

SUMMARY
Directed evolution, artificial selection toward designed objectives, is routinely used to develop new molecular tools and therapeutics. Successful directed molecular evolution campaigns repeatedly test diverse sequences with a designed selective pressure. Unicellular organisms and their viral pathogens are exceptional for this purpose and have been used for decades. However, many desirable targets of directed evolution perform poorly or unnaturally in unicellular backgrounds. Here we present the first system for facile directed evolution in mammalian cells. Using the RNA alphavirus Sindbis as a vector for heredity and diversity, we achieved 24-hour selection cycles surpassing 10−3 mutations/base. Selection is achieved through genetically actuated sequences internal to the host cell, thus the systems name: Viral Evolution of Genetically Actuating Sequences or ‘VEGAS’. Using VEGAS, we evolve transcription factors, GPCRs, and allosteric nanobodies toward functional signaling end-points each in under one week’s time.
Keywords: mammalian directed evolution, artificial selection, RNA virus, transcription factor, G-protein coupled receptors, molecular pharmacology
Graphical Abstract
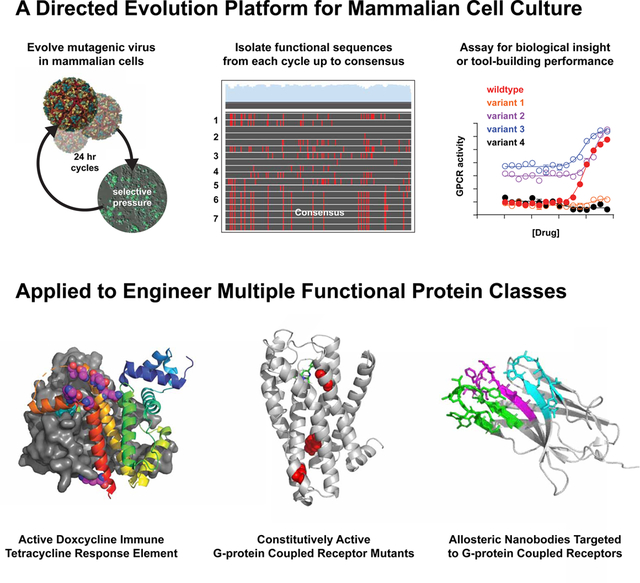
The VEGAS system is a platform for directed evolution, a method for engineering DNA sequences, in mammalian cells. The system is highly mutagenic, facile and self-contained, requiring no in vitro handling during evolution cycles. As a result, robust evolution campaigns can be run within the context of a mammalian cell signaling environment. We perform 3 such campaigns as a proof-of-concept -evolving a transcription factor, a G-protein coupled receptor, and llama-derived nanobodies toward specific in vivo activities.
INTRODUCTION
Spontaneous genetic mutations diversify traits among a population of organisms while selective pressure culls diverse populations. This enrichment of ultimately advantageous traits is a process known as evolution by means of natural selection (Darwin and Bynum, 2009; Wallace, 1855, 1871). Humans can accelerate the development of organisms with desirable traits by guiding evolution through artificial selection. This technique can be traced back to the earliest agricultural crops (Diamond, 2002; Wright et al., 2005) and domesticated animals (Driscoll et al., 2009). As a biomedical laboratory tool, artificial selection has been instrumental in understanding myriad processes ranging from the cell cycle (Hartwell et al., 1970) to bacterial antibiotic resistance (Albert et al., 2005; Baym et al., 2016; Toprak et al., 2012). Artificial selection of targeted DNA sequences, rather than whole organisms, is called directed evolution (Arnold, 1998; Chen and Arnold, 1993). Directed evolution has been used to create novel binding proteins (Hanes and Plückthun, 1997; Xu et al., 2002), enzymes (Chen and Arnold, 1993; Kuchner and Arnold, 1997), chemogenetic tools (Armbruster et al., 2007), and fluorescent reporters (Campbell et al., 2002; Crameri et al., 1996) with broad scientific and industrial utility.
Directed evolution approaches typically use peptide display or microorganisms to screen large-scale DNA libraries that encode mutant proteins. ‘Hits’ from these systems are isolated, mutagenized, and rescreened in an interrupted or iterative fashion. Iterative systems minimize evolution cycle time and omit user-biased “winner” selection by combining mutagenesis, selection, and heredity in parallel. Iterative systems have been improved using uninterrupted facile (McMahon et al., 2018) and continuous methods (Badran and Liu, 2015; Carlson et al., 2014; Esvelt et al., 2011). Although both methods have produced excellent results, these systems have been developed outside the context of the mammalian cell signaling environment. Consequently, incompatibility of function when transferring evolved products from unicellular to mammalian systems frequently occurs (see Armbruster et al., 2007 for examples), wherein additional rounds of selection and focused mutagenesis must be performed. Additionally, the currently available directed evolution systems omit classes of proteins that are usually incompatible with non-mammalian host systems – including G-protein coupled receptors (GPCRs).
GPCRs comprise one of the largest protein families in the human genome with greater than 900 unique protein coding genes (Fredriksson et al., 2003; Wacker et al., 2017a). GPCRs represent the largest class of druggable targets and are known to regulate most biological processes (Hauser et al., 2017). Despite their importance, GPCRs are largely omitted from directed evolution studies due to their functional incompatibilities with non- mammalian systems (although see Armbruster et al., 2007; Sarkar et al., 2008; Schütz et al., 2016). GPCRs are seven-transmembrane receptors that transduce extracellular signals into biological responses via heterotrimeric G proteins and ȕ-arrestins (Gilman, 1987; Pierce et al., 2002). GPCR signal transduction is accomplished via a network of interacting molecular switches (Wacker et al., 2017a), yielding an isomerizing landscape of conformations that evoke unique cellular signaling cascades (De Lean et al., 1980; Kobilka and Deupi, 2007; Onaran and Costa, 2009). GPCR-directed pharmaceuticals stabilize subsets of this signaling landscape leading to stabilization of active (e.g. agonism) or inactive (Neubig, 2003; Wacker et al., 2017a) states. A directed evolution system capable of targeting these states, and the signaling pathways downstream of such targets, could provide key insights necessary to advance cell signaling biology and drug development.
Here we present a system for the Viral Evolution of Genetically Actuating Sequences, we dub ‘VEGAS’, using a facile directed evolution platform in mammalian cells. Using the RNA alphavirus Sindbis for parallel mutagenesis, selection, and heredity we demonstrate the robust, directed, and functional evolution of both GPCRs and allosteric GPCR intrabodies in mammalian cell culture in under one week.
RESULTS
Sindbis Virus for Directed Evolution in Mammalian Cell Culture
Mammalian cell-based directed evolution has had many successes (Armbruster et al., 2007; Buchholz et al., 1998; Chan et al., 2017; Hess et al., 2016; Maheshri et al., 2006), albeit using time consuming, costly, and specialized screening platforms. The use of engineered viruses has advanced the field, serving as vectors for library storage, delivery, and heredity; the use of viruses, however, has been limited to conventional iterative systems involving panning, ‘winner-picking’, and ex vivo mutagenesis. Such iterative directed evolution approaches sabotage the powerful evolutionary principles at play in competitive genetic populations (Huston, 1979). Here we aimed to develop a mammalian directed evolution system where viral mutagenesis, selection, and heredity could operate simultaneously.
