

Abstract
A successful chemotherapy-immunotherapy solid-tumor protocol should accomplish the following goals: debulk large tumors, release tumor antigen for cross-presentation and cross-priming, release cancer-suppressive cytokines and enhance anti-tumor immune cell populations. Thermally-activated drug delivery particles have the potential to synergize with immunotherapeutics to accomplish these goals; activation can release chemotherapy within bulky solid tumors and can enhance response when combined with immunotherapy. We set out to determine whether a single protocol, combining locally-activated chemotherapy and agonist immunotherapy, could accomplish these goals and yield a potentially translational therapy. For effective delivery of free doxorubicin to tumors with minimal toxicity, we stabilized doxorubicin with copper in temperature-sensitive liposomes that rapidly release free drug in the vasculature of cancer lesions upon exposure to ultrasound-mediated hyperthermia. We found that in vitro exposure of tumor cells to hyperthermia and doxorubicin resulted in immunogenic cell death and the local release of type I interferons across murine cancer cell lines. Following intravenous injection, local activation of the liposomes within a single tumor released doxorubicin and enhanced cross-presentation of a model antigen at distant tumor sites. While a variety of protocols achieved a complete response in more than 50% of treated mice, the complete response rate was greatest (90%) when 1 week of immunotherapy priming preceded a single activatable chemotherapeutic administration. While repeated chemotherapeutic delivery reduced local viable tumor, the complete response rate and a subset of tumor immune cells were also reduced. Taken together, the results suggest that activatable chemotherapy can enhance adjuvant immunotherapy; however, in a murine model the systemic adaptive immune response was greatest with a single administration of chemotherapy.
Keywords: CpG, αPD-1, Doxorubicin, Temperature-Sensitive Liposome, Ultrasound, Immunotherapy, Breast Cancer
1. Introduction
Immunotherapy has emerged as a robust and durable treatment option for a subset of patients; however, therapeutic efficacy has been limited to tumors with high mutational burdens and T-cell infiltration [1–5]. While immune checkpoints and co-stimulatory molecules have been effective for overcoming the inhibitory mechanisms largely dictated by the PD-1/PD-L1 axis, IDO, and regulatory T-cells in the T-cell-inflamed tumor micro-environment (TME), it has become apparent that new strategies may be required to treat non-T-cell-inflamed tumors [5]. For example, local adjuvant therapy can create an innate immune response that enhances efficacy [6–8]. The addition of chemotherapy to the protocol can further facilitate reaching the following goals: debulk large tumors, release tumor antigen for cross-presentation and cross-priming, release cancer-suppressive cytokines and retain circulating immune cell populations. Thermally-activated drug delivery particles have the potential to accomplish these goals as ultrasound or other treatment modalities can release the chemotherapy within bulky solid tumors to maximize the local response and minimize off-target effects. We set out to determine whether a single protocol could accomplish these goals and yield a potentially translational therapy.
Chemotherapeutics have long been the clinical standard for the treatment of solid tumors, yet the overwhelming majority of patients with metastatic disease do not have durable responses [9,10]. Chemotherapy is generally considered immune suppressive as it is cytotoxic to dividing cells in lymphoid tissue, however, recent work has demonstrated that a fraction of these drugs can induce cancer cells to undergo immunogenic cell death (ICD) [11]. ICD mediates tumor immunity through the spatiotemporal release of soluble factors that recruit leukocytes, support antigen processing and presentation, and stimulate tumor specific cytotoxic lymphocytes (CTL) [12,13]. The cumulative effect of ICD in response to anti-neoplastic agents has been reported to dramatically alter the composition of the tumor immune infiltrate towards an effector T-cell phenotype, which has a positive prognostic value [14–17]. Specifically, anthracyclines are a class of cytotoxic chemotherapeutics that are known to induce a potent ICD response [18], and as a result are of particular interest for the treatment of non-T-cell-inflamed-tumors. However, the dosing and duration of anthracyclines for treating metastatic cancer is limited due to the acute and cumulative cardiotoxicity incurred to the patient throughout treatment, and can be insufficient for overcoming tumor-mediated immune suppression [19].
Doxorubicin-loaded liposomes have been employed to deliver a localized dose of drug and circumvent the cardiotoxicity observed with anthracycline administration [20–23]. Previously, we formulated novel temperature-sensitive liposomes (TSL) loaded with a pH-sensitive complex between doxorubicin (Dox) and copper (CuDox) and demonstrated the efficient delivery of drug to a tumor in a murine model of multi-focal murine cancer [24,25]. This CuDox complex confers a highly-stable particle at physiological pH, thus minimizing the concentration of circulating free Dox [24–26]. The local release of Dox is triggered by insonifying a tumor to mild-hyperthermic temperatures (~42°C) via ultrasound (US). The incorporation of a toll-like receptor (TLR) 9 agonist, CpG, into a treatment protocol with CuDox-TSL and US (activatable drug delivery, ADD) enhanced the local response, where all directly-treated tumors were eliminated after 3 treatments over a one-week treatment protocol [26]. However, this treatment protocol was insufficient for overcoming tumor-mediated immune suppression in distant solid tumors. Similarly, we have previously shown that local administration of CpG combined with anti-PD-1 (αPD-1) could achieve a complete response in ~70 % of mice with multisite disease; however, such a protocol is less effective when a greater tumor burden is present and therefore clinical management will require the incorporation of chemotherapy, surgery, or focal therapy [27].
In this study, we combine activatable chemotherapy with a TLR9 agonist (CpG) and PD-1 blockade. We employed enzyme-linked immunosorbent assays (ELISAs), flow cytometry, immunohistochemistry (IHC), and tumor growth analysis to assess the efficacy of treatment protocols where immunotherapy begins before and after ADD administration and for single and multiple dosing of ADD to a single tumor. These combined protocols were evaluated in three murine models of primary and distant site cancer: the B16-F10/B16-Ovalbumin (OVA) model of melanoma with known antigen [28], the syngeneic neu exon deletion line (NDL) of HER2+ mammary adenocarcinoma [29], and the MMTV-PyMT transgenic model of breast cancer [30]. Given the importance of combination therapy for improving the treatment of non-T-cell-inflamed tumors, these studies may inform future human trials of activatable chemotherapy.
2. Materials and Methods
2.1. Materials
Copper (II) gluconate, triethanolamine (TEA), and doxorubicin hydrochloride (USP grade) were purchased from Sigma (St. Louis, MO). All lipids including 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (MPPC), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxypolyethyleneglycol-2000 (DSPE-PEG2k) were from Avanti Polar Lipids Inc. (Alabaster, AL). CpG-ODN 1826 (5’-tccatgacgttcctgacgtt-3’; total backbone phosphorothioated) was obtained from InvivoGen (San Diego, CA). The checkpoint inhibitor, rat anti-mouse PD-1 antibody (clone RMP1–14) was from Bio X Cell (West Lebanon, NH). The monoclonal antibody PE-anti-H-2Kb-SIINFEKL (25-D1.16) was purchased from eBioscience (San Diego, CA). The neu deletion (NDL) metastatic mammary carcinoma cell line was obtained from the Alexander Borowsky Laboratory (UC Davis) [31,32]. B16-F10 and 4T1 cells were purchased from American Type Culture Collection (ATCC, #CRL-6475 and #CRL-2539, respectively). B16-OVA cells, genetically modified to express chicken ovalbumin (OVA), were a generous gift from the William Murphy Laboratory (UC Davis, Davis, CA). The mT4 syngeneic mouse KPC pancreatic cancer cell line was isolated from KPC tumors derived from KPC-B6 background mice (Kras +/LSL-G12D; p53 +/LSL-R172H; PDX-Cre) and was a generous gift from Dr. David Tuveson (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). All above cell lines were cultured in high-glucose DMEM (Gibco, #11995) supplemented with 10% FBS and 1% penicillin-streptomycin in a 37°C humidified CO2 incubator. Syngeneic murine MC-38 colon cancer cells (kindly provided by Dr. David Colcher, Small Animal Imaging Core, City of Hope) were cultured in MEM Earle’s medium with 2mM L-glutamine (#11095, Gibco) supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin, 1 mM sodium pyruvate, and 0.1 mM non-essential amino acids (# 13–114E, Lonza). For implantation of B16 and B16-OVA cells in mice, cells were collected once they reached 85–90% confluency and resuspended in 1:1 Matrigel (Corning, #356234): PBS without calcium and magnesium (PBS−/−).
2.2. Preparation of liposomes and Dox loading
Temperature-sensitive liposomes (TSL) were made from DPPC:DSPE-PEG2k:MPPC (86:4:10, molar ratio). Liposomes were prepared by the hydration and extrusion method as described previously. Lipids, at the indicated ratios, were dissolved in chloroform. The chloroform was removed under a gentle stream of nitrogen gas and, subsequently, the residual solvent was removed under vacuum overnight. The dried lipid was hydrated in 0.3 mL of 100 mM copper (II) gluconate including triethanolamine at 540 mM (pH 8.4) to prepare copper-TSL (Cu-TSL). The multi-lamellar lipid solution at a final concentration of 50 mg/mL was extruded above the phase transition temperature of the lipid mixture through a polycarbonate membrane with a pore diameter of 100 nm. Cu-TSL was separated from non-encapsulated copper/TEA by passing the extruded liposomal suspension through a spin column of Sephadex G-75 (5 × 1 cm, GE Healthcare, Biosciences, Piscataway, NJ) equilibrated with saline (0.9% sodium chloride). The hydrodynamic size and zeta potential values were measured using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). The average size and the polydispersity index (PDI) of the purified Cu-TSL were 108 nm and 0.078, respectively, and the zeta value was −25 ± 10 mV. Lipid concentration was measured using the Phospholipids C assay kit (Wako Chemicals USA, Richmond, VA) according to manufacturer’s instructions.
Following separation of Cu-TSL using a Sephadex G-75 column, a transmembrane gradient was created across the membrane that allows highly membrane permeable TEA to diffuse to the external liposomal environment, leaving Cu (P < 10−11 cm/s) entrapped within the core of the liposomes. Upon incubation of Cu-TSL with Dox at a drug-to-lipid ratio of 0.2:1 (wt:wt), TEA diffuses through the lipid bilayer into the external liposomal environment and facilitates active loading of Dox into the core of the liposomes where it forms a complex with Cu at neutral pH and results in CuDox-TSL. A loading of 100% was achieved when Dox was added to Cu-TSL at a drug-to-lipid ratio of 0.2:1 (wt:wt) and incubated at 37°C for 1.5 h [25]. The resulting CuDox-TSL were then separated from non-encapsulated Dox using Sephadex G-75 spin columns. Loading of Dox into Cu-TSL did not affect the size and surface charge of the liposomes. The purified CuDox-TSL exhibited a rapid release of ~90% of Dox within 2 min at 42°C as assayed by fluorescence in a reduced pH buffer [25,33]. Additional details are provided in the supporting information.
