

Abstract
Damage Associated Molecular Patterns, including mitochondrial DNA (mtDNA) are released during hemorrhage resulting in the development of endotheliopathy. Tranexamic acid (TXA), an anti-fibrinolytic drug used in hemorrhaging patients, enhances their survival despite the lack of a comprehensive understanding of its cellular mechanisms of action. The present study is aimed to elucidate these mechanisms, with a focus on mitochondria. We found that TXA inhibits the release of endogenous mtDNA from granulocytes and endothelial cells. Furthermore, TXA attenuates the loss of the endothelial monolayer integrity induced by exogenous mtDNA. Using the Seahorse XF technology, it was demonstrated that TXA strongly stimulates mitochondrial respiration. Studies using Mitotracker dye, cells derived from mito-QC mice, and the ActivSignal IPAD assay, indicate that TXA stimulates biogenesis of mitochondria and inhibits mitophagy. These findings open the potential for improvement of the strategies of TXA applications in trauma patients and development of more efficient TXA derivatives.
Introduction
Tranexamic acid (TXA), a cyclic analog of lysine, has been used for many years as an efficient anti-fibrinolytic agent to treat blood loss resulting from trauma [Jennings et al., 2016; Rappold and Bochicchio, 2016], cardiac surgery [Ng et al., 2015] and postpartum hemorrhage [Simonazzi et al., 2016]. The CRASH-2 study has demonstrated the life-saving effect of early TXA application in trauma patients [Morrison et al., 2012]. A similar result was obtained in the MATTERs study of military patients wounded in combat [collaborators et al., 2010]. The anti-fibrinolytic effect of TXA is based on its binding to the kringle-1 domain of plasminogen that results in competitive inhibition of the proteolytic maturation of plasmin [Hoover et al., 1993].
Early treatment of bleeding trauma patients with TXA has been shown to decrease mortality, which may be due to the enhancement of clot formation, as well as to pronounced anti-inflammatory effect of TXA [Levy, 2007]. TXA has also successfully been used to treat hereditary angioedema which suggests its potential to protect the integrity of the endothelial cell (EC) monolayer [van den Elzen et al., 2016]. Furthermore, TXA has been shown to suppress the loss of intestinal glycocalyx in a rat hemorrhagic trauma model [Peng et al., 2016]. Considering the wide variety of beneficial effects of TXA, we suggest that the mechanisms underlying its activity may not be limited to the inhibition of plasmin maturation. Understanding the complex therapeutic range of TXA requires a grasp of its cellular and molecular mechanisms. We hypothesize that TXA can suppress the release of Damage Associated Molecular Patterns (DAMPs) and enhance endothelial cell metabolism, thus increasing the resistance of endothelial cells to trauma-induced stress. To substantiate this hypothesis, we performed studies in an in vitro setting where the results of TXA treatment can be evaluated while avoiding the complexity of the systemic inflammatory response (SIRS) of in vivo injury models.
We studied the effect of TXA upon the release of mitochondrial DNA, a major DAMP [Srikrishna and Freeze, 2009] known to induce a SIRS response [Zhang et al., 2010]), from human umbilical vein endothelial cells (HUVECs) and from neutrophils, which represent the most abundant type of granulocytes in human peripheral blood. Neutrophils are highly sensitive to various damaging agents, and have been shown to be a source of DAMPs during traumatic hemorrhagic shock [Srikrishna and Freeze, 2009]. Given that blood-derived granulocytes are unstable with a short lifespan post isolation in in vitro setting [Monceaux et al., 2016], we used neutrophils obtained as a result of retinoic acid (RA) induced differentiation of human promyelocytic leukemia-derived HL60 cell culture [Rovera et al., 1979]. These cells are a well-established model for the study of neutrophils [Birnie, 1988]. The used HL60/S4 subculture is characterized by accelerated granulocytic differentiation [Campbell et al., 1995].
The release of mitochondrial DAMPs, in particular mtDNA may be a consequence of energy metabolism dysregulation provoked by hemorrhagic trauma [Van Way et al., 2003]. Indeed the sharp decrease of mitochondrial respiration usually leads to mitophagy that can eventually result in the excretion of mitochondrial content [Ney, 2015]. To assess the effects of TXA upon energy metabolism and mitophagy, we used the Seahorse XF system technology and cells derived from reporter mito-QC mice. Additionally, we studied the protective effects of TXA on the monolayer of EC, the major cell type involved in the vascular response to trauma, using HUVEC, which are highly sensitive to damaging agents such as DAMPs [Sun et al., 2013].
Materials and Methods
1. Cell cultures
Human umbilical vein endothelial cell (HUVEC) and human arterial endothelial cell (HAEC) cultures were obtained from ATCC (Leesburg, VA) and grown in full EBM-2 medium (Lonza, Portsmouth, NH). Cells were cultured at 37°C in the atmosphere of 5% CO2. Cells at passages 5 to 15, at 90–100% confluency were used for TXA studies. HL60/S4 cell culture was a gift of Ada and Don Olins (University of New England, Portland, ME), and grown in RPMI medium (Sigma-Aldrich, St. Louis, MO) with 10% fetal calf serum (VWR, Radnor, PA) at 37°C in the atmosphere of 5% CO2. To differentiate HL60 cells to granulocytes, they were treated for 72 h with 1 µM all-trans-retinoic acid (Sigma-Aldrich, St Louis, MO). The differentiation of HL60 cells was confirmed by the analysis of nuclear morphology after DAPI staining (development of nuclear segmentation), and by the positive staining with nitroblue tetrazolium. NIH3T3 cells were obtained from ATCC and cultured in DMEM with 10% fetal calf serum. Mito-QC mice were a gift from Anyonya Guntur (MMCRI, Scarborough, ME). Lung EC were isolated from 7-days-old pups of mitoQC mice using two cycles of magnetic separation based on CD31 and ICAM cell surface positivity, as described [Sobczak et al., 2010] . Lung EC were cultured in full EBM-2 medium. Immunofluorescence staining demonstrated that at least 90% of the isolated EC were CD31-positive.