We required a mutagenic virus that could replicate freely at titers sufficient for constant reinfection of naïve cells in culture and for this we focused on obligate RNA viruses, the most mutagenic viral class (Drake and Holland, 1999). Due to concerns related to laboratory safety and utility only a handful of RNA viruses are feasible for routine use. Of those available, we focused our efforts on the Alphavirus Sindbis, from the Togaviridae family (Strauss et al., 1984; Xiong et al., 1989). Sindbis virus is a single stranded RNA virus encoding an RNA-dependent RNA replicase targeted to the viral genome by cis-acting, conserved 5–3’ sequences (Frolov et al., 2001). These sequences are required to initiate replication and RNA templates, even those from related viral families, cannot be replicated by the Sindbis virus replicase, resulting in high selectivity between the replicase and the Sindbis virus genome (Frolov et al., 2001), which functions simultaneously as a replication template and coding strand for viral protein translation. Sindbis virus has been engineered as a transgene delivery vector (Agapov et al., 1998; Schlesinger, 1993; Strauss and Strauss, 1994; Xiong et al., 1989); here we further engineered Sindbis virus to control the packaging process using mammalian expression vectors.
We first determined that Sindbis virus can be continuously packaged in mammalian cell culture using an expression plasmid encoding the Sindbis virus structural genome (Figure 1A, also refer to VEGAS Supplemental Resource and VEGAS Extended Protocol for additional details). Transgenic Sindbis virus plasmid harboring green fluorescent protein (pTSin-EGFP, see Methods) was packaged and titered at 5.45×1011 genomes/mL (Figure 1B) as determined by qRT-PCR targeting the Sindbis virus packaging signal sequence (see Methods). This initial titer was applied to 1×107 cells transfected with pCMV-SSG (Sindbis structural genome, see methods) at an MOI (multiplicity of infection) of 1. Harvesting and subsequent analysis of the culture media from these cells revealed high viral titer production, with 6.64×108 genomes/mL produced after 4 hrs and 5.57×1010 genomes/mL produced after 24 hrs (Figure 1B). The 24 hr sample from round 1 was transferred to naïve cells transfected with or without pCMV-SSG at an MOI=1. After 24-hrs pCMV-SSG transfected cells produced 6.37×108 genomes/mL while untransfected control cells produced 2.6×105 genomes/mL (Figure 1B). Fluorescent imaging of the infected culture over time confirmed passage of the EGFP transgene (Figure S1A). Transgene expression is rapid, with EGFP detectable in as few as 4 hrs post infection. These experiments demonstrate that Sindbis virus can be used for sustained transgene packaging in mammalian cell culture using a plasmid- borne structural genome.
Figure 1. Sindbis Virus for Facile Directed Evolution in Mammalian Cell Culture.

Development of Sindbis virus for facile, mutagenic viral propagation in mammalian cell culture. A. Design of plasmids used for facile directed evolution with Sindbis virus. Artificial Sindbis genome; Girdwood, MF459683.1. pSSG plasmid; capsid, E3, E2, E1 and 3’ UTR moved to a mammalian expression vector. pTSin plasmid; The structural genome elements of the artificial Sindbis genome replaced by any transgene sequence (pTSin). Propagation and selection can then be performed in mammalian cell culture using pTSin packaged virus applied to cells transfected with pSSG. B. qRT-PCR quantification of Sindbis virus production from cell culture. Data are represented as mean of individual biological replicates, N>3. C. Mutations observed from Illumina paired-end sequencing of Sindbis packaged EGFP transgene over time. Mutation frequency is plotted as mutations observed per read at each nucleotide position across the transgene. Data are plotted for each individual replicate (N=3; 24HR and VECTOR, N=2) around mean ± 95% confidence interval. D. Base changes observed from sequencing of Sindbis packaged EGFP transgene over time. A, adenine, T, thymine, G, guanine, C, cytosine. Statistical comparison tested within base groups between each time point. E. Calculation of Sindbis mutation rate from sequencing of Sindbis packaged EGFP transgene over time. Data are represented as mean ± SEM and as linear regression, dotted line highlights the 99% confidence interval band. See also Figure S1.
RNA viruses, such as Sindbis, are highly mutagenic, with no known proof-reading capability. Approximations of RNA virus mutation rates range from 10−5 to 10−3 mutations/base replicated (Drake et al., 1998; Morley and Turner, 2017; Sanjuan et al., 2010; Schnell et al., 1996; Strauss and Strauss, 1994). As no prior study quantified the genetic stability of a non-essential transgenic gene during Sindbis virus replication, we next determined the mutation frequency of our directed evolution system. We initiated packaging of pTSin-EGFP in pCMV-SSG transfected cells and collected supernatant after 3, 6, 12, 24, and 36 hrs. The EGFP transgenic segment, as well as the vector template and initially packaged RNA, was amplified and sequenced using an Illumina NextSeq500. The sequences were aligned (see Methods) and quantified for positional sequence integrity of EGFP (Figure S1B–C). A significant (p<0.0001, see Methods) time-dependent increase in average mutation frequency was observed when comparing the 0HR (initial RNA) sample vs. the 6, 12, 24, and 36 hour samples (Figure 1C). The number of observed insertions and deletions also increased with time (Figure S1D). Nucleotide substitution rates was relatively even, with the exception of a modest (p<0.05) C>G preference in samples 6–36 hours (Figure 1D). Linear regression analysis of mutation frequency vs. time (Figure 1E) yielded an estimate of 1.0×10−4 ± 3.7×10−5 mutations base−1/hour -- approximately 1 mutation per 1000 bases replicated. However, viral propagation accumulates, rather than evenly distributes, mutations. We postulated that a sub-population of genomes with high mutation density should appear over time. These subpopulations were identified by quantifying the number of mutations observed per individual read. Our analysis revealed a time-dependent increase in total mutation (Figure S1E), insertion (Figure S1F), and deletion (Figure S1G) density per read. This high mutation rate, coupled to accumulating insertions and deletions infrequently accessible to rational design platforms, makes Sindbis virus an ideal candidate for developing a mammalian directed evolution platform.
Directed Evolution of Transcription Factors with Sindbis Virus
To yield a robust directed evolution platform which leverages the replicative and mutagenic potential of Sindbis virus, artificial selective pressure must be applied. Each Sindbis viral particle requires 240 copies of each of the structural proteins E1, E2, and capsid to form a functional viral particle that can mature and propagate (Tang et al., 2011) and without this envelope the virus is unable to mature and propagate. By engineering restrictions on structural genome transcription we developed a system to apply selective pressure on transgenic Sindbis virus. As proof of concept for this method we placed the Sindbis virus structural genome under control of the tetracycline operator sequence (Das et al., 2004; Gossen et al., 1995; Orth et al., 2000) (pTETO7-SSG, see methods) and packaged transgenic Sindbis virus with tetracycline transactivator (pTSin-tTA, Gossen and Bujard, 1992, see methods). We infected cells +/− TETO7-SSG with viral pTSin-tTA or pTSin-EGFP and then treated cells with either the tTA inhibitor doxycycline (DOX, 1μM) or vehicle at the time of infection. Virus was packaged at 3.53×1010 genomes/mL in the vehicle+TETO7-SSG cell line, while < 106 genomes/mL were detected for all other conditions (Figure S2A).
