

ABSTRACT
Objectives
When the 2017 Movement Disorders Society Criteria for progressive supranuclear palsy is applied, patients may appear to have multiple phenotypes. The maximum allocation extinction rules were developed to provide a consistent method for applying the criteria and reaching a single diagnostic label. In this study, we apply both to a neuropathologic cohort of progressive supranuclear palsy and other parkinsonian conditions.
Methods
An autopsy cohort of 54 patients with progressive supranuclear palsy and 56 patients with other neuropathologic diseases was selected. Clinical data were retrospectively abstracted, and the diagnostic criteria for progressive supranuclear palsy were applied. All possible phenotypes applicable were listed and maximum allocation extinction rules were applied to assess reduction in the number of phenotypes ascribed per patient.
Results
In the progressive supranuclear palsy group, 52 patients met the criteria for multiple phenotypes, with an average of 7 phenotypes per patient. In the nonprogressive supranuclear palsy group, all 56 patients had features of more than one phenotype, up to 3 per patient. After application of maximum allocation extinction rules, the majority of the patients in both groups had a single predominant phenotype. Freezing of gait, supranuclear gaze palsy, and frontal behavioral syndrome were more common in the progressive supranuclear palsy group.
Conclusions
The diagnostic criteria for progressive supranuclear palsy identify many clinical features, thereby leading to assignment of multiple phenotypes per patient. We demonstrate that the maximum allocation extinction rules can effectively lead to a single consensus phenotype, maintaining a uniform diagnostic label for clinical and research applications.
Keywords: MDS‐PSP criteria, PSP diagnostic criteria, progressive supranuclear palsy, diagnosis
Over the past several years, it has become apparent that PSP is a far more heterogenous condition than previously thought. This clinical diversity was recognized in the 2017 International Parkinson and Movement Disorder Society diagnostic criteria for PSP (MDS‐PSP), which operationalized the diagnosis of seven phenotypes. In contrast, the National Institute of Neurological Diseases and Society for Progressive Supranuclear Palsy (NINDS‐SPSP) criteria, published in 1996,1 recognized only one phenotype (“Richardson Syndrome”). The expanded PSP framework outlined in the MDS‐PSP criteria is based on several clinicopathological studies, which demonstrated that a wide range of motor, cognitive, and behavioral symptoms may occur as a result of PSP neuropathology. In addition to Richardson syndrome, characterized by vertical supranuclear gaze palsy and postural instability, other described phenotypes include corticobasal syndrome,2, 3 partially levodopa responsive parkinsonism,4 agrammatic/nonfluent aphasia and apraxia of speech,5, 6 progressive gait freezing,7 frontal behavioral syndrome,8, 9 cerebellar ataxia,10 and corticospinal tract degeneration presenting with a primary lateral sclerosis (PLS)‐like upper motor neuron syndrome.11 To systematically account for this diversity, the MDS‐PSP criteria defined four main clinical domains: ocular motor (O), postural instability (P), akinesia (A), and cognitive (C; hereafter summarized as OPAC).12, 13, 14 Based on the specific combinations of symptoms in each domain, a number of distinct PSP phenotypes are assigned across a range of certainties (suggestive, possible, and probable).14 Phenotypes recognized by the criteria include Richardson syndrome (PSP‐RS), parkinsonism (PSP‐P), speech and language abnormalities (PSP‐SL) including progressive aphasia and apraxia of speech, corticobasal syndrome (PSP‐CBS), postural instability (PSP‐PI), ocular motor abnormalities (PSP‐OM), frontal syndrome (PSP‐F), and progressive gait freezing (PSP‐PGF). The criteria do not include categories to account for the cerebellar ataxia and PLS‐like phenotypes.
We have previously analyzed the sensitivity and specificity of the MDS‐PSP criteria and found that it was indeed highly sensitive (84.4–100%) secondary to the inclusion of more PSP‐related syndromes that would have been excluded by the NINDS‐SPSP criteria, without much loss of specificity (81.6–91.4%).15 Although the new criteria appear to enhance detection of PSP, and were designed for research and clinical practice, there are some challenges in its application to clinical research and utilization in clinical practice. Patients often have a multitude of PSP features, especially later in the disease course, and may meet criteria for multiple PSP phenotypes. This problem was subsequently recognized by the MDS‐PSP task force and was addressed by the Multiple Allocation eXtinction (MAX) rules: four principles which put forward a structured way of assigning a single phenotypic label, usually based on features that were shown to be most strongly predictive of PSP neuropathology.16 In this article, we apply the MAX rules to an independent neuropathological cohort and shed light on challenges when it comes to the diagnosis of PSP.