2. Quantitative Polymerase Chain Reaction (qPCR) of mtDNA
In preparation for qPCR, mtDNA was isolated from cell culture medium using a ZR Plasmid Miniprep kit (Zymo Cat#D4016) (Zymo Research, Irvine, CA). Briefly, the medium was centrifuged for 45 min at 13000 rpm, and 50 μl of supernatant was added to P1 buffer and the rest of the procedure was performed exactly according to manufacturer’s protocol. In the last step, 50µl of elution buffer was added to extract the DNA from the column.
mtDNA levels in the medium were measured by SYBR-green dye based qPCR assay using a Bio-Rad-CFX Connect Real Time System (Bio-Rad, Hercules, CA). Primers (IDT, Coralville, IA) specific for human mtDNA used were: forward CACCCAAGAACAGGGTTTGT, reverse TGGCAATGGGTATGTTGTTA [Rooney et al., 2015]. DNA levels were calculated as Arbitrary Units percent of control (untreated cells) by subtracting experimental qCT values of mtDNA from control DNA qCT values.
3. Preparation of mouse mtDNA
The mtDNA used in this study for treatment of granulocytes and HUVEC was isolated from mouse NIH3T3 cell culture using the Mitochondrial DNA isolation kit KA0895 from Abnova (Taipei, Taiwan) according to manufacturer’s protocol. Concentration of mtDNA was measured spectrophotometrically based on absorption at 260 nm.
4. Studies of oxidative phosphorylation
HUVEC and HAEC were assayed using the Seahorse XF 96 extraflux analyzer (Agilent Technologies, Lexington, MA) according to manufacturer’s instructions. Briefly the cells were plated at 10,000 cells/well and allowed to adhere overnight. The next day, the cells were pretreated for 2h with 0, 20 or 100 µg/ml TXA (Mylan Technologies, St Albans city, VT) in full cell culture medium. Then, the medium was changed over to Seahorse XF DMEM base medium, without phenol red (Agilent, cat # 103335–100) supplemented with 10mM Glucose, 2mM Glutamine and 1mM Sodium Pyruvate pH 7.4, and the same TXA concentration as during the pretreatment, and then metabolic parameters were measured using the Mitochondrial Stress Test (Agilent, Cat # 103015–100). Analysis of oxygen consumption rate (OCR) was done using the following protocol after calibration and equilibration: 3 cycles of mix 3 min, wait 2 min, measure 3 min, starting with basal values, and then after each of the following injections: 1.25µM Oligomycin (final concentration), 1µM FCCP (final), and 0.5µm Rotenone and Antimycin (final). Post assay wells were washed, lysed, and a standard BSA assay was performed for protein content. Data presented are normalized for protein content/well and are representative of 3 independent experiments with at least 10 replicate wells/treatment within each experiment.
5. ActivSignal IPAD assay
HUVEC were incubated for 24h in full medium with 0, 20 or 100 µg/ml TXA. The cells were then lysed with PBS + 1%NP40 lysis buffer. The lysates were sent to the ActivSignal company (http://www.activsignal.com, Watertown, MA) for further processing. ActivSignal IPAD (Immuno-Paired-Antibody Detection assay) platform is a proprietary technology for analyzing the activity of multiple signaling pathways in one reaction. Activities of greater than twenty signaling pathways are monitored simultaneously in a single well through assessing expression or protein phosphorylation of 70 target human proteins. The technology allows detection of targets with high specificity and sensitivity due to the combination of two distinct antibodies per target. Multiple targets were utilized to cover each pathway.
6. Immunofluorescence confocal microscopy
HUVEC were plated on fibronectin-coated glass coverslips to assess the possible rescuing effect of TXA upon their adherence junctions and actin cytoskeleton. After treatment with mtDNA and TXA, cells were formalin fixed, blocked in 2% bovine serum albumin and stained with anti-β-catenin rabbit antibodies (Santa Cruz, Santa Cruz, CA) followed by Alexa 546-conjugated anti-rabbit IgG secondary antibodies, Alexa 488-conjugated phalloidin and Hoechst (all from Invitrogen, Carlsbad, CA). Fluorescent preparations were embedded in Vectashield (Vector Laboratories, Burlingame, CA), and images were taken by utilizing a Leica SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany) at 60X objective magnification. Confocal microscopy was also utilized to study the effect of TXA upon the mitophagy in mito-QC cells, and upon the morphology of mitochondria in HUVEC stained for 5 min with 1 µg/ml Mitotracker Deep Red (Thermo Fisher Scientific, Haverhill, MA), followed by formalin fixation. Image G program was used to measure mitochondria lengths and to calculate the numbers of mitopagies (red-only cytoplasmic structures) per cell.