Using the TS-tTA system we sought to benchmark the capabilities of VEGAS by evolving tTA to be doxycycline insensitive. To accomplish this, we packaged TS-tTA virus under non-selective conditions (R0) and exposed it to constant rounds of selection using increasing concentrations of doxycycline (Figure 2A). Seven selection rounds, encompassing just 7 days of evolution, produced a large number of full length tTA sequences (see Table S1). By Round 6 a consensus sequence dominated the observed coding sequence pool that was carried through to Round 7. This consensus sequence, dubbed “R7”, was completely resistant to DOX (Figure 2B). R7 possessed twenty-two coding mutations spanning all functional domains of the protein (Figure 2C). We had predicted that mutations directly involved in ligand interaction (see Figure 2C–D, Kisker et al., 1995; Orth et al., 1999a, 1999b) would be enriched in the final consensus. To our surprise, none of these residues were mutated in R7. Instead, mutations accumulated primarily adjacent to key interacting residues for each functional domain (Figure 2C, S2B), many of which have been previously identified to reduce the effect of DOX on TETR-TETO interaction (Berens et al., 1992; Hecht et al., 1993; Müller et al., 1995; Orth et al., 1998; Scholz et al., 2004; Schubert et al., 2001; Smith and Bertrand, 1988; Urlinger et al., 2000; Wissmann et al., 1991, see Figure 2C and Table S2 for details). In addition, a cluster of negatively charged residues comprising helices 8 and 9 residing over the ligand binding pocket spanning Q149-H179 (Figure 2D) were converted to primarily positively charged residues (Figure 2C, Table S2). The net charge, but not specific residues of this loop, are conserved across TETR variants and this conserved charge landscape has been proposed to attract the tetracycline-Mg2+ inducer to the ligand binding pocket (Orth et al., 1998). The mutations observed in R7 increase the net charge of this loop by +3.19, concentrated near the ligand entry tunnel. This gain in local charge presumably repels the positively charged doxycycline-Mg2+.
Figure 2. Directed Evolution of Transcription Factors with Sindbis.

Sindbis was used as a directed evolution platform to generate a Doxycycline-resistant variant of the transcription factor tTA. A. Nucleotide sequence alignment of TETR clones isolated from each round of selection to the wildtype TETR(B) sequence. Each round is outlined in yellow or blue, applied concentration of Doxycycline (DOX) to the left, name of individual clones to the right. Gray DOX values indicate no clones were isolated from the round. Red lines in the alignment denote a sequence mismatch from wildtype. B. TETO7-Rluc reporter assay with increasing concentrations of Doxycycline (DOX). Dotted lines are selection round DOX concentrations, for reference. Data are represented as mean ± SEM of individual biological replicates. C. Peptide sequence alignment of TETR(B) and the R7 consensus. Matching residues are shaded, mutations are unshaded. Alpha helices (α) are labeled and color coded to match with palettes in Figures 2D and S2B. Exact residue (Δ), position (@), or subtype (D) substitutions previously published to enhance tTA activity in the presence of Doxycycline as per Table S2. Residues (*) with direct involvement in DNA binding (green), ligand binding (magneta), and ligand entry (cyan) as per (Orth et al., 2000; Schubert et al., 2004) D. Crystal structure PDB 4AC0 of TETR(B) in complex with Minocycline-Mg2+. Helix 8–9 ligand enclosure spanning Q149-H179 is displayed with spheres highlighting the residues for mutations Q149R, Q152R, K155R, R158G, T160A (no density), D178G, and H197R observed in R7. See also Figure S2, Table S1, and Table S2.
Interestingly, in addition to augmenting the peptide sequence through directed evolution our analysis of the nucleotide sequences from each round revealed codon usage optimizations as well (Figure S2C–D). Non-synonymous mutations acquired through tTA evolution converted rarely used codon sequences for BHK21, derived from Mesocricetus auratus, to the more frequently used GAC (D, +13%), GAG (E, +29%), AAG (K, +43%), TTG (L, 30%), and CAG (Q, +19%).
Augmenting TETR ligand sensitivity has been attempted previously using mammalian directed evolution (Das et al., 2004), wherein 2 mutations were identified in 114 days. Our evolution of tTA generated an order of magnitude more functional mutations in 7 days thereby illustrating how our Sindbis virus system can be used for successful directed evolution of a transcription factor in mammalian cell culture. Key to the evolutionary component of this method is the actuation of a genetically encoded circuit to unlock expression of the Sindbis structural proteins, capsid, E1, and E2. Consequently, we gave the Sindbis virus system the name ‘VEGAS’ for Viral Evolution of Genetically Actuating Sequences.
VEGAS for the Evolution of GPCRs
With VEGAS in hand to perform directed evolution in mammalian cells we focused our efforts on GPCRs, a superfamily of transmembrane receptors with substantial pharmacological and physiological importance (Hauser et al., 2017; Wacker et al., 2017a). Critical to the GPCR field is the mapping of interacting residues associated with the transition from an inactive to active receptor. Mapping these motifs can provide anchor points for homology modeling, evolutionary sequence analysis, and ligand design (Fan et al., 2009; Michino et al., 2015; Roth et al., 2017). Even among the best studied receptors, using extensive mutation campaigns and high resolution crystal structures of inactive and active receptor conformations, the field has struggled to consistently identify key residues involved in state transition (Dror et al., 2011; Huang et al., 2015a; Latorraca et al., 2017). Class A GPCRs possess conserved binding pockets and trigger motif residues involved in the inactive to active state transition. However, many Class A GPCRs lack conservation within these motifs, a disproportionate number of which are classified as understudied or orphan receptors (Figure S3A). Here we used VEGAS to identify previously unknown constitutively active mutations for the understudied receptor MRGPRX2; our approach demonstrates how VEGAS can illuminate the complex conformational changes involved in GPCR activation even in the absence of structural information.
MRGPRX2 is a primate-exclusive GPCR recently identified as an atypical opioid- recognition receptor (Lansu et al., 2017). MRGPRX2 possesses limited homology to other opiate receptors (see Table S3) and minimal conservation of classic interacting residues. The curious composition of MRGPRX2 hampers de novo prediction of functional motifs. We therefore used VEGAS to develop constitutively active mutations (CAMs) of MRGPRX2. As VEGAS requires an activity-coupled transcriptional response to gate selection, we screened MRGPRX2 activated by (+)-morphine, across a panel of transcription factor reporters driving luciferase (luc2P, FLuc) expression (Figure S3B). The serum response element (SRE) and serum response factor (SRF) minimal promoters gave 5-fold responses 4 hrs post-ligand addition and persisted for 24 hrs. We chose SRE for its lower total basal signal in unstimulated conditions and replaced luc2P with the Sindbis virus structural genome (SRE-SSG). Cells transfected with SRE-SSG were infected with transgenic Sindbis virus harboring MRGPRX2 (pTSin-MRGPRX2) and selected with diminishing amounts of (+)-morphine over 3 days (Figure 3). Resultant viral genomes were isolated and their MRGPRX2 transgenes were tested in subsequent assays.
Figure 3. VEGAS for the Evolution of GPCRs.

