

SUMMARY
The structural diversity of glycans on cells – the glycome – is vast and complex to decipher. Glycan arrays display oligosaccharides and are used to report glycan hapten binding epitopes. Glycan arrays are limited resources and present saccharides without context of other glycans and glycoconjugates. We used maps of glycosylation pathways to generate a library of isogenic HEK293 cells with combinatorially engineered glycosylation capacities designed to display and dissect the genetic, biosynthetic and structural basis for glycan binding in a natural context. The cell-based glycan array is self-renewable and reports glycosyltransferase genes required (or blocking) for interactions through logic sequential biosynthetic steps, which is predictive of structural glycan features involved and provides instructions for synthesis, recombinant production, and genetic dissection strategies. Broad utility of the cell-based glycan array is demonstrated and we uncover higher order binding of microbial adhesins to clustered patches of O-glycans organized by their presentation on proteins.
Keywords: microarray, glycan array, glycoengineering, glycosylation, glycosyltransferase, siglec, galectin, adhesin, lectin, carbohydrate
Graphical Abstract
eTOC Blurb
Narimatsu and colleagues display the diversity of human sugars on the surface of a library of cells by genetically engineering the cellular glycosylation machinery. Sugars on the cell surface play important roles in interactions with the environment, and the cell library developed opens for studies of biological interactions with sugars.
INTRODUCTION
Great structural diversity and complexity of the glycome of cells pose huge challenges for analytic and functional studies to extract and define specific biological roles and the underlying molecular bases. Glycan arrays have played a pivotal role in surveying and mapping the informational content of complex glycans. Different strategies have been undertaken to immobilize and display libraries of glycans in printed array formats drawing parallels to DNA arrays (Rillahan and Paulson, 2011), and approaches to produce comprehensive oligosaccharide libraries vary from chemical and chemoenzymatic synthesis to isolation of natural oligosaccharides and glycoconjugates. Different immobilization strategies have been employed with the most prevalent being coupling to NHS-activated slides and the neoglycolipid approach utilizing reductive amination to link released oligosaccharides to an amino-phospholipid (Blixt et al., 2004; Fukui et al., 2002; Palma et al., 2014; Puvirajesinghe and Turnbull, 2016). Two larger academic initiatives host these resources and offer services for the scientific community (www.functionalglycomics.org; www.imperial.ac.uk/glycosciences/). These glycan arrays are utilized with great success to probe glycan binding specificities of lectins and microbial adhesins, and in particular the Consortium for Functional Glycomics arrays and expanded sialoside microarrays have been instrumental in dissecting the subtle binding specificities of different influenza hemagglutinins and their role in cross-species transmission (Padler-Karavani et al., 2012; Peng et al., 2017; Rillahan and Paulson, 2011; Wang et al., 2013). Glycan array technology depends on continued synthesis and/or isolation of glycans, and often requires larger community-supported efforts. There are limitations in size and complexity of glycans that can be synthesized and in variable impact of the linkers used (Padler-Karavani et al., 2012; Rillahan and Paulson, 2011). Perhaps the most important limitation is the unnatural context in which the oligosaccharides or neoglycolipids are displayed in high densities without context of the protein or lipid backbone to which the glycans are normally attached (Blixt et al., 2004), as well as interactions with adjacent glycans that have been suggested to generate “clustered saccharide patches” (Cohen and Varki, 2014; Varki, 1994). Printed glycan arrays do not display glycans as they are found on the cell surface and they most often report a complex collection of glycan hapten structures with limited guidance for further studies.
While the human glycome is vast the cellular glycosylation machinery producing this diversity is simpler, and arguably better understood and approachable. The human genome contains over 200 distinct genes encoding glycosyltransferases, and the knowledge of their properties of these and their roles in the 15 known distinct glycosylation pathways in human cells is relatively advanced (Joshi et al., 2018a). Clearly, orchestration of the human glycome involves a large number of additional enzymes, including those that modify glycans such as sulfotransferases and epimerases, transporters and other proteins, and the glycosylation processes involve overlapping, competing, and intertwined reactions in particular by families of isoenzymes that may be difficult to predict. However, to a large extent the general scaffolds and structures of the glycome may be predicted from the repertoire of only 154 glycosyltransferase genes as proposed in Figures 1, S1 and S2. The glycosylation pathway maps organize glycosyltransferase genes into pathway-specific and pathway-nonspecific steps in the biosynthesis of distinct glycoconjugates, and illustrates potential redundancies for individual biosynthetic steps provided by isoenzymes and alternative competing biosynthetic steps. These maps provide predictions of structural glycan features affected by the presence or absence of individual genes and whether global or differential subtle outcomes are expected. Combined with the recent introduction of the facile nuclease-based gene-editing tools (Steentoft et al., 2014), we posit that it is now timely and conceivable to take an entirely genetic approach to dissect and display the structural diversity of the glycome of a human cell. Clearly, the glycosylation pathway maps require continuous refinement with increased insight into nonredundant and competing functions of isoenzymes, which requires further studies in isogenic cell models with combinatorial engineering to evaluate outcome. This has been a fruitful strategy for discovery of functions of the large family polypeptide GalNAc-transferase isoenzymes controlling O-glycosylation (Schjoldager et al., 2015a). To begin to facilitate such comprehensive gene targeting we previously generated a validated CRISPR/Cas9 gRNA library for highly efficient knockout (KO) targeting of all human glycosyltransferase genes (Narimatsu et al., 2018b), and strategies for stable site-specific knockin (KI) of non-expressed genes (Yang et al., 2015).
Figure 1. An Atlas of Human Glycosylation Pathway Maps With Assigned Functions of 170 Glycosyltransferase Genes.

Rainbow depiction of the 15 distinct human glycosylation pathways includes from left to right are: GPI-anchor, glycolipids (two pathways), N-linked glycans, O-GalNAc mucin-type, O-Fuc type (two pathways), O-GlcNAc type (EGF), O-Man type (POMT-directed), O-Man type (TMTC-directed), C-Man type, O-Glc type, O-Xyl type (proteoglycans), O-Gal type (collagen) and O-GlcNAc type (cytosolic). The basic structural features of oligosaccharides for most glycosylation pathways are shown above each rainbow segment, and the common glycan structures found on these 15 different types of glycosylation with the predicted functions of individual GTf-genes in biosynthetic steps are illustrated in Figure S2. Glycosyltransferase genes are arranged in the pathway-specific initiation and core extention steps (117 genes) and in pathway-nonspecific elongation and capping steps (53 genes). Genes circled by green are predicted to be expressed in HEK293 cells based on RNAseq analysis, and the predicted basic glycan features missing in HEK293 cells are faded out. Glycan symbols are drawn according to the SNFG format(Varki et al., 2015a).
Genetic approaches studying the biology of glycans have a long history. A large collection of Chinese hamster ovary (CHO) cell lines was originally developed through chemical mutagenesis and selection for loss of lectin binding or expression of cell surface proteins (Conzelmann and Kornfeld, 1984; Kingsley et al., 1986; Patnaik and Stanley, 2006), and most of these cells have been characterized to have defects in a distinct glycosyltransferase gene, donor sugar nucleotide synthase gene, sugar nucleotide transporter or sugar nucleotide epimerase gene with resulting loss/gain of global glycan features such as complex N-glycans, complete loss of sialylation or fucosylation of all glycans, or complete loss of glycosaminoglycan structures. These mutant cells have been important tools for determining requirement for global glycan features, but they do not enable deeper insight into glycan structures involved in biological interactions. Studies of deficiencies in glycosylation genes with rodents have also illustrated the wide biological importance of glycans and the glycosylation process (Lowe and Marth, 2003), while in humans a large number of congenital disorders of glycosylation (CDGs) have contributed substantially to our understanding of the importance of many different types of glycosylation (Ng and Freeze, 2018). In many cases the complexities of whole-organism level analyses have precluded identification of specific structure-function relationships. Past studies with mutant cell lines and organisms clearly demonstrate the power of genetic approaches to probe the glycome, and perhaps one of the best illustrations hereof is the complete unraveling of the many genes necessary for biosynthesis of the extraordinarily complex O-mannose glycan structure required for laminin binding to α-dystroglycan (Jae et al., 2013). This and more recent studies using precise gene engineering to delineate and reprogram glycosylation pathways in mammalian cell lines demonstrate the ability to dissect biological roles of glycosylation genes and their contribution to the glycome (Lavrsen et al., 2018; Schulz et al., 2018; Stolfa et al., 2016; Yang et al., 2015). These studies also show that genetic reprogramming of glycosylation can be performed with a high degree of predictability.
Here, we apply a fairly comprehensive genetic “tree-pruning” approach - glycotopiary - to engineer and reprogram the glycosylation capacities of a human cell line in order to construct a cell-based glycan array that covers a large part of the structural diversity of the human glycome. We used a rational combinatorial approach to eliminate and/or introduce de novo glycosylation capacities to develop sublibraries of stably engineered HEK293 isogenic cells that individually display loss or gain of distinct features of the human glycome. Importantly, combinatorial engineering of isoenzyme families with poorly understood functions enabled dissection and display of uniquely regulated glycan features. We demonstrate performance of the array with a series of plant, microbial and human lectins. We confirmed the hypothesis that the glycoconjugate and cellular context of glycans provide additional and necessary diversity in structural permutations of the human glycome. Cell-based array analysis of avian and human Influenza virus hemagglutinins (HAs) fully recapitulated the known selective binding to α2-3/α2-6 linked sialic acids (SA) (Rillahan and Paulson, 2011), and the added context of the cell provided evidence for binding selectivities beyond the simple SA linkage. Analysis of streptococcal serine-rich repeat adhesins produced refinement of the recognized O-glycan structures compared to information derived from printed glycan arrays, providing evidence for recognition of clusters or patterns of O-glycans created by the protein carrier. Thus, the cell-based glycan array fully complements the traditional printed glycan arrays, and further provides insight into the genetic and biosynthetic regulation of glycan recognition events with broader context of glycoconjugate nature and higher order presentation.