Methods
We used a neuropathological cohort of PSP and other parkinsonian conditions described previously.15 Clinical signs and symptoms were recorded for the clinical domains (O, P, A, C) as defined by the MDS‐PSP criteria,14 and only patients who had complete data available for all four domains were included. These data were used to determine all phenotypes per the MDS‐PSP criteria applicable to each patient. Data were collected for two separate time points: within 3 years of disease onset and after 3 years from onset. We then applied the MAX rules to the list of multiple phenotypes for each patient as described by Grimm et al.16 The first rule requires picking the phenotype with the highest level of diagnostic certainty. The second rule proposes that the phenotype that developed first in the disease course should be retained as the diagnostic label, until another phenotype becomes clinically predominant. The third rule sets a phenotypic hierarchy based on highest to lowest likelihood of predicting PSP pathology, where PSP‐RS is considered more predictive than ocular motor impairment or postural instability alone, which are, in turn, more predictive than all other phenotypes listed in the criteria. We assessed the utility of MAX rules, the relative likelihood of various clinical symptoms occuring in the PSP versus non‐PSP groups, and their ability to predict underlying neuropathology.
Results
Fifty‐four patients with PSP and 56 patients with other neurodegenerative neuropathologies had complete data and were included in the analyses. The non‐PSP group was comprised of 33 corticobasal ganglionic degeneration, 15 cases with Lewy body disease manifesting clinically as Parkinson's disease, 4 cases of MSA, 3 globular glial tauopathy,11 and 1 case of frontotemporal lobar degeneration from transactive DNA‐binding protein 43. Before the application of the MAX rules, 52 of the 54 definite PSP patients had more than one phenotype (mean, 7.00 phenotypes per patient). After application of MAX rules, only 1 patient had multiple phenotypes (mean, 1.02 phenotypes per patient). In the non‐PSP pathology group, all 56 patients had multiple phenotypes (mean, 3.41 phenotypes per patient), which was reduced to 7 patients (mean, 1.04 phenotypes per patient) after application of the MAX rules. Ocular motor abnormalities (O1, O2), postural instability (P1), and symmetric akinetic rigid parkinsonism with or without freezing (A1, A2) were more common in PSP as compared to the non‐PSP group (Fig. 1). For the cognitive domain, patients with PSP were more likely to have a frontal behavioral syndrome (C2) versus a speech/language disorder.
Figure 1.
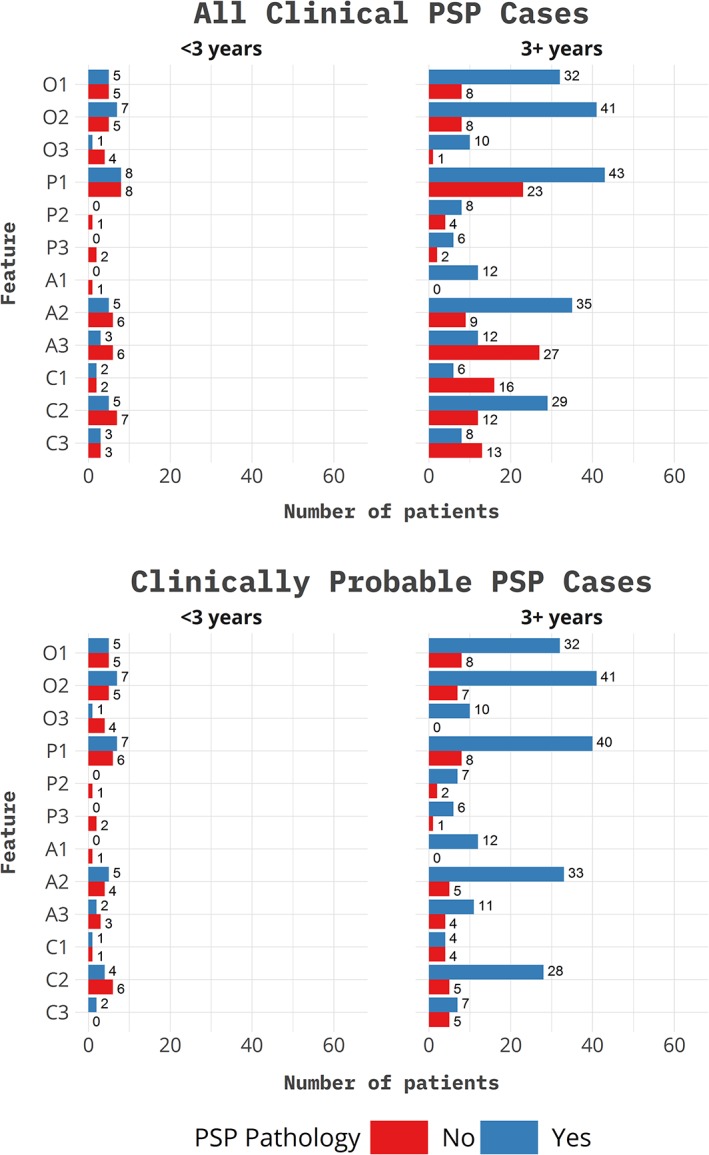
Symptoms by neuropathological category and time of evaluation. All patients with clinical features meeting PSP criteria are divided into PSP and non‐PSP neuropathology. At evaluation <3 years from onset, 21 patients (9 PSP pathology and 12 with non‐PSP pathology) met clinical criteria for PSP, of which 14 met probable criteria (7 PSP pathology and 7 non‐PSP pathology). At evaluation, >3 years from onset 82 patients (44 PSP pathology and 38 non‐PSP pathology) met criteria for PSP, of which 50 met criteria for probable PSP (41 had PSP pathology and 9 had non‐PSP pathology). OPCA designations as defined by the MDS‐PSP criteria.