8. Statistical analysis
Experiments were triplicated with consistent results and representative experiments are shown. Quantitative data were analyzed and graphs were produced with Prism 7 statistical software (GraphPad Software, Inc., La Jolla, CA, USA). We used the unpaired t-test to determine the significance of differences between TXA-treated and control levels of mtDNA. We compared the means of repeated RT-PCR measurements (mtDNA), which are normally distributed and in most cases show similar standard deviations that satisfies the requirements for the application of the unpaired t-test. Same applies to the distribution of mitochondria lengths and mitophagy numbers per cell. When compared data exhibited uneven standard deviations, we used the unpaired t-test with Welch correction. A one-way ANOVA was performed to test the effects of TXA on mitochondrial stress test parameters; significance was set at p<0.05.
Results
1. TXA suppresses the release of endogenous mtDNA in granulocytes and endothelial cells.
After RA-induced differentiation, HL60 granulocytes were treated with TXA and the release of mtDNA to the medium was determined. We found that the incubation of HL60 granulocytes with 100 µg/ml TXA significantly decreased the amount of spontaneous release of endogenous mtDNA to the medium (Figure 1A). Treatment of HL60 granulocytes with exogenous mouse mtDNA enhanced the release of endogenous mtDNA, and TXA suppressed this effect (Figure 1B). Interestingly, the similar effect of TXA was observed in non-differentiated HL60 cells treated with mouse mtDNA (Supplementary Figure 1). We have shown that similarly to HL60, TXA (20 or 100 µg/ml) suppresses the spontaneous release of mtDNA from HUVEC (Figure 1C), however, unlike HL60 granulocytes, HUVEC did not exhibit a consistent increase of endogenous mtDNA release in response to exogenous mtDNA (Figure 1D).
Figure 1. TXA suppresses the spontaneous and induced mtDNA release from HL60 granulocytes, and spontaneous mtDNA from HUVEC.
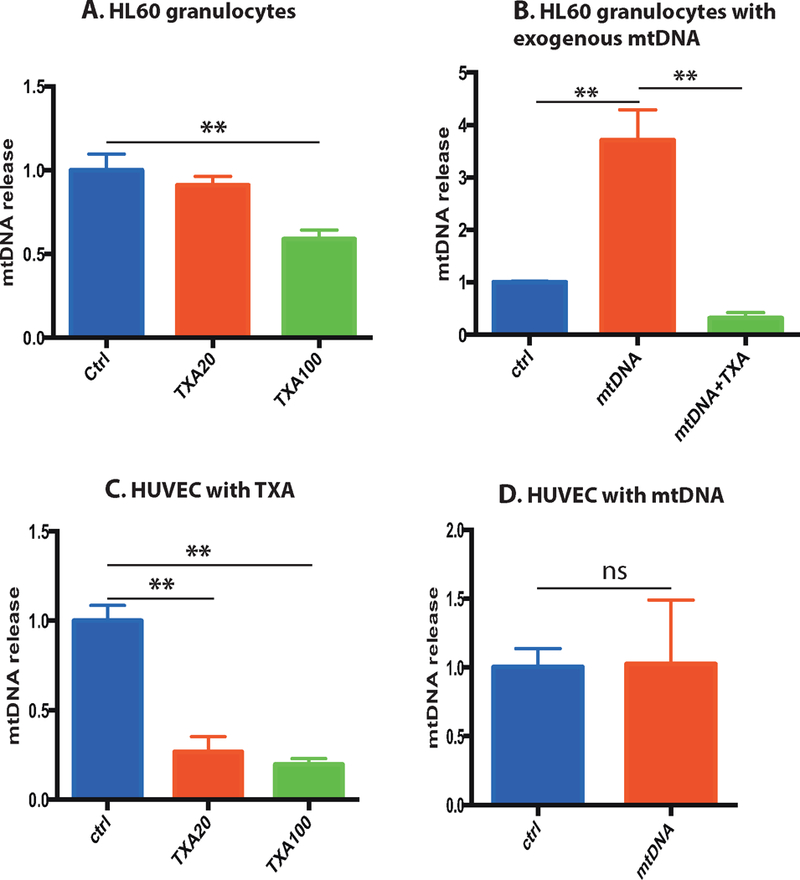
A) Differentiated HL60 granulocytes were incubated for 24 h with or without 20 or 100 μg/ml TXA. Cells were centrifuged, and released mtDNA in the supernatant was determined by qPCR using specific primers to human mtDNA. Mean mtDNA release levels normalized to untreated control, with standard deviations are shown. B) HL60 granulocytes were incubated for 4 h in presence of 0.3 μg/ml mouse mtDNA, with or without 100 μg/ml TXA, released mtDNA was determined. C) HUVEC were incubated for 4 h with or without 20 or 100 μg/ml TXA, released mtDNA was determined. D) HUVEC were incubated for 4 h in presence of 0.3 μg/ml mouse mtDNA, released mtDNA was determined.
2. TXA protects endothelial monolayer from damage induced by exogenous mtDNA.
Endothelial cells are highly sensitive to DAMPs that are released during hemorrhagic shock. DAMPs induce the loss of adhesions between endothelial cells resulting in leakiness of the endothelial monolayer [Sun et al., 2013]. We evaluated morphologic changes using immunofluorescent confocal and phase contrast microscopy, and found that exogenous mtDNA treatment disrupts the structure of cultured HUVEC monolayers (Figure 2A). This resulted in the loss of β-catenin positive adherens junctions (Figure 2B) and disorganization of the actin cytoskeleton and cell contraction (Figure 2C). TXA treatment attenuated the negative effects of exogenous mtDNA (Figure 2A–C).