Using VEGAS multiple constitutively active mutants of the GPCR MRGPRX2 were produced in 3 days through application of decreasing concentrations of the MRGPRX2 agonist (+)-Morphine. Mutations acquired in each round were tested functionally. Mutations are listed with their receptor residue position and Ballesteros-Weinstein annotation. A. Serum response element (SRE) reporter assay. RLuc production equates to relative receptor activation. ND, no drug. Data are represented as mean ± SEM, N=3. B. TANGO reporter assay. RLuc production equates to receptor-mediated β-arrestin2 activation. ND, no drug. Data are represented as mean ± SEM, N=3. C. Phosphoinositide hydrolysis assay. Accumulation of [3H] inositol equates to receptor-mediated Gαq activation. ND, no drug. Data are represented as mean ± SEM, N=3. See also Figure S3.
We presumed these clones would be CAMs, however none of the isolated mutants mapped to classic sites of constitutive activity modulation identified in other GPCRs (Figure S3C). We screened each mutant for activity in SRE-luc2P (Figure 3A), ȕ - arrestin recruitment (Figure 3B), and phosphoinositide (PI) hydrolysis (Figure 3C) functional assays. We also quantified surface receptor expression via ELISA to ensure proper trafficking and expression (Figure S3D). For SRE and TANGO assays, basal activity across the variants increased at each evolutionary generation. TANGO basal activity reached 100% of wild type (+)-morphine stimulation for three independent mutants: L210P, Y67H+L210P+V265A, and L42P+D306G. PI hydrolysis, a proxy for GĮq activity, detected decreased basal activity, ligand potency, and efficacy for all mutants. Decreases in maximum agonist-induced GĮq activity correlated with increases in constitutive TANGO and SRE activity.
VEGAS for the Evolution of Active-State Nanobodies
GPCR ligands stabilize signal-state specific receptor conformations (Kobilka and Deupi, 2007; Onaran and Costa, 2009; Strachan et al., 2014) and the development of novel ligands is enhanced by signal-state specific GPCR crystal structures (Che et al., 2018; Manglik et al., 2016; Wang et al., 2017). Nanobodies, genetically encodable antigen recognition domains from dromedaries (Muyldermans et al., 2001), can be used to obtain these stabilized active state structures (Che et al., 2018; Manglik et al., 2016; Rasmussen et al., 2007; Staus et al., 2016). The nanobodies developed in these studies mimic the GĮ protein, displacing it. More desirable would be a nanobody which stabilizes the complex between GPCR and its transducer GĮ protein. These nanobodies would be allosteric modulators capable of enhancing GPCR-GĮ coupling. Using VEGAS we create allosteric nanobodies for multiple GPCR-GĮ pairings in less than a week.
To create GPCR nanobodies using VEGAS we first generated a GPCR-targeted nanobody library by immunizing a llama against the serotonin 2A (5-HT2A) GPCR bound to the high affinity agonist lysergic acid diethylamide (LSD). We isolated single- domain antibodies from the peripheral blood mononuclear cells of the immunized llama through amplification of the variable region “VHH” of IgG (Pardon et al., 2014, see Methods). The VHH amplicon was used to generate a cDNA library of approximately 1×107 colonies, which was subsequently packaged in Sindbis virus. This library was then used to evolve intracellular targeting nanobodies against 5-HT2A, as well as the dopamine-D2 (DRD2) and pH-sensing GPR68 (Huang et al., 2015b) receptors. Each of these receptors couples canonically to a different GĮ protein (2A, GĮq; D2, GĮi, 68, GĮs). Developing nanobodies toward each receptor serves to demonstrate the broad applicability of the VEGAS system.
To evolve active state-stabilizing nanobodies for each GPCR we first screened each receptor for transcription factor coupling (Figure S4A–F). All three receptors were determined to activate SRE with varying efficacy and we therefore chose to develop nanobodies that engage the SRE-signaling state for each receptor. Cells transfected with SRE-SSG and a GPCR were infected with an MOI=1 of the viral nanobody library. To select for SRE activating nanobodies 5-HT2A and DRD2 cultures were incubated in the absence of ligand, while GPR68 was incubated at its inactive pH 8 (Huang et al., 2015b). Day 1 viral particles were harvested, the selection was repeated, and individual nanobody clones were isolated from the Day 1 and 2 titers and sequenced. Clones with N>2 identity in the subcloned population were selected and screened for GPCR- dependent SRE activation (Figure 4B). Each evolution series produced nanobodies capable of SRE activation only in the presence of the intended GPCR target, with the exception of VGS-Nb1 which appears to constitutively activate SRE even under GPCR- free conditions. To determine if the nanobodies obtained using VEGAS came directly from the originating library, or were evolved, we deep-sequenced the clonal library using a NextSeq500 (see methods). Stringent end-to-end alignment of the entire sequence pool (total reads equaled 20 million) was assessed over 3 independent score cut-offs. Reads aligning to the VEGAS derived nanobodies are displayed in Figure 4A. Each VEGAS derived nanobody possessed sequences that were not detected within or outside the complementarity-determining regions (CDRs). In addition, we compared reference nanobodies (REF_Nbs) cloned from the parent library and VEGAS isolates to the amino acid frequency distribution of 1,346 deposited Llama glama VhH sequences from >50 animals (Table S4). Five positions with >99% sequence conservation across populations were conserved in the REF_Nb sequences, but were mutated in the VEGAS evolved sequences (Figure 4C). Both sequence analysis methods demonstrate that the VEGAS-derived nanobodies were not original to the library but evolved from that initial pool of nanobody cDNAs.
Figure 4. VEGAS for Evolution of Active-State Nanobodies.

VEGAS was used to develop nanobodies that selectively activate diverse GPCR targets from a single cDNA library. A. Deep sequencing of the nanobody cDNA library used for VEGAS. 20 million reads were aligned to VEGAS derived clones and plotted as % mismatch. Data was analyzed with score cut-offs (CO) of 25, 50, and 100 (see methods). Grey blocks are gaps in alignment as per Figure 4C. Black blocks are regions with mapped reads < 2000 counts (< 0.0001%). Symbols ^ and * mirror those on Figure 4C. Bottom histogram, percent total mapped reads for each alignment. B. Serum-response element (SRE) reporter assay. Nanobody:receptor:reporter transfection ratio of 5:1:1. RLuc production equates to relative receptor activation. Data are represented as mean ± SEM, N=3. C. Amino acid sequence alignment of library (REF_NB#) and VEGAS derived clones. Shading; 100% (red), > 75% (yellow), < 75% (white). Variations identified in VEGAS, but not reference sequence, derived clones at positions of high genetic conservation (see Table S4) are annotated, ^ >99% conserved, * >95% conserved. Nanobody secondary structure annotated above, retrieved from PDB 3P0G, Chain B. Complementarity determining regions (CDRs) annotated below. β, beta sheet. TT, strict β-turn. See also Figure S4.
Positive Allosteric Modulation of GPCRs by VEGAS-Evolved Nanobodies
Using VEGAS, we produced 8 nanobodies targeted against 3 independent GPCRs in under one week. Here we interrogate their physical and molecular interactions with each target and provide a detailed characterization of the mechanism of VGS-Nb2, a positive allosteric modulator of the 5-HT2A serotonin receptor.
