

Abstract
Presentation of membrane proteins to host immune systems has been a challenging problem due to complexity arising from the poor in vivo stability of the membrane-mimetic media often used for solubilizing the membrane proteins. We report the use of functionalized, biocompatible nanoparticles as substrates to guide the formation of proteoliposomes that can present many copies of membrane proteins in a unidirectional manner. The approach was demonstrated to present the membrane-proximal region of the HIV-1 envelope glycoprotein. These nanoparticle-supported liposomes are broadly applicable as membrane antigen vehicles for inducing host immune responses.
Keywords: membrane antigen presentation, nanoparticle, proteoliposome formation, unidirectional, HIV-1 MPER
Graphical Abstract
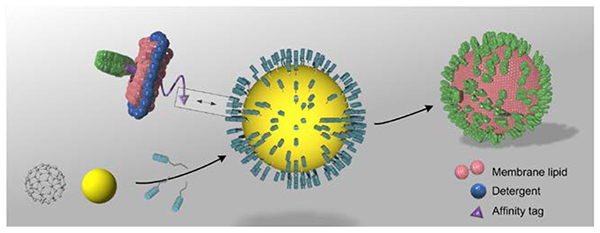
The solid and hallow nanoparticles can both be used as core substrates to recruit membrane proteins in bicelles through affinity interaction. As detergent is removed, the bicelles will merge and form proteoliposomes encapsulating the nanoparticles, presenting high copy numbers of membrane proteins in a unidirectional manner.
The large majority of antibodies currently being developed for therapeutic use target membrane-anchored or integral membrane proteins. But presenting these proteins to immune systems to induce antibodies is a difficult problem, in particular, for those whose structural integrity can only be preserved on the membrane. Several approaches have been developed to address this problem. Liposomes are often used as membrane protein carriers for inducing immune responses[1]. During liposome formation, however, the protein orientation is random. The unstable nature of liposome, i.e., its tendency to fuse with other cellular vesicles, could be another source of risk in its application. Greater stability in serum could be achieved using interbilayer-crosslinked multilamellar vesicle (ICMV)-coated particles[2], but the technique has not been demonstrated to incorporate transmembrane proteins. Lipid nanodisc is another popular medium for membrane proteins[3], and has been used previously in phage display[4]. But, nanodisc samples are generally difficult to make in large quantities. Moreover, regular nanodiscs usually can only contain 1–2 copies of protein due to its small size (10–15 nm in diameter)[5], and are thus not ideal for inducing strong immunogenic responses in vivo. It is also possible to use mammalian cells to produce virus-like particles (VLP) incorporating membrane proteins on the VLP membrane[6]. The success of this approach, however, depends on the efficiency of protein incorporation into VLP, which needs laborious optimization for each target and often cannot be controlled manually.
We sought to use nanoparticles as substrates to guide proteoliposome assembly. By modifying nanoparticles with functional moieties that specifically recruit affinity-tagged membrane proteins in bicelles, we could form proteoliposome around the nanoparticle where the proteins are presented in a unidirectional manner. We have demonstrated this approach, named Supported ProteoLiposome for Antigen Directed Display (SPLAnDiD), for a membrane fragment of the HIV-1 envelope glycoprotein (Env) encompassing the transmembrane (TM) domain and the membrane-proximal external region (MPER).
The design concept, illustrated in Fig. 1a–d, is to specifically recruit membrane proteins solubilized in lipid/detergent bicelles (that closely mimic a lipid bilayer) to the surface of a globular shaped nanoparticle, also referred to as the substrate. As detergent is removed, the protein-containing bicelles will grow on the substrate surface to form proteoliposomes. Nanoparticles can take on various forms and compositions. For biological or medical applications, it is important that the particles are biocompatible with no major toxic or immunogenic risk to biological organisms. We experimented with two different types of nanoparticles (Fig. 1a). One is polyphenol-stabilized gold nanoparticle (AuNP), which is solid and can be fabricated to various sizes ranging from 3 to 50 nm by exploiting the coordination and stabilization of gold ions with polyphenol moieties[7]. AuNPs are biocompatible and have been approved by FDA as Generally Recognized as Safe (GRAS) and for drug use[8]. Polyphenol moieties can be further functionalized via their hydroxyl groups and active carbon sites[9]. Another form of nanoparticle is the hollow DNA buckyball assembled by DNA origami[10]. DNAs are generally non-immunogenic[11]. A variety of modifications can be incorporated into DNA during synthesis for functionalization.
