

Abstract
There is a growing interest in materials that can dynamically change properties in the presence of cells to study mechanobiology. Here, we exploit 365 nm light-mediated [4+4] photodimerization of anthracene groups to develop cytocompatible PEG-based hydrogels with tailorable initial moduli that can be further stiffened. A hydrogel formulation that can stiffen from 10 to 50 kPa, corresponding to the stiffness of the healthy and fibrotic heart, respectively, was prepared. This system was used to monitor the stiffness-dependent localization of NFAT, a downstream target of intracellular calcium signaling using a reporter in live cardiac fibroblasts (CFbs). NFAT translocates to the nucleus of CFbs on stiffening hydrogels within 6 hours, whereas it remains cytoplasmic when the CFbs are cultured on either 10 or 50 kPa static hydrogels. This finding demonstrates how dynamic changes in mechanical properties of a material can reveal the kinetics of mechanoresponsive cell signaling pathways that may otherwise be missed in cells cultured on static substrates.
Keywords: Hydrogels, Photochemistry, Matrix Mechanics, Signal Transduction, Mechanobiology
Graphical Abstract
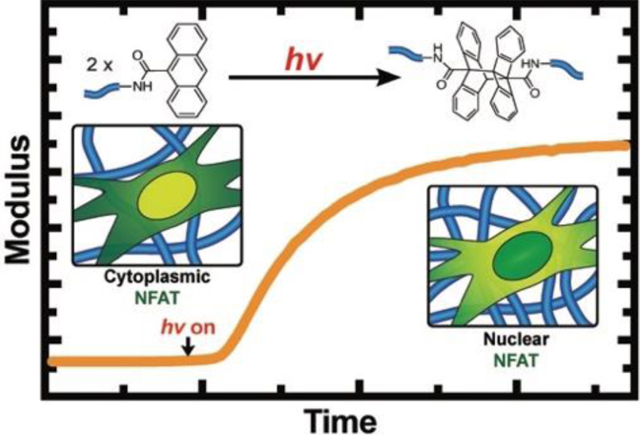
Materials with on-demand tunable properties allow to study changes in cellular behavior in response to their microenvironments. Anthracene photodimerization enables a simple, but well-defined and cytocompatible strategy to stiffen PEG-based hydrogels. Using these hydrogels, we found that NFAT, a downstream target of calcium signaling, responds to changes in hydrogel modulus in short timescales (80 min. to <24h.) in cardiac fibroblasts.
The interactions of cells with their microenvironment are complex and dynamic, and in many tissues, the extracellular matrix (ECM) undergoes major remodeling after injury or disease over short and long time scales, respectively. The ECM remodeling often changes the physical stiffness of the local microenvironment. For example, following a heart attack, the indentation Young’s modulus of cardiac muscle can change from E’ ~10 kPa to 50 kPa in just 2 weeks.[1] This change in the tissue stiffness can influence the behavior of resident cells via a process known as mechanotransduction.[2] As one example, cardiac fibroblasts (CFbs) can respond to increased ECM stiffness and transform from a quiescent phenotype to an activated myofibroblast.[3] The wound-healing myofibroblast exhibits enhanced secretion of ECM proteins, such as collagen, that can further stiffen the cardiac tissue, perpetuating further myofibroblast activation and eventually leading to long-term cardiac fibrosis.[4] While CFbs respond to changes in the tissue modulus, cellular mechanisms involved in sensing and responding to stiffness remain elusive.