RESULTS
The Glycotopiary Strategy
We organized current knowledge of 170 glycosyltransferase genes directing the human glycome into a rainbow diagram that organizes these genes into the 15 distinct glycosylation pathways symbolized by the color used for the first monosaccharide (Figure 1) (Joshi et al., 2018a; Joshi et al., 2018b; Narimatsu et al., 2018b), with the predicted functions in biosynthetic steps and pathways as shown in Figure S2. 45 genes can be assigned to pathway-specific functions in the initiation of glycosylation of different types of glycoconjugates, 16 genes assigned to assembly of the lipid-linked oligosaccharide precursor and oligosaccharyltransferase dedicated to N-glycosylation, and 56 genes can be assigned to pathway-specific functions in immediate core extension and branching steps. Thus, 120 of the 170 genes are assignable to distinct glycosylation pathways, and several of these predictions were previously validated with CHO mutant cells (Patnaik and Stanley, 2006), targeted CHO KO cells (Yang et al., 2015), and other mammalian cell lines (Stolfa et al., 2016). We classified 18 genes to pathway-nonspecific elongation/branching and another 35 genes to pathway-nonspecific capping, including sialylation and fucosylation. While it is possible to reliably assign most of the glycosyltransferases that belong to the large isoenzyme families to general biosynthetic steps, it is important to note that for most of these isoenzymes our understanding of their specific non-redundant functions is still very limited. We previously demonstrated how genetic KO/KI dissection of isoenzyme genes can be used to identify non-redundant functions of isoenzymes (Schjoldager et al., 2015a), and this is clearly the strategy needed to dissect the large β3/4Gal-transferase, β3GlcNAc-transferase, and α2-3/6sialyltransferase isoenzyme families. We previously also classified human glycosyltransferase genes grossly into regulated and non-regulated based on organ transcriptome data (Joshi et al., 2018a), and this provides indications of differentially regulated glycosylation steps and pathways that contribute to the diversity of the glycome.
We selected the human embryonic kidney HEK293 cell line as the platform for construction of the cell-based glycan display, because structural analyses of different types of glycans suggest a high degree of complexity in glycosylation (Fujitani et al., 2013; Termini et al., 2017; Yang et al., 2012), and this cell line is widely used for recombinant expression and characterization of glycoproteins (Thomas and Smart, 2005). We used RNAseq transcriptomics as a rough prediction of the glycosylation capacity of HEK293 cells, and 123 of the 170 glycosyltransferase genes had detectable transcripts (FPKM≥1), while 47 were not or poorly detectable (FPKM<1) (Figure S1). Figure 1 illustrates the glycosyltransferase genes predicted to be expressed and their proposed functions, and the interpretation largely correlates with reported structural analysis (Yang et al., 2012). Thus, HEK293 cells are predicted to have capacity for all types of lipid and protein glycosylation, comprehensive elaboration of pathway-specific elongation and branching features, type2 chain LacNAc and LacDiNAc core chains, and both α2-3 and α2-6SA capping. SA in HEK293 is primarily Neu5Ac unless cells are cultured in bovine serum from where Neu5Gc can be scavenged, and acetylation has been reported (Wasik et al., 2017). The limited glycan features not predicted to be produced in HEK293 cells are globoseries glycosphingolipids (A4GALT), core1 extended (B3GNT3) and core3/4 branched GalNAc-type O-glycans (B3GNT6, GCNT3, GCNT4), type1 chain N-Acetyl-lactosamine (LacNAc) structures (B3GALT1, T2, T4, T5), and capping by blood group ABH, Sda and Lewis antigens (ABO, B4GALNT2, FUT1, FUT2, FUT3). Moreover, the capacity for α3-fucosylation and α2-8-sialylation is predicted to be low or absent due to the limited expression of members of the large isoenzyme FUT and ST8SIA families.
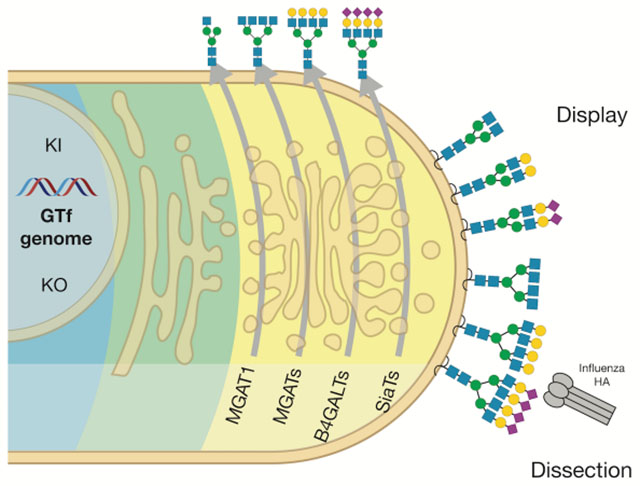
The basic glycotopiary concept to add and remove branches of glycan complexity by genetic KO/KI of glycosyltransferase genes, in order to generate isogenic cells displaying loss/gain of particular glycosylation features, is presented in Figure 2A. This Illustrates how combinatorial CRISPR/Cas9 KO targeting of the genes controlling the earliest essential steps in elongation or elaboration of glycans found on glycosphingolipids (B4GALT5/6), N-glycoproteins (MGAT1), and GalNAc-type O-glycoproteins (C1GALT1/COSMC) results in isogenic cells differentially displaying glycan features on one or more of these glycoconjugates. The performed KO/KI targeting is indicated by red/green dots, respectively, with lines between dots representing combinatorial gene engineering.
Figure 2. Design and Construction of a Cell-Based Display Platform for the Human Glycome.

(A) Depiction of the KO/KI gene engineering strategy for development of isogenic HEK293 cells with selective loss/gain of glycosylation capacities. Illustrated is how early combinatorial KO (red dots combined by black line) of the glycosyltransferase genes controlling the early steps in glycosphingolipid glycosylation (B4GALT5/6), N-glycosylation (MGAT1), or GalNAc-type O-glycosylation (C1GALT1/COSMC), generate cells with and without elaborated glycan features found on these glycoconjugates. The depicted glycan structures and outcomes of the engineering are simplified to illustrate the truncation effects by greying out.
(B) Depiction of a sublibrary cell strategy for display of glycan features with sublibraries (1-8) covering the principal pathway-specific and pathway-nonspecific glycosylation steps that enable differential display of the following key features: 1) type of glycoconjugates; 2) type of glycosphingolipids; 3) branching and core fucose of N-glycans; 4) core of GalNAc-type O-glycans; 5) chondroitin/dermatin (CS/DS) and heparan sulfate (HS) GAGs; 6) core structures LacNAc and LacDiNAc; 7) capping by α2-3 and α2-6SA; and 8) sialylation by α2-6SA. Red dots in circles of sublibraries represent KO of a glycosyltransferase gene with combinatorial KO shown by lines between genes. Green dots represent KI of a glycosyltransferase gene not endogenously expressed in HEK293 cells.
(C) Design of sublibrary connectivities for display of distinct complex glycan features. Illustrated is the design strategy used to differentially display homogenous N-glycans with LacNAc/LacDiNAc elongation and α2-3/α2-6SA capping. A biantennary N-glycan design (sublibrary 3) was combined with LacNAc or LacDiNAc designs (sublibrary 6) and α2-3SA, α2-6SA, or no SA designs (sublibrary 7). A complete summary of current status of sublibraries and connectivities generated is presented in Table S1.
(D) Graphic depiction of experimental workflows using the cell-based array. Libraries of isogenic cells with known genetic engineering and display of distinct subsets of the human glycome may be used in diverse bioassays and for recombinant production of reporter glycoconjugates for further studies and validation. The readout of these assays generate patterns of interactions (loss/gain), e.g. binding intensities quantified by flow cytometry, and these patterns are translated using the atlas of glycosylation maps (Figure 1) into structural glycan motifs and types of glycoconjugates. Interaction patterns typically include multiple informative datapoints due to the sequential nature of oligosaccharide biosynthesis to strengthen the structural interpretations. Sequential use of sublibraries and additional connectivity engineering are used to simplify workflow and for refinement. See also Figures S3,5 and S6.
A Sublibrary Strategy to the Cell-Based Glycan Array
We designed sublibraries with the capacity to differentially display defined glycan features grouped according to the rainbow biosynthetic scheme (Figure 2B). The sublibraries consist of groups of isogenic cells with reprogrammed glycosylation capacities for the major steps in glycosylation. Sublibrary 1 was designed to differentially display the major types of glycoconjugates by eliminating the earliest pathway-specific elongation steps for one or more glycoconjugates resulting in the display of N-glycans (KO MGAT1), GalNAc-type O-glycans (KO COSMC), and/or glycosphingolipids (KO B4GALT5/6), as well as independently Man-type O-glycans (KO POMGNT1, POMGNT2, and TMTC1-4) and glycosaminoglycans (GAGs) (KO B4GALT7). Sublibraries 2-5 differentially display most pathway-specific glycan features separately for glycosphingolipids, N-glycan branching, GalNAc-type O-glycan branching, and GAG core structures, respectively. Sublibrary 6 differentially displays pathway-nonspecific elongation by type2 chain LacNAc and/or LacDiNAc and poly-LacNAc. Sublibrary 7 and 8 differentially display pathway-nonspecific Gal capping by α2-3 and/or α2-6SA, as well as GalNAc capping by α2-6SA. Importantly, the individual and combinatorial targeting of isoenzyme genes enables display of the contribution of individual isoenzymes to the glycome and dissection of their unique non-redundant functions and interpretation of the underlying structural glycan features (Schjoldager et al., 2015b).
We used site-directed KI integration of human glycosyltransferase genes to introduce glycan features not endogenously expressed in HEK293 cells. These cells do not express the complex GalNAc-type core3/4 O-glycans, which are generally poorly expressed in cancer cell lines. The core3 pathway competes with core1, so we generated stable KI of the core3 synthase (B3GNT6) in HEK293 cells with KO of the core1 (COSMC). The important cancer-associated STn O-glycan is not endogenously displayed in HEK293 cells even after KO of COSMC (Steentoft et al., 2011), so we used KI of ST6GALNAC1 in cells with KO of COSMC to induce homogenous display of STn. We also used KI of dominating glycosyltransferases (ST3GAL4, ST6GAL1, GCNT1) to enhance the corresponding glycan features.