Figure 1. Schematic illustration of nanoparticle-supported proteoliposome for unidirectional presentation of membrane proteins.

(a) The solid nanoparticle (e.g., polyphenol-stabilized gold nanoparticle) and the hollow DNA buckyball can both be used as core substrates for liposome formation. (b) Membrane proteins with cytoplasmic affinity tag are reconstituted in bicelles that mimic a lipid bilayer. (c) Functional moieties for interacting with the protein affinity tag are covalently linked to the nanoparticles. (d) High copy numbers of membrane proteins are presented in a unidirectional manner in a nanoparticle-supported liposome.
In our assembly method, membrane proteins are purified and solubilized in bicelles composed of 1,2-dimyristoyl-sn-Glycero-3-Phosphocholine (DMPC) and 1,2-dihexanoyl-sn-Glycero-3-Phosphocholine (DH6PC) (Experimental Procedures) (Fig. 1b). An affinity tag is placed at the cytoplasmic end of the protein for directing the protein to the nanoparticles. In the current study, this affinity tag is a polyhistidine tag (His6-tag), and the functional moieties on the nanoparticles are Ni-NTAs (Fig. 1c). Only bicelles containing the membrane protein of interest will specifically “glue” to the nanoparticle surface via (Ni-NTA)-(His6-tag) interaction, and since the His6-tag is on the cytoplasmic side of the protein, the protein-bicelle complex will be “glued” in a unidirectional manner. As DH6PC is removed, the bicelles will merge and form proteoliposome encapsulating the nanoparticle (Fig. 1d).
AuNPs were synthesized by mixing the gold precursor, chloroauric acid (HAuCl4), and tannic acid (TA) solution as described previously[7a]. Phenolic hydroxyl group (-OH) of TA attaches to the surface of the gold along with the rest of polyphenol to act as surfactant to stabilize the metal ion with electrostatic interaction (Fig. S1a). The size of the AuNPs can be precisely controlled by the ratio of chloroauric acid to TA and their mixing speed. We first prepared AuNPs with average diameter of ~47 nm, as shown by transmission electron microscopy (TEM) (Fig. 2a) and dynamic light scattering (DLS) (Fig. S2a). The AuNPs were then functionalized with lysine-NTA (Experimental Procedures) by conjugating the amine groups of lysine-NTA and the nucleophilic carbons of the polyphenol groups via glutaraldehyde (Fig. 2a, S1b). The NTA-AuNPs were charged with Ni2+. The surface charges of AuNPs, evaluated as the zeta potential (ξ), were monitored during the functionalization process. As shown in Fig. S2b, the polyphenol-stabilized AuNPs are negatively charged (ξ = −52 mV) due to deprotonation of the hydroxyl groups. The ξ increased to −36 mV after conjugation with lysine-NTA and increased further to −20.1 mV after addition of Ni2+, as a result of modification of polyphenol groups and coordination with positively charged Ni2+, respectively. Finally, to examine the ability of the Ni-NTA functionalized AuNPs (Ni-NTA-AuNPs) to specifically interact with His6-tag, we recorded 1D 1H NMR spectrum of the Foldon protein containing C-terminal His6-tag (Foldon-His6) before and after mixing with the Ni-NTA-AuNPs (Methods). The Foldon NMR signals were essentially ablated upon the addition of Ni-NTA-AuNPs (Fig. 2b), indicating strong binding of Foldon-His6 to the nanoparticles that caused NMR signals to decay rapidly.
Figure 2. Nanoparticle-supported proteoliposome formation using bicelle-reconstituted MPER-TMD.

