFM images of [5]triangulene.
Fig. S2. STS spectra collected over the [5]triangulene molecule on the Au(111) surface.
Fig. S3. Constant current dI/dV maps of P1 to P4 with the corresponding STM images.
Fig. S4. Calculated energy diagram and spin density distribution of [5]triangulene with different magnetic configurations.
Fig. S5. Wave function patterns and orbital densities of ψ4 ↑ ( ↓ ) to ψ7 ↑ ( ↓ ).
Fig. S6. Wave functions and charge densities of molecular orbitals in the singlet closed-shell, singlet open-shell, and quintuplet states.
Fig. S7. The calculated energy diagrams of a free [5]triangulene via DFT, HSE06, and GW methods.
Fig. S8. Simulated dI/dV maps of [5]triangulene taken at an energetic position of the P2 state contributed by different weights of orbitals ψ4↓ to ψ7↓.
Table S1. Total system energies of quintuplet (Q), triplet (T), singlet open-shell (SO), and singlet closed-shell (SC) states with respect to that of the SC state.
Table S2. The calculated energy gap of [3]triangulene and [5]triangulene.
Appendix S1. 1H-13C nuclear magnetic resonance spectra and mass spectra for all new compounds