Synthetic hydrogels with tissue-like elastic properties offer an attractive platform to study stiffness-associated changes in cell behavior in vitro. While numerous hydrogels have enabled researchers to study mechanotransduction in vitro,[5] there is a growing interest in materials that not only mimic the modulus of various tissues, but ones that allow mechanical properties to be changed in situ to simulate the dynamic changes taking place in ECM in vivo. To date, a variety of softening hydrogels that rely on photocleavage of crosslinks have been reported,[6] but reactions that enable hydrogel stiffening are more limited and often involve exogenous introduction or release of soluble reactive components that need to be orthogonal and cytocompatible.[7] In one example, radical mediated thiol-ene reaction was used to stiffen PEG hydrogels upon introduction of a photoinitiator and exposure to light.[6a] Alternatively, complementary DNA strands and enzymes have also been used to fabricate stiffening hydrogels;[8] however, these strategies necessitate careful selection of bioactive molecules that are inert to the cellular pathways of interest. While these collective strategies have advanced in vitro cell culture models, there is an unmet need for material chemistries that would provide simple, one-step strategies for hydrogel stiffening in the presence of cells while simultaneously achieving a broad range of moduli, such as the large stiffness transitions that can take place during cardiac fibrosis.
Here, we explore [4+4] photocyclodimerization of anthracenes by irradiation with 365 nm light as a simple but robust strategy to fabricate on-demand stiffening hydrogels. The photodimerization reaction does not require exogenous addition of any catalyst or reactive monomers, and the reaction does not generate any radical species that might damage cells in a concentration dependent manner. This work uses 365 nm light for the photocyclodimerization of anthracenes,[9] a wavelength commonly used for many photoinitiated cell encapsulations, but this can be shifted to >400 nm by incorporating nucleophilic moieties to the 9-position of anthracene, such as triazoyl groups,[10] or alternatively by exploiting the [2+2] photocycloaddition of styrylpyrene groups.[11]
Specifically, anthracene functionalized poly(ethylene glycol) (PEG) precursors (PEG-Ant) were prepared by addition of 9-anthracenecarboxylic acid to 8-arm 20,000 g/mol PEG-NH2 (Figure 1a and Scheme S1). The PEG-Ant was soluble in PBS up to 25 wt%, and hydrogels were readily achieved upon exposure of dilute PEG-Ant formulations to low intensity and cytocompatible 365 nm light. In situ photo-rheology shows that for a 13 wt% PEG-Ant formulation, the modulus reaches its final plateau (E’max = 98 ± 4 kPa) within 15 min. at 10 mW/cm2, and it can be achieved via continuous or stepwise 365 nm light irradiation by shuttering the light on and off (Figure 1b). By varying the PEG-Ant concentration between 2–13 wt%, hydrogels with a broad range of plateau modulus between 0.1 to 100 kPa were achieved (Figure 1c). Presumably due to the hydrophobicity of the anthracene end groups, the volumetric swelling ratios of in situ prepared 4–6 wt% PEG-Ant hydrogels did not change >10% after equilibration in PBS at 37 °C for 16 h., and the difference between the E’max of the swollen and in situ prepared gels were comparable (Figure S4). This result indicates that the cells seeded on PEG-Ant gels will “sense” a comparable modulus to the values measured by in situ rheology.
Figure 1.

(A) Structure of the 8-arm, 20000 g/mol PEG-Ant used in this study. (B) Gel evolution of 13 wt% PEG-Ant as a function of time either with continuous (–) or stepwise (–) irradiation. Yellow rectangles represent the periods that the 365 nm light was turned on during stepwise irradiation. (C) E’max scales exponentially with wt% in PEG-Ant hydrogels. (D) E’max of 5wt% PEG-Ant hydrogels prepared in 10% fetal bovine serum (FBS) was comparable to those synthesized in PBS, highlighting the bioorthogonality of the anthracene photodimerization in a complex protein solution. In these experiments, Io = 10 mW/cm2, gap = 100 μm, γ = 5% and ω = 1 rad·s−1 were used.