2.3. Animals and in vivo procedures
All animal studies were conducted according to guidelines approved by the University of California, Davis, Animal Care and Use Committee (IACUC) and additional details of the study are provided in the supporting information. Female FVB/n mice, female C57BL/6 mice, C57BL/6-Tg(CAG-OVA)916Jen/J (CAG-OVA) mice, and female FVB/n-Tg(MMTV-PyMT) transgenic mice were purchased from Jackson Laboratory (Bar Harbor, ME). To generate the bilateral syngeneic neu-deletion (NDL) tumor model, FVB/n mice were orthotopically transplanted with NDL tumor biopsies into the #4 and #9 inguinal mammary fat pads as previously described [34]. A B16-F10/B16-OVA dual tumor model was used to quantify SIINFEKL-presenting immune cells at a 48-hour time point (Day 15 post tumor cell injection). On Day 0, mice were subcutaneously (s.c.) injected with B16-F10 (3 × 105 cells per 50 μL Matrigel:PBS−/−) and B16-OVA cells (6 × 105 cells per 50 μL Matrigel:PBS−/−) in the left and right flanks, respectively. SIINFEKL-negative control mice, bearing bilateral B16-F10 control tumors, were injected s.c. with 2.5 × 105 B16-F10 cells per 50 μL Matrigel:PBS−/− into the left and right flanks. C57BL/6-Tg(CAG-OVA)916Jen/J (CAG-OVA) mice (6 weeks old, 15–25 g), which constitutively express the OVA protein, were used as SIINFEKL-positive controls. FVB/n-Tg(MMTV-PyMT) transgenic mice were used to evaluate the systemic response to the chemo-immunotherapy in a mouse model of breast cancer metastasis.
A total of 139 bilateral NDL-tumor bearing mice, 12 MMTV-PyMT mice, and 13 bilateral B16 melanoma tumor mice were randomly distributed among several groups including: CuDox+US+CpG+αPD-1-prime (IT-ADD, n = 17), CuDox+US+CpG+αPD-1 (ADD-IT, n = 21), CuDox+US+CpG (ADD-CpG, n = 8), US+CpG+αPD-1 (US-IT, n = 2), CpG+αPD-1-prime (IT, n = 16), CpG+αPD-1(IT, n = 4), CuDox+US (ADD, n = 15), US+CpG (US-CpG, n= 7), CpG (n = 12), αPD-1 (n = 4), CuDox-TSL (n = 4), and no-treatment control (n = 54). Tumor diameters were measured with ultrasound and treatment started when tumors reached ~3 to 5 mm (~ 20 – 60 mm3) in longitudinal diameter. In the treatment cohorts involving ADD, mice were injected via the tail vein with CuDox-TSL (~6 mg Dox/kg body weight and ~30 mg lipid/kg body weight). Non-drug treated control mice received a saline injection; 150 µL of 0.9% sodium chloride intravenously and/or 50 µL intratumorally were each evaluated and combined into the control group as no significant difference was observed. For treatments involving CpG, a dose of 100 µg in 50 µL of endotoxin-free water was administered intratumorally in a single injection (CpG only and IT) or to the treated tumor immediately following US hyperthermia (ADD-IT and ADD-CpG, US+CpG). αPD-1 (200 µg) was delivered intraperitoneal (i.p.) in 50 µl volume of PBS (−/−). For all insonified mice, one tumor per mouse was insonified for 5 min at 42°C prior to intravenous administration of drug and saline in the drug treatment with US and US-only groups, respectively. Tumor insonation was continued for an additional 20 min at 42°C after injection, and CpG was injected into the insonified tumor. Hyperthermia was performed using a programmable US system combining imaging and therapy (Vantage 256, Verasonics, Kirkland, WA). A custom 128-element 1.5 MHz therapeutic array was used to heat the tumor [35]. Heating was performed with bursts of 2.5 MPa peak negative pressure, with a pulse repetition frequency of 100 Hz and burst duration ranging from 0 to 7 ms as controlled by a proportional integral derivative (PID) controller (duty cycle ranging from 0 to 0.7) set to maintain the tumor temperature at 42 °C.
2.4. Antibodies
The following fluorochrome-conjugated monoclonal antibodies (mAbs) were purchased from BioLegend (San Diego, CA): Pacific blue (PB)-anti-CD45 (30-F11), fluorescein isothiocyanate (FITC)-anti-F4/80 (BM8), phycoerythrin (PE)-anti-NK1.1 (PK136), PE-Cy7-anti-CD3 (145–2C11), PE-Cy7-anti-CD11c (N418), allophycocyanin (APC)-CD206 (C068C2), APC-Cy7-anti-CD11b (M1/70), APC-Cy7-anti-CD25 (PC61), Alexa Fluor (AF)-700-anti-CD8 (53–6.7), AF-700-anti-Ly6G/Ly6C (Gr-1, RB6–8C5); from BD Biosciences (San Jose, CA): FITC-anti-CD4 (GK1.5), PE-anti-CD86 (GL1); and from eBioscience (San Diego, CA): PE-Cy5-anti-MHCII (M5/114.15.2), PE-anti-H-2Kb-SIINFEKL (25-D1.16). Isotype-matched mouse, rat and hamster IgG mAbs were used as negative staining controls. To block FcγIII/II receptor-mediated unspecific binding, the anti-CD16/CD32 antibody (2.4G2) from BD Biosciences was used.
2.5. Cell preparation and flow cytometry
Bilateral tumor-bearing mice were sacrificed on days 28 and 35, a week after one or two complete treatments of ADD-IT, for immune cell profiling via flow cytometry. Tumors and the tumor-draining inguinal lymph nodes were collected separately and processed for these analyses. Single-cell suspensions were obtained by mechanical disruption of the tissue followed by enzymatic digestion with 1mg/mL collagenase IV (Sigma Aldrich, St. Louis, MO) and filtration through a 70 μm cell strainer (BD Biosciences, San Jose, CA). Cell suspensions were stained using the LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions to exclude dead cells from analysis. Cells were then incubated with 2.4G2 mAb for 10 min to block nonspecific antibody binding and finally stained with combinations of the indicated fluorochrome-conjugated anti-mouse antibodies for 25 min at 4°C. Antibody combinations used to distinguish immune cell populations were CD45+ (leukocytes) plus the following: CD3+, CD4+ (CD4+ T-cells), CD3+, CD8+ (CD8+ T-cells), CD4+, CD25+, Foxp3+ (regulatory T-cells), CD11b+, F4/80+, Gr-1− (macrophages), CD11b+, F4/80+, Gr-1−, CD86+, MHCIIhi (M1 macrophages), CD11b+, F4/80+, Gr-1−, CD206+, MHCIIlow (M2 macrophages), CD11c+, MHCII+, F4/80− (dendritic cells), CD3−, NK1.1+ (natural killer cells), and CD11b+, Gr-1+ (myeloid derived suppressor cells). To identify immune cells interacting with tumor antigens, we used H-2Kb-SIINFEKL antibody that reacts with the OVA peptide, SIINFEKL, bound to H-2Kb of the MHC class I on antigen-presenting cells. Antibody panel combinations were CD45+ (leukocytes) plus the following: SIINFEKL+ (SIINFEKL+ leukocytes); SIINFEKL+, CD11b+, F4/80+, Gr-1− (SIINFEKL+ macrophages); and SIINFEKL+, CD11c+, MHCII+, F4/80− (SIINFEKL+ dendritic cells).
All cell preparations were fixed in Cytofix buffer (BD Biosciences) diluted to 1% paraformaldehyde (PFA) in phosphate buffered saline (PBS). Stained cells were analyzed within 24 h on a LSRII flow cytometer (BD, San Jose, CA) and all datasets were analyzed using FlowJo software vX (TreeStar).
IFN-γ secretion from CD4+ and CD8+ T-cells was quantified using the Mouse IFN-γ Secretion Assay Cell Enrichment and Detection Kit (130–090-517, Miltenyi Biotec, San Diego, CA) according to the manufacturer’s instructions, except cells were not magnetically labeled and enriched over a MACS column. Instead, after addition of the IFN-γ catch reagent to cells, followed by incubation in the recommended culture medium for 45 min at 37°C, cells were washed and incubated with PE-anti-IFN-γ detection antibody in combination with the helper and cytotoxic T-cell fluorochrome-conjugated antibodies indicated above for 15 min at 4°C. Finally, cells were washed, fixed in 1% PFA Cytofix buffer and run on a LSR II cytometer.
2.6. In vitro release of HMGB1
NDL tumor cells were plated at 3.5 × 105 cells/well in 12-well tissue culture treated plates 24 hours prior to experiments. Cell plates were placed in a water bath at 42°C for 1 minute and allowed to cool to room temperature for 5 minutes. Doxorubicin (5 µg/mL final concentration) or media alone was then added directly to the cell wells to bring the total volume to 500 µL. Cells were incubated continuously with treatments for 24 hours in a 37°C humidified CO2 incubator. After 24 hours, cell culture media was collected and centrifuged at 300 × g for 5 minutes. Debris-free culture supernatant was transferred to a fresh tube, and HMGB1 content in undiluted samples was measured via ELISA (MBS722248, MyBioSource) according to the manufacturer’s instructions. HMGB1 concentration was normalized to total cell number (live + dead cells, counted via hemocytometer) for each sample. All treatments were analyzed in triplicate.
2.7. In vitro IFN-alpha and IFN-beta ELISA studies
Tumor cells were plated in 12-well tissue-culture treated plates at a concentration of 3.5 × 105 cells/well. Cells were cultured overnight in 1 mL complete media in a 37°C humidified chamber containing 5% CO2. The next day, the cell media was removed and replaced with 500 μL of pre-warmed media per well. The biological groups were the following: 37°C (n=3), 42°C (n=3), 42°C+Free Dox (n=6). The latter two samples were placed in a 42°C water bath for 5 min, immediately followed by addition of either media or a solution of free Dox in media (5 μg/mL) which were preincubated separately at 42°C, to the appropriate wells and incubation for another 5 min at 42°C. All cells were then incubated continuously for 24 hours in a 37°C humidified chamber containing 5% CO2. After 24 hours, the cell culture medium was collected, centrifuged (300 × g, 10 min, 4°C), and the supernatant was collected for IFN-α and IFN-β ELISA quantification (#42115 and 42410, respectively, PBL Assay Science, Piscataway, NJ). IFN-α and IFN-β supernatant concentrations were normalized to total cell number for each sample.