Figure 2.
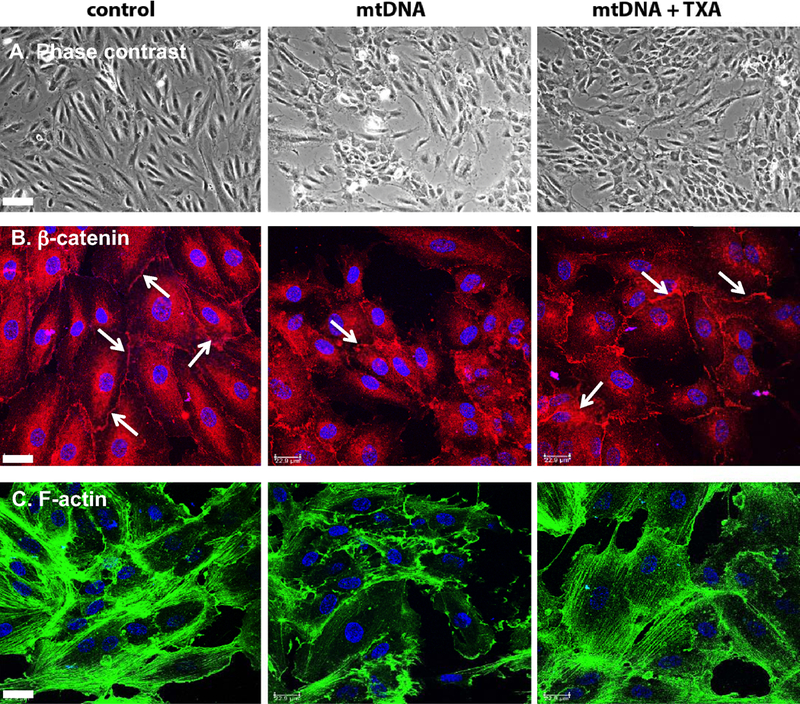
TXA protects endothelial monolayer from damage induced by exogenous mtDNA. HUVEC growing on fibronectin coated glass coverslips were incubated for 1 h in serum-free medium with 0.3 μg/ml mouse mtDNA, in presence or absence of 100 μg/ml TXA. Cells were formalin fixed, fluorescently stained for β-catenin, actin cytoskeleton and nuclear DNA as described in Material and Methods and studied using a phase contrast microscope (A) or a confocal microscope (B and C). Bar in A – 70 μm. Bars in B and C – 23 μm.
3. TXA enhances oxidative phosphorylation in endothelial cells.
Next we investigated the effects of TXA treatment on mitochondrial function through assessing energy expenditure. Our results show that TXA treatment significantly increased basal oxidative phosphorylation levels in HUVEC cells (Figure 3A). Maximal respiration, was also found to be increased with TXA treatment (Figure 3A), alongside spare respiratory capacity (Figure 3B) suggesting that TXA improves the cells flexibility and ability to meet energy demands under times of stress (TXA100: 10.92 ± 0.66 vs. TXA20: 12.16 ± 0.61 vs. CON 7.29 ± 0.23 ; p<.0001). In addition, TXA increased ATP production in the cell (TXA100: 3.86 ± 0.1 vs. TXA20: 3.77 ± 0.12 vs. CON 2.56 ± 0.25; p<.0001). Similar effects were observed when TXA was applied to human arterial endothelial cells (HAEC) (Supplementary Figure 2).
Figure 3. TXA Increases the cells ability to meet energy demands.
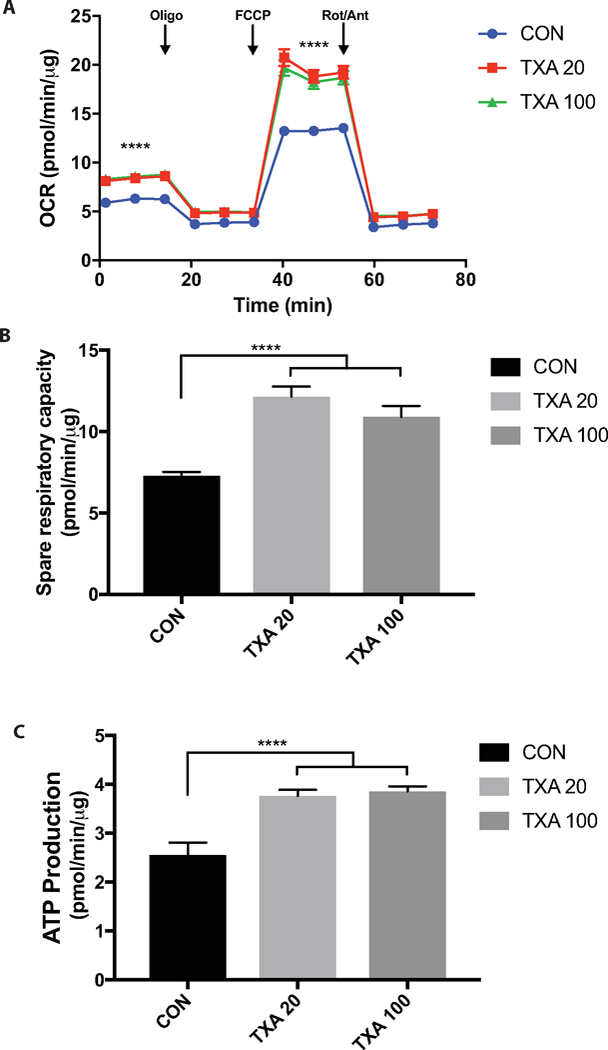
A) Mitochondrial Stress Test profile revealing increased basal and maximal respiration with TXA treatment (**** = P <0.0001, ANOVA effect of Treatment). B) Spare respiratory capacity of cells (maximal respiration minus basal respiration) suggestive of the cells increased flexibility to meet energetic demands. C) ATP production indicative of the amount of ATP being produced by the cell through oxidative phosphorylation pathways.