Connectivity of Sublibraries for Fine Dissection
The sublibraries display glycan features independently of each other, and further engineering is required to connect results obtained by individual libraries. As an example Figure 2C illustrates how a specific design from the N-glycan branching sublibrary 3 (biantennary N-glycan) is connected with designs from the LacNAc/LacDiNAc sublibrary 6 (biantennary N-glycan with LacNAc or LacDiNAc), and ultimately with the α2-3/2-6SA sublibrary 7, to display biantennary LacNAc N-glycans capped with and without α2-3 or α2-6SA. This strategy for connecting sublibraries enabled display of biantennary N-glycans without LacDiNAc capped with only α2-3SA, as validated by N-glycan profiling of total cell lysates and with a N-glycoprotein reporter construct (Figure S3B). Such combinatorial connectivity between sublibraries will be needed to cover and dissect the complete glycoconjugate context, and this should be carried out during pursuit of interesting specific biological interactions where multiple positive/negative data points are used to build and validate the entire glycoconjugate structure required for biological interactions, as illustrated later. The current state of the cell library including combinatorial engineering across sublibraries is listed in Table S1.
Figure 2D illustrates the principles, workflow and some applications of the cell-based glycan array. Individual cells from sublibraries of isogenic HEK293 cells with reprogrammed glycosylation are used in different biological assays amenable with live or fixed cells. Here we used flow cytometry to interrogate the glycosyltransferase gene(s) that affect the outcome of the assay used. The cell-based array will report patterns of reactivities (positive, negative, or none) with libraries of engineered isogenic cells, and these data points together with the atlas of glycosylation pathways are used to interpret the structural features of glycans and potential specific glycoconjugates involved (Figure 1). In striking contrast to the traditional printed glycan arrays the cell-based array provides information of the genetic and biosynthetic regulation of the target glycan as well as instructions for production of the relevant target and a ready engineered host cell for this. The approach will generally bring multiple independent data points that support the structural interpretation of the involved glycan because glycosylation is a step-by-step process. It is important to recognize the power of reinforcement provided by the complementarity and plurality of data points from sequential steps in the glycosylation pathways, as well as the discovery power of seemingly common glycan hapten structures that may be differentially regulated by individual isoenzymes on specific scaffold structures or types of glycoconjugates.
Validating the Cell-Based Glycan Array
The glycosylation capacities of HEK293WT cells have been probed previously by N and O-glycan profiling of total cell lysates and recombinant expressed glycoproteins (Fujitani et al., 2013; Termini et al., 2017; Yang et al., 2012), demonstrating expression of glycan features in excellent agreement with the predictions made from transcriptome analysis (Figure S1). These included high-Man and multiantennary complex type N-glycans with and/without LacNAc/LacDiNAc/core Fuc and with both α2-3/α2-6SA, while O-glycans included core1 and core2 structures with SA. Here we confirmed and extended this by N-glycan and GAG profiling of total cell lysates from HEK293WT as well as site-specific analysis of recombinant expressed N- and O-GalNAc glycoprotein reporters (Figure S3, Table S2). It is a daunting, largely unnecessary, and to some extent impossible task to undertake detailed structural analysis of the outcomes of all the extensive glycoengineering performed. In particular, it is currently essentially impossible to capture the fine structural features involved in particular biological interactions at the cell membrane through analysis of the glycome of whole cells. The cell-based array is in contrast ideal for this task. The array is designed to report the genetic and biosynthetic basis for interactions with glycan features on the cell surface, and the positive/negative signals provided by loss/gain of interactions by KO/KI of glycosyltransferase genes are used to interpret the structural features of the glycans and the glycoconjugates involved. Arguably, we know more about glycosylation pathways and the general roles of glycosyltransferases than the structure of glycans found on specific glycoconjugates and at specific sites in proteins. The genetic engineering design enables attribution of plausible structural features of glycans and the involved types of glycoconjugates, and the availability of appropriate engineered cells enables structural validation and further studies of interactions through production of targets (Figures 1 and 2).
To test the outcome of the engineering performed we analyzed a limited set of HEK293 engineered cells (Figure S3). For N-glycans we compared profiling of total cell lysates of HEK293WT and isogenic cells engineered to eliminate LacDiNAc (KO B3GALNT3/4) or the combination of LacDiNAc, α2-6SA (KO ST6GAL1), and multiantennary complex type N-glycans (KO MGAT3/4A/4B/5), which as predicted resulted in loss of these glycan features. We further compared these results with site-specific analysis of the N-glycan reporter protein FcγRIIa expressed in the same cells, and this correlated well and confirmed that FcγRIIa faithfully reported the features in question. To evaluate engineering of GalNAc-type O-glycosylation we stably expressed a secreted MUC1 tandem repeat (TR) reporter construct (Figure S3C), since endo-Asp digestion releases 20-mer TR glycopeptides amenable for O-glycan site and structure analysis by LC-MS/MS analysis. The MUC1 reporter expressed in cells with truncated O-glycans (KO COSMC) had TRs with 3-5 O-glycosites occupied with HexNAc, while the reporter expressed in cells without capacity for α2-3SA capping of core1 and core2 synthesis (KO ST3GAL1/2 and GCNT1) produced TRs with 2-4 O-glycosites with core1 structures (Hassan et al., 2000). To evaluate the GAG biosynthesis capacity we used disaccharide analysis of total cell lysates. We eliminated GAG biosynthesis entirely (KO B4GALT7) or selectively biosynthesis of chondroitin/dermatan sulfate (CS/DS) (KO CHSY1/3) or heparan sulfate (HS) (KO EXTL3) (Figure S5). The KO of B4GALT7 was in accordance with a classical CHO mutant cell line (Esko et al., 1987), and further in agreement with a comprehensive genetic deconstruction of GAG biosynthesis in CHO cells with focus on the complex epimerization and sulfation patterns (Chen et al., 2018).
To specifically probe the outcome at the cell surface of the gene engineering performed we undertook a series of validation studies. We first tested the performance using plant lectins and antibodies with known glycan specificities. Testing N-glycan specific features with GNL (high/paucimannose) and the PHA-E and PHA-L lectins (bisecting GlcNAc and tetraantennary N-glycans) on the glycoconjugate sublibrary 1, confirmed that KO of MGAT1 induced GNL binding and high-Man on the cell surface, while this conversely abrogated PHA labelling and complex-type N-glycans as predictable (Figures 3A, 3B and S4A). Similarly, the binding of RCA-1, ECL, and DSL (Galβ1-4GlcNAc; core βGlcNAc) were largely dependent on capacity for elaborated N-glycosylation (KO MGAT1), while capacity for elaborated glycolipid biosynthesis also slightly influenced binding (KO B4GALT5/6). Testing GalNAc-type O-glycan specific features with VVA and GSL-1 (αGalNAc, Tn) as well as Jacalin and MPL (Galβ1-3GalNAc, T; αGalNAc, Tn) revealed strong induction of binding only when O-glycans were truncated to Tn (KO COSMC) (Figures 3C and S4A). The WFL lectin (α/βGalNAc, LacDiNAc and Tn) binding to WT cells was eliminated by truncation of N-glycans (KO MGAT1) predicted to be due to loss of LacDiNAc on N-glycans, while truncation of O-glycans (KO COSMC) enhanced binding presumably due to binding to Tn (Figure S4A). These results all corroborated the predicted outcome of the engineering designs.
Figure 3. Flow Cytometry Analysis of the Glycoconjugate Sublibrary With Plant Lectins to N-Glycan Specific Features.

(A-C) Sublibrary 1 analyzed with lectins at different concentrations (color coded). Radar charts show mean fluorescence intensities (MFI) with engineered isogenic HEK293 cell as indicated. The KO engineering (Δ) and predicted display of glycoconjugate structures (solid glycan symbols) are illustrated. Charts represent single experiments and independent experiments were performed at least three times with similar results.
(D) Sublibrary 1 analyzed with a mAb NUH2. Note minor enhancement in binding of NUH2 after truncation of N-glycosylation (KO MGAT1) and both N and O-glycosylation (KO MGAT1/COSMC) presumably due to unmasking of glycosphingolipids. See also Figure S4.
To specifically probe glycosphingolipids we tested a monoclonal antibody (mAb) NUH2 with sperm immobilizing activity, previously shown to bind α2-3SA capped symmetric I-branched glycolipids (Nudelman et al., 1989), as an illustrative example for use of multiple data points from different sublibraries to predict detailed structure information of a binding epitope. NUH2 binding was first tested on the glycoconjugate library 1, and the binding was exclusively dependent on glycolipids (KO B4GALT5/6) (Figure 3D), which demonstrated that elaborated lactoseries glycolipids are readily detectable. Testing sublibrary 7 confirmed that NUH2 requires α2-3SA (Figure S4B), but also interestingly demonstrated an absolute requirement for the ST3GAL6 isoform suggesting that this isoenzyme has unique specificity for at least I-branched lactosamine. We confirmed requirement for the I-branching structure (KO of GCNT2) which completely abrogated NUH2 binding. KO/KI of ST3GAL4 did not affect NUH2 binding showing that only the ST3Gal-VI sialyltransferase can produce this epitope on glycolipids. A mAb 1B2 directed to LacNAc (Young et al., 1981), confirmed the effects of eliminating the α2-3SA and/or α2-6SA sialylation capacities for LacNAc based substrates (Figure S4C). This also demonstrated that capping of LacNAc in HEK293WT cells is primarily based on α2-3SA, although α2-6 capping is detectable as demonstrated by SNA lectin binding (Figure 5). We also tested the efficiency of blocking uncapped LacNAc by KI of ST6GAL1 or ST3GAL4 in the combinatorial ST6GAL1/2 and ST3GAL3/4/6 KO cells, and ST3GAL4 was slightly more efficient than ST6GAL1. These results also corroborated the predicted outcome of the engineering designs, and added knowledge of non-redundant functions of the ST3Gal isoenzymes.
Figure 5. Flow Cytometry Analysis With Influenza Hemagglutinins.

(A) Analysis of a modified α2-3/α2-6SA sublibrary 7 at different concentrations of four HAs and control lectins. Radar charts show MFIs.
(B) Analysis of glycoconjugate sublibrary 1. The charts represent single experiments and independent experiments were performed at least three times and when available with multiple clones with similar results.
Throughout the study we used live cells for flow cytometry analyses, which requires growing and maintaining the cell libraries in real time. While this clearly is possible for ongoing experiments, we also demonstrated feasibility of using fixed and frozen cell stocks for subsequent flow cytometry analyses directly after rethawing with lectins and antibodies (Figure S6). This strategy obviously enables distribution of vials or 96-well plates with sublibraries for ease of analysis in cell binding experiments, however, the cell-based array can be used in many other assay formats where viable cells may be needed.