(a) The TEM images of gold-polyphenol nanoparticles (AuNPs) functionalized with NTA (NTA-AuNP). (b) 1H NMR spectra the Foldon protein with C-terminal His6-tag in the absence and presence of Ni-NTA-AuNPs. Blue: 450 μl of 30 μM Foldon-His6; Red: 450 μl of 30 μM Foldon-His6 mixed with 100 μl of Ni-NTA-AuNP (OD530 = 0.1) (the volume of the mixture adjusted to 450 μl before NMR measurement). (c) Schematic illustration of unidirectional coating of bicelle-reconstituted MPER-TMD onto Ni-NTA-AuNP. (d) Negative staining EM (nsEM) image (wide view) of Ni-NTA-AuNP supported MPER-TMD proteoliposome (see text). (e) nsEM images of the Ni-NTA-AuNP-supported MPER-TMD proteoliposomes at two magnifications. (f) nsEM images of DNA buckyball-supported MPER-TMD proteoliposomes at two magnifications. (g) Analysis of FLAG-MPER-TMD-His6 orientation in Ni-NTA-AuNP-supported liposomes by antibody resin pull-down and SDS-PAGE. Lane 1: Ni-NTA-AuNP-supported MPER-TMD liposome; Lane 2: flow-through from anti-His6 resin after 30 minutes incubation; Lane 3: elution from anti-His6 resin; Lane 4: flow-through from anti-FLAG resin after 30 minutes incubation; Lane 5: elution from anti-FLAG resin.
Having generated Ni-NTA-AuNPs, we next tested the above scheme of proteoliposome formation using a fragment of HIV-1 Env that contains the membrane-proximal external region (MPER) and the transmembrane domain (TMD). This fragment (residues 660–710) is derived from a clade D HIV-1 isolate 92UG024.2 (designated MPER-TMD)[12]. The MPER is one of the most conserved regions of HIV-1 Env and bears epitopes of broadly neutralizing antibodies from infected individuals[13]. Therefore, methods for presenting the MPER to human immune system are of strong interest to HIV vaccine development.
We introduced a His6-tag at the C-terminus of the MPER-TMD to interact with the Ni-NTA-AuNPs. The MPER-TMD was expressed, purified, and reconstituted in bicelles with q = 0.5 as described previously[12, 14]. The Ni-NTA-AuNP solution (OD530 = 0.1) and the solution of bicelle-reconstituted MPER-TMD-His6 (0.3 mM) were mixed at a volume ratio of 3:1 to allow coating of protein-containing bicelles onto the nanoparticle surface (Fig. 2c). After complete removal of DH6PC by dialysis, relatively uniformly sized liposomes were formed around the nanoparticles (Fig. 2d, 2e). The diameter of the liposomes is 75±5 nm, which is consistent with the predicted size of the complex, including the Ni-NTA-AuNP (~50 nm), the lipid bilayer (~6 nm × 2), and the space between Ni-NTA-AuNP and lipid envelope. To provide additional evidence that the formation of stable nanoparticle-supported liposome is due to specific interaction between Ni-NTA and His6-tag, we performed a series of control experiments, i.e., forming liposomes 1) with empty bicelles in the absence of AuNPs (Fig. S3a), 2) by mixing Ni-NTA-AuNPs with empty bicelles (Fig. S3b), and 3) by mixing non-functionalized AuNPs with bicelle-reconstituted MPER-TMD (Fig. S3c). As shown by negative staining EM (nsEM), liposomes formed under these conditions were mostly broken and highly inhomogeneous, indicating that direct recruitment of bicelles onto the nanoparticles via the membrane protein affinity tag is crucial to achieving homogeneous proteoliposome assemblies.
To achieve the highest proteoliposome assembly efficiency, different ratios of Ni-NTA-AuNP to MPER-TMD-His6 were tested. When the volume ratio between the solution of Ni-NTA-AuNP (OD530 = 0.1) and the solution of bicelle-reconstituted MPER-TMD-His6 (0.3 mM) was set at 1:1, in addition to the expected liposome size (~75 nm), much smaller liposomes (< 20 nm) were observed (Fig. S3d), likely formed with excessive MPER-TMD-His6 and DMPC lipid without the support of Ni-NTA-AuNP. When the ratio was set to 3:1 and 6:1, the smaller liposomes mostly disappeared and very similar liposome populations were observed (Fig. S3e, S3f), suggesting that all protein-containing bicelles were recruited to Ni-NTA-AuNPs at the two ratios. Excessive Ni-NTA-AuNPs could not be observed due to incompatibility with negative staining.