4. TXA treated cells contain longer mitochondria and exhibit decreased mitophagy.
For morphological assessment of mitochondrial status, we used live staining of HUVEC with fluorescent Mitotracker followed by formalin fixation. We found that cells treated for 24 h with TXA contain longer mitochondria than control cells (Figure 4A), indicating that the overall level of mitochondrial metabolism in the cell could be enhanced. Mitophagy, which involves engulfment of mitochondria by autophagosomes and their consecutive elimination is known to regulate metabolism [Farmer et al., 2018; Woodall and Gustafsson, 2018]. In order to assess the effect of TXA on mitophagy we used the mito-QC mouse model, which has been shown as an effective tool for measuring mitophagy in previous studies [McWilliams et al., 2016]. These mice have inserted into the Rosa26 locus a functionally inert, tandem mCherry-GFP tag fused to the mitochondrial targeting sequence of the outer mitochondrial membrane protein, FIS1. Under steady-state conditions, the mitochondrial network fluoresces both red and green, and during mitophagy the mitochondria are taken up in lysosomes where mCherry fluorescence remains stable, but GFP fluorescence becomes quenched by the acidic microenvironment. This results in the appearance of punctate mCherry-only foci that can be readily used as an index of mitophagy. Using lung endothelial cells isolated from mito-QC mice we found that treatment with TXA results in the reduction of mitophagy (Figure 4B).
Figure 4. Effects of TXA on mitochondria.
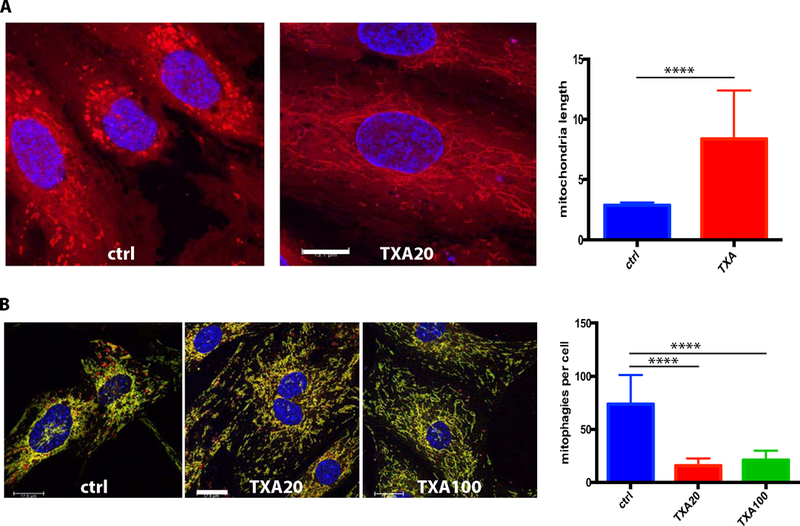
A) HUVECs were treated for 24 h with 0 or 20 μg/ml TXA, stained with Deep Red Mitotracker, formalin fixed and studied using a confocal microscope. The mitochondria lengths were measured using the Image J program, and the mean mitochondria length and SD were calculated. TXA treated cells display longer mitochondria. Bar – 13.1 μm. B) Lung endothelial cells obtained from mito-QC mice were incubated for 24h in serum free medim with 0, 20 or 100 μg/ml TXA to assess mitophagy using confocal microscopy. The mean number of mitophagies (red only structures) per cell and SD were calculated. TXA decreases the presence of mitochondia that have underwent mitophagy (red only fluorescence). Bar – 18 μm.
5. The unbiased analysis of TXA effects on signaling pathways in endothelial cells indicates that TXA regulates biogenesis of mitochondria and mitophagy.
To assess the effects of TXA on intracellular signaling in HUVEC, we used a novel array (ActivSignal, Watertown, MA) based on a large set of antibodies against signaling proteins and their activated forms (see the full results of analysis in Supplementary Table and Figure). We found that TXA significantly suppresses the expression of protein SQSTM1 (Figure 5A), which is needed for clustering of mitochondria that precedes and enables mitophagy [Narendra et al., 2010]. Conversely, TXA enhances the phosphorylation of IGF1R (Figure 5B), which is known to stimulate the biogenesis of mitochondria [Lyons et al., 2017].
Figure 5.