Applying the Cell-Based Glycan Array for Dissecting Glycan-Binding Specificities
Siglecs –
Siglecs are a group of SA-binding immunoglobulin-like lectins with important immunoregulatory functions (Macauley et al., 2014). The specificity of human Siglec-7 is reported to be broad with binding to α2-8SA capped sulfated oligosaccharides, gangliosides and bacterial polysaccharides. Testing sublibrary 1 with Siglec-7 revealed a clear requirement for O-glycans, while sublibrary 7 revealed that elimination of α2-3SA specifically on LacNAc destroyed binding, suggesting that siglec-7 recognizes core2 O-glycans with SA capping (Figures 4A and 4C). Interestingly, re-engineering sialylation capacity through combined KO of α2-3SA and α2-6SA capping on LacNAc/LacDiNAc and reintroduction of either ST3GAL4 or ST6GAL1 reinstated binding. Siglec-7 was shown to bind both α2-3 and α2-6SA, and we predict that core2 O-glycans in HEK293WT primarily is capped by α2-3SA. The effects of α2-8SA was not investigated, but HEK293 cells express a limited number of ST8SIAs.
Figure 4. Flow Cytometry Analysis With Siglecs.

(A-B) Analysis of glycoconjugate sublibrary 1 with Siglec-7 and 9 at different concentrations. Radar charts show MFIs.
(C-D) Analysis of a modified α2-3/α2-6SA sublibrary 7 with genes introduced by KI shown in green, and two clones of each KI design were tested (indicated by line and #1/2). We included combinatorial KO of α2-3 and α2-6 sialylation capacities for LacNAc (KO ST3GAL3/4/6 and ST6GAL1/2) with KI of ST3GAL4 or ST6GAL1 to ensure efficient presentation of α2-3 or α2-6SA capping, because KO of ST3GAL3/4/6 only slightly enhanced binding of SNA suggesting that ST6GAL1 did not fully compensate the loss of α2-3 sialylation of LacNAc (See Figure 5A). The charts represent single experiments and independent experiments were performed at least three times with similar results.
Human Siglec-9 is reported to bind α2-3SA capped LacNAc with flexibility of substitutions on the internal HexNAc residue (6-sulfation and α3-fucose) (Yu et al., 2017). Testing sublibrary 1 revealed that Siglec-9 binding was dependent primarily on N-glycans, and sublibrary 7 revealed strict requirement for α2-3SA on LacNAc (Figures 4B and D). Siglec-9 is reported to also bind MUC1 with α2-3SA capped core1 (Beatson et al., 2016), but we were unable to demonstrate substantial binding to the MUC1 reporter and studies with printed glycan arrays also showed no or low binding to sialylated core1 (Padler-Karavani et al., 2012; Yu et al., 2017). A recent study demonstrated that the Siglec-7 ligands were sensitive to a mucin-specific protease, while ligands for Siglec-9 were not, further supports our findings (Malaker et al., 2019).
Influenza Virus Hemagglutinin –
The receptor specificity of influenza virus is essential for virus transmission in different species (Paulson and de Vries, 2013). The influenza surface hemagglutinin (HA) binds SA receptors leading to membrane fusion, while the neuraminidase (NA) cleaves SA from the receptor enabling release of the virus (Skehel and Wiley, 2000). Extensive studies with different binding assays and printed glycan arrays have established that human pandemic influenza viruses from over a century (1918 (H1N1), 1957 (H2N2), 1968 (H3N2), 2009 (H1N1)) have preference for α2-6SA receptors (human-type), while HA from avian viruses have preference for α2-3SA receptors (avian-type) (Paulson and de Vries, 2013; Rillahan and Paulson, 2011). Testing sublibrary 7 with representative HAs, including two avian-origin (Duck/Ukraine/1968 H3 and Vietnam/2004 H5), and human (Hongkong/1968 H3 and Texas/2010 H1) recapitulated the selective α2-3/2-6SA receptor specificities (Figure 5A). While the α2-6SA binding lectin SNA reacted weakly with HEK293WT and retained binding after eliminating α2-3SA capping of LacNAc/LacDiNAc (KO ST3GAL3/4/6), the two human HAs mainly reacted after KI of ST6GAL1, which also induced higher SNA binding. Interestingly, a low but detectable binding of the Hongkong/1968 H3 to α2-3SA was demonstrated with KI of ST3GAL4 while simple KO of ST6GAL1 did not reveal this. This has been predicted based on the close evolutionary relationship with the avian virus, but it has not been possible to demonstrate this previously using printed glycan arrays.
Further testing of glycoconjugates involved in binding revealed that the avian HAs bound equally well to N-glycans, GalNAc-type O-glycans and glycolipids, and only KO of all three types resulted in abrogation of binding (Figure 5B). Interestingly, the Hongkong/1968 HA showed strong preferential dependence on N-glycans, resembling the binding pattern of SNA, suggesting that the early HA had preference for N-glycoproteins. In contrast, the later Texas 2010 HA accepted both N-glycans and glycolipids. The α2-3SA binding lectin MAL-I showed strong preference for both N-glycoproteins and glycolipids, while O-glycans alone did not support binding. The ST6Gal-I sialyltransferase is known to transfer to terminal LacNAc core structures on all types of glycoconjugates, but studies have also demonstrated competitive advantages of ST3Gals over ST6Gal-I (Weinstein et al., 1982).
Streptococcal Serine-Rich Adhesins -
The serine-rich repeat (SRR) adhesins of commensal and pathogenic Gram-positive bacteria have highly divergent ligand-binding regions and bind to different ligands (Bensing et al., 2016). A group of these adhesins expressed by oral streptococci contains “Siglec-like” binding regions, and bind to α2-3SA on the human salivary mucin MUC7 and the platelet membrane glycoprotein GPIbα (Bensing et al., 2016). The binding to MUC7 is believed to be important for natural oral colonization and serendipitous binding to GPIbα for adherence to platelets in the pathogenesis of endocarditis (Deng et al., 2014). The Siglec-like adhesins have shown preference for α2-3SA on O-glycans (Deng et al., 2014; Bensing et al., 2018), but the selectivity for just a small subset of mucins or mucin-like proteins is not fully understood. We first tested binding to HEK293WT cells by the binding regions derived from Streptococcus mitis (10712BR) and Streptococcus gordonii (HsaBR). Reported oligosaccharide ligands for 10712BR include α2- 3SA on LacNAc structures (NeuAcα2-3Galβ1-4(Fucα1-3)+/−GlcNAc) and for HsaBR α2-3SA on LacNAc as well as on Galb1–3GalNAc found with GalNAc-type O-glycans. Despite that HEK293WT cells display substantial α2- 3SA we found only low binding. We considered the reported selectivity for GP1bα and MUC7, and designed reporter constructs containing the high density O-glycan regions derived from the stem region of GP1ba and TRs of different mucins (Table S2). We transiently expressed these reporters of mucin domains in HEK293WT and evaluated binding (Figure 6A). Surprisingly, the 10712BR and HsaBR adhesins showed markedly enhanced but different binding selectivity to cells displaying the mucin reporter constructs. Both adhesins bound cells expressing the GP1bα and MUC7 reporters, but there were striking differences in binding to other mucins, and e.g. the only mucin with characterized O-glycan occupancy, MUC1 (Hassan et al., 2000; Muller and Hanisch, 2002), did not support binding by either adhesins. As shown in Figure S3C the MUC1 reporter clearly displays the uncapped truncated Tn or core1 O-glycans as predicted from the engineering designs.
Figure 6. Flow Cytometry Analysis With Streptococcus Adhesins.

(A) Analysis of HEK293WT cells transiently expressing 21 human mucin and mucin-like (GP1bα) reporter constructs at one concentration. MFIs were quantified for the cell population positive for the reporter ECFP tag.
(B-C) Analysis of HEK293 cells stably expressing the GP1bα mucin reporter construct with engineering designs from sublibraries 1, 4, and 7 as indicated.
(D) Analysis of sublibrary 8. The charts represent single experiments performed at different concentrations of adhesins as indicated. Experiments were performed at least three times.
(E-F) Depiction of the distinct O-glycan structures demonstrated to be important for binding of the HsaBR and 10721BR adhesins with the positive (arrows) and negative (faded) datapoints obtained and used for the structural interpretation.
We therefore stably expressed the GP1bα reporter containing the densely O-glycosylated stem region in HEK293WT cells, and superimposed the gene engineering for the glycoconjugate sublibrary 1 design (Figures 6B, 6C and 6D). This modified library demonstrated that HsaBR and 10712BR binding was selectively abolished by loss of elongated O-glycans (KO COSMC). Applying the O-glycan branching sublibrary 4 further interestingly showed that 10712BR binding was also abolished by loss of core2 O-glycans (KO GCNT1), whereas HsaBR binding was unaffected. Thus, HsaBR binding does not require core2, and it may not be impeded by a core2 branch (Bensing et al., 2018). Further analysis of sialyltransferases with sublibrary 7 revealed that HsaBR binding was abrogated by loss of α2-3SA on core1 O-glycans (KO ST3GAL1/2), and specifically activated by loss of α2-6SA on core1 (KO ST6GALNAC3). These results indicate that the minimum binding epitope of HsaBR is a monosialylated core1 O-glycan (NeuAcα2-3Galβ1-3GalNAcα1-O-Ser/Thr), while the disialylated structure (NeuAcα2-3Galβ1-3(NeuAcα2-6)GalNAca1-O-Ser/Thr) is not (Figure 6E). 10712BR binding was abolished by loss of α2-3SA on core1 (KO ST3GAL1) as well as on LacNAc (KO ST3GAL4/6), and the preferred binding epitope is therefore predicted to be a complex core2 O-glycan with two terminal α2- 3SA residues (NeuAcα2-3Galβ1-3(NeuAcα2-3Galβ1-4GlcNAcβ1-6)GalNAca1-O-Ser/Thr) (Figure 6F). It is important to note that the current library does not include α1-3Fuc residues. The different effects of KO of ST3GAL1/2 and ST6GALNAC2/3/4 for binding of the adhesins indicate interesting fine specificities of these isoenzymes that need further exploration.