Interestingly, Figure 1c shows that E’max of the PEG-Ant hydrogels scaled exponentially with wt%, which is typically linear for step growth networks prepared using two complementary functional groups.[12] One explanation for this trend is the increased probability of intermolecular photodimerization of anthracene groups over intramolecular ones with increased polymer concentration owing to decreased interpolymer distance, which has been previously observed in other one-component step-growth networks.[11] The presence of intramolecular bonds that did not participate in network formation was more pronounced when a 4-arm, 20,000 g/mol PEG-Ant was used instead of an 8-arm PEG-Ant (Figure S5). In the former case, at least 8 wt% of 4-arm PEG-Ant was required to form a network, and E’max did not exceed 2 kPa even when 20 wt% of PEG-Ant was used. Therefore, we exclusively focused on the 8-arm PEG-Ant precursors for future experiments. The one-component nature of the gel formulation also permitted facile photopatterning with well-defined kinetics, which was demonstrated using a heterobifunctional PEG functionalized with an anthracene and rhodamine at different ends (Figure S6). This patterning method could be particularly useful to localize the presentation of bioactive cues in the presence of cells. Furthermore, the anthracene photodimerization was carried out in cell culture media (DMEM + 10% FBS) in the presence of −NH2, −COOH and −SH groups and resulted in comparable E’max and gel evolution kinetics as hydrogels prepared in PBS (Figure 1d). This orthogonality allows predictable stiffening of the PEG-Ant hydrogels in a complex cell culture milieu with minimal side reactions that might influence the final modulus.
An advantage of using anthracene groups to prepare hydrogels is the ability to measure directly the functional group conversion kinetics during gel evolution. This was achieved by monitoring the decrease in the intensity of the anthracene absorption peaks between 346, 364 and 384 nm as a function of time and light intensity (Io) (Figure 2a). The rate of disappearance of the 384 nm peak followed second order kinetics with an apparent rate constant (k’) of 0.73 – 0.92 M−1·sec −1 at 10 mW/cm2, which was comparable to typical click reactions used to prepare hydrogels (Figure 2b).[13] One potential explanation for the relatively fast kinetics is an aggregation-induced alignment of the hydrophobic anthracene groups in water, which would be further facilitated by the flexibility of the PEG chains.[14] When the reaction was carried out in DMF, a good solvent for both anthracene and PEG, the reaction was significantly slower (k’ = 0.37 M−1·sec−1). Next, we irradiated 5 wt% PEG-Ant formulations with Io between 2.5–20 mW/cm2 and determined that the anthracene photodimerization followed second order kinetics irrespective of Io (Figure 2c). Plotting k’ as a function of Io revealed that the reaction is a one-photon process with a rate constant of 0.087 M−1·cm2·mJ−1 (Figure 2d). Figure 2e shows that the anthracene conversion was linearly proportional to the gel evolution kinetics for 4–6 wt% PEG-Ant hydrogels, but that the average gel percolation threshold (pc,av) was reached only after 54% anthracene conversion. The theoretical prediction for pc,th of an 8-arm-8-arm system (92 mol% functionalization of the arms) is 0.16 according to Flory-Stockmayer.[15] This significant discrepancy may be due to intramolecular dimerization, which amounted to 60% of the bonds formed (Equations 4–6, SI), indicating that on average, 2.8 arms out of 8 participated in network formation. Thus, determining the kinetics of anthracene conversion and linking it to gel evolution for a selected PEG-Ant wt% (e. g., a predefined t and Io), enables precise tunability of initial and final moduli in the presence of the cells.
Figure 2 .

(A) Absorbance spectra of 5 wt% PEG-Ant hydrogels as a function of irradiation time in PBS, pH 7.4. (B) Anthracene photodimerization follows second order kinetics as 1/[C] scales linearly with irradiation time for different PEG-Ant formulations. (C) 1/[C] vs time for 5 wt% PEG-Ant at different I0. (D) Second order kinetic rate constant (k’) was linearly proportional to Io, indicating that the reaction is a one-photon process. (E) Plotting E’/E’max vs. anthracene conversion shows that the hydrogel percolation point significantly deviates from the theoretical prediction due to intramolecular photodimerization. In these experiments, Io = 10 mW/cm2 of 365 nm light and a path length of 100 μm were used unless otherwise described.