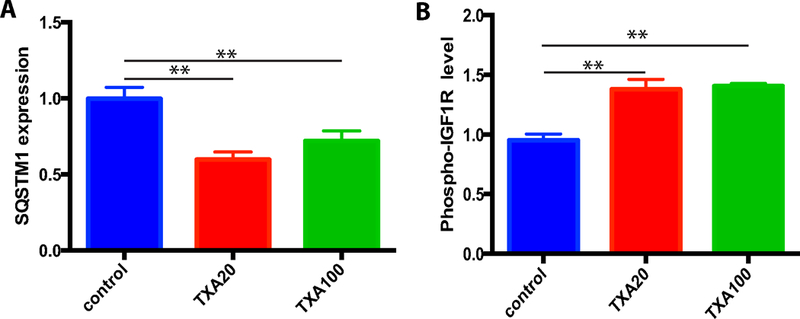
Analysis of signaling pathways in TXA-treated HUVEC indicates that TXA stimulates mitochondrial biogenesis and suppresses mitophagy. HUVEC were incubated for 24h with 0, 20 or 100 μg/ml TXA. Cell lysates were prepared and activity of signaling pathways was analyzed used the ActivSignal IPAD platform. TXA suppresses the expression of SQSTM1 (A) and enhances the phosphorylation of IGF1R (B).
Discussion
Injury and hemorrhagic shock result in a number of key cell and molecular processes that induce vascular damage. Those processes could be targeted as opportunities to understand TXA’s effects mechanistically, expanding the effectiveness of TXA and developing next-generation strategies for the management of hemorrhagic shock. In general, vascular damage involves the following critical events: (i) the release of DAMP molecules [Pandolfi et al., 2016] from damaged blood and vessel wall cells [Nie et al., 2016]; (ii) the loss of the barrier glycocalyx layer localized on the luminal membrane of ECs [Chappell et al., 2008]; (iii) a drastic increase of EC monolayer permeability followed by intensive transendothelial migration of inflammatory cells and fluids [Nagy et al., 2008]; and (iv) the enhanced production and release of proinflammatory cytokines [Portou et al., 2015]. One of the earliest signaling events following major trauma is the release of DAMPs into the circulation [Srikrishna and Freeze, 2009]. DAMPs are signaling molecules that bind to various pattern recognition receptors, such as Toll-like receptors (TLRs) and receptor for advanced glycation end products (RAGE) [Srikrishna and Freeze, 2009]. DAMPs include mitochondrial mtDNA, ATP, uric acid, HMGB1 protein and S100 family proteins [Rock and Kono, 2008; Srikrishna and Freeze, 2009]. They induce various signaling pathways converging on proinflammatory NFκB signaling and enhancing the production of proinflammatory cytokines [Srikrishna and Freeze, 2009] resulting in systemic endotheliopathy manifested by the increase of endothelial monolayer permeability, glycocalyx degradation, tissue edema and vasodilation [Srikrishna and Freeze, 2009]. These trauma-related pathologies have been shown to be exacerbated by the transfusion of crystalloid solutions commonly administered to replace lost intravascular volume [Chappell et al., 2008] in the setting of hemorrhagic shock. Protecting the integrity of the EC monolayer and EC junctions is needed to promote success of the current clinical treatment of hemorrhagic shock that consists of the administration crystalloids, colloids, blood products and vasoconstrictors.
The beneficial effects of TXA in hemorrhagic trauma patients are not limited to its anti-fibrinolytic activity; rather they include the suppression of inflammation [Levy, 2007] and overall increase of well-being [Morrison et al., 2012]. To better understand the mechanisms of TXA action, we have started to study its cellular and molecular effects in vitro using two types of cells that are important in response to hemorrhage: granulocytes and ECs. We have demonstrated that TXA strongly suppresses the release of one important DAMP (mtDNA) from granulocytes and EC. Previous in vitro studies have shown that mtDNA induces the leakiness of endothelial monolayer [Sun et al., 2013]. We have demonstrated for the first time, that exogenous mtDNA stimulates the release of endogenous mtDNA from granulocytes indicating the existence of a feed forward phenomenon of enhanced DAMP release causing the exacerbation of hemorrhage-induced endovascular pathology. Additionally, we have found that the enhancement of endogenous mtDNA release from granulocytes was abrogated by TXA in vitro. Moreover, suppression of spontaneous mtDNA release by TXA was demonstrated in both ECs and granulocytes. Together with platelets, granulocytes are a major potential source of mitochondrial DAMPs in hemorrhagic shock [Rock and Kono, 2008; Srikrishna and Freeze, 2009].
Our experiments revealed that TXA also stabilized the actin cytoskeleton and prevented the loss of adherens junctions induced by exogenous mtDNA in endothelial monolayer. These data are in agreement with the results of Diebel et al who have shown that TXA decreases the permeability of endothelial monolayers treated with hydrogen peroxide [Diebel et al., 2017a]. Interestingly, although cell-cell adherence of HUVEC was sensitive to mtDNA, the latter did not significantly enhance the release of mtDNA from these cells unlike what we had observed in granulocytes. That difference between granulocytes and EC could be due to different rates of the export of damaged mitochondria from these two cell types. Although the cell protective effects of TXA could be at least partially explained by the inhibition of the activity of extracellular and cell surface proteases [Diebel et al., 2017b], the effect upon the intracellular events remained to be determined. Using the Seahorse XF system, we found that TXA enhances the mitochondrial reserve respiratorycapacity and ATP production in HUVEC and HAEC. This observation was strengthened by the data of confocal microscopy and the unbiased analysis of signaling pathways using the ActivSignal IPAD platform which both indicated that TXA stimulated mitochondrial biogenesis and suppresses mitophagy. The specific molecular mechanisms underlying the effects TXA upon mitochondria remain to be deciphered. However, we can suggest that the stimulating effect of TXA upon mitochondrial respiration, and the suppression of mitophagy could both increase vascular cell and leukocyte survival in trauma patients and inhibit the release of mitochondrial DAMPs thus decreasing the development of inflammation.