First, we established whether the VEGAS-evolved nanobodies directly associated with their intended GPCR targets via bioluminescence resonance energy transfer (BRET). For BRET, GPCR-RLuc fusions and increasing concentrations of mVenus nanobody (mVenus-Nb) fusion proteins were co-transfected in to HEK293T cells. We observed a strong association between mVenus-VGS-Nb2 and 5-HT2A-RLuc, but no association to the closely related serotonin 2B (5-HT2B) receptor (Figure 5A). Addition of the agonist serotonin (5-HT) at 1μM or above (Figure 5A and S5A) had no effect on VGS-Nb2 association to either 5-HT2A or 5-HT2B. We also observed association between mVenus-VGS-Nb6 and GPR68-RLuc at pH 8, but no association of the nanobody to 5- HT2A, the protein used to develop the initial nanobody library for directed evolution (Figure 5B). GPR68 activity increases with increasing pH (Huang et al., 2015b), we therefore stimulated our BRET assay with a pH 6 buffer and observed an increased association between mVenus-VGS-Nb6 and GPR68-RLuc (Figure 5B). Low, non- specific interaction of the DRD2 targeted nanobodies VGS-Nb7 and VGS-Nb8 was also observed (Figure S5C and S5D). However, VGS-Nb7 and VGS-Nb8 both increase SRE activity in the presence of DRD2 (Figure S5J), through an unknown mechanism.
Figure 5. Positive Allosteric Modulation of GPCRs by VEGAS-Evolved Nanobodies.

VEGAS derived nanobodies were tested for direct association and allosteric modulation of their targets. A. Bioluminescence resonance energy transfer (BRET) association assay between 5HT2A-RLuc or 5HT2B-RLuc and mVenus-VGS- Nb2 at increasing transfection ratios of nanobody. Data are represented as mean ± SEM, N=3. Symbols for 5HT2B-Rluc data underlie those for the +1uM 5HT data. B. Bioluminescence resonance energy transfer (BRET) association assay between GPR68-RLuc or 5HT2A-RLuc and mVenus-VGS-Nb6. Data are represented as mean ± SEM, N=3. C. Serum response element (SRE) reporter assay. RLuc production equates to relative receptor activation. Data are represented as mean ± SEM, N=3. D. Saturation radioligand binding assay. 5HT2A-Gaq membrane treated with vehicle or 5uM VGS- Nb2. 5HT2A; Kd = 0.30 nM, BMax = 1333 fmol/mg. 5HT2A+VGS-Nb2; Kd = 0.566 nM, BMax = 1993fmol/mg. Data are represented as mean ± SEM, N=3, * P<0.05. E. Competitive radioligand binding assay. 5HT2A and 5HT2A-Gaq membrane treated with vehicle or 7.5μM VGS-Nb2 labeled with 1nM [3H] Ketanserin and increasing concentrations of DOI. Data are represented as total-count normalized means ± SEM, N=3. See also Figure S5.
The serotonin 2A (HTR2A, 5-HT2A) receptor is a GPCR of significant importance to mental health, disease, pharmacology, and homeostatic biology (McCorvy and Roth, 2015). Structures of 5-HT2A and closely related 5-HT2-family receptors 5-HT2B (Wacker et al., 2013) and 5-HT2C (Peng et al., 2018) have yet to be obtained for their active states. Using VEGAS we have identified a nanobody that binds active 5-HT2A, but not 5-HT2B. We therefore further characterized the 5-HT2A nanobody VGS-Nb2.
We first confirmed the interaction between 5-HT2A and VGS-Nb2 via co-IP, as analyzed by Western blot (Figure S5B) and mass spectrometry (Figure S5E). These assays confirmed a stable interaction between the receptor and nanobody. These assays were performed in the absence of ligand, confirming our previous BRET observation that the interaction is ligand-independent. This is consistent with our directed evolution selection paradigm, which required a nanobody capable inducing receptor activity in the absence of ligand.
5-HT2A couples to Gαq and β-arrestin to transduce its signal in cells (Wacker et al., 2017b). We tested whether VGS-Nb2 association to 5-HT2A was GĮ dependent using GαqΔ/11Δ/sΔ knock-out cell lines (Alvarez-Curto et al., 2016) in BRET recruitment assays (Figure S5A), co-IP by Western (Figure S5B), and mass spectrometry (Figure S5E). In all three studies, no appreciable difference in VGS-Nb2 association was detected.
Notably, as with all epistasis experiments, knockout cell lines frequently adapt to gene loss by augmenting signaling pathways (Duncan et al., 2012; Luttrell et al., 2018). However, from the additional proteins identified in HEK-T and GαqΔ/11Δ/sΔ cells (Table S5), no canonical GPCR transducers were identified. We concluded from this analysis that VGS-Nb2 does not stabilize, or lock, a transducer-coupled state. This aligns with the evolved purpose of this nanobody to act as a positive allosteric modulator (PAM) of 5-HT2A rather than inhibit transducer cycling.
To further validate VGS-Nb2 as a 5-HT2A PAM we first assessed its ability to positively modulate SRE signaling downstream of 5-HT2A. At Nb ratios demonstrated to bind less than 50% of 5-HT2A (Figure 5A), VGS-Nb2 increased the agonist-mediated SRE response by up to 2-fold (Figure 5C). This SRE signal could originate from Gαq and/or β-arrestin pathways. We assessed the effect of the nanobody using calcium and arrestin recruitment assays, respectively. VGS-Nb2 allosterically enhanced 5-HT2A calcium release (Figure S5F), a Gαq mediated signal response. Conversely, VGS-Nb2 diminished mVenus- ȕ-arrestin2 recruitment to the 5-HT2A-RLuc fusion protein as a function of time (Figure S5G) and agonist concentration (Figure S5H–I). From these experiments we hypothesized that VGS-Nb2 stabilizes the high-affinity Gαq -coupled state of the receptor.
Unliganded receptors are rarely found in their active, or high-affinity, conformational state (Manglik et al., 2015). However, radiolabeled ligands can be used to probe and quantify high-affinity receptor sites. The number of these sites increases when allosteric effectors, such as Gαq or nanobodies, are bound to the receptor (Che et al., 2018; Staus et al., 2016; Strachan et al., 2014). To test whether VGS-Nb2 stabilizes the 5- HT2A Gαq -coupled active state, as predicted from our functional data, we first employed radioligand saturation binding using the partial agonist [3H]LSD (Figure 5D). Membranes from cells transfected with 5-HT2A fused to its transducer Gαq (5-HT2A-Gq) were incubated with increasing concentrations of [3H]LSD +/− 5μM purified VGS-Nb2. As shown in Figure 5D, VGS-Nb2 increased labeled agonist binding sites by 50%. This increase in high affinity agonist binding sites was additionally confirmed through competitive radioligand binding wherein 5-HT2A and 5-HT2A-Gαq membranes were incubated with ± 7.5μM purified VGS-Nb2 (Figure 5E). In competition with the radiolabeled 5-HT2 antagonist [3A H]Ketanserin, the selective agonist DOI bound the 5- HT2A receptor with a half maximal inhibitory concentration (IC50) equal to 550nM. Neither a local excess of Gαq (DOI IC50=307nM, p=0.1264) nor the addition of purified VGS-Nb2 (DOI IC50=746nM, p=0.3957) significantly affected DOI binding. However, in the presence of both Gαq and VGS-Nb2 approximately 50% of the available ligand binding sites were stabilized in the high affinity conformation that bound DOI with an IC50 = 0.15nM.
We have therefore demonstrated the directed evolution of multiple functionally distinct nanobody sequences against GPCRs using VEGAS. Of these, we have characterized VGS-Nb2 as a GĮq-dependent positive allosteric modulator of 5-HT2A.