We next tested the use of hollow DNA nanoparticles to guide proteoliposome assembly. We used a previously designed DNA buckyball formed with three different DNA strands (long, medium, and short)[10]. To functionalize the DNA buckyballs with NTA moieties, we used the TriNTA with modified primary amine[15], which can be covalently linked to thiol-modified DNA via an amine-to-sulfhydryl crosslinker (Fig. S4a). As such, the long strand was synthesized with the dithiol group at the 3’ end, and TriNTA was covalently linked to the long strand via a bi-functional crosslinker, PEG2-SMCC (Experimental Procedures). When charged with Ni2+, the TriNTA has high binding affinity for the His6-tag (20 ± 10 nM)[15]. The TriNTA-linked DNA strand was purified and mixed with the other two strands to form DNA buckyballs with an average diameter of ~80 nm (Fig. S4b, S5). As in the AuNP application above, the bicelle-reconstituted MPER-TMD (with C-terminal His6-tag) was mixed with the DNA buckyballs, and the ratio of MPER-TMD trimer to TriNTA (or long strand) was kept approximately at 1:1 to achieve ~60 MPER-TMD trimers per buckyball (there are 60 long strands in the assembly[10]). Upon removal of DH6PC, spherical liposomes with diameter of 95±15 nm were formed (Fig. 2f, S6). We note that a fraction of liposomes are smaller than estimated size, probably because some DNA buckyballs were not completely assembled (thus smaller size) but could still catalyze liposome formation. More robust buckyball assembly can be achieved by using additional supportive DNA strands[16].
To examine whether the MPER-TMD was unidirectionally presented on Ni-NTA-AuNP-supported liposomes, we introduced a FLAG-tag and a His6-tag at the N- and C- termini of the MPER-TMD, respectively, and examined their relative accessibility to antibodies using an antibody resin pull-down assay. The Ni-NTA-AuNP-supported proteoliposomes containing the FLAG-MPER-TMD-His6 were prepared using the same protocol as described above. Equal amounts of the assembled proteoliposomes were incubated with anti-FLAG and anti-His6 resins separately. The flow-throughs from the resins were collected as readout of MPER-TMDs that do not bind the antibodies. Further, a 0.1 M glycine solution was used to elute the resin-bound fraction. The samples from flow-through and elution were analyzed by SDS-PAGE. For the anti-His6 resin, 97.1% of MPER-TMDs were found in the flow-through (Fig. 2g, S7; lanes 2, 3), whereas 94.8% of the MPER-TMDs were retained by the anti-FLAG resin (Fig. 2g, S7; lanes 4, 5). The results indicate that the MPER-TMD was incorporated in the nanoparticle-supported liposomes in a unidirectional manner.
Finally, to test whether the nanoparticle-supported proteoliposomes are immunogenic, five guinea pigs were immunized with Ni-NTA-AuNP-supported MPER-TMD proteoliposome with Adju-Phos adjuvant. Animal sera from different time points (Fig. 3a) were used to assess the ability of the vaccination regimen to elicit antibodies that can bind MPER-TMD reconstituted in regular liposome. The results from the enzyme-linked immunosorbent assay (ELISA) show that all of the sera after the first vaccination already contained MPER-TMD binding antibodies, and that sera from the second immunization elicited antibodies that bind much stronger and more robust to MPER-TMD (Fig. 3b). As a negative control, no binding to the empty liposome was observed (Fig. 3c).
Figure 3. Immunogenicity of Ni-NTA-AuNP-supported MPER-TMD proteoliposomes.

(a) Immunization schedule of 5 guinea pigs. Serum samples were collected at weeks 4 and 12. (b) ELISA analysis of serum for anti-MPER-TMD antibodies. ELISA plates were coated with MPER-TMD in regular liposomes. Sera were added in serial dilutions (2700, 900, 300, 100) and detected with a horseradish peroxidase (HRP)-conjugated rabbit anti-guinea pig secondary antibody for total IgG ELISAs. (c) ELISA plate incubated with empty liposomes.