We performed our studies in an in vitro setting where the results of TXA treatment could be evaluated without the complexity of SIRS of in vivo injury models. Accordingly, in this in vitro analysis, there are limitations as to how our results would relate to in vivo models and to the clinical setting. Any study at the in vitro stage suffers from this limitation but it may be overcome through following research endeavors. In a recently published parallel study [Carter et al., 2018], we found that TXA suppresses the release of mtDNA to blood plasma of severely burned mice and decreased the burn-induced invasion of macrophages to the lungs of these animals. In the future, we plan to evaluate our findings through in vivo studies in different systemic shock contexts, such as severe hemorrhage and sepsis, first utilizing murine and porcine animal models, and subsequently, based on our results, in the clinical setting. Understanding these mechanisms in the context of the complex response to injury will be critical to investigating the potential therapeutic range of TXA in attenuating the deleterious effects of trauma induced SIRS on endothelial barrier function.
In conclusion, our results indicate that TXA may have a potential to attenuate the effects of severe trauma by enhancing mitochondrial respiration, suppressing DAMP release and improving capillary integrity. However, to exploit this potential, a better mechanistic understanding of the effects of TXA in the hemorrhagic trauma setting is warranted.
Supplementary Material
Supplementary Figure 1. TXA suppresses the induced mtDNA release from nondifferentiated HL60 cells. Nondifferentiated HL60 granulocytes were incubated for 4 h in presence of 0.3 µg/ml TXA, released mtDNA was determined
Supplementary Figure 2. TXA enhances the spare respiratory capacity of mitochondria and ATP production in arterial EC. A) Mitochondrial Stress Test profile of HAEC revealing increased basal and maximal respiration with TXA treatment (*= P <0.05, ANOVA effect of Treatment). B) Spare respiratory capacity of cells (maximal respiration minus basal respiration) suggestive of the cells increased flexibility to meet energetic demands. C) ATP production indicative of the amount of ATP being produced by the cell through oxidative phosphorylation pathways.
Supplementary Table and Figure 3. Analysis of TXA effects upon the signaling pathways activity using the ActivSignal IPAD platform. HUVEC were incubated for 24 h with 0, 20 and 100 µg/ml TXA. The cells were lysed PBS + 1%NP40 lysis buffer, and the lysates were analyzed using the ActivSignal IPAD platform.
Acknowledgments
We are grateful to Christine Duarte for advice about statistical analysis, to Anyonya Guntur for providing mito-QC mice and to Don and Ada Olins for providing HL60/S4 cells.
Funding: The study was funded by the intramural MMCRI support, a pilot research grant from the Maine Medical Center Cardiovascular Institute to RK, JR and IP, and a research grant from the American Association for the Surgery of Trauma (AAST) to DC. Support was also provided by the Molecular Phenotyping Core of MMCRI (funded by COBRE NIGMS P30GM106391) and the Confocal Imaging Core of MMCRI (funded by COBRE NIGMS P30GM103391).
Footnotes
No conflict of interest to declare.
References
- Birnie GD. 1988. The HL60 cell line: a model system for studying human myeloid cell differentiation. Br J Cancer Suppl 9:41–5. [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Lovell MA, Gorbsky GJ. 1995. Stability of nuclear segments in human neutrophils and evidence against a role for microfilaments or microtubules in their genesis during differentiation of HL60 myelocytes. J Leukoc Biol 58:659–66. [DOI] [PubMed] [Google Scholar]
- Carter DW, Prudovsky I, Kacer D, Soul T, Kumpel C, Pyburn K, Palmeri M, Kramer R, Rappold J. 2018. Tranexamic Acid Suppresses the Release of Mitochondrial Damps and Reduces Lung Inflammation in a Murine Burn Model. J Trauma Acute Care Surg [DOI] [PMC free article] [PubMed]
- Chappell D, Jacob M, Hofmann-Kiefer K, Conzen P, Rehm M. 2008. A rational approach to perioperative fluid management. Anesthesiology 109:723–40. [DOI] [PubMed] [Google Scholar]
- collaborators C-t,Shakur H, Roberts I, Bautista R, Caballero J, Coats T, Dewan Y, El-Sayed H, Gogichaishvili T, Gupta S, Herrera J, Hunt B, Iribhogbe P, Izurieta M, Khamis H, Komolafe E, Marrero MA, Mejia-Mantilla J, Miranda J, Morales C, Olaomi O, Olldashi F, Perel P, Peto R, Ramana PV, Ravi RR, Yutthakasemsunt S. 2010. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 376:23–32. [DOI] [PubMed] [Google Scholar]
- Diebel LN, Martin JV, Liberati DM. 2017a. Early tranexamic acid administration ameliorates the endotheliopathy of trauma and shock in an in vitro model. J Trauma Acute Care Surg 82:1080–1086. [DOI] [PubMed] [Google Scholar]
- Diebel ME, Martin JV, Liberati DM, Diebel LN. 2017b. The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J Trauma Acute Care Surg [DOI] [PubMed]
- Farmer T, Naslavsky N, Caplan S. 2018. Tying trafficking to fusion and fission at the mighty mitochondria. Traffic 19:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover GJ, Menhart N, Martin A, Warder S, Castellino FJ. 1993. Amino acids of the recombinant kringle 1 domain of human plasminogen that stabilize its interaction with omega-amino acids. Biochemistry 32:10936–43. [DOI] [PubMed] [Google Scholar]