2.8. Statistical analyses
Statistical analyses were performed using Prism 6 software (GraphPad Software Inc.). Data are expressed as mean ± SEM, unless otherwise indicated. For analysis of three or more groups, a one-way ANOVA test was performed with Tukey’s post-hoc test as stated. Analysis of differences between two normally-distributed test groups was performed using an unpaired t test assuming unequal variance. For t tests comparing only two groups within a larger data set, the mean of each treatment group was compared only to the mean of the control group and the Welch correction for multiple comparisons was applied. Differences between groups in the Kaplan-Meier plot were determined using Log-rank statistics. P-values less than 0.05 were considered significant.
3. Results
3.1. Dox mediates type I interferon (IFN) release, HMGB1 release, and cross-presentation of tumor antigen
Type I IFN induces a viral-like immune response, including the elevation of interferon-stimulated genes (ISGs) involved in polynucleotide sensing and the binding of non-self RNA [36]. We first quantified the in vitro release of type I IFNs from murine mammary carcinoma cells (4T1 and NDL), murine Kras-driven pancreatic cancer cells (mT4), murine colon adenocarcinoma cells (MC-38) and murine melanoma cells (B16). Cells were heated to 42°C for 1 minute and then incubated with Dox for 24 hours at 37°C to mimic the local release of Dox in vivo from temperature-sensitive particles via ultrasound-mediated hyperthermia. Release of IFN-α and IFN-β was enhanced in cells exposed to Dox and hyperthermia (42°C) as compared to the no treatment (37°C) and hyperthermia only (42°C) controls in all cell lines tested except MC-38, which did not provide a significant release of IFN-β (Fig. 1 A–B).
Figure 1. Doxorubicin increased type I interferon production in murine cancer cell lines and immunogenic cell death in NDL cells.
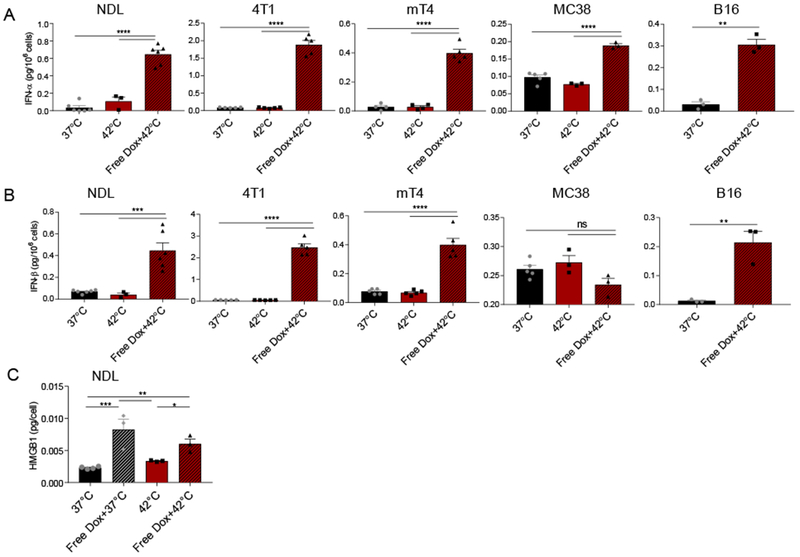
In vitro release of IFN-α (A), IFN-β (B) in NDL and 4T1 murine mammary carcinoma, mT4 murine pancreatic cancer, MC-38 murine colon cancer cells, B16 murine melanoma, and HMGB 1 in NDL cell culture 24 h post treatment, respectively. Cells were preincubated for 5 min (type I IFN) or 1 min (HMGB 1) at 42°C prior to addition of media only (42°C) or a solution of 5 µg/mL Dox in media (Free Dox+42°C) at 42°C for another 5 min. * p < 0.05, ** p< 0.01, *** p< 0.001, **** p< 0.0001.
Dox is known to achieve ICD in many cancers [37–39] and we confirmed HMGB1 release in our tumor model. Dox only and Dox plus pre-treatment with hyperthermia (42°C) elevated the release of the ICD factor, high-mobility group box protein-1 (HMGB1), suggesting Dox alone is sufficient to promote ICD of tumor cells (Fig. 1C).
We next set out to assess tumor antigen release and cross-presentation after ultrasonic delivery of Dox from TSL in vivo. To do this, we used a model of primary and distant melanoma, where B16 tumor cells expressing chicken ovalbumin (B16-OVA) were injected into one site, and B16-F10 cells, not expressing the cognate antigen, were injected into a distant site in the same wildtype immune-competent C57BL/6 mouse. A single treatment of CuDox-TSL was administered intravenously and B16-OVA tumors were heated with ultrasound (US) to 42°C to promote the local release of Dox approximately 12 days after tumor cell transplantation (Fig. 2A). Tumor antigen release and cross-presentation was assessed with flow cytometry, where the fraction of antigen presenting cells (APCs) displaying the OVA peptide fragment, SIINFEKL, bound to the MHC-I molecule was quantified in tumors, lymph nodes, spleen and blood 48 hours after treatment. ADD increased the total fraction of leukocytes (Fig. 2B), dendritic cells (Fig. 2C), and macrophages (Fig. 2D) displaying the MHC-I-SIINFEKL complex (SIINFEKL+) in the distant, or untreated, tumors, whereas this effect was attenuated in the directly-treated tumor and only an upward trend was observed. Further, a single treatment of ADD was sufficient to significantly enhance circulating SIINFEKL+ leukocytes in the blood of treated mice (Fig. 2E), whose antigen presenting cell populations (CD11c+, MHCII+ cells and CD11b+, F4/80+ cells) were largely SIINFEKL+ (SI Fig. S1A–B). Taken together, we find synergies between Dox and hyperthermia, with enhanced type I IFN, markers of ICD and systemic antigen cross-presentation by APCs 48 hours after treatment.
Figure 2. Tumor-specific antigen presentation increased in tumor and blood after treatment.
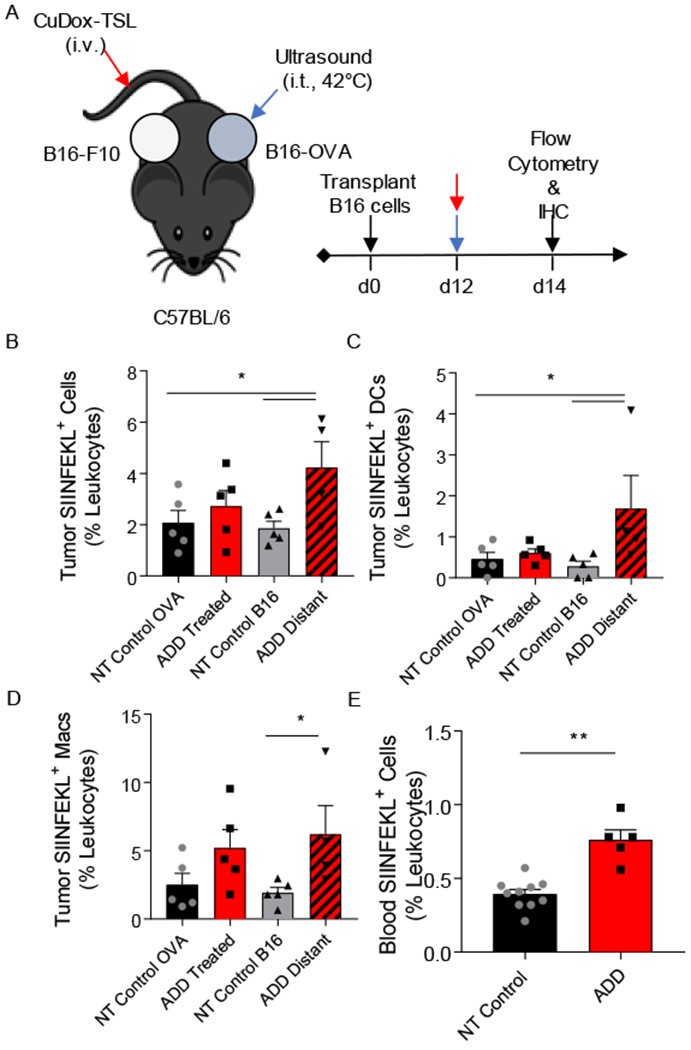
A) Schematic diagram illustrating the treatment protocol of CuDox+US (ADD) in C57BL/6 mice (n = 13) bearing one B16-OVA tumor (locally treated tumor) and one control B16-F10 tumor on the opposite side (distant tumor). B-D) Frequency of SIINFEKL-labeled immune cells, and specifically DCs, and macrophages as a percentage of leukocytes in tumors directly treated with ADD or in the distant tumor of the treated mice (n = 5) compared to B16-OVA and B16-F10 tumors of untreated control mice (n = 5), respectively. E) Frequency of SIINFEKL-labeled blood immune cells as a percentage of leukocytes in blood of mice treated with ADD compared to no-treatment (NT) control mice (n = 3). * p < 0.05, ** p< 0.01.