Collectively, these results suggest that the Siglec-like adhesins recognize specific O-glycan structures in ways influenced by their presentation on the protein or mucin backbone. While further studies are needed, the likely explanation for this is recognition of clustered patches or patterns of multiple O-glycans. Simple multivalent presentation of O-glycans does not seem to rule since all the reporter constructs studied are predicted to present multiple closely spaced O-glycans. Mucin TRs and mucin-like domains such as the stem region of GP1bα are characterized by being densely decorated with O-glycans, and the positions and patterns of the O-glycan decoration is determined by the peptide sequence (distribution of Ser/Thr residues) and the specificities of the available polypeptide GalNAc-transferases (GALNT1-20) that control initiation of O-glycosylation (Bennett et al., 2012). Analysis of the sequences used for the reporter constructs derived from the mucin TRs and GP1bα did not reveal simple common sequence motifs shared among those providing binding for the Siglec-like adhesins, and thus, the data do not enable us to define the recognition motifs in further detail. Recognition of “clustered saccharide patches” orchestrated by positions, spacing and direct interactions of multiple glycans in a protein was earlier proposed to provide expanded binding specificities and high affinity interactions, and evidence in support of this has been found with all types of glycoconjugates (Cohen and Varki, 2014; Varki, 1994).
DISCUSSION
Here, we developed a sustainable platform for cell-based display of a substantial part of the human glycome that enables wide surveying of the informational content of human glycans. Use of the cell-based glycan display is fundamentally different than printed glycan arrays in that the primary read-out is consequences of loss/gain of glycosyltransferase genes that are used to predict structural glycan features. Printed glycan arrays provide direct information of glycan haptens involved in interactions, while the cell-based array provides comprehensive knowledge of genes regulating the expression of glycan features. By organizing sublibraries designed to independently query the glycoconjugate status, features specific for individual glycosylation pathways and features more commonly found on glycoconjugates, we reached manageable size cell libraries that can be used broadly by the community. We illustrate the utility and discovery potential of the cell-based platform by applying it to dissect the sialic acid dependent adhesion of Influenza hemagglutinins and streptococcal Siglec-like adhesins. These studies fully recapitulated previous results, provided further structural refinements of binding motifs glycan structures, and led to discovery of importance of the glycoconjugate context for binding. The cell-based array reports the genetic and biosynthetic regulation of glycan binding motifs, and the strategy inherently generates engineered cells that can be used for production of reporter glycoproteins and further structural analysis, validation and other biological assays. The current design of the array covers the human glycome quite widely, but continuous efforts are needed to custom-design sublibraries (connectivities) for specific applications, which is facilitated by the atlas of glycosylation pathways and the library of validated gRNA targeting constructs (Narimatsu et al., 2018b).
Printed glycan arrays have transformed the field of glycosciences and served as essential tools for exploring the interactome of glycans and proteins (Rillahan and Paulson, 2011); however, studies have also resulted in the emergence of an interesting conundrum in explaining diversity of pathogen interactions and their host tropism in nature (Cohen and Varki, 2014; Cummings, 2009). Results from printed glycan arrays indicate that relatively few distinct glycan motifs serve as common ligands for many microbial adhesins and glycan-binding proteins (Cummings, 2009). The core structural motifs recognized, typically only 3-5 monosaccharides, are even more limited since glycans are built on common scaffolds with units such as N-Acetyllactosamine (LacNAc) (Blixt et al., 2004; Cummings, 2009). Many host-pathogen interactions identified involve terminal sialic acid residues on common structural motifs, and while the glycosidic linkages, the underlying core structures, and numerous modifications of sialic acids (Varki et al., 2015b), do vary to a degree, the overall structural permutations are still somewhat limited. It therefore seems likely that additional features, such as the context of these binding motifs with respect to the glycoconjugate carrying the glycans, interactions with adjacent glycans, as well as their overall presentation at the cell membrane may further enhance specificity. Traditional printed glycan arrays display different oligosaccharides without the context of the overall glycoconjugate and often in high densities, disproportionate to those normally found at the cell surface (Blixt et al., 2004), and the immobilization strategies may also affect lectin binding results (Padler-Karavani et al., 2012).
The cell-based array format can overcome some of these issues and provide further refinements of complex binding motifs, and this was clearly demonstrated with the streptococcal adhesins where we uncovered a higher order diversity in binding to specific O-glycans presented on different mucin peptide TR backbones (Figure 6). Mucins are ideally designed to present O-glycans in high densities and distinct patterns with their diverse TR sequences (Hollingsworth and Swanson, 2004), and the decoration with O-glycans is differentially regulated by the many polypeptide GalNAc-transferases that enables cell-specific glycosylation patterns (Bennett et al., 2012). The complex interplay of positions and structures of O-glycans on distinct mucin TRs is not addressable by traditional printed glycan arrays and structural analysis of mucin TRs is largely impossible. The results obtained here with the cell-based array strongly suggest that the informational content of mucin TRs is far greater than expected and essentially yet unexplored. Mucins are suggested to be poorly conserved in evolution because of high divergence in their TRs, however, an alternative interpretation relevant to our findings here may be that divergence in TRs has evolved under selective pressure in response to recognition of specific patterns and clusters of O-glycans. The panel of human mucin TR reporters and the cell-based glycan display platform now opens for wider studies of how clusters and patterns of O-glycans on mucins are recognized by microbial adhesins.
Analysis of influenza HAs with the cell-based array also pointed to new insights into the difference in sialic acid receptors for avian and human influenza HAs (Figure 5). The α2-3SA binding specificity of avian HAs and α2-6SA specificity of human HAs were clearly recapitulated. However, importantly, low binding to α2-3SA was seen with the HongKong/1968 (H3) HA, consistent with the fact this virus is derived from the avian (duck) Ukraine/1963 (H3) precursor and had just entered the human population (Ha et al., 2003). The analysis also alluded to differences in the types of glycoconjugates recognized by avian and human HAs, with avian HAs showing binding to all three of the major α2-3SA carrying glycoconjugates and human HAs showing variable dependence on expression of α2-6SA on N-glycoproteins and glycolipids. Further dissection combining the sialylation and glycoconjugate sublibraries can now be performed to explore these interesting findings in greater detail.
In summary, the cell-based array differentially displays the human glycome in a natural glycoconjugate and cell surface context, and is likely to generate a new and deeper understanding of the many biological roles of the glycome. Importantly, the cell-based array provides specific information of regulatory genes that can be directly used to further explore and validate findings by any cell biologist without detailed insight into glycosciences and access to advanced glycan analytics. These studies should be facilitated by the classification and overview of glycosylation pathways (Figure 1) and the library of validated CRISPR/Cas9 gRNA targeting constructs for all human glycosyltransferase genes (Narimatsu et al., 2018a). Limitations with the cell-based array design are the current size of the cell libraries, connectivity between sublibraries, and glycan features still missing, but these can be improved with future expansion. The array can be used when substantial changes in cell binding and/or other assay read-outs are obtainable in response to a gene KO/KI design, but it is important to consider that intensity of signals may vary due to overlapping functions and competition of enzymes, which may not always yet be predictable from the presented pathways. It is especially important to consider potential requirement for specific proteins not expressed in HEK293 to obtain optimal and biological relevant signals. Finally, the interpretation of structural glycan features involved in observed interactions relies on predictions based on the current understanding of glycosylation pathways, and it is important to consider unpredictable outcomes and use data points from multiple logical biosynthetic steps to strengthen predictions. The array will be made broadly available to the community, and we hope that the simple cellular platform and matrix for interpretation provided will help engage use beyond the glycoscience field.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests from resources and reagents should be directed to and will be fulfilled by the lead Contact, Henrik Clausen (hclau@sund.ku.dk).
METHOD DETAILS
Cell Culture
HEK293 cells (female, SIGMA) were maintained in DMEM (SIGMA) media supplemented with 10% fetal bovine serum (SIGMA) and 4 mM GlutaMAX (Gibco) at 37°C and 5% CO2. HEK293-6E were grown in suspension in serum-free F17 culture media (Invitrogen) supplemented with 0.1% Kolliphor P188 (SIGMA) and 4 mM GlutaMax. Cultures were grown at 37°C and 5% CO2 under constant agitation (120 rpm).
Gene constructs and other reagents
Genes encoding both avian and human hemagglutinin (HA) were amplified from synthesized DNA templates, and HA ectodomains cloned into a customized DNA vector for expression in mammalian tissue culture. Final HA expression constructs with N-terminal CD5 signal peptide for secretion, a C-terminal leucine zipper (GCN4) motif to promote native trimer formation, and HIS8-tag for immobilized metal-affinity chromatography (IMAC) purification. Constructs were transfected into HEK293S (GnTI−/−) cells and recombinant HA trimers were purified directly from the culture medium (72 hrs) by IMAC. Purified HAs were concentrated and buffer-exchanged to approximately 0.2 - 0.5 mg ml−1 in PBS prior to cell-binding assays. GST-tagged ligand-binding regions (GST-BRs) were overexpressed in E. coli and purified using Glutathione-Sepharose as described (Bensing et al., 2016).
Transcriptome analysis in HEK293 cell line
RNAseq of HEK293WT cells was performed by deep RNA sequencing using total RNA isolated by standard methods followed by sequencing on an Illumina Hiseq platform using standard protocols (BGI Co, Copenhagen). The RNAseq data resulted in total 4.39 Gb clean bases, which corresponded to 43.9 Mb clean reads. The clean reads were mapped with a total ration of 93.8% to the human reference sequence employing Bowtie2 and RSEM calculation of gene expression levels. A total of 17,364 known genes were detected.
CRISPR/Cas9 targeted KO in HEK293 cells
Knock-out (KO) was performed using a validated gRNA library for all human glycosyltransferases (GlycoCRISPR) (Narimatsu et al., 2018), as previously described (Lonowski et al., 2017). Briefly, HEK293 cells were seeded in 6 well plates (NUNC, Denmark), after one day 1 μg of gRNA was co-transfected with 1 μg of GFP-tagged Cas9-PBKS using Lipofectamine 3000. Cells were harvested after 24 h, and bulk sorted for GFP expression by FACS (SONY SH800). After culturing for 1 week, the sorted cell pool was further single-sorted into 96 well plates and screened by Indel Detection by Amplicon Analysis (IDAA) (Yang et al., 2015), and select clones were further verified by Sanger sequencing.
ZFN-mediated KI in HEK293 cells
For site-directed knock-in (KI) a modified ObLiGaRe targeted KI strategy utilizing two inverted ZFN binding sites flanking the full coding human glycosyltransferase genes in donor plasmids was used, as described previously (Pinto et al., 2017). KI was performed as described for targeted KO with 1 μg of each ZFN tagged with GFP/Crimson and 3 μg donor plasmid. KI clones were screened by PCR with primers specific for the junction area between the donor plasmid and the AAVS1 locus, and a primer set flanking the targeted KI locus was used to characterize the allelic insertion status.