DISCUSSION
Here we demonstrate the development of asystem for facile directed evolution in mammalian cells: VEGAS. Leveraging the alpha virus Sindbis as a vector for heredity, mutagenesis, and selection we succeeded in evolving novel, context-dependent functions for three independent classes of proteins. Our evolution targets were primary (tTA), secondary (GPCR), and tertiary (Nbs) interactors to downstream outputs, demonstrating the ability of VEGAS to provide tools at multiple levels of cell signaling. Our primary system evolved tTA to engage with TETO7 in the presence of >1μM doxycycline. Our secondary system evolved the GPCR MRGPRX2 to constitutively activate the serum response element via endogenous signaling pathways. Our tertiary system evolved nanobodies to selectively activate GPCRs, which in turn activated the serum response element via endogenous signaling pathways. Together, these applications showcase the ease and power of the VEGAS system as a tool for enabling directed evolution campaigns across a broad range of potential mammalian applications.
Directed evolution allows genetic sequences to evolve under selective pressure in an appropriate context. Through this process we are able to guide solutions to otherwise intractable biological problems (Hammer et al., 2017; Kan et al., 2016; Matsumoto et al., 2015; Shapiro et al., 2010). However, powerful systems for directed evolution in a mammalian cell context have lagged behind unicellular systems. VEGAS offers three major advantages for the directed evolution of biomedical tools and therapies.
First, VEGAS evolves within the signaling framework of the host cell. Signaling proteins never operate in isolation, but as interacting heteromeric complexes, to transfer information through the cell (Garrington and Johnson, 1999; Pawson and Scott, 1997; Purvis and Lahav, 2013; Varnaitơ and MacNeill, 2016). The timing, location, and kinetics of these interactions is critical to performance and cannot be easily replicated in non-native environments. In addition, we can take advantage of the negative feedback (Amit et al., 2007; Behar et al., 2007; English et al., 2015; Ferrell, 2002; Howell et al., 2012; Subramaniam et al., 1989) mechanisms built in to endogenous signaling pathways to encode viral selection – as was done for both MRGPRX2 and nanobody directed evolution in this study.
Second, VEGAS is wholly dependent on the host cell for transgene maturation (Garciã Moreno et al., 2013). Directed evolution performance can falter when transferring tools evolved in one context to another (Armbruster et al., 2007). This may be a consequence of improper trafficking, failed compartmentalization, incorrect protein maturation, or absence of non-native co-factors. With VEGAS, mammalian translation is a requirement of the evolved product.
Third, VEGAS selection is constant and highly mutagenic, enabling it to overcome many of the pitfalls inherent to complex fitness landscapes (Romero and Arnold, 2009; Tracewell and Arnold, 2009). To avoid dead-ends and early fitness bias, directed evolution systems must sample toward saturation whenever possible. This directed evolution paradigm helps to maintain diversity by preserving even poor performing early evolution variants, which may ultimately rise to the highest fitness peaks. This is achieved with VEGAS, in part, because each host cell operates as a closed system. This allows evolved solutions derived in each cell to compete in the subsequent rounds – even when vastly superior solutions may have arisen elsewhere within the same selection cycle.
There are many potential applications of the VEGAS system. Sindbis virus has a transgene packaging capacity of >6kb (Huang and Summers, 1991), placing few limits on the potential targets for directed evolution. High value targets would include: Cas9 variants evolved to engage endogenous coding sequences (Doudna and Charpentier, 2014; Kleinstiver et al., 2015; Lee et al., 2018), fluorescent protein variants evolved for maturity time, photostability, brightness, or wavelength specificity in human tissue (Drobizhev et al., 2011; Piatkevich et al., 2017; Shaner et al., 2004, 2008), or designer receptors exclusively activated by designer drugs (DREADDs) for the chemogenetic control of cell signaling (Armbruster et al., 2007; Roth, 2016).
There also remains significant room for improvement. VEGAS has performed well with positive genetic selection and can be engineered for AND and OR gated selection paradigms. It cannot at this time generate exclusive or NOT gated selection paradigms. Developing a dominant negative selective pressure for VEGAS will make these modes possible. In addition, a method to tune the speed of replication would be advantageous for developing slower phase gene circuits including those coupled to the cell cycle, metabolism, or other slow-maturing signal classes. Adapting VEGAS for mammalian cell bioreactors would also allow it to be adapted as a continuous system (Badran and Liu, 2015). Here we add VEGAS to the growing toolbox of synthetic biology, filling an essential need for the facile directed evolution in a mammalian context.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Justin English (jenglis@email.unc.edu).
Molecular Biology & Plasmid Construction
All standard plasmids were constructed via PCR amplification of the desired amplicons using PrimeSTAR Max DNA polymerase (Takara Bio, #R045) and primers (Table S6, Eton Biosciences). Ligation of backbones and amplicons was performed using NEBuilder HiFi DNA Assembly Master Mix (NEB, #E2621). Clones were isolated by transformation of ligated DNA to One Shot Stbl3 Chemically Competent E. Coli (Thermofisher, #C737303) and selected on LB agar plates supplemented with 100μg/mL carbenicillin (Teknova L1010). Individual colonies were grown shaking at 37°C overnight in liquid LB broth (ThermoFisher, 10855001) supplemented with 100μg/mL carbenicillin (GoldBio, C-103–25). Plasmids were purified with QIAprep Spin Miniprep Kits (Qiagen, #27104) or PowerPrep HP Plasmid Maxiprep Systems (OriGene, #NP100010), dependent on downstream application. For construction of viral-sequence containing vectors see specific methods sections. All constructs were designed and confirmed via Sanger sequencing alignment (Eton Biosciences) using Benchling (Benchling.com). The list of plasmids used in this study can be found on Table S6, those necessary to perform VEGAS directed evolution have been made available at Addgene.org.
General Cell Culture
All cells were grown in a humidified 37°C incubator with 5% CO2 using media supplemented with 100 I.U./mL penicillin and 100μg/mL streptomycin (ThermoFisher, #15140122), unless otherwise indicated. The human cell lines HEK293T (ATCC, #CRL- 3216), HTLA (kindly provided by Dr. Richard Axel, Columbia Univ.), HEK-GĮq/11/sΔ, and HEK parental (HEKp, both kindly provided by Asuka Inoue, Tohoku Univ.) were maintained in DMEM (VWR, #45000) containing 10% fetal bovine serum (FBS, VWR, #89510–186). HTLA cells were additionally supplemented with 5 mg/mL Puromycin (Gemini, 400–128P) and 100 mg/mL Hygromycin B (KSE, 98–923). The hamster cell line BHK21 (ATCC, #CCL-10) was maintained in MEM-Į with nucleosides (ThermoFisher, #32571036) containing 5% FBS, 100 I.U./mL penicillin, 100ug/mL streptomycin, and 10% Tryptose Phosphate Broth (ThermoFisher, #T8159).
Sindbis Virus Production
Construct Design
pTSin (pTransgenicSindbis, Table S6), a transgene-free variant of pSinRep5 (kindly provided by Mark Heise, UNC Chapel Hill, see Bredenbeek et al., 1993), was used as the base plasmid for the construction of all transgenic Sindbis virus packaging experiments. Each transgene of interest was subcloned to pTSin via PCR amplification adding 5’-NotI & 3’-ClaI cut sites or double NotI sites. The amplicon and pTSin were both digested overnight at 37°C with NotI-HF (NEB, #R3189) and ClaI (NEB, #R0197).