3.2. Single dosing with activatable drug delivery and immunotherapy or immunotherapy alone enhanced tumor infiltrating CD8+ T-cells and myeloid recruitment in treated and distant lesions
In order to elucidate a protocol for coupling chemotherapy with immunotherapy (CpG+αPD-1, abbreviated here as IT), we first surveyed the immune environment one week after a single administration of ADD, CpG, and αPD-1 (ADD-IT) in a murine model of primary and distant site mammary adenocarcinoma. NDL tumors were orthotopically transplanted into the fourth and ninth mammary fat pads of FVB mice, where a single tumor was treated 21 days after transplantation. Dox was delivered to the tumor via ultrasound-mediated release from CuDox-TSL and CpG was injected intratumorally; αPD-1 was then administered intraperitoneally 3 days after treatment of a single tumor (Fig. 3A). One week after the start of treatment, ADD-IT significantly reduced the total fraction of live cells in the treated tumor (pooled tumor and draining lymph node) compared to the no-treatment (NT) control (Fig. 3B). While IT and ADD-IT enhanced the fraction of leukocytes (CD45+ cells) in the treated tumor (Fig. 3C), ADD-IT increased the frequency of CD8+ T-cells in both the treated and distant tumor compared to the NT control (Fig. 3D). Despite the presence of tumor infiltrating CD8+ T-cells observed with ADD-IT treatment, the fraction of activated CD8+ T-cells secreting IFN-ɣ showed only an insignificant upward trend but was significantly enhanced in tumors directly treated with IT compared to NT control tumors (Fig. 3E). Although the frequency of antigen-presenting dendritic cells (DCs) was significantly increased by αPD-1 alone and not with other treatments (Fig. 3F), the number of infiltrating macrophages increased in the treated and distant tumors of the ADD-IT cohort on flow cytometry (Fig. 3G). IHC verified the infiltration of CD8+ T-cells in mice treated with the ADD-IT protocol (Fig. 3H). This was accompanied by a massive recruitment of myeloid cells (F4/80+ cells) into the tumor periphery and pockets of infiltration into non-viable tumor tissue. Foxp3 expression as a marker of regulatory T-cells was not increased by the ADD-IT treatment.
Figure 3. Combining ADD with CpG and αPD-1 increased cytotoxic T lymphocytes in local and distant NDL tumors.
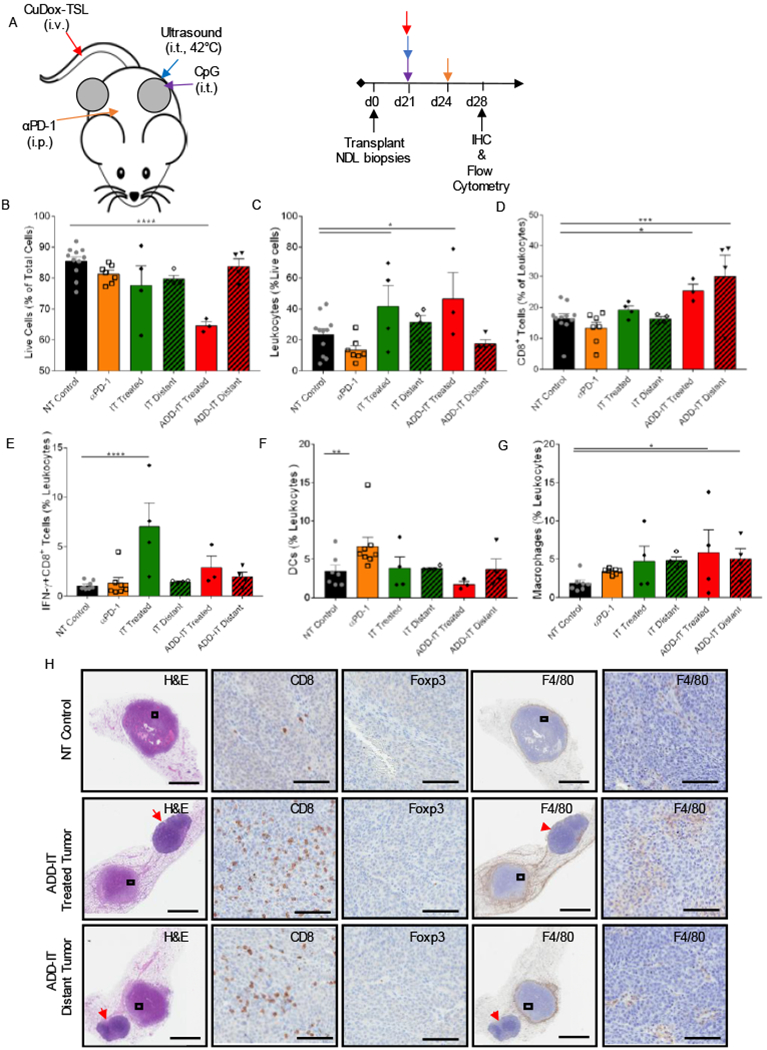
A) Representative treatment protocol and timeline for treatment of NDL tumor-bearing mice. One tumor of bilateral tumors of NDL orthotopically transplanted into FVB/n mice was treated with one complete administration of each component of CuDox+US+CpG (ADD-CpG) followed by αPD-1 post three days. B-G) On day 28 after one complete treatment of ADD-IT (n = 4) or IT (n = 4) or a single administration of αPD-1 (n = 4), tumors/inguinal lymph nodes of treated mice were stained with the antibody cocktail as listed below and analyzed via flow cytometry and compared with untreated control tumors. Fraction of live cells given as a percentage of total isolated cells (B), frequency of leukocytes (CD45+ cells) as a percentage of live cells (C) and frequency of cytotoxic T lymphocytes (CD3+CD8+ cells) as a percentage of total leukocytes (D), populations of IFN-γ producing CD8+ (CD3+CD8+IFN-γ+ cells) T-cells as a percentage of total leukocytes (E), fraction of DCs (CD11c+MHCII+ F4/80− cells) as a percentage of leukocytes (F), fraction of macrophages (CD11b+F4/80+Gr-1− cells) as a percentage of leukocytes (G) across various treatments. H) Histological sections of the no-treatment control, the tumor locally treated with CuDox+US+CpG+αPD-1 (ADD-IT) and the distant tumor on day 28 were stained with H&E (the left first column), CD8 (second column), Foxp3 (third column), F4/80 (fourth column) and the magnified images (fifth column), respectively. CD8, F4/80, and Foxp3 staining (brown) identify CD8+ T-cells, macrophages, and T regs, respectively. Red arrowheads denote the inguinal lymph nodes. Scale bars are 3 mm for whole tumor image and 100 µm for the magnified image. * p < 0.05, ** p< 0.01, *** p< 0.001, **** p< 0.0001.
We previously reported that leukopenia was not observed in mice treated with eight repeated administrations of ADD over a 4-week treatment course [25]. However, to thoroughly evaluate the effect of multiple administrations and activation of CuDox-TSL (ADD) on peripheral blood, we treated NDL tumor-bearing mice with 4 doses of ADD or CuDox-TSL (no US-activation) on days 21, 24, 28 and 31 post NDL transplants (SI Fig. S2A). Three days following the last treatment, frequencies of circulating live cells, leukocytes, total T-cells, and CD4+ and CD8+ T-cell subsets were not significantly altered for mice treated with ADD compared to those receiving CuDox-TSL without activation or saline-treated control groups (SI Fig. S2B–E). Circulating CD11c+, MHCII+ cells were significantly elevated in mice treated with CuDox-TSL and showed an insignificant trend to enhancement in those treated with ADD (SI Fig. S2F). Both CuDox-TSL and ADD treatments significantly elevated circulating macrophages (CD11b+, F4/80+ cells) (SI Fig. S2G). Circulating natural killer (NK) cells were significantly elevated only with the ADD treatment protocol (SI Fig. S2H)). These results clearly demonstrate that multiple administrations of ADD do not adversely affect the circulating immune cell counts and expansion of circulating macrophages and NK cells.
3.3. Repeated dosing of ADD-IT reduces local tumor burden and locally enhances T-cell activation
We next sought to characterize the impact of repeating the ADD-IT and IT protocols, as presented in Fig. 4A. Immunocytes from treated and distant tumors were harvested two weeks after the start of treatment (day 35) and analyzed with flow cytometry and IHC. Cell viability further decreased in the treated tumor and draining lymph node after 2 complete treatments as compared to a single dose of ADD-IT (Fig. 4B compared to Fig. 3B). Further, repeated ADD-IT dosing increased the fraction of leukocytes in both locally-treated and distant tumors (Fig. 4C). Immune cells from pooled tumor and draining lymph node at the day 35 time-point demonstrated that two doses of ADD-IT significantly increased both the frequency of CD8+ T-cells and the fraction of activated, or IFN-ɣ secreting CD8+ T-cells in directly-treated tumors (Fig. 4D–E). However, the IFN-ɣ secreting CD8+ T-cell fraction was on average reduced after two ADD-IT treatments (Fig. 4E) as compared with one treatment (Fig. 3E) The frequencies of DCs for both IT and ADD-IT treatments decreased to NT control group levels (Fig. 4F). Macrophages (as a fraction of leukocytes), however, trended toward increasing in both local and distant tumors of ADD-IT treated mice and in tumors directly treated with IT compared to the NT control group (Fig. 4G). In ADD-IT treated and distant tumors, the fraction of macrophages displaying an M1-phenotype dominated that of an M2-phenotype (SI Fig. S3A). Moreover, repeated ADD-IT treatment also decreased the fraction of intratumoral myeloid-derived suppressor cells (MDSCs) in both the treated and distant tumors (SI Fig. S3B). Hematoxylin and Eosin (H&E) staining confirmed the enhanced tumor cell death resulting from two cycles of ADD-IT treatments (Fig. 4H). IHC staining additionally demonstrated the sustained infiltration of CD8+ T-cells into the tumor following two treatments (Fig. 4H). This repeated dosing also elicited an increased recruitment of myeloid cells in the periphery of both the treated and distant tumors compared to the NT control (Fig. 4H).
Figure 4. Two treatments of ADD-IT increased IFN-ɣ-producing T-cells in the treated NDL tumor.