Stable expression of the GP1bα reporter in HEK293
HEK293WT cells were transfected with an expression construct containing the O-glycosylated stem region of human GP1bα using the same protocol as for KO, and stable clones were selected after G418 selection. A pool was single cell sorted into 96 well plates, and high expressing clones selected by FACS (ECFP) and ICC with an anti-FLAG antibody. Stable clones were used further for KO engineering as listed in Table S1.
Human mucin reporter constructs
A cell membrane reporter construct (MUC-surf-reporter) designed with exchangeable inserts of 150-200 amino acids derived from tandem repeats (TRs) of human mucins was generated by fusion of human MUC1 signal peptide (aa1-62, Uniprot P15921) with 6xHis, Flag-tag, EGFP, multiple cloning site, and the membrane anchoring domain of human MUC1 (aa1042-1138) (Table S2). The multiple cloning site contained EcoRV/PmeI/BamHI/BamHI/NotI/PmeI restriction sites and mucin inserts were synthesized as TrueValue constructs with in-frame BamHI and NotI sites (Genewiz, USA). The MUC1 secreted reporter construct was synthesized with NotI/XhoI and a 10xHis STOP encoding ds oligo (5’-GCGGCCGCCCATCACCACCATCATCACTGATAGCGCTCGAG-3’, NotI/XhoI restriction sites underlined). All constructs were Sanger sequence verified. Transient expression in HEK293 cells was performed one day after seeding cells into 24 wells, and transfected cells were harvested 48 h post-transfection and used for binding studies.
Cell binding assays
Cell binding assays were performed on ice with mAbs, lectins, precomplexed glycan-binding proteins, Siglecs and influenza virus HAs. Biotinylated lectins (Vector Laboratories, Burlingame, CA) were incubated at different concentrations for 1 h, followed by washing and incubation with Streptavidin-Alexa Fluor488 (Invitrogen) for 1 h. Recombinant human Siglec Fc-chimera (R&D systems, Minneapolis, MN) were precomplexed with Alexa Fluor647 goat anti-human IgG antibody (Life Technologies) for 1 h followed by incubation with cells for 1 hr. His-tagged HAs were precomplexed for 45 min with anti-6xHis mouse IgG (Thermo) and Alexa Fluor647 goat anti-mouse IgG (Invitrogen) in 4:2:1 ratio followed by incubation with cells for 3 h. HEK293 cells stably expressing GP1bα or transiently expressing mucin reporter constructs were incubated with GST-BRs at different concentrations for 1 h, followed by washing, and incubation with rabbit polyclonal anti-GST (Thermo) for 1 h, and further incubated with Alexa Fluor647 goat anti-rabbit IgG antibody (Life Technologies) for 1 h. Washing was performed with 1% BSA/PBS and cells were resuspended for flow cytometry analysis (SONY SA3800). Testing of different concentrations of precomplexed reagents was performed by diluting the complexes in 1% BSA/PBS.
Production of N- and O-glycosylation reporter proteins
A reporter for N-glycosylation was designed as a secreted construct of the FcγRIIa N-glycoprotein, encoding the extracellular domain (aa1-217, Uniprot P12318) fused with a 10xHis tag. Secreted FcγRIIa was transiently produced in suspension HEK293-6E cells. Briefly, 30 ml of HEK293-6E cells were seeded at a cell density of 0.5 × 106 cells/ml and transfected the next day with 30 μg of the secrete FcγRIIa construct and 90 μg of PEI (1:3 ratio) incubated at room temp. for 15 min and cultured for 72 h before harvest. A reporter for GalNAc-type O-glycosylation was designed as a secreted construct of the mucin MUC1 encoding TRs (also used for the mucin display). The reporter was stable expressed in HEK293-6E after G418 selection, and a stable pool of cells were seed at a cell density of 1.0 × 106 cells/ml and cultured 72 h. Secreted FcγRIIa and MUC1 were purified from culture medium by nickel affinity chromatography. Media was mixed 3:1 (v/v) in 4x binding buffer (100 mM sodium phosphate, pH 7.4, 2 M NaCl) and applied to self-packed nickel-nitrilotriacetic acid (Ni-NTA) affinity resin column (Qiagen), pre-equilibrated in washing buffer (25 mM sodium phosphate, pH 7.4, 500 mM NaCl, 20 mM imidazole). After washing, bound protein was eluted with 200 mM imidazole in washing buffer. The protein containing fractions were identified by SDS-PAGE, and further purified by reverse-phase HPLC with a Jupiter 5 μ C4 300A column (Phenomenex, Torrence, CA), using 0.1% trifluoroacetic acid (TFA) and a gradient of 10-100% acetonitrile.
Sample preparation for site-specific N-glycopeptide analysis
20 μg of the purified FcγRIIa was dissolved in 50mM ammonium bicarbonate (AmBic) buffer, reduced in 10 mM dithiothreitol (DTT) at 60°C for 45 min, alkylated (20 mM iodoacetamide (IAA), in the dark at RT for 30 min), reduced again (10 mM DTT, 20°C, RT) and digested with 0,8U chymotrypsin (1:25 ratio chymotrypsin:protein) (Roche) (25°C, 4h). The proteolytic digest was desalted by in-house produced modified StageTip columns containing 3 layers of C18 membrane (3M Empore disks from Sigma Aldrich) (Rappsilber et al., 2007). Samples were eluted with 50% methanol in 0.1% formic acid (FA), dried in SpeedVac and re-solubilized in 0.1% FA and submitted for LC MS/MS analysis.
Sample preparation for total cell lysate N-glycan analysis
The packed cell pellets (1×107 cells) were lysed in 1 μg Rapigest SF Surfactant (Waters), in 50 mM AmBic and homogenized by sonication. Cleared lysates were heated (95°C, 15 min), diluted in 50 mM AmBic to a final concentration of 0.2% Rapigest and sonicated again followed by reduction (10 mM DTT, 60°C, 45 min), alkylation (20 mM iodoacetamide, in the dark at RT for 30 min), reduction (10 mM DTT, RT, 20 min) and digested with 25U trypsin (Roche) in 50mM AmBic (37°C, 12 h, 650 rpm). Each tryptic digest was acidified with TFA and incubated at 37°C for 30 min before centrifuging (max speed, RT, 15 min). Sep-Pak C18 columns (Waters) where washed with 100% methanol, 50% methanol 0.1% FA and equilibrated with 0.1% trifluoroacetic acid (TFA) before loading the supernatants containing the tryptic digests. After reloading the flow through, the column was washed with 0.1% TFA, twice with 0.1% FA and eluted twice with 50% methanol 0.1% FA. 10 ug of Freeze-dried samples were resuspended in 50 mM AmBic with 8U PNGaseF (Roche) and incubated at 37°C for 16 h. The digest was applied to Sep-pak C18 column, as described above, and flow through containing the released N-glycans was collected for further labeling with Rapifluor-MS glycan kit (Waters) as described by manufacturers.
Sample preparation for O-glycopeptide analysis
Approximately 50 μg of the purified MUC1 reporter was reduced, alkylated and digested with 10 μg endo-AspN (Roche) (37°C, O/N). The digested sample was fractionated by C18 HPLC, and the MUC1 20mer TR glycopeptide identified and analyzed by LC-MS/MS.
Mass Spectrometry
LC MS/MS analysis was performed on EASY-nLC 1200 UHPLC (Thermo Scientific) interfaced via nanoSpray Flex ion source to an Orbitrap Fusion Lumos MS (Thermo Scientific). Briefly, the nLC was operated in a single analytical column set up using PicoFrit Emitters (New Objectives, 75 μm inner diameter) packed in-house with Reprosil-Pure-AQ C18 phase (Dr. Maisch, 1.9-μm particle size, 19-21 cm column length). Each sample was injected onto the column and eluted in gradients from 3 to 32 % B for glycopeptides, and 10 to 40% for released and labeled glycans in 45 min at 200 nL/min (Solvent A, 100% H2O; Solvent B, 80% acetonitrile; both containing 0.1 % (v/v) formic acid). A precursor MS1 scan (m/z 350-2,000) of intact peptides was acquired in the Orbitrap at the nominal resolution setting of 120,000, followed by Orbitrap HCD-MS2 and at the nominal resolution setting of 60,000 of the five most abundant multiply charged precursors in the MS1 spectrum; a minimum MS1 signal threshold of 50,000 was used for triggering data-dependent fragmentation events. Targeted MS/MS analysis was performed by setting up a targeted MSn (tMSn) Scan Properties pane. A target list was composed from top 30 most abundant glycans or glycopeptides from the proposed compositional list (Table S3).
Data Analysis
Glycan and glycopeptide compositional analysis was performed from m/z features extracted from LC-MS data using in-house written SysBioWare software (Vakhrushev et al., 2009). For m/z feature recognition from full MS scans LFQ Profiler Node of the Proteome discoverer 2.2 (Thermo Scientific) was used. The list of precursor ions (m/z, charge, peak area) was imported as ASCII data into SysBioWare and compositional assignment within 3 ppm mass tolerance was performed. The main building blocks used for the compositional analysis were: NeuAc, Hex, HexNAc, dHex and the theoretical mass increment of the most prominent peptide corresponding to each potential glycosites. The most prominent peptide sequences related to N-glycosites of interest were determined experimentally by comparing the yield of deamidated peptides with and without PNGase F treatment. One or two phosphate groups were added as building blocks for assignment. To generation the potential glycopeptide list, all the glycoforms with an abundance higher than 10% of the most abundant glycoform were used for glycan feature analysis. All high mannose and hybrid structures were excluded in the TCL data.