Upon detailed characterization of the photodimerization reaction, we used PEG-Ant hydrogels to study the mechanobiology of cardiac fibroblasts (CFbs). First, we screened for PEG-Ant hydrogels that could be stiffened from 10 to 50 kPa to match the reported moduli of the healthy and fibrotic rat heart, respectively (Figure 3a).[1] In parallel, 10 and 50 kPa PEG hydrogels that would not further stiffen were also prepared and irradiated with identical light dose and used as controls. CFb attachment was promoted by incorporation of 2 mM of an Ant-functionalized integrin binding sequence (Ant-GRGDS, Scheme S4). Prior to the cellular experiments, we tested if anthracene groups and dimers could alone promote cell adhesion without cell adhesion motifs. This could confound mechanobiology studies, as increased Ant concentration on stiffer substrates would increase the number of cell-adhesion sites, and therefore would not allow discriminating the role of the stiffness and adhesion sites on the cellular output. Figure S7a shows that myoblast cells can only adhere to PEG-Ant hydrogels in the presence of Ant-GRGDS, indicating that hydrophobic anthracene groups do not act as cell adhesion sites. Furthermore, nuclear localization of Yes-associated protein (YAP), a key mechanosensor, increases with increased RGD concentration, indicating that cells sense PEG-Ant hydrogels through integrins (Figure S7b). Myoblasts were also highly proliferative on PEG-Ant hydrogels as evidenced by the percentage of EdU+ cells (Figure S7c).
Figure 3.

(A) Rheology of the PEG-Ant hydrogels used to culture CFbs. For the stiffening hydrogel, I0 = 5 mW/cm2 was used to reach 10 kPa, whereas in situ stiffening was carried out using 10 mW/cm2. Static hydrogels of 5.5 wt% and 10 wt% PEG-Ant (10 and 50 kPa, respectively), were prepared using I0 = 5 mW/cm2 and t = 15 min. for the cell experiments but were exposed to I0 = 10 mW/cm2 to control for changes resulting from light exposure. In situ stiffening of the anthracene hydrogels from 10 to 50 kPa (B) increased the cell area and decreased the nuclear roundness within 2–5 days, whereas both remained constant on 10 kPa static hydrogels. Cell area (top) and nuclear morphology (bottom) were determined using HCS CellMask Orange Stain (H32713) and DAPI staining, respectively. In situ stiffening was carried out at Day 0. Error bars represent the 95% CI and *** indicates p < 0.001 in one-way ANOVA test. (C) Representative images of CFbs on 10 kPa and stiffening hydrogels.
Substrate stiffness is known to influence cell morphology in a variety of cell types, including CFbs.[3] To elucidate whether CFbs would respond to stiffening PEG-Ant hydrogels over the selected range, we first measured cell area and nuclear roundness up to 5 days post-stiffening. Figure 3b shows that in situ stiffening from 10 to 50 kPa resulted in a gradual increase in cell area from 1000 to 3200 μm2 and a decrease in nuclear roundness from 0.92 to 0.73, whereas neither of these cell features changed significantly on the 10 kPa static hydrogels. It’s well known that on stiff hydrogels, cells tend to become more spread over the surface of the hydrogel, resulting in an increase in cell area, and the cell and nuclear morphology becomes more elongated, causing a decrease in nuclear roundness. Collectively, these results indicate the cytocompatibility of the dimerization/hydrogel stiffening reaction, and that the CFbs respond to changes in the hydrogel modulus over the selected range.
Increased substrate modulus can activate CFbs and lead to their transformation into myofibroblasts;[3] however, the pathways that initiate this transformation remain elusive. It was previously proposed that cardiac cellular mechanotransduction can be mediated via mechanosensitive calcium channels.[16] A downstream target of intracellular calcium signaling is Nuclear Factor of Activated T cells (NFAT), which can translocate to the nucleus from the cytoplasm with increased intracellular calcium levels within minutes,[17] resulting in the transcription of genes associated with fibroblast activation.[18] Since mechanosensitive calcium signaling leads to NFAT nuclear localization, we sought to characterize whether a dynamic increase in matrix modulus alone could lead to NFAT nuclear localization.