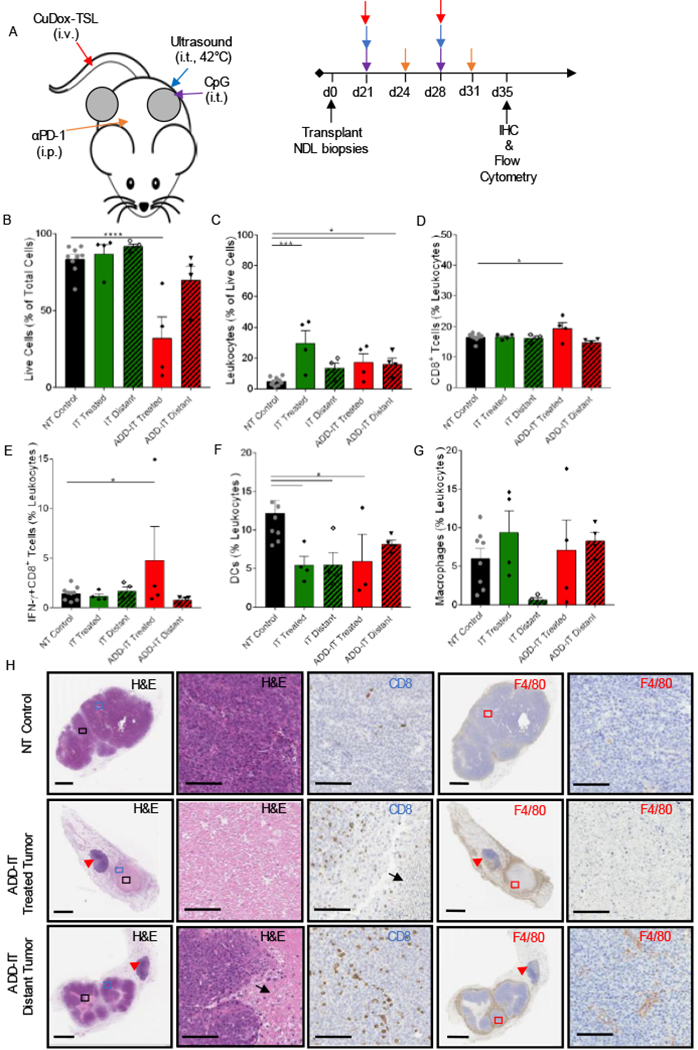
A) Representative treatment protocol and timeline for two treatments of ADD-IT administered to NDL tumor-bearing mice. B-G) After two sequential treatments of CuDox+US+CpG+ αPD-1 (ADD-IT, n = 4), the entire inguinal fat pads containing tumor and lymph node were harvested from treated mice and stained for CD45, CD3, CD4, CD8, F4/80, CD11b, CD11c, MHCII, Gr-1, and IFN-γ on day 35 and compared to untreated control mice (n = 4) via flow cytometry analysis. Number of live cells given as a percentage of total isolated cells (B), frequency of leukocytes (CD45+ cells) as a percentage of live cells (C), frequency of cytotoxic T lymphocytes (CD3+ CD8+ cells) as a percentage of total leukocytes (D), fraction of IFN-γ producing CD8+ (CD3+CD8+IFN-γ+) T-cells given as a percentage of total leukocytes (E), fraction of DCs (CD11c+ MHCII+ F4/80− cells) as a percentage of leukocytes (F), and fraction of macrophages (CD11b+ F4/80+ Gr-1− cells) as a percentage of leukocytes (G). H) Histological sections of no-treatment control, local and distant tumors of mice treated with two administrations of ADD-IT on day 35 and stained for H&E (the left first column, whole tumor view) and (second column, magnified view), CD8 (third column), F4/80 (fourth column, whole tumor) and the magnified panels (fifth column), respectively. CD8 and F4/80 staining (brown) identify CD8+ T-cells and macrophages, respectively. Black arrows and red arrowheads denote the necrotic cells and inguinal lymph nodes, respectively. Scale bars are 3 mm for whole tumor image and 100 µm for the magnified image. * p < 0.05, *** p< 0.001, **** p< 0.0001
To assess the effects of ADD-IT dosing in a transgenic model, we applied the protocol shown in Fig. 4A in the MMTV-PyMT (PyMT) transgenic model of breast cancer (SI Fig. S4A). Since this model provides a larger and more rapidly progressing tumor burden than the NDL model, we administered two doses of ADD combined with IT to the left axillary tumor (SI Fig. S4A, #3) followed by two doses to the right axillary tumor (SI Fig. S4A, #8). We monitored the untreated left and right inguinal tumors (SI Fig. S4A, #4 and #9, respectively) as the distant tumors. The ADD-IT protocol was then compared to the IT protocol in which the right cervical tumor was directly treated with a combination of CpG and αPD-1 as depicted in SI Fig. S4A, #6) and the untreated cervical tumor (#1) was considered as the distant tumor. Histological sections of tumors isolated following ADD-IT and IT treatment protocols showed extensive areas of necrosis and discohesive cells with pyknotic nuclei and ghost cells that were more pronounced throughout directly-treated tumors compared to distant tumors (SI Fig. S4B). IHC staining of CD8+ T-cells confirmed strong CD8+ T-cell and myeloid cell infiltration in mice treated with ADD-IT, indicating enhanced CD8+ T-cell and macrophage recruitment with the ADD-IT, as compared to the IT, treatment protocol.
3.4. Combining priming with a single dose of ADD expedited the anti-tumor response and enhanced survival
Motivated by the enhanced IFN-γ CD8+ T-cell fraction resulting from immunotherapy alone (Fig. 3E), we assessed the implementation of immunotherapy priming prior to ADD dosing (IT-ADD) in the NDL tumor model (Fig. 5A). A robust antitumor response was observed in nine of ten mice in the IT-ADD group and seven of nine mice in the IT group (Fig. 5B). Significantly greater regression occurred in both directly-treated and distant tumors at day 38 for mice treated with IT-ADD as compared to the IT protocol (Fig. 5C). Tumor regression was faster in the responding IT-ADD distant tumors, compared to those in the IT group (Fig. 5B), achieving a complete response 15 days earlier than the distant tumors in the IT protocol (44 ± 3 days vs 59 ± 9 days, respectively) (Fig. 5D, p = 0.002). Tumor regrowth and recurrence were not observed over the course of 101 days (Fig. 5B). While both IT and IT-ADD treatments significantly improved the survival, higher survival rates were achieved for mice treated with IT-ADD compared to IT, 90% versus 78%, respectively; both significantly improved survival compared to no-treatment control mice (p < 0.0001, Fig. 5E). NDL tumors in the no-treatment control group (NT Control) grew rapidly and approached the humane end point 36–38 days post NDL biopsy transplant (Fig. 5B–C). A single dose administration of ADD slowed the tumor growth in both treated and distant sites and slightly extended survival to 39–46 days post transplantation, with no noticeable difference in tumor growth between the treated and distant tumors (Fig. 5B,E).
Figure 5. Administration of CpG and αPD-1 prior to a combined ADD and immunotherapy protocol reduced tumor viability in both local and distant tumors of NDL-tumor bearing mice.
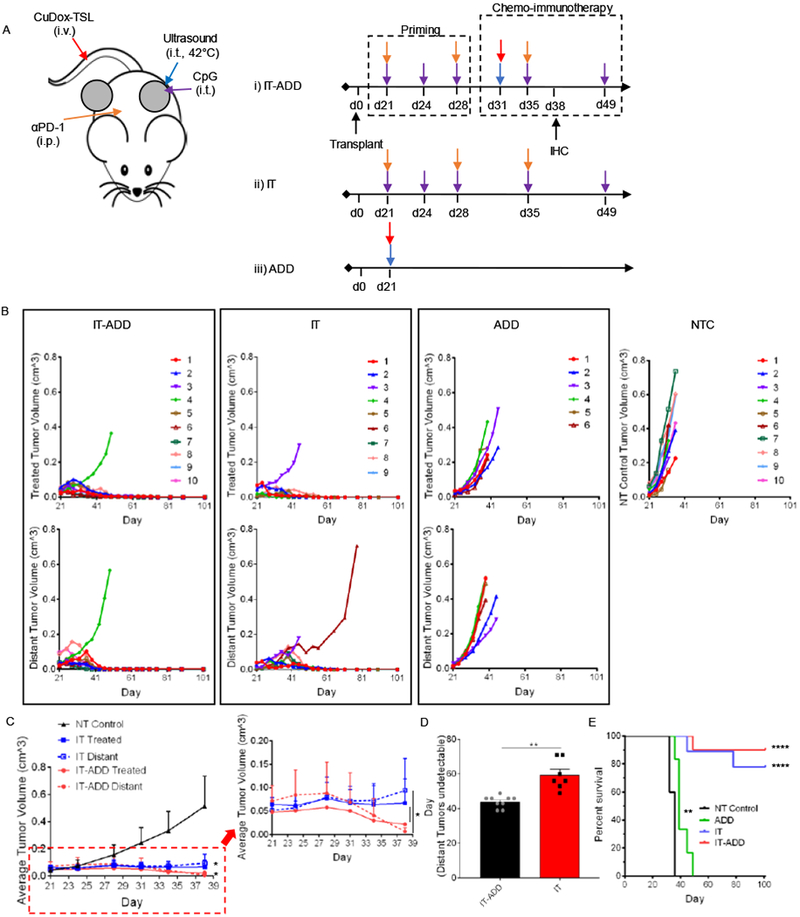
A) Treatment protocols of an immunopriming sequence with and without chemotherapy and chemotherapy alone. One tumor was treated directly with either CpG intratumorally in CpG+αPD-1 Prime (IT) or with CuDox+US (ADD) in CuDox+US+CpG+αPD-1 Prime (IT-ADD) and in CuDox+US (ADD) treatment groups. B) Growth of directly treated and distant tumors in bilateral NDL-tumor bearing mice treated with IT-ADD (n = 10), IT (n = 9), and ADD (n = 6) (B) compared to no-treatment control tumors (n = 10). C) Average growth of NDL tumors treated with either IT-ADD (n = 3) or IT (n = 5) treatment protocols compared to no-treatment control group over 38 days. The inset shows a representative magnified area. D) Temporal response of distant tumors to IT-ADD and IT treatments. E) The overall survival over the course of 101 days. In D, the survival curves for both primed & non-primed ADD-IT (p<0.0001) and ADD (p<0.01) were found statistically significant compared to NT Control group as evaluated by Log-rank (Martel-Cox) test. * p< 0.05, ** p< 0.01, **** p<0.0001).
3.5. IT-ADD treatment protocol revealed histological clearance of cancer cells in both directly treated and distant tumors
Further, IHC analysis from H&E staining on day 38 confirmed that a single dose of ADD combined with immunotherapy priming (IT-ADD) more effectively reduced viability and enhanced infiltration of CD8+ T-cells in both directly treated and distant tumors by day 38 (Fig. 6A). Upon termination of the study on day 101, the surviving mice were euthanized, and inguinal fat pads were evaluated by histology. H&E histological sections revealed a tumor cell-free fat pad embedding the lymph node in both treated and distant sites of mice treated with either IT-ADD or IT (Fig. 6B). Therefore, a single dose of ADD enhances the treatment efficacy when combined with immunotherapy.
Figure 6. Histological assessment of local and distant tumors confirmed antitumor efficacy of the ADD-IT treatment protocol.