Disaccharide analysis of GAGs
Disaccharide composition analysis was performed with a panel of bacterial polysaccharide lyases (R&D Systems) and 2-aminoacridone (AMAC) labeling on a Waters Acquity UPLC system equipped with a fluorescence (FLR) detector as described previously (Ref Nature method). The 500 ul packed cell pellets from 5 million cells were resuspended in a lysis buffer containing 50 mM Tris-HCl buffer, pH 7.6, 10 mM CaCl2, 0.1% Triton X-100, and 1 μg/ml Pronase (Roche) in a total volume of 1 ml. Reactions were incubated at 37 °C overnight with end-to-end rotation, and heated at 100 °C for 10 min to inactivate Pronase; after centrifugation, supernatants were adjusted to 2 mM MgCl2, mixed with 250 U of Benzonase (Sigma), and incubated at 37 °C for 2 h. Reactions were acidified with acetic acid to pH 5.0 and loaded onto a Q-Sepharose column (0.5 ml) (Sigma) equilibrated with 20 mM sodium acetate, pH 5.0, 100 mM NaCI, and 0.1% Triton X-100. The column was washed first with the equilibration buffer and then with the same buffer without Triton X-100 to remove the detergent. The bound GAG fraction was eluted with 1.5 ml of buffer containing 20 mM sodium acetate, pH 5.0, and 1 M NaCl and pelleted by ethanol precipitation and dissolved in de-ionized water. For evaluation of total CS/DS, chondroitinase ABC (10 mU) was used for digestion in 40 mM sodium acetate and 1 μ M CaCl2 at 37 °C overnight. For the evaluation of HS, a mixture of heparinases I, II, and III, or of II and III only (10 mU of each), was used in 40 mM sodium acetate and 5 mM CaCl2 at 37 °C overnight. Released disaccharides were dissolved in 5 μl of 0.1 M AMAC solution in glacial acetic acid-DMSO (vol/vol 3:17) and incubated at room temperature for 15 min, followed by mixing with 5 μl of 1 M NaCNBH3 and further incubation at 45°C for 3 h. Excess AMAC was removed by acetone precipitation. Labeled disaccharides corresponding to 0.5 million cells were subsequently analyzed on a Waters Acquity UPLC system equipped with a fluorescence (FLR) detector. The separation was optimized on a BEH C18 column (2.1 Å ~ 150 mm, 1.7 μm; Waters) at 30°C for CS/DS and 40°C for HS, with 80 mM ammonium acetate as mobile phase A (pH 5.5) for CS/DS and 150 mM ammonium acetate as mobile phase A (pH 5.6) for HS, and with 100% acetonitrile as mobile phase B for CS/DS and HS. Separation of the disaccharides was performed with a gradient of mobile phase B increasing from 3 to 13% over 30 min at a flow rate of 0.2 ml/min. Each series of HPLC runs was preceded with standards (20 pmol AMAC-labeled disaccharides; Iduron).
DATA AND SOFTWARE AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE repository with the dataset identifier PXD013676.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti 6xHis tag | Thermo | Cat#MA1-21315; RRID:AB557403 |
| Goat anti-mouse IgG (H+L), Alexa Fluor™ 647 | Invitrogen | Cat#A-21235 |
| Streptavidin, Alexa Fluor™ 488 conjugate | Invitrogen | Cat#S32354 |
| Goat anti-Human IgG (H+L), Alexa Fluor™ 647 | Invitrogen | Cat#A-21445 |
| GST tag polyclonal antibody | Invitrogen | Cat#71-7500 |
| Goat anti-Rabbit IgG (H+L), Alexa Flouor™ 647 | Invitrogen | CatA-21245 |
| NUH2 antibody | Nudelman et al., 1989 | N/A |
| 1B2 antibody | Young et al. 1981 | N/A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Influenza hemagglutinins | Paulson and Vries, 2013 | N/A |
| Streptococcus | Bensing et al., 2016 | N/A |
| Recombinant Human Siglec-7/CD328 Fc Chimera | R&D systems | Cat#1138-SL |
| Recombinant Human Siglec-9 Fc Chimera | R&D systems | Cat#1139-SL |
| Biotinylated Lens Culinaris Agglutinin (LCA) | Vector Laboratories | Cat#B-1045 |
| Biotinylated Concanavalin A (Con A) | Vector Laboratories | Cat#B-1005 |
| Biotinylated Jacalin | Vector Laboratories | Cat#B-1155 |
| Biotinylated Galanthus Nivalis Lectin (GNL) | Vector Laboratories | Cat#B-1245 |
| Biotinylated Maclura Pomifera Lectin (MPL) | Vector Laboratories | Cat#B-1345 |
| Biotinylated Vicia Villosa Lectin (VVL, VVA) | Vector Laboratories | Cat#B-1235 |
| Biotinylated Griffonia Simplicifolia Lectin I (GSL I, BSL I) | Vector Laboratories | Cat#B-1105 |
| Biotinylated Datura Stramonium Lectin (DSL) | Vector Laboratories | Cat#B-1185 |
| Biotinylated Ricinus Communis Agglutinin I (RCA I, RCA120) | Vector Laboratories | Cat#B-1085 |
| Biotinylated Erythrina Cristagalli Lectin (ECL, ECA) | Vector Laboratories | Cat#B-1145 |
| Biotinylated Phaseolus Vulgaris Erythroagglutinin(PHA-E) | Vector Laboratories | Cat#B-1125 |
| Biotinylated Phaseolus Vulgaris Leucoagglutinin(PHA-L) | Vector Laboratories | Cat#B-1115 |
| Biotinylated Wisteria Floribunda Lectin (WFA, WFL) | Vector Laboratories | Cat#B-1355 |
| Biotinylated Maackia Amurensis Lectin I (MAL I) | Vector Laboratories | Cat#B-1315 |
| Biotinylated Sambucus Nigra Lectin (SNA, EBL) | Vector Laboratories | Cat#B-1305 |
| DMEM-high glucose | Sigma | Cat#D-5796 |
| Fetal Bovine Serum (heat activated) | Sigma | Cat#F9665 |
| Glutamax | Gibco | Cat#35050061 |
| TrypLE™ Express Enzyme (1X), phenol red | Gibco | Cat#12605028 |
| F17 medium | Invitrogen | Cat#A13835-01 |
| Kolliphor P188 | Sigma | Cat#K4894-500g |
| Polyethylenimine (linear, 25 kDa) | Polysciences | Cat#23966 |
| Ni-NTA Agarose | QIAGEN | Cat#30210 |
| Jupiter® 5 μm C4 300 Å, LC Column 250 x 4.6 mm | Phenomenex | Cat#00G-4167-E0 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human cells: HEK293 | Sigma | Cat#85120602 |
| Human cells: HEK293S (GnTI−/−) | ATCC | CRL3022™ |
| Human cells: HEK293-6E | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| CAS9PBKS | Lonowski et al., 2017 | Addgene Plasmid#68371 |
| Secrete FcγRIIa reporter construct | This paper | N/A |
| Secrete MUC1 reporter construct | This paper | N/A |
| Software and Algorithms | ||
| SysBioWare software | Vakhrushev et al., 2009 | N/A |
| Proteome discoverer 2.2 | Thermo Scientific | OPTON-30795 |
| Other | ||
Highlights.
Human glycosyltransferases (170 GTf genes) organized in glycosylation pathway maps.
The human glycome displayed in a natural context on the cell surface.
Sustainable cell-based array resource to dissect biological functions of glycans.
Microbial adhesins may bind to clustered patches of O-glycans.
ACKNOWLEDGMENTS
This work was supported by the Lundbeck Foundation, Novo Nordisk Foundation, Kirsten og Freddy Johansen Fonden, A.P. Møller Fonden, Læge Sophus Carl Emil Friis og hustru Olga Doris Friis’ Legat, European Commission (GlycoImaging H2020-MSCA-ITN-721297, BioCapture H2020-MSCA-ITN-722171), the Danish National Research Foundation (DNRF107), the National Institutes of Health (AI114730 and R01AI41513, R01AI106987, U01OD024857), and Kuang Hua Educational Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
University of Copenhagen has filed a patent application on the cell-based display platform. GlycoDisplay Aps, Copenhagen, Denmark, has obtained a license to the field of the patent application. Y.N., Z.Y., E.P.B., and H.C. are co-founders of GlycoDisplay Aps and hold ownerships in the company as well as served as unpaid consultants.
SUPPLEMENTAL INFORMATION
Document S1. Figures S1–S6
Table S1. Summary of Engineered HEK293 Isogenic Cell Library Generated to Date, Related to Figure 2
Table S2. Amino Acid sequences of Reporter Constructs Used, Related to Figures 6 and S3
Table S3. MS List of N-glycan Profiling of Total Cell Lysates and Site-specific N-glycan Analysis of Recombinatn FcgRIIa, Related to Figure S3
(A,B) MS List of N-glycan Profiling of HEK293WT and Gene KO Engineered Total Cell Lysates as Indicated. (C,D) Site-specific N-glycan Analysis of N97 and N178 Sites of Recombinant FcgRIIa Expressed in HEK293WT and KO Engineered Cells as Indicated.