The digested DNA fragments were purified using the QIAquick Gel Extraction Kit (Qiagen, #28115) and ligated with T7 DNA ligase (NEB, #M0318). Clones were selected and confirmed as described previously.
Preparation of mRNA
Our preparation of RNA for Sindbis virus packaging is modified from previous methods (Bredenbeek et al., 1993). pTSin plasmids containing transgene, as well as pSinHelper and pSinCapsid (both kindly provided by Mark Heise, UNC Chapel Hill), must be linearized before converting them to mRNA for viral packaging. Linearization was accomplished by mixing 3μg of plasmid with 2μL XbaI (NEB, #R0145), 8μL CutSmart Buffer (NEB, #B7204), raised to 80μL with nuclease-free water (hereafter H2O, NEB, #B1500) and incubated at 37°C for 24 hrs. The linearized DNA was extracted by adding 20μL H2O to the digestion mix, followed by 100μL UltraPure Phenol:Chloroform:Isoamyl Alcohol (25:24:1 w/v, ThermoFisher, #15593031). The mixture was vortexed for 15 sec, centrifuged at 13,000 x g for 5 min, and the top aqueous phase was transferred to a new tube. The extraction was then repeated. The extracted DNA was then treated with 10μL 3M sodium acetate (pH 5.2), mixed by inversion, and precipitated with 220μL 100% ethanol. The precipitant was kept at −80°C for a minimum of 20 min (or held indefinitely for future use). The DNA:ethanol mixture was then centrifuged at 13,000 x g for 10 min at 4°C. The liquid was gently aspirated from the pellet and the retained pellet was washed with 300μL 75% ice-cold ethanol prior to centrifugation at 13,000 x g for 3 min at 4°C. The wash was aspirated and the pellet was resuspended in 10μL RNAsin (Promega, #N2111) treated H2O (1:10, v:v). DNA was then immediately in vitro transcribed to mRNA.
Conversion of linearized pTSin transgene plasmids to mRNA was performed using the Ambion mMessage mMachine in vitro mRNA transcription kit (Fisher, #AM1340). Reagents were added in the following order at room temperature: 1μL reaction buffer, 5μL CAP/dNTP mix, 1μL GTP, 10μL linearized pTSin vector, and 0.7μL enzyme. The reaction was incubated at 37°C for 1 hr and then used immediately or stored at −80°C until needed.
Packaging to Sindbis Viral Particles
To package pTSin in to viral particles pTSin, pSinHelper, and pSinCapsid mRNA must be electroporated in to BHK21 cells. One day prior to electroporation BHK21 cells were split to canted-neck culture flasks seeded at 5×105 cells/dish. One flask is prepared per desired electroporation. On the day of electroporation cells were washed with 10mL Ca2+/Mg2+ free DPBS (ThermoFisher, #14190144), disassociated with trypsin (VWR, #45000–660), rinsed with 7mL ice-cold DPBS, and held on ice. Cells were centrifuged at 500 x g for 5 min at 4°C. Cell pellet was resuspended in 10mL ice-cold DPBS and centrifuged at 500 x g for 5 min 4°C. The DPBS wash was repeated 2 additional times. Before the final wash, cells were counted by hemocytometer. The washed cell pellet was then resuspended in ice cold Neon E1 electroporation buffer (ThermoFisher, #MPK10096) to 50M cells/mL and aliquoted 115μL for each transgene to be packaged. To each cell aliquot 10μL of pTSin, pSinHelper, and pSinCapsid mRNA were added, pipette mixed, and electroporated with a Neon Transfection System (ThermoFisher, #MPK5000) set to 1400V, 10 width, 3 pulses. Electroporated cells were incubated at room temperature for 10 minutes and then applied to 25mL pre-incubated serum-free MEM-Į media (ThermoFisher, #32571036) supplemented with 10% tryptose phosphate broth and pen/strep in canted neck culture flasks (Sigma-Aldrich, #CLS430641U). After 24 hrs media was decanted, 0.45μm filtered (EMD Millipore, #SCGP00525), and stored at 4°C for no longer than 1 month for use in downstream applications.
Sustained Passage of Sindbis for Directed Evolution and Transgene Isolation
Construct Design
For sustained passage and selection by directed evolution the Sindbis virus structural genome (pG100, SSG; 7662–11718 of full length Sindbis virus genome NCBI Accession MF459683.1, kindly provided by Mark Heise, UNC Chapel Hill) was subcloned using PCR/HiFi assembly as described above to mammalian expression plasmids possessing either CMV (pCDNA3.1), TETO7 (pTRE3G-BI-ZsGreen1), or SRE (pGL4.33) promoters (see Table S6 for source and mapping information).
Cell Culture and Selection
One day prior to selection 5×105 BHK21 cells were plated in canted neck culture flasks (Sigma-Aldrich, #CLS430641U). The following day 10μg of the appropriate SSG plasmid was transfected in to the cells using the TransIT-2020 Transfection Reagent (VWR, MIR5400). The transfected cells were incubated for six hrs before removing all media, rinsing the cells with PBS, and then applying the appropriate titer of virus diluted to 2mL in serum-free MEM-Į media supplemented with 10% TPB, pen/strep, and either doxycycline HCl (Sigma-Aldrich, #D3447), (+)-morphine (NIDA Drug Supply, 9300–012), or vehicle. For GPR68-targeted selection, media was additionally supplemented with 2mg/L sodium bicarbonate and equilibrated to pH 8.0 in 5% CO2 overnight prior to use.
After incubating 1 hr with intermittent rocking 23mL of additional supplemented media was added and the cells were incubated for 24 hrs. Following culturing and viral propagation the cell culture media is decanted, 0.45μm filtered, and stored at 4°C for no longer than 1 month.
Transgene Isolation
Using the MagMax Viral RNA Isolation Kit (ThermoFisher, AM1939) a 1mL aliquot of the viral stock was processed to collect a viral RNA sample. The purified RNA was maintained, precipitated in 100% EtOH at −80°C, until use in downstream applications unless noted otherwise. Isolating only translated transgenes from the viral pool is critical for the success of VEGAS. To isolate positively selected transgene sequences we used a forward primer annealing to the 26S promoter (26S-F, 5’-atctctacggtggtcctaaatagt-3’) alongside 8 reverse primers (pooled as “SinRev”) annealing to the conserved RNA structural components of the viral 3’ UTR (See Table S6). Production of a cDNA library from the Sindbis virus RNA genome with SinRev and subsequent PCR amplification with 26S-F & SinRev were performed in series using the SuperScript IV One-Step RT- PCR System (Invitrogen, #12594025). Amplicons and pCDNA3.1 were digested with NheI-HF (NEB, #R3131) and BamHI-HF (NEB, #R3136), gel purified, and ligated together. Colonies were selected, cultured, plasmids were purified, and identified by sequencing as described above. Any clone appearing >2 times out of 12 clones was selected for functional screening.
RNA Deep Sequencing
Sample Processing
Samples were prepared as follows. For EGFP mutation analysis, the pTSin-EGFP construct was independently packaged via NEON electroporation into three separate cell populations as described above, using fresh mRNA prepared on the day. During packaging, viral samples were decanted and 0.45μM filtered at 3, 6, 12, 24, and 36 hrs from each of the three independently grown cell flasks. At each collection time point the cells were washed with PBS and returned to incubate with fresh media. Viral RNA for all time points and replicates was collected simultaneously using the MagMax Viral RNA Isolation Kit (ThermoFisher, AM1939) from a 1mL aliquot of each viral stock. For nanobody library analysis, the pTSin-Nb library mRNA used for subsequent directed evolution experiments was purified and used for sequencing.