- Jennings JD, Solarz MK, Haydel C. 2016. Application of Tranexamic Acid in Trauma and Orthopedic Surgery. Orthop Clin North Am 47:137–43. [DOI] [PubMed] [Google Scholar]
- Levy JH. 2007. Anti-inflammatory strategies and hemostatic agents: old drugs, new ideas. Hematol Oncol Clin North Am 21:89–101. [DOI] [PubMed] [Google Scholar]
- Lyons A, Coleman M, Riis S, Favre C, O’Flanagan CH, Zhdanov AV, Papkovsky DB, Hursting SD, O’Connor R. 2017. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J Biol Chem 292:16983–16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG. 2016. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214:333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monceaux V, Chiche-Lapierre C, Chaput C, Witko-Sarsat V, Prevost MC, Taylor CT, Ungeheuer MN, Sansonetti PJ, Marteyn BS. 2016. Anoxia and glucose supplementation preserve neutrophil viability and function. Blood 128:993–1002. [DOI] [PubMed] [Google Scholar]
- Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. 2012. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) Study. Arch Surg 147:113–9. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. 2008. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11:109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. 2010. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney PA. 2015. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 1853:2775–83. [DOI] [PubMed] [Google Scholar]
- Ng W, Jerath A, Wasowicz M. 2015. Tranexamic acid: a clinical review. Anaesthesiol Intensive Ther 47:339–50. [DOI] [PubMed] [Google Scholar]
- Nie Y, Yang, Oppenheim JJ. 2016. Alarmins and Antitumor Immunity. Clin Ther [DOI] [PMC free article] [PubMed]
- Pandolfi F, Altamura S, Frosali S, Conti P. 2016. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin Ther [DOI] [PubMed]
- Peng Z, Ban K, LeBlanc A, Kozar RA. 2016. Intraluminal tranexamic acid inhibits intestinal sheddases and mitigates gut and lung injury and inflammation in a rodent model of hemorrhagic shock. J Trauma Acute Care Surg 81:358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portou MJ, Baker D, Abraham D, Tsui J. 2015. The innate immune system, toll-like receptors and dermal wound healing: A review. Vascul Pharmacol 71:31–6. [DOI] [PubMed] [Google Scholar]
- Rappold JF, Bochicchio GV. 2016. Surgical adjuncts to noncompressible torso hemorrhage as tools for patient blood management. Transfusion 56 Suppl 2:S203–7. [DOI] [PubMed] [Google Scholar]
- Rock KL, Kono H. 2008. The inflammatory response to cell death. Annu Rev Pathol 3:99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, Meyer JN. 2015. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol 1241:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovera G, Santoli D, Damsky C. 1979. Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc Natl Acad Sci U S A 76:2779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonazzi G, Bisulli M, Saccone G, Moro E, Marshall A, Berghella V. 2016. Tranexamic acid for preventing postpartum blood loss after cesarean delivery: a systematic review and meta-analysis of randomized controlled trials. Acta Obstet Gynecol Scand 95:28–37. [DOI] [PubMed] [Google Scholar]
- Sobczak M, Dargatz J, Chrzanowska-Wodnicka M. 2010. Isolation and culture of pulmonary endothelial cells from neonatal mice. J Vis Exp [DOI] [PMC free article] [PubMed]
- Srikrishna G, Freeze HH. 2009. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11:615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. 2013. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 8:e59989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Elzen M, Go MF, Knulst AC, Blankestijn MA, van Os-Medendorp H, Otten HG. 2016. Efficacy of Treatment of Non-hereditary Angioedema. Clin Rev Allergy Immunol [DOI] [PMC free article] [PubMed]
- Van Way CW 3rd, Dhar A, Morrison DC, Longorio MA, Maxfield DM. 2003. Cellular energetics in hemorrhagic shock: restoring adenosine triphosphate to the cells. J Trauma 54:S169–76. [DOI] [PubMed] [Google Scholar]
- Woodall BP, Gustafsson AB. 2018. Autophagy-A key pathway for cardiac health and longevity. Acta Physiol (Oxf) 223:e13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. 2010. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. TXA suppresses the induced mtDNA release from nondifferentiated HL60 cells. Nondifferentiated HL60 granulocytes were incubated for 4 h in presence of 0.3 µg/ml TXA, released mtDNA was determined
Supplementary Figure 2. TXA enhances the spare respiratory capacity of mitochondria and ATP production in arterial EC. A) Mitochondrial Stress Test profile of HAEC revealing increased basal and maximal respiration with TXA treatment (*= P <0.05, ANOVA effect of Treatment). B) Spare respiratory capacity of cells (maximal respiration minus basal respiration) suggestive of the cells increased flexibility to meet energetic demands. C) ATP production indicative of the amount of ATP being produced by the cell through oxidative phosphorylation pathways.
Supplementary Table and Figure 3. Analysis of TXA effects upon the signaling pathways activity using the ActivSignal IPAD platform. HUVEC were incubated for 24 h with 0, 20 and 100 µg/ml TXA. The cells were lysed PBS + 1%NP40 lysis buffer, and the lysates were analyzed using the ActivSignal IPAD platform.