We have shown that nanoparticles with functionalized surfaces can serve as effective guide for the formation of proteoliposomes with unidirectional presentation of membrane proteins. Since the protein affinity tag drives uniform coating of bicelles, which are essentially solubilized membrane patches, around the nanoparticles, proteoliposome formation upon detergent removal is highly robust. Moreover, varying sizes of proteoliposomes are achievable for different applications, as the size of the nanoparticle substrate can be accurately controlled.
We believe the nanoparticle-supported liposomes can be effective vaccine carriers. First, potentially high copy number of membrane proteins can be incorporated. The unidirectional presentation further increases the amount of effective antigens for the immune system. Second, the AuNP used in the current study is highly biocompatible and inexpensive to produce. The presence of nickel inside the liposome may be a safety concern but its toxicity is expected to be greatly reduced when chelated by NTA[17]. Finally, the nanoparticle-supported liposome is structurally more stable than the regular liposomes owing to the nanoparticle-protein interaction, and such enhanced stability is important for application in vivo.
Previous attempts at presenting MPER in immunogens have not been successful in inducing neutralizing antibodies in vivo[18]. The failure could be due to the conformation nature of the epitopes or their limited accessibility on the membrane surface. Recent studies suggest lipid bilayer also accounts for the neutralizing potency of MPER-specific antibodies[19]. The reported method allowed unidirectional presentation of many MPER-TMD trimers on a single particle in a lipid bilayer environment. Indeed, the new immunogen elicited MPER-specific antibodies in the guinea pigs, though the neutralizing potential of these antibodies remains to be investigated.
In conclusion, the use of functionalized nanoparticles to guide proteoliposome formation offers many distinct advantages, including the improved efficiency and uniformity of liposome formation, the unidirectional presentation of transmembrane proteins, the preservation of membrane protein native structure, the greater control of protein incorporation number per liposome, and the greater stability of the nanoparticle-supported liposomes. While these advantages are particularly important for vaccine development, they are equally useful for developing therapeutic antibodies against membrane proteins such as GPCRs, transporters, and ion channels.
Supplementary Material
Acknowledgements
We thank Liqiang Pan for the insightful discussion. This work was supported by NIH grant AI127193 (to B.C. and J.J.C.), and GM116898 (to J.J.C.).
Footnotes
Conflict of interest
W.C., J.G., B.C., J.J.C. declare competing financial interests. A provisional patent was filed on behalf of the Harvard Medical School.
Contributor Information
Wen Chen, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115, United States.
Junling Guo, Department of Biomass Science and Engineering, Sichuan University, 252 Shuncheng Street, Chengdu, Sichuan 610065, China.
Yongfei Cai, Division of Molecular Medicine, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, 3 Blackfan Street, Boston, MA 02115, United States.
Qingshan Fu, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115, United States.
Bing Chen, Division of Molecular Medicine, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, 3 Blackfan Street, Boston, MA 02115, United States.
James J. Chou, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115, United States.