Discussion
The MDS‐PSP criteria classify symptoms into four clinical domains, namely ocular motor, postural instability, akinesia, and cognitive (OPAC), combinations of which define various phenotypes that can result from PSP neuropathology. The criteria are a milestone in formal recognition of these diverse phenotypes. PSP‐RS is indeed the most widely recognized phenotype; however, it accounts for only a fraction of pathologically defined PSP cases. It is important for clinicians to be mindful of other phenotypes when evaluating patients clinically or for recruitment to natural history studies and tau‐directed treatment trials. Inclusion of these patients, who may have been excluded previously, will greatly add to our understanding of PSP pathobiology.
When applying the MDS‐PSP criteria, it is important to note certain practical points. The symptoms and signs within in each clinical domain are not listed in the order of disease progression, but rather in a descending order of predictive power for PSP pathology. For example, the ocular motor domain includes O1 (vertical supranuclear gaze palsy), O2 (slowing of vertical saccades), and O3 (apraxia of eyelid opening or square wave jerks) where apraxia of eyelid opening is a late finding if and when present, whereas vertical supranuclear gaze palsy is most predictive of PSP.17, 18 In our cohort, O1 (59% PSP vs. 20% non‐PSP), O2 (76% PSP vs. 18% non‐PSP), P1 (74% PSP vs. 20% non‐PSP), A2 (61% PSP vs. 13% non‐PSP), and C2 (52% PSP vs. 13% non‐PSP) were most predictive of PSP pathology. According to the criteria, P1 is defined as repeated unprovoked falls in the first 3 years, A2 is levodopa‐unresponsive akinetic rigid parkinsonism, whereas C2 frontal cognitive/behavioral syndrome.
Because of the multifocal nature of signs and symptoms, most patients may fulfill criteria for multiple phenotypes at a given point in time (e.g., a patient with PSP‐RS may also have features of PSP‐SL or PSP‐P), as we demonstrated previously.15 Furthermore, as the neurodegenerative process progresses, the phenotypes may evolve. The MAX rules were created to address this issue.16 We applied these rules as detailed in the Methods section and found them to be very helpful in reducing the number of diagnostic allocations. A unifying diagnostic label will help maintain standardization during clinical practice and research encounters. In doing so, however, it is apparent that important clinical information may be lost. For example, a patient who presents with language difficulties/apraxia of speech and years later eventually develops PSP‐P will be labeled as PSP‐P only. When applying these criteria, it is important to document all clinical signs and symptoms and their evolution given that early features may have diagnostic implications as we show in this study. For example, patients who had a frontal behavioral/cognitive syndrome (C2) were more likely to have PSP pathology as compared to patients to had speech/language disorders (C1) or corticobasal syndrome (C3) as shown in Figure 1. Even if a standardized diagnostic label is used, it will be important to consider disease heterogeneity when enrolling and assessing response to therapy in clinical trials. Prospective application of the MDS‐PSP criteria and MAX rules in clinical practice and research setting will lead to an evolution in the way we classify these patients. In the meantime, MAX rules provide a helpful way to standardize application of the MDS‐PSP criteria.