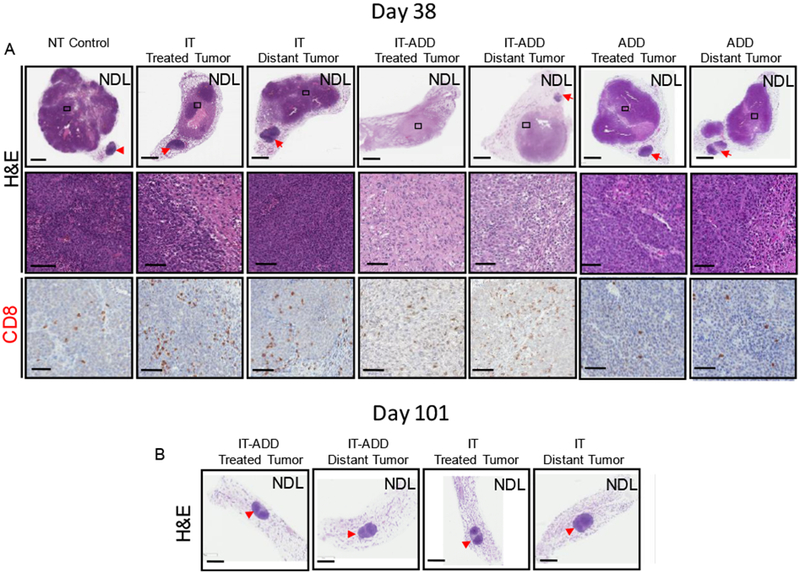
A) On day 38, a subset of mice was euthanized and tumors were isolated for histology and IHC. Histological sections stained for H&E (upper row, whole tumor view) and (middle row, magnified view), CD8 (lower row, magnified view). Whole tumor sections and the magnified views enclosed by black boxes are shown. B) H&E stained histological sections of mice treated with either IT-ADD or IT and survived 101 days. Red arrowheads denote the inguinal lymph nodes. Scale bars correspond to 3 mm (whole tumor panels) and 100 µm (magnified panels).
3.6. Repeating ADD reduced survival
We finally assessed whether repeated ADD cycles with and without one-week immune priming would enhance further survival in the NDL tumor model (SI Fig. S5A, S6A). With three repeated ADD treatments incorporated into an immunotherapy protocol, only 50% of treated NDL mice achieved a complete response 100 days after transplantation. For this repeated chemotherapy administration, survival was similar for protocols where immunotherapy or ADD were administered first (SI Fig. S5B,D and S6B). Taken together, we found that the rate of survival in this mouse model was greatest with a protocol where immunotherapy was administered for 1 week before ADD and ADD was administered only once.
4. Discussion
Combining locally-activated drug delivery with immunotherapy has the potential to enhance efficacy in bulky solid tumors. We demonstrated that a combined chemotherapy-immunotherapy protocol can rapidly reduce viable tumor, release tumor antigen for cross-presentation, release cancer-suppressive cytokines and result in increases in cytotoxic T-cells. In vitro experiments verified that type I IFN production by tumor cells was dependent on the cancer lineage, where murine breast cancer cells (NDL and 4T1) generated the highest levels. Type I IFN viral-like signaling pathways are known to recruit myeloid cells through downstream chemokines and cytokines, which provides the innate immune system with an opportunity to improve tumor antigen processing. This may provide insight into the efficacy of doxorubicin (Dox) for the treatment of various cancers and should be investigated further. We next confirmed that Dox induces NDL cells to secrete the immunogenic cell death (ICD) factor, HMGB1. This soluble factor is a well-known TLR4 agonist involved in DC maturation for efficient antigen engulfment and processing. Taken together, these data suggest that debulking a tumor with Dox should mediate cross-presentation of tumor antigen.
We verified with flow cytometry that the delivery of Dox from activatable liposomes to a single tumor mediates tumor antigen release, uptake, and cross-presentation by dendritic cells and macrophages. Interestingly, cross-presentation of tumor antigen was only observed in distant tumors 48-hours after treatment compared to the no-treatment controls. Moreover, the fraction of SIINFEKL+ leukocytes was significantly elevated in the blood at this timepoint. Our findings demonstrate that Dox induces the release of immunocompetent tumor antigen and this antigen is presented systemically. In the absence of immunotherapy priming, the reduced cross-presentation at the treated tumor site may be due to the large volume of locally-released drug and the resulting chemotherapy-mediated death of APCs or the rapid death of tumor cells that would otherwise shed antigen.
We found that the timing of the protocol for chemo-immunotherapy greatly impacts efficacy. Applying one week of the CpG adjuvant and αPD-1 prior to chemotherapy enhanced CD8+ T-cells and IFN-γ CD8+ T-cells. Similarly, a single dose of ADD enhanced CD8+ T-cells and reduced viable tumors. Combining these treatments with a priming dose of immunotherapy followed by one dose of ADD and additional immunotherapy resulted in a complete response in 90% of mice studied. The response was more rapid and consistent than that obtained with immunotherapy alone. In fact, the tumor volume rapidly decreased after drug release when release followed immunotherapy.
Alternatively, we found that the rate of complete response was lower for protocols that incorporated multiple doses of chemotherapy, regardless of whether the immunotherapy or chemotherapy began the treatment regimen. We further demonstrated that the activatable drug delivery methodology did not reduce the circulating immune cell numbers or fraction of circulating T cells. Therefore, the reduction in immunotherapy efficacy with repeated chemotherapy does not appear to result from a direct effect of chemotherapy on circulating immune cells. Instead, the data suggest that the rapid tumor cell death that results from the release of a large volume of drug within mouse tumors may blunt the impact of the local antigen and cytokine release [40]. This could in part result from the treatment of relatively small tumors (<1 cm) in mice. In human therapy, the treatment of larger tumor volumes will likely result in continued antigen and cytokine release over a longer time frame and therefore repeated administration could continue to enhance the immune response.
4.1. Activatable drug delivery with ultrasound
Debulking solid tumors with Dox offers the potential to generate ICD; however, delivering a therapeutic dose is challenging due to the rapid clearance of free drug and its associated cardiotoxicity. While the lipid-shelled drug carriers, Doxil® and Thermodox®, seek to circumvent these barriers to efficacious delivery, the low bioavailability and leakiness of these nanoparticles, respectively, have limited their clinical implementation [21,41–43]. Previously, we demonstrated the use of a novel activatable liposomal particle, where Dox was stabilized with copper [24,25]. CuDox liposomes have temperature-sensitive shells that provide localized delivery of CuDox through the application of transdermal ultrasonic energy. More importantly, these CuDox complexes improve drug bioavailability and minimize toxicity, as Dox remains chelated at physiological pH, preventing free Dox from disseminating into the blood and untargeted tissues [33,44]. Although treatments with ADD alone (8 treatments over 4 weeks) or combined with CpG to expedite the treatment response (3 treatments over one week), successfully cured local cancer, they were unfortunately insufficient for eliminating tumor at distant locations [25,26] . The faster response of the adjuvant-containing protocol suggests that combined therapy is beneficial. Thus, to better understand how to combine adjuvant therapy with chemotherapy, we investigated the impact of Dox on the anti-tumor immune response.
Ultrasound provides noninvasive image-guided and highly focused means of applying heat with growing application in clinical cancer treatment [45–47]. In addition, as an adjuvant, mild hyperthermia provides positive benefits to cancer therapy. Raising tumor temperature to a few degrees above the body temperature improves drug availability and efficacy, and induces immune responses. In addition, we reported previously that application of US hyperthermia 5 minutes prior to administration of CuDox-TSL sensitizes both vascular endothelium and tumors cells to drug by inducing hemorrhage that triggers infiltration of immune cells and augments the treatment outcome [24,25,48]. Intravascular release of Dox from circulating liposomal drug is triggered by ultrasound-mediated mild hyperthermia (US) applied locally on the entire tumor.
4.2. Challenges in the development of protocols for human translation
While a single dose of chemotherapy was optimal to achieve a complete response in mouse models with tumor diameters on the order of 5 mm, larger tumor volumes in patients may require additional cycles of treatment. Such activatable delivery systems result in rapid immunogenic cell death and antigen presentation. Therefore, enhancing the proportion of leukocytes, and creating a systemic immune response prior to locally-activated chemotherapy is a rational choice. The development of non-invasive methods to assess and track macrophage and T-cell population and activation in patients spatiotemporally over the course of treatment could provide important information for the timing of ADD [49–51]. A major challenge for this field is to translate the use of temperature-sensitive liposomes to the treatment of large animals and humans. The cost of such translational studies required for commercialization has limited the translation and availability of the these activatable nanotherapies.
5. Conclusions
We demonstrated that administration of a single dose of activatable chemotherapy effectively reduces tumor viability and induces release of tumor antigens that are effectively presented by macrophages and DCs. In addition, focal delivery of Dox, which mediates local release of ICD and production of type I IFN, amplifies the signaling pathways to release curative chemokines and cytokines. The resulting local and systemic immune stimulation was combined with an immune priming protocol to elicit potent antitumor T-cell responses, thereby exerting robust antitumor efficacy in a murine model of breast cancer. Tumor eradication was observed in both treated and distant tumors of mice treated with the chemo-immunotherapy protocol and 90% of treated mice were tumor-free for 101 days.
Supplementary Material
Figure S1. Tumor antigens released by CuDox+US treatment were presented by circulating immune cells. A-B) Flow cytometric analysis of blood samples collected from mice treated with CuDox+US (ADD) and no-treatment control mice 48 h post treatment. Frequency of SIINFEKL-labeled blood immune CD11b+, F4/80+ macrophages (A) and CD11c+, MHCII+ cells (B) as a percentage of total blood leukocytes. Statistical analysis was performed using an unpaired, one-tailed t test assuming unequal variance * p < 0.05.
Figure S2. Repeated administration of CuDox+US did not reduce circulating immune cells. A) Representative treatment protocol. B-H) On day 34 after four complete treatments of ADD (n = 4) or CuDox-TSL (n = 4), blood collected from the treated mice were stained with the antibody cocktail as listed in the Materials and Methods, analyzed via flow cytometry and compared with untreated control tumors (n = 4). Frequency of blood live cells given as a percentage of total isolated cells (B), frequency of leukocytes (CD45+ cells) as a percentage of live cells (C), frequency of all T-cells as a percentage of total leukocytes (D), CD4+ and CD8+ T-cell subsets as a percentage of total leukocytes (E), fraction of CD11c+MHCII+ cells as a percentage of leukocytes (F), fraction of macrophages (CD11b+F4/80+ cells) as a percentage of leukocytes (G), and fraction of natural killer (NK) cells as a percentage of leukocytes (H) across treatments. * p < 0.05, ** p< 0.01.