To determine whether NFAT is mechanosensitive in CFbs, cells were transduced with a GFP-NFAT reporter[19] and cultured on static 10 and 50 kPa as well as 10 → 50 kPa stiffening hydrogels. For all conditions, CFbs were cultured for 24 h. before irradiation and in situ measurement of GFP. Figure 4A and B and Supplementary Video 1 show that stiffening increased NFAT Nuc:Cyto ratio within 80 minutes, whereas it remained cytoplasmic in CFbs cultured on either 10 or 50 kPa static hydrogels up to 6 h. This result suggests that a change in microenvironmental modulus promotes nuclear translocation of NFAT. The initial kinetics of NFAT nuclear localization was comparable to that of a key mechanosensitive protein, YAP. YAP has been reported to localize in the nucleus within 60 min. upon stiffening of a methacrylated hyaluronic acid (MeHA) hydrogel from E’ = 1.75 to 33 kPa in hepatic stellate cells.[20] While YAP Nuc:Cyto ratio is known to scale with the substrate modulus,[21] NFAT changes were only observed on a short time scale and in response to a modulus change. A possible mechanism of this transient NFAT nuclear localization may first require a change in membrane tension followed by the opening of mechanosensitive calcium channels. Following the adaptation to the new stiffness, a feedback loop may inhibit calcium uptake by inhibiting certain channels,[22] leading to nuclear exit of NFAT.
Figure 4.

Upon in situ stiffening from 10 to 50 kPa, NFAT localizes to the nucleus in CFbs. (A) Representative images of CFbs transduced with a GFP-NFAT adenovirus within the first 5 hours after stiffening. (B) Average NFAT Nuc:Cyto ratio as a function of time within the first 6 hours in 10, 50 kPa and stiffening hydrogels. **, *** and **** represents p < 0.01, 0.001 and 0.0001 in a t-test comparing the stiffened and 10 kPa hydrogel at each time point (N = 93). (C) Three distinct trends observed in the evolution of NFAT Nuc:Cyto ratio and (D) their abundance in CFbs cultured on 10, 50 kPa and stiffening PEG-Ant hydrogels. The quantification is described in the supporting information.
In conclusion, PEG-Ant hydrogels offer a simple, robust and highly controlled platform to prepare stiffening hydrogels in the presence of cells. This reaction allowed us to achieve a broad range of elastic moduli that are comparable to many soft tissues. The temporal control of dynamic material properties should allow innovative experiments to track real time cellular responses related to outside-in mechanobiology signaling. The reaction itself is efficient, proceeds without the addition of any exogenous molecules, and even in the presence of the complex protein milieu found in cell culture environments. Key results demonstrate that NFAT nuclear localization is sensitive to changes in substrate modulus in cardiac fibroblasts over short time scales (e.g., 80 min. to 6 h.), but on longer time scales, not to the magnitude of the modulus of the microenvironment (e.g., 10 to 50 kPa). This finding highlights the usefulness of hydrogels with dynamically tunable properties as a tool to explore mechanosensitive pathways.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (RO1 HL132353). Tova Ceccato was funded through the AHA Predoctoral Fellowship. We would like to thank Dr. Tobin Brown for his help and expertise in photopatterning and confocal microscopy imaging and Tim McKinsey’s group for the GFP-NFAT reporter and Steve Langer for helping to grow the adenovirus reporter.