A few other practical points were raised during application of the MDS‐PSP criteria and MAX rules. Certain PSP phenotypes may bring to mind other neurodegenerative diseases with competing criteria such as corticobasal syndrome, behavioral variant frontotemporal dementia, and primary progressive aphasia. In such instances, we suggest that clinicians look for signs and symptoms highly associated with PSP, observe evolution over time, and ensure that the patient does not fit a competing criterion more precisely when considering research enrollment. Second, rare variants of PSP that were not formally defined in the MDS‐PSP criteria because of lack of sufficient clinicopathological correlation data, including PSP with predominant cerebellar ataxia10, 19 and PSP‐PLS,20 are important to retain in clinical practice as we learn more about these phenotypes.
In summary, it is important for clinicians to be aware of the diverse phenotypes and clinical heterogeneity of PSP. Application of the MDS‐PSP criteria and MAX rules in everyday clinical practice poses some challenges, but provides a good framework for standardizing assignment of phenotypes, which will be essential to prospective research.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
F.A.: 1A, 1B, 1C, 3A
H.B.: 2A, 2B, 3B
J.L.W.: 2C, 3B
K.A.J.: 1A, 2C, 3B
Disclosures
Ethical Compliance Statement: This study was performed with the approval of the Mayo Clinic Institutional Review Board (IRB). Since no identifiable information is reported, the study obtained a waiver for individual patient consent through the IRB. All authors state: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: Patient data included in this study were acquired as part of research activity under NIH grants R01‐NS89757, R01‐DC12519, and NIH R01‐NS89757. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months: Drs. Josephs, Whitwell, and Botha had NIH funding support during the past 12 months.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP International Workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 2. Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48. [DOI] [PubMed] [Google Scholar]
- 3. Tsuboi Y, Josephs KA, Boeve BF, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord 2005;20:982–988. [DOI] [PubMed] [Google Scholar]
- 4. Morris HR, Gibb G, Katzenschlager R, et al. Pathological, clinical and genetic heterogeneity in progressive supranuclear palsy. Brain 2002;125(Pt 5):969–975. [DOI] [PubMed] [Google Scholar]
- 5. Josephs KA, Boeve BF, Duffy JR, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase 2005;11:283–296. [DOI] [PubMed] [Google Scholar]
- 6. Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, Shoji S. Progressive supranuclear palsy presenting with primary progressive aphasia—clinicopathological report of an autopsy case. Acta Neuropathol 2003;105:610–614. [DOI] [PubMed] [Google Scholar]
- 7. Owens E, Josephs KA, Savica R, et al. The clinical spectrum and natural history of pure akinesia with gait freezing. J Neurol 2016;263:2419–2423. [DOI] [PubMed] [Google Scholar]
- 8. Hassan A, Parisi JE, Josephs KA. Autopsy‐proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase 2012;18:478–488. [DOI] [PubMed] [Google Scholar]
- 9. Donker Kaat L, Boon AJW, Kamphorst W, Ravid R, Duivenvoorden HJ, van Swieten JC. Frontal presentation in progressive supranuclear palsy. Neurology 2007;69:723–729. [DOI] [PubMed] [Google Scholar]
- 10. Kanazawa M, Shimohata T, Toyoshima Y, et al. Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord 2009;24:1312–1318. [DOI] [PubMed] [Google Scholar]
- 11. Josephs KA, Katsuse O, Beccano‐Kelly DA, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol 2006;65:396–405. [DOI] [PubMed] [Google Scholar]
- 12. Respondek G, Höglinger GU. The phenotypic spectrum of progressive supranuclear palsy. Parkinsonism Relat Disord 2016;22(Suppl 1):S34–S36. [DOI] [PubMed] [Google Scholar]
- 13. Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29:1758–1766. [DOI] [PubMed] [Google Scholar]
- 14. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ali F, Martin PR, Botha H, et al. Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord 2019. Feb 6. doi: 10.1002/mds.27619. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grimm MJ, Respondek G, Stamelou M, et al.; Movement Disorder Society‐endorsed PSP Study Group. How to apply the Movement Disorder Society criteria for diagnosis of progressive supranuclear palsy. Mov Disord 2019. Mar 18. doi: 10.1002/mds.27666. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osaki Y, Ben‐Shlomo Y, Lees AJ, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord 2004;19:181–189. [DOI] [PubMed] [Google Scholar]
- 18. Litvan I, Campbell G, Mangone CA, et al. Which clinical features differentiate progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome) from related disorders? A clinicopathological study. Brain 1997;120(Pt 1):65–74. [DOI] [PubMed] [Google Scholar]
- 19. Koga S, Josephs KA, Ogaki K, et al. Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of PSP‐C. Mov Disord 2016;31:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagao S, Yokota O, Nanba R, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci 2012;323:147–153. [DOI] [PubMed] [Google Scholar]