REFERENCES
- Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, Hillier M, Maher J, Noll T, Crocker PR, et al. (2016). The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol 17, 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, and Tabak LA (2012). Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Khedri Z, Deng L, Yu H, Prakobphol A, Fisher SJ, Chen X, Iverson TM, Varki A, and Sullam PM (2016). Novel aspects of sialoglycan recognition by the Siglec-like domains of streptococcal SRR glycoproteins. Glycobiology 26:1222–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensing BA, Li Q, Park D, Lebrilla CB, and Sullam PM (2018). Streptococcal Siglec-like adhesins recognize different subsets of human plasma glycoproteins: implications for infective endocarditis. Glycobiology 28, 601–611.Blix [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, et al. (2004). Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U.S.A 101, 17033–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Narimatsu H, Clausen TM, Gomes C, Karlsson R, Steentoft C, spliid CB, Gustavsson T, Salanti A, Persson A, et al. (2018). The GAGOme: a cell-based library of displayed glycosaminoglycans Nat. Methods 15:881–888. [DOI] [PubMed] [Google Scholar]
- Cohen M, and Varki A (2014). Modulation of glycan recognition by clustered saccharide patches. Int. Rev. Cell Mol. Biol 308, 75–125. [DOI] [PubMed] [Google Scholar]
- Conzelmann A, and Kornfeld S (1984). Beta-linked N-acetylgalactosamine residues present at the nonreducing termini of O-linked oligosaccharides of a cloned murine cytotoxic T lymphocyte line are absent in a Vicia villosa lectin-resistant mutant cell line. J. Biol. Chem 259, 12528–12535. [PubMed] [Google Scholar]
- Cummings RD (2009). The repertoire of glycan determinants in the human glycome. Mol. BioSys 5, 1087–1104. [DOI] [PubMed] [Google Scholar]
- Deng L, Bensing BA, Thamadilok S, Yu H, Lau K, Chen X, Ruhl S, Sullam PM, and Varki A (2014). Oral streptococci utilize a Siglec-like domain of serine-rich repeat adhesins to preferentially target platelet sialoglycans in human blood. PLoS pathog 10, e1004540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Weinke JL, Taylor WH, Ekborg G, Roden L, Anantharamaiah G, and Gawish A (1987). Inhibition of chondroitin and heparan sulfate biosynthesis in Chinese hamster ovary cell mutants defective in galactosyltransferase I. J. Biol. Chem 262, 12189–12195. [PubMed] [Google Scholar]
- Fujitani N, Furukawa J, Araki K, Fujioka T, Takegawa Y, Piao J, Nishioka T, Tamura T, Nikaido T, Ito M, et al. (2013). Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. U.S.A 110, 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui S, Feizi T, Galustian C, Lawson AM, and Chai W (2002). Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat. Biotechnol 20, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Ha Y, Stevens DJ, Skehel JJ, and Wiley DC (2003). X-ray structure of the hemagglutinin of a potential H3 avian progenitor of the 1968 Hong Kong pandemic influenza virus. Virology 309, 209–218. [DOI] [PubMed] [Google Scholar]
- Hassan H, Reis CA, Bennett EP, Mirgorodskaya E, Roepstorff P, Hollingsworth MA, Burchell J, Taylor-Papadimitriou J, and Clausen H (2000). The lectin domain of UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase-T4 directs its glycopeptide specificities. J. Biol. Chem 275, 38197–38205. [DOI] [PubMed] [Google Scholar]
- Hollingsworth MA, and Swanson BJ (2004). Mucins in cancer: protection and control of the cell surface. Nat. Rev. Cancer 4, 45–60. [DOI] [PubMed] [Google Scholar]
- Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P et al. (2013). Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi HJ, Hansen L, Narimatsu Y, Freeze HH, Henrissat B, Bennett E, Wandall HH, Clausen H, and Schjoldager KT (2018a). Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology 28:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi HJ, Narimatsu Y, Schjoldager KT, Tytgat HLP, Aebi M, Clausen H, and Halim A (2018b). SnapShot: O-Glycosylation Pathways across Kingdoms. Cell 172, 632–632 e632. [DOI] [PubMed] [Google Scholar]
- Lavrsen K, Dabelsteen S, Vakhrushev SY, Levann AMR, Haue AD, Dylander A, Mandel U, Hansen L, Frodin M, Bennett EP, et al. (2018). De novo expression of human polypeptide N-acetylgalactosaminyltransferase 6 (GalNAc-T6) in colon adenocarcinoma inhibits the differentiation of colonic epithelium. J. Biol. Chem. 293, 1298–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonowski LA, Narimatsu Y, Riaz A, Delay CE, Yang Z, Niola F, Duda K, Ober EA, Clausen H, Wandall HH, et al. (2017). Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protocols 12, 581–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JB, and Marth JD (2003). A genetic approach to Mammalian glycan function. Ann. Rev. Biochem 72, 643–691. [DOI] [PubMed] [Google Scholar]
- Macauley MS, Crocker PR, and Paulson JC (2014). Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, Yu J, Woods EC, Kramer JR, Westerlind, Uv et al. (2019). The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, and Hanisch FG (2002). Recombinant MUC1 probe authentically reflects cell-specific O-glycosylation profiles of endogenous breast cancer mucin. High density and prevalent core 2-based glycosylation. J. Biol. Chem 277, 26103–26112. [DOI] [PubMed] [Google Scholar]
- Narimatsu Y, Joshi HJ, Zhang Y, Gomes C, Chen YH, Lorenzetti F, Furukawa S, Schjoldager K, Hansen L, Clausen, Hv et al. (2018). A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology 28:295–305. [DOI] [PubMed] [Google Scholar]
- Ng BG, and Freeze HH (2018). Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet 34, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman ED, Mandel U, Levery SB, Kaizu T, and Hakomori S (1989). A series of disialogangliosides with binary 2--−-3 sialosyllactosamine structure, defined by monoclonal antibody NUH2, are oncodevelopmentally regulated antigens. J. Biol. Chem 264, 18719–18725. [PubMed] [Google Scholar]
- Padler-Karavani V, Song X, Yu H, Hurtado-Ziola N, Huang S, Muthana S, Chokhawala HA, Cheng J, Verhagen A, Langereis MA, et al. (2012). Cross-comparison of protein recognition of sialic acid diversity on two novel sialoglycan microarrays. J. Biol. Chem 287, 22593–22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma AS, Feizi T, Childs RA, Chai W, and Liu Y (2014). The neoglycolipid (NGL)-based oligosaccharide microarray system poised to decipher the meta-glycome. Curr. Opin. Chem. Biol 18, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik SK, and Stanley P (2006). Lectin-resistant CHO glycosylation mutants. Methods Enzymol 416, 159–182. [DOI] [PubMed] [Google Scholar]
- Paulson JC, and de Vries RP (2013). H5N1 receptor specificity as a factor in pandemic risk. Virus Res 178, 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, de Vries RP, Grant OC, Thompson AJ, McBride R, Tsogtbaatar B, Lee PS, Razi N, Wilson IA, Woods RJ, et al. (2017). Recent H3N2 Viruses Have Evolved Specificity for Extended, Branched Human-type Receptors, Conferring Potential for Increased Avidity. Cell Host Microbe 21, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R, Hansen L, Hintze J, Almeida R, Larsen S, Coskun M, Davidsen J, Mitchelmore C, David L, Troelsen JT, et al. (2017). Precise integration of inducible transcriptional elements (PrIITE) enables absolute control of gene expression. Nucl. Acids Res 45, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvirajesinghe TM, and Turnbull JE (2016). Glycoarray Technologies: Deciphering Interactions from Proteins to Live Cell Responses. Microarrays (Basel) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protocols 2, 1896. [DOI] [PubMed] [Google Scholar]
- Rillahan CD, and Paulson JC (2011). Glycan microarrays for decoding the glycome. Ann. Rev. Biochem 80, 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjoldager KT, Joshi HJ, Kong Y, Goth CK, King SL, Wandall HH, Bennett EP, Vakhrushev SY, and Clausen H (2015). Deconstruction of O-glycosylation-GalNAc-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep 16, 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz MA, Tian W, Mao Y, Van Coillie J, Sun L, Larsen JS, Chen YH, Kristensen C, Vakhrushev SY, Clausen, Hv et al. (2018). Glycoengineering design options for IgG1 in CHO cells using precise gene editing. Glycobiology 28:542–549. [DOI] [PubMed] [Google Scholar]
- Skehel JJ, and Wiley DC (2000). Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Ann. Rev. Biochem 69, 531–569. [DOI] [PubMed] [Google Scholar]
- Steentoft C, Bennett EP, Schjoldager KT, Vakhrushev SY, Wandall HH, and Clausen H (2014). Precision genome editing: A small revolution for glycobiology. Glycobiology 24, 663–680. [DOI] [PubMed] [Google Scholar]
- Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, and Clausen H (2011). Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered Simple Cell lines. Nat. Methods 8, 977–982. [DOI] [PubMed] [Google Scholar]
- Stolfa G, Mondal N, Zhu Y, Yu X, Buffone A Jr., and Neelamegham S (2016). Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and Glycosphingolipids to human leukocyte-endothelium adhesion. Sci. Reports 6, 30392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termini JM, Silver ZA, Connor B, Antonopoulos A, Haslam SM, Dell A, and Desrosiers RC (2017). HEK293T cell lines defective for O-linked glycosylation. PLoS One 12, e0179949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, and Smart TG (2005). HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods 51, 187–200. [DOI] [PubMed] [Google Scholar]
- Vakhrushev SY, Dadimov D, and Peter-Katalinic J (2009). Software platform for high-throughput glycomics. Analyt. Chem 81, 3252–3260. [DOI] [PubMed] [Google Scholar]
- Varki A (1994). Selectin ligands. Proc. Natl. Acad. Sci. U.S.A 91, 7390–7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Aebi M, Packer NH, Seeberger PH, Esko JD, Stanley P, Hart G, Darvill A, Kinoshita T, et al. (2015a). Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 25, 1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Schnaar RL, and Schauer R (2015b). Sialic Acids and Other Nonulosonic Acids In Essentials of Glycobiology, rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, et al. , eds. (Cold Spring Harbor (NY)), pp. 179–195. [PubMed] [Google Scholar]
- Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, and Boons GJ (2013). A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik BR, Barnard KN, Ossiboff RJ, Khedri Z, Feng KH, Yu H, Chen X, Perez DR, Varki A, and Parrish CR (2017). Distribution of O-Acetylated Sialic Acids among Target Host Tissues for Influenza Virus. mSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J, de Souza-e-Silva U, and Paulson JC (1982). Sialylation of glycoprotein oligosaccharides N-linked to asparagine. Enzymatic characterization of a Gal beta 1 to 3 (4)GlcNAc alpha 2 to 3 sialyltransferase and a Gal beta 1 to 4GlcNAc alpha 2 to 6 sialyltransferase from rat liver. J. Biol. Chem 257, 13845–13853. [PubMed] [Google Scholar]
- Yang X, Tao S, Orlando R, Brockhausen I, and Kan FW (2012). Structures and biosynthesis of the N- and O-glycans of recombinant human oviduct-specific glycoprotein expressed in human embryonic kidney cells. Carbohydrate Res 358, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Steentoft C, Hauge C, Hansen L, Thomsen AL, Niola F, Vester-Christensen MB, Frodin M, Clausen H, Wandall HH, et al. (2015a). Fast and sensitive detection of indels induced by precise gene targeting. Nucl. Acids Res 43:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang S, Halim A, Schulz MA, Frodin M, Rahman SH, Vester-Christensen MB, Behrens C, Kristensen C, Vakhrushev SY, et al. (2015b). Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol 33, 842–844. [DOI] [PubMed] [Google Scholar]
- Young WW Jr., Portoukalian J, and Hakomori S (1981). Two monoclonal anticarbohydrate antibodies directed to glycosphingolipids with a lacto-N-glycosyl type II chain. J. Biol. Chem 256, 10967–10972. [PubMed] [Google Scholar]
- Yu H, Gonzalez-Gil A, Wei Y, Fernandes SM, Porell RN, Vajn K, Paulson JC, Nycholat CM, and Schnaar RL (2017). Siglec-8 and Siglec-9 binding specificities and endogenous airway ligand distributions and properties. Glycobiology 27, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE repository with the dataset identifier PXD013676.