References
- [1].a Russell DG, Alexander J, Journal of immunology 1988, 140, 1274–1279; [PubMed] [Google Scholar]; b Rigaud JL, Levy D, Methods Enzymol 2003, 372, 65–86; [DOI] [PubMed] [Google Scholar]; c Jespersen LK, Kuusinen A, Orellana A, Keinanen K, Engberg J, Eur J Biochem 2000, 267, 1382–1389. [DOI] [PubMed] [Google Scholar]
- [2].a Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Nat Mater 2011, 10, 243–251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pejawar-Gaddy S, Kovacs JM, Barouch DH, Chen B, Irvine DJ, Bioconjug Chem 2014, 25, 1470–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Civjan NR, Bayburt TH, Schuler MA, Sligar SG, Biotechniques 2003, 35, 556–560, 562–553. [DOI] [PubMed] [Google Scholar]
- [4].a Pavlidou M, Hanel K, Mockel L, Willbold D, PloS one 2013, 8, e72272; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dominik PK, Kossiakoff AA, Methods Enzymol 2015, 557, 219–245; [DOI] [PubMed] [Google Scholar]; c Dominik PK, Borowska MT, Dalmas O, Kim SS, Perozo E, Keenan RJ, Kossiakoff AA, Structure 2016, 24, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a Bayburt TH, Sligar SG, FEBS letters 2010, 584, 1721–1727; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schuler MA, Denisov IG, Sligar SG, Methods in molecular biology 2013, 974, 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a Grgacic EV, Anderson DA, Methods 2006, 40, 60–65; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kushnir N, Streatfield SJ, Yusibov V, Vaccine 2012, 31, 58–83; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lua LH, Connors NK, Sainsbury F, Chuan YP, Wibowo N, Middelberg AP, Biotechnology and bioengineering 2014, 111, 425–440. [DOI] [PubMed] [Google Scholar]
- [7].a Huang X, Wu H, Liao X, Shi B, Green Chemistry 2010, 12, 395–399; [Google Scholar]; b Guo J, Ping Y, Ejima H, Alt K, Meissner M, Richardson JJ, Yan Y, Peter K, von Elverfeldt D, Hagemeyer CE, Angewandte Chemie 2014, 126, 5652–5657. [DOI] [PubMed] [Google Scholar]
- [8].Ejima H, Richardson JJ, Caruso F, Nano Today 2017, 12, 136–148. [Google Scholar]
- [9].Guo J, Wang X, Miao P, Liao X, Zhang W, Shi B, Journal of Materials Chemistry 2012, 22, 11933–11942. [Google Scholar]
- [10].Frey G, Peng H, Rits-Volloch S, Morelli M, Cheng Y, Chen B, Proc Natl Acad Sci U S A 2008, 105, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bastian D, Borel H, Sasaki T, Steinberg AD, Borel Y, Journal of immunology 1985, 135, 1772–1777. [PubMed] [Google Scholar]
- [12].Fu Q, Shaik MM, Cai Y, Ghantous F, Piai A, Peng H, Rits-Volloch S, Liu Z, Harrison SC, Seaman MS, Chen B, Chou JJ, Proc Natl Acad Sci U S A 2018, 115, E8892–E8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Ruker F, Katinger H, J Virol 1993, 67, 6642–6647; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H, AIDS Res Hum Retroviruses 2001, 17, 1757–1765; [DOI] [PubMed] [Google Scholar]; c Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M, Nature 2012, 491, 406–412; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Williams LD, Ofek G, Schätzle S, McDaniel JR, Lu X, Nicely NI, Wu L, Lougheed CS, Bradley T, Louder MK, McKee K, Bailer RT, O’Dell S, Georgiev IS, Seaman MS, Parks RJ, Marshall DJ, Anasti K, Yang G, Nie X, Tumba NL, Wiehe K, Wagh K, Korber B, Kepler TB, Alam SM, Morris L, Kamanga G, Cohen MS, Bonsignori M, Xia SM, Montefiori DC, Kelsoe G, Gao F, Mascola JR, Moody MA, Saunders KO, Liao HX, Tomaras GD, Georgiou G, F. HB, Science Immunology 2017, 2, eaal2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dev J, Park D, Fu Q, Chen J, Ha HJ, Ghantous F, Herrmann T, Chang W, Liu Z, Frey G, Seaman MS, Chen B, Chou JJ, Science 2016, 353, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lata S, Reichel A, Brock R, Tampe R, Piehler J, J Am Chem Soc 2005, 127, 10205–10215. [DOI] [PubMed] [Google Scholar]
- [16].Tian C, Li X, Liu Z, Jiang W, Wang G, Mao C, Angew Chem Int Ed Engl 2014, 53, 8041–8044. [DOI] [PubMed] [Google Scholar]
- [17].Flora SJ, Pachauri V, Int J Environ Res Public Health 2010, 7, 2745–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a Zwick MB, Aids 2005, 19, 1725–1737; [DOI] [PubMed] [Google Scholar]; b Hinz A, Schoehn G, Quendler H, Hulsik DL, Stiegler G, Katinger H, Seaman MS, Montefiori D, Weissenhorn W, Virology 2009, 390, 221–227. [DOI] [PubMed] [Google Scholar]
- [19].Chen J, Frey G, Peng H, Rits-Volloch S, Garrity J, Seaman MS, Chen B, J Virol 2014, 88, 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.