Figure S3. Two treatments of CuDox+US+CpG+αPD-1 altered macrophage phenotype and reduced MDSCs. A-B) After two sequential treatments of CuDox+US+CpG+αPD-1 (ADD-IT), the entire inguinal fat pads containing tumor and lymph node were harvested from treated and untreated control mice on day 35 and analysis of immune cells was performed using flow cytometry. Frequency of macrophages (CD11b+F4/80+Gr-1−) with M1 and M2 phenotypes as a percentage of total macrophages (A) and fraction of myeloid derived suppressor cells (MDSCs, CD11b+Gr-1+) as a percentage of leukocytes (B). ** p < 0.01.
Figure S4. Combinatorial CuDox+US+CpG+αPD-1 protocol increased tumor infiltration of cytotoxic T lymphocytes and macrophages in local and distant tumors of transgenic PyMT mice. A) Treatment protocols applied to PyMT transgenic mice. i) The axillary lymph node tumor (#3) was treated with two sequential administrations of CuDox+US+CpG+αPD-1 (ADD-IT) for two weeks, then the same two-treatment protocol was applied to the axillary lymph node tumor (#8) for another 2 weeks (n = 5). ii) The cervical lymph node tumor (#6) was treated with CpG+αPD-1 (IT) for 15 days (n = 2) and all treatment groups were compared to no-treatment (NT) control mice (n = 5). Mice were euthanized and all lymph node tumors were isolated for histology and immunohistochemistry. B) histological sections of NT control tumor, local and distant tumors of mice treated with either ADD-IT or IT and stained for H&E (the left first column, whole tumor view) and (second column, magnified view), CD8 (third column, magnified view), and F4/80 (fourth column, magnified view). Whole tumor sections and the magnified views enclosed by black or colored boxes are shown. Scale bars correspond to 3 mm (whole tumor panels) and 100 µm (magnified panels).
Figure S5. Repeated CuDox+US+CpG+αPD-1 protocol with immunopriming induced complete local and systemic responses with 50% long term survival. A) Treatment protocol integrating immunopriming sequence prior to chemo-immunotherapy. B-C) Growth of directly treated and distant tumors in bilateral NDL-tumor bearing mice treated with primed (IT-ADD, n = 4) (B) compared to no-treatment control (NT Control) tumors (C). D) Open-ended survival achieved with IT-ADD and ADD-IT compared with tri-combinatorial treatment of CuDox+US+CpG (ADD-CpG), bi-combinatorial treatment of US+CpG, and CpG only treatment. The survival curves for both IT-ADD and ADD-IT (p<0.0001), ADD-CpG (p<0.01), and CpG (p<0.01) were found statistically significant compared to the NT Control group as evaluated by Log-rank (Martel-Cox) test. ** p< 0.01, **** p<0.0001.
Figure S6. Three administrations of CuDox+US+CpG+αPD-1 without immunopriming achieved 50% long term survival. A) Treatment protocol starting with three cycles of ADD-IT without immunopriming. B) Growth of directly treated and distant tumors in bilateral NDL-tumor bearing mice treated with three administrations of (ADD-IT, n = 4).
Acknowledgements
This work was supported by funding from National Institute of Health grants NIHR01CA112356, NIHR01CA134659, NIHR01CA199658, NIHR01CA210553 and NIHR01CA211602.
Footnotes
Supplementary Information Available: Additional information for Methods, Supplementary information (SI) Figures S1–S6, and Table S1.
Conflict of interest
The authors declare no completing financial of interest.
References
- [1].Topalian SL, Drake CG, Pardoll DM, Immune checkpoint blockade: a common denominator approach to cancer therapy, Cancer Cell. 27 (2015) 450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science. 348 (2015) 124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA, Genomic correlates of response to CTLA-4 blockade in metastatic melanoma, Science. 350 (2015) 207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C, Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade, Science. 351 (2016) 1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gajewski TF, The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment, Semin Oncol. 42 (2015) 663–671. doi: 10.1053/j.seminoncol.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baird JR, Monjazeb AM, Shah O, McGee H, Murphy WJ, Crittenden MR, Gough MJ, Stimulating Innate Immunity to Enhance Radiation Therapy–Induced Tumor Control, Int. J. Radiat. Oncol. Biol. Phys. 99 (2017) 362–373. doi: 10.1016/j.ijrobp.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].van Aalst S, Ludwig IS, van Kooten PJS, van der Zee R, van Eden W, Broere F, Dynamics of APC recruitment at the site of injection following injection of vaccine adjuvants, Vaccine. 35 (2017) 1622–1629. doi: 10.1016/j.vaccine.2017.02.005. [DOI] [PubMed] [Google Scholar]
- [8].Coffman RL, Sher A, Seder RA, Vaccine adjuvants: Putting innate immunity to work, Immunity. 33 (2010) 492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Turcotte S, Rosenberg SA, Immunotherapy for metastatic solid cancers, Adv Surg. 45 (2011) 341–360. https://www.ncbi.nlm.nih.gov/pubmed/21954698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen G, Emens LA, Chemoimmunotherapy: reengineering tumor immunity, Cancer Immunol. Immunother. 62 (2013) 203–216. doi: 10.1007/s00262-012-1388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nowak AK, Robinson BW, Lake RA, Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors, Cancer Res. 63 (2003) 4490–4496. https://www.ncbi.nlm.nih.gov/pubmed/12907622. [PubMed] [Google Scholar]
- [12].Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Immunogenic cell death in cancer therapy, Annu Rev Immunol. 31 (2013) 51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- [13].Garg AD, Dudek-Peric AM, Romano E, Agostinis P, Immunogenic cell death, Int J Dev Biol. 59 (2015) 131–140. doi: 10.1387/ijdb.150061pa. [DOI] [PubMed] [Google Scholar]
- [14].Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G, Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer, J Clin Oncol. 28 (2010) 105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- [15].Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B, Feyen N, Martens UM, Beckhove P, Gnjatic S, Schirmacher P, Herpel E, Weitz J, Grabe N, Jaeger D, Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy, Cancer Res. 71 (2011) 5670–5677. doi: 10.1158/0008-5472.CAN-11-0268. [DOI] [PubMed] [Google Scholar]
- [16].Ladoire S, Mignot G, Dabakuyo S, Arnould L, Apetoh L, Rebe C, Coudert B, Martin F, Bizollon MH, Vanoli A, Coutant C, Fumoleau P, Bonnetain F, Ghiringhelli F, In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival, J Pathol. 224 (2011) 389–400. doi: 10.1002/path.2866. [DOI] [PubMed] [Google Scholar]
- [17].West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH, Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer, Breast Cancer Res. 13 (2011) R126. doi: 10.1186/bcr3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, Spisek R, Human tumor cells killed by anthracyclines induce a tumor-specific immune response, Cancer Res. 71 (2011) 4821–4833. doi: 10.1158/0008-5472.CAN-11-0950. [DOI] [PubMed] [Google Scholar]
- [19].Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A, Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials, BMC Cancer. 10 (2010) 337. doi: 10.1186/1471-2407-10-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sparano JA, Makhson AN, Semiglazov VF, Tjulandin SA, Balashova OI, Bondarenko IN, V Bogdanova N, Manikhas GM, Oliynychenko GP, Chatikhine VA, Zhuang SH, Xiu L, Yuan Z, Rackoff WR, Pegylated liposomal doxorubicin plus docetaxel significantly improves time to progression without additive cardiotoxicity compared with docetaxel monotherapy in patients with advanced breast cancer previously treated with neoadjuvant-adjuvant anthracyclin, J Clin Oncol. 27 (2009) 4522–4529. doi: 10.1200/JCO.2008.20.5013. [DOI] [PubMed] [Google Scholar]
- [21].de Smet M, Langereis S, van den Bosch S, Grull H, Temperature-sensitive liposomes for doxorubicin delivery under MRI guidance, J Control Release. 143 (2010) 120–127. doi: 10.1016/j.jconrel.2009.12.002. [DOI] [PubMed] [Google Scholar]
- [22].Chuang E, Wiener N, Christos P, Kessler R, Cobham M, Donovan D, Goldberg GL, Caputo T, Doyle A, Vahdat L, Sparano JA, Phase I trial of ixabepilone plus pegylated liposomal doxorubicin in patients with adenocarcinoma of breast or ovary, Ann Oncol. 21 (2010) 2075–2080. doi: 10.1093/annonc/mdq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wolff AC, Wang M, Li H, Pins MR, Pretorius FJ, Rowland KM, Sparano JA, Davidson NE, Phase II trial of pegylated liposomal doxorubicin plus docetaxel with and without trastuzumab in metastatic breast cancer: Eastern Cooperative Oncology Group trial E3198, Breast Cancer Res Treat. 121 (2010) 111–120. doi: 10.1007/s10549-010-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kheirolomoom A, Mahakian LM, Lai CY, Lindfors HA, Seo JW, Paoli EE, Watson KD, Haynam EM, Ingham ES, Xing L, Cheng RH, Borowsky AD, Cardiff RD, Ferrara KW, Copper-Doxorubicin as a Nanoparticle Cargo Retains Efficacy with Minimal Toxicity, Mol. Pharm. 7 (2010) 1948–1958. doi: 10.1021/mp100245u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kheirolomoom A, Lai CY, Tam SM, Mahakian LM, Ingham ES, Watson KD, Ferrara KW, Complete regression of local cancer using temperature-sensitive liposomes combined with ultrasound-mediated hyperthermia, J. Control. Release. 172 (2013) 266–273. doi: 10.1016/j.jconrel.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kheirolomoom A, Ingham ES, Mahakian LM, Tam SM, Silvestrini MT, Tumbale SK, Foiret J, Hubbard NE, Borowsky AD, Murphy WJ, Ferrara KW, CpG expedites regression of local and systemic tumors when combined with activatable nanodelivery, J. Control. Release. 220 (2015) 253–264. doi: 10.1016/j.jconrel.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Monjazeb AM, Hsiao HH, Sckisel GD, Murphy WJ, The role of antigen-specific and non-specific immunotherapy in the treatment of cancer, J. Immunotoxicol. 9 (2012) 248–258. doi: 10.3109/1547691x.2012.685527. [DOI] [PubMed] [Google Scholar]