References
- [1].Berry MF, Engler AJ, Woo J, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL, Am. J. Physiol. Heart Circ. Physiol 2006, 290, 2196–2203. [DOI] [PubMed] [Google Scholar]
- [2].a) Ingber DE, FASEB J. 2006, 20, 811–827. [DOI] [PubMed] [Google Scholar]; b) Orr AW, Helmke BP, Blackman BR, Schwartz MA, Dev. Cell 2006, 10, 11–20. [DOI] [PubMed] [Google Scholar]; c) Eyckmans J, Boudou T, Yu X, Chen CS, Dev. Cell 2011, 21, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mol. Biol. Cell 2017, 14, 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bochaton-Piallat M-L, Gabbiani G, Hinz B, F1000, 2016, 5, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677–689. [DOI] [PubMed] [Google Scholar]; b) Lutolf MP, Gilbert PM, Blau HM, Nature, 2009, 462, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Caliari SR, Burdick JA, Nat. Met 2016, 13, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science 2009, 324, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fairbanks BD, Singh SP, Bowman CN, Anseth KS, Macromolecules 2011, 44, 2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Azagarsamy MA, McKinnon DD, Alge DL, Anseth KS, ACS Macro Lett. 2014, 3, 515–519. [DOI] [PubMed] [Google Scholar]; d) Rosales AM, Mabry KM, Nehls EM, Anseth KS, Biomacromolecules, 2015, 16, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Brown TE, Marozas IA, Anseth KS, Adv. Mater 2017, 29, 1605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Guvendiren M, Burdick JA, Nat. Commun 2012, 3, 792–799. [DOI] [PubMed] [Google Scholar]; b) Brown TE, Silver JS, Worrell BT, Marozas IA, Yavitt FM, Günay KA, Bowman CN, Anseth KS, J. Am. Chem. Soc 2018, 140, 11585–15588. [DOI] [PubMed] [Google Scholar]; c) Stowers RS, Allen SC, Suggs LJ, Proc. Natl. Acad. Sci. U.S.A 2015, 112, 1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Liu H-Y, Greene T, Lin T-Y, Dawes CS, Korc M, Lin C-C, Acta Biomaterialia 2017, 48, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rammensee S, Kang MS, Georgiou K, Kumar S, Schaffer DV, Stem Cells 2017, 35 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu L, Shadish JA, Arakawa CK, Shi K, Davis J, DeForest CA, Adv. Biosys 2018, 2, 1800240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Zheng Y, Micic M, Mello SV, Mabrouki M, Fotios M, Andreopoulos FM, Konka V, Pham SM, Leblanc RM, Macromolecules. 2002, 35, 5228–5234. [Google Scholar]; b) Wells LA, Brook MA, Sheardown H, Macromol. Biosci 2011, 11, 988–998. [DOI] [PubMed] [Google Scholar]
- [10].Truong VX, Li F, Forsythe JS, ACS Macro Lett. 2017, 6, 657–662. [DOI] [PubMed] [Google Scholar]
- [11].Truong VX, Li F, Ercole F, Forsythe JS, ACS Macro Lett. 2018, 7, 464–469. [DOI] [PubMed] [Google Scholar]
- [12].Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS, Adv. Mater 2010, 22, 3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Lutolf M, Hubbell J, Biomacromolecules, 2003, 4, 713–722. [DOI] [PubMed] [Google Scholar]; b) Zheng J, Callahan LAS, Hao J, Guo K, Wesdemiotis C, Weiss RA, Becker ML, ACS Macro Lett. 2012, 1, 1071–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tazuke S, Banba F, J. Polym. Sci. Polym. Chem 1976, 14, 2463–2478. [Google Scholar]
- [15].a) Flory PJ, J. Am. Chem. Soc 1941, 63, 3083–3090. [Google Scholar]; b) Stockmayer WH, J. Chem. Phys 1943, 11, 45–55. [Google Scholar]
- [16].Sharma S, Goswami R, Zhang DX, Rahaman SO, J. Cell Mol. Med 2018, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Oh-hora M, Rao A, Microbes Infect. 2009, 11, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shin S-Y, Yang HW, Kim J-R, Heo WD, Cho K-H, J. Cell Sci 2011, 124, 82–90. [DOI] [PubMed] [Google Scholar]
- [18].Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD, Dev. Cell 2012, 23, 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA, J. Biol. Chem 2006, 281, 33487–33496. [DOI] [PubMed] [Google Scholar]
- [20].Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA, Sci. Rep 2016, 6, 21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Digabel JL, Forcato M, Bicciato S et al. , Nature, 2011, 474, 179–183. [DOI] [PubMed] [Google Scholar]
- [22].Gopal S, Søgaard P, Multhaupt HAB, Pataki C, Okina E, Xian X, Pedersen ME, Stevens T, Griesback O, Park PW et al. , J. Cell Biol 2015, 210, 1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.