- [28].Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP, Combination CTLA-4 blockade and 4–1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production, PLoS One. 6 (2011) e19499. doi: 10.1371/journal.pone.0019499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cardiff RD, Hubbard NE, Engelberg JA, Munn RJ, Miller CH, Walls JE, Chen JQ, Velasquez-Garcia HA, Galvez JJ, Bell KJ, Beckett LA, Li YJ, Borowsky AD, Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers, Lab. Investig. 93 (2013) 480–497. doi: 10.1038/labinvest.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fluck MM, Schaffhausen BS, Lessons in signaling and tumorigenesis from polyomavirus middle T antigen, Microbiol Mol Biol Rev. 73 (2009) 542–63, Table of Contents. doi: 10.1128/MMBR.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Miller JK, Shattuck DL, Ingalla EQ, Yen LL, Borowsky AD, Young LJT, Cardiff RD, Carraway KL, Sweeney C, Suppression of the Negative Regulator LRIG1 Contributes to ErbB2 Overexpression in Breast Cancer, Cancer Res. 68 (2008) 8286–8294. doi: 10.1158/0008-5472.can-07-6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Siegel PM, Ryan ED, Cardiff RD, Muller WJ, Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer, Embo J. 18 (1999) 2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kheirolomoom A, Ingham ES, Commisso J, Abushaban N, Ferrara KW, Intracellular trafficking of a pH-responsive drug metal complex, J. Control. Release. 243 (2016) 232–242. doi: 10.1016/j.jconrel.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Borowsky AD, Namba R, Young LJT, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff R, Gregg JP, Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior, Clin. Exp. Metastasis. 22 (2005) 47–58. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- [35].Liu JF, Foiret J, Stephens DN, Le Baron O, Ferrara KW, Development of a spherically focused phased array transducer for ultrasonic image-guided hyperthermia, Phys. Med. Biol. 61 (2016) 5275–5296. doi: 10.1088/0031-9155/61/14/5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF, Type I interferon response and innate immune sensing of cancer, Trends Immunol. (2013). doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gebremeskel S, Johnston B, Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies, Oncotarget. 6 (2015) 41600–41619. doi: 10.18632/oncotarget.6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Showalter A, Limaye A, Oyer JL, Igarashi R, Kittipatarin C, Copik AJ, Khaled AR, Cytokines in immunogenic cell death: Applications for cancer immunotherapy, Cytokine. 97 (2017) 123–132. doi: 10.1016/j.cyto.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Inoue S, Setoyama Y, Odaka A, Doxorubicin treatment induces tumor cell death followed by immunomodulation in a murine neuroblastoma model, Exp. Ther. Med. 7 (2014) 703–708. doi: 10.3892/etm.2014.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM, Ilovitsh A, Monjazeb AM, Murphy WJ, Hubbard NE, Davis RR, Tepper CG, Borowsky AD, Ferrara KW, Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation, Theranostics. 8 (2018) 3611–3628. doi: 10.7150/thno.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li L, Ten Hagen TLM, Hossann M, Süss R, Van Rhoon GC, Eggermont AMM, Haemmerich D, Koning GA, Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy, J. Control. Release. 168 (2013) 142–150. doi: 10.1016/j.jconrel.2013.03.011. [DOI] [PubMed] [Google Scholar]
- [42].Kong G, Braun RD, Dewhirst MW, Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size, Cancer Res. 60 (2000) 4440–4445. [PubMed] [Google Scholar]
- [43].Kong G, Braun RD, Dewhirst MW, Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature, Cancer Res. 61 (2001) 3027–3032. [PubMed] [Google Scholar]
- [44].Fite BZ, Kheirolomoom A, Foiret JL, Seo JW, Mahakian LM, Ingham ES, Tam SM, Borowsky AD, Curry FRE, Ferrara KW, Dynamic contrast enhanced MRI detects changes in vascular transport rate constants following treatment with thermally-sensitive liposomal doxorubicin, J. Control. Release. 256 (2017) 203–213. doi: 10.1016/j.jconrel.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Unga J, Hashida M, Ultrasound induced cancer immunotherapy, Adv. Drug Deliv. Rev. 72 (2014) 144–153. doi: 10.1016/j.addr.2014.03.004. [DOI] [PubMed] [Google Scholar]
- [46].Liu F, Hu ZL, Qiu L, Hui C, Li C, Zhong P, Zhang JP, Boosting high-intensity focused ultrasound-induced anti-tumor immunity using a sparse-scan strategy that can more effectively promote dendritic cell maturation, J. Transl. Med. 8 (2010) 12. doi: 10.1186/1479-5876-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xu ZL, Zhu XQ, Lu P, Zhou Q, Zhang J, Wu F, Activation of tumor-infiltrating antigen presenting cells by high intensity focused ultrasound ablation of human breast cancer, Ultrasound Med. Biol. 35 (2009) 50–57. doi: 10.1016/j.ultrasmedbio.2008.08.005. [DOI] [PubMed] [Google Scholar]
- [48].Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC, Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer, Clin. Cancer Res. 11 (2005) 728–734. [PubMed] [Google Scholar]
- [49].Mayer AT, Levy R, Johnson EM, Czerwinski DK, Sagiv-Barfi I, Alam IS, James ML, Gambhir SS, Wang K, Vermesh O, Imaging activated T cells predicts response to cancer vaccines, J. Clin. Invest. 128 (2018) 2569–2580. doi: 10.1172/jci98509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Seo JW, Tavaré R, Mahakian LM, Silvestrini MT, Tam S, Ingham ES, Salazar FB, Borowsky AD, Wu AM, Ferrara KW, CD8+ T-cell density imaging with 64Cu-labeled cys-diabody informs immunotherapy protocols, Clin. Cancer Res. 24 (2018) clincanres.0261.2018. doi: 10.1158/1078-0432.CCR-18-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Khurana A, Nejadnik H, Gawande R, Lin G, Lee S, Messing S, Castaneda R, Derugin N, Pisani L, Lue TF, Daldrup-Link HE, Intravenous Ferumoxytol Allows Noninvasive MR Imaging Monitoring of Macrophage Migration into Stem Cell Transplants, Radiology. 264 (2012) 803–811. doi: 10.1148/radiol.12112393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tumor antigens released by CuDox+US treatment were presented by circulating immune cells. A-B) Flow cytometric analysis of blood samples collected from mice treated with CuDox+US (ADD) and no-treatment control mice 48 h post treatment. Frequency of SIINFEKL-labeled blood immune CD11b+, F4/80+ macrophages (A) and CD11c+, MHCII+ cells (B) as a percentage of total blood leukocytes. Statistical analysis was performed using an unpaired, one-tailed t test assuming unequal variance * p < 0.05.
Figure S2. Repeated administration of CuDox+US did not reduce circulating immune cells. A) Representative treatment protocol. B-H) On day 34 after four complete treatments of ADD (n = 4) or CuDox-TSL (n = 4), blood collected from the treated mice were stained with the antibody cocktail as listed in the Materials and Methods, analyzed via flow cytometry and compared with untreated control tumors (n = 4). Frequency of blood live cells given as a percentage of total isolated cells (B), frequency of leukocytes (CD45+ cells) as a percentage of live cells (C), frequency of all T-cells as a percentage of total leukocytes (D), CD4+ and CD8+ T-cell subsets as a percentage of total leukocytes (E), fraction of CD11c+MHCII+ cells as a percentage of leukocytes (F), fraction of macrophages (CD11b+F4/80+ cells) as a percentage of leukocytes (G), and fraction of natural killer (NK) cells as a percentage of leukocytes (H) across treatments. * p < 0.05, ** p< 0.01.
Figure S3. Two treatments of CuDox+US+CpG+αPD-1 altered macrophage phenotype and reduced MDSCs. A-B) After two sequential treatments of CuDox+US+CpG+αPD-1 (ADD-IT), the entire inguinal fat pads containing tumor and lymph node were harvested from treated and untreated control mice on day 35 and analysis of immune cells was performed using flow cytometry. Frequency of macrophages (CD11b+F4/80+Gr-1−) with M1 and M2 phenotypes as a percentage of total macrophages (A) and fraction of myeloid derived suppressor cells (MDSCs, CD11b+Gr-1+) as a percentage of leukocytes (B). ** p < 0.01.
Figure S4. Combinatorial CuDox+US+CpG+αPD-1 protocol increased tumor infiltration of cytotoxic T lymphocytes and macrophages in local and distant tumors of transgenic PyMT mice. A) Treatment protocols applied to PyMT transgenic mice. i) The axillary lymph node tumor (#3) was treated with two sequential administrations of CuDox+US+CpG+αPD-1 (ADD-IT) for two weeks, then the same two-treatment protocol was applied to the axillary lymph node tumor (#8) for another 2 weeks (n = 5). ii) The cervical lymph node tumor (#6) was treated with CpG+αPD-1 (IT) for 15 days (n = 2) and all treatment groups were compared to no-treatment (NT) control mice (n = 5). Mice were euthanized and all lymph node tumors were isolated for histology and immunohistochemistry. B) histological sections of NT control tumor, local and distant tumors of mice treated with either ADD-IT or IT and stained for H&E (the left first column, whole tumor view) and (second column, magnified view), CD8 (third column, magnified view), and F4/80 (fourth column, magnified view). Whole tumor sections and the magnified views enclosed by black or colored boxes are shown. Scale bars correspond to 3 mm (whole tumor panels) and 100 µm (magnified panels).
Figure S5. Repeated CuDox+US+CpG+αPD-1 protocol with immunopriming induced complete local and systemic responses with 50% long term survival. A) Treatment protocol integrating immunopriming sequence prior to chemo-immunotherapy. B-C) Growth of directly treated and distant tumors in bilateral NDL-tumor bearing mice treated with primed (IT-ADD, n = 4) (B) compared to no-treatment control (NT Control) tumors (C). D) Open-ended survival achieved with IT-ADD and ADD-IT compared with tri-combinatorial treatment of CuDox+US+CpG (ADD-CpG), bi-combinatorial treatment of US+CpG, and CpG only treatment. The survival curves for both IT-ADD and ADD-IT (p<0.0001), ADD-CpG (p<0.01), and CpG (p<0.01) were found statistically significant compared to the NT Control group as evaluated by Log-rank (Martel-Cox) test. ** p< 0.01, **** p<0.0001.
Figure S6. Three administrations of CuDox+US+CpG+αPD-1 without immunopriming achieved 50% long term survival. A) Treatment protocol starting with three cycles of ADD-IT without immunopriming. B) Growth of directly treated and distant tumors in bilateral NDL-tumor bearing mice treated with three administrations of (ADD-IT, n = 4).