The purified RNA samples, synthesized mRNA, and pTSin-EGFP vector template were immediately amplified as described above with the SuperScript IV One-Step RT-PCR System (Invitrogen, #12594025) using 26S-F and the SinRev primer pool.
For mutation rate analysis the EGFP transgene amplicons were processed using a KAPA HyperPlus kit (KAPABiosystems, KK8512) with a 30’ digestion period followed by standard size selection with KAPA Pure Beads (KAPABiosystems, KK8001). The resulting fragments were barcoded with the Illumina compatible SeqCAP Adapater Kit A & B (KAPABiosystems, 7141530001 and 07141548001). Samples were normalized, pooled, and processed using a NextSeq NSQ 500/550 Hi Output KT v2.5 300 cycle kit (Illumina, 200249808) calibrated to acquire dual-indexed, 2 × 150 bp reads, yielding an average of 4.3 × 108 reads per sample.
For nanobody library analysis the amplicons were >100bp size-selected using Agencourt AMPure magnetic beads (Beckman Coulter, #A63881). The purified DNA was diluted to 10ng/μL in 10mM Tris, 1mM EDTA, pH 7.5 sonication buffer and sheared to 150bp fragments on a Bioruptor Pico sonication device (Diagenode, #B01060010) using a 30”/30” cycle program for 30 cycles at 4°C. The sheared DNA was then used for library construction using a KAPA Hyperprep Kit (Roche, #KK8500) and Illumina TruSeq indexed adapters (IDT, HPLC purified) following the manufacturer’s recommended protocol. Samples were normalized and processed using 12-plex, single-indexed, 1 × 150 bp Illumina NextSeq500 sequencing.
Data Analysis
Data are deposited on the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo), GEO accession # GSE123269.
The EGFP experiment sequence data was initially processed for exact matching paired- end reads >25bp in length. The longest perfect match from each read pair was isolated and the remaining non-matching sequences were omitted from further analysis. The truncated reads were aligned to sense and anti-sense EGFP and Sindbis genome reference sequences using a Smith-Waterman algorithm (Smith and Waterman, 1981) on the UNC Longleaf Linux Cluster (UNC ITS). Alignment score weighting was m3, x1, o5, e1 for correct match (m), mismatch penalty (x), insert/deletion penalty (o), and gap extension penalty (e). Reads were associated with their high scoring reference sequence. The sequences aligning to anti-sense EGFP were reverse-complemented. All EGFP aligning reads were then re-aligned to the EGFP reference sequence and a sequence where EGFP was excised (TGDEL). Sequences scoring best for the sense EGFP reference were then used to retrieve individual base position, deletion, and insertion counts for the EGFP transgene. No score cut-offs were implemented.
The nanobody experiment sequence data was aligned to the cloned nanobody reference sequence using a Smith-Waterman algorithm (Smith and Waterman, 1981) on the UNC Longleaf Linux Cluster (UNC ITS). Alignment score weighting for all nanobodies was m1, x5, o5, e5 for correct match (m), mismatch penalty (x), insert/deletion penalty (o), and gap extension penalty (e). Sequence reads containing the 3’ barcode and vector backbone were trimmed and the final alignments were designated with varying score cut-offs as described in Figure 4A.
Data were analyzed on Graphpad Prism 8. The mutation frequency data per time point (Figure 1C) statistics were calculated using One-Way ANOVA with Geisser-Greenhouse correction and Sidak multiple comparisons correction. Multiplicity adjusted P values for each comparison are p<0.0001 for all significant comparisons, using family-wise significance and confidence level set to 99.9%. The mutation type per base reads data (Figure 1D) was plotted as individual replicates. The values represent the number of total substitutions divided by the number of total reads for the mutated base per replicate and time point. Statistics were calculated within each base substitution group using Two-Way ANOVA and Tukey’s multiple comparisons correction with one family per column (substitution type). A minimum multiplicity adjusted P value (Į=0.05) was applied for each comparison. The mutations base−1/time (Figure 1E) data was plotted with a linear regression model for mean±SD of N=715 measures with 99% confidence band displayed. Data in Figure S1B–D are residuals of the data displayed in Figure 1 and no statistical analysis was performed. For Figures S1E–G the number of mutations observed per total reads at the indicated read length are plotted with a linear regression model beginning at the observed linear range of read length 50. 95% confidence bands are displayed for each time point.
Quantification of Viral RNA via qRT-PCR
Quantification of Sindbis viral genomic RNA by TaqMan qRT-PCR was performed as described previously (Sane et al., 2012) with modification. We designed a probe and primer pair specific for the packaging signal sequence spanning g.138-a.269 of nsP1 in pTSin; Probe: 5’-/5HEX/ATTTTGGAC/ZEN/ATAGGCAGCGCACC/3IABkFQ/−3’, Forward: 5’-GTTCCTACCACAGCGACG-3’, Reverse: 5’- GGTACTGGTGCTCGGAAAAC-3’ (IDT) (see Table S6). Sindbis virus containing media was diluted across 3 × 10-fold serial dilutions, 4 replicates each, mixed directly with TaqMan® Fast Virus 1-Step Master Mix (ThermoFisher, #4444432) and cycled on a BioRad CFX96 Touch RT-PCR machine (BioRad). Serially diluted (10−12-10−6 M) in vitro transcribed Sindbis virus RNA reference samples were present on each sample plate. Standard curves were calculated on CFX Manager (BioRad) and used to calculate and report genomes/mL for each sample.
tTA Reporter Assay
The wildtype and R7_G8 tetracycline transactivator (tTA) sequences were subcloned in pCDNA3.1. The reporter was constructed by subcloning luc2p from SRE-luc2p (Promega, pGL4.33) over the ZsGreen1 CDS of pTRE3G-BI-ZsGreen1 (Takara, 631339) to create pTRE3G-BI-luc2p. The day before transfection, HEK293T cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 10μg DNA per 15-cm, 5μg of pTRE3G-BI-luc2p and either 5μg of WT or R7 constructs using TransIT-2020. Six hours after transfection media and transfection reagents were removed, cells were washed with PBS, dissociated using versene (ThermoFisher, 15040066), centrifuged and resuspended in DMEM supplemented with 1% dialyzed FBS. Transfected cells were then plated onto poly-L- lysine-coated 384-well white clear bottom cell culture plates at a density of 10,000 cells/well in a total of 20μl. Doxycycline HCl solutions were prepared in plating media at 2 x and added to cells (20 μl per well) for overnight incubation. After 20–22 hr overnight incubation, media and drug solutions were removed from plates and 20μl per well of 1:20 diluted Bright-Glo reagent (Promega, E2620) was added per well. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration and analyzed using “log(inhibitor) vs. response” in GraphPad Prism 8.0.
Transcription Factor Reporter Primary Screen
The MRGPRX2, DRD2, 5-HT2A, and GPR68 constructs originated from their respective PRESTO-TANGO plasmids (Kroeze et al., 2015), from which the C-terminal V2Tail- TEV-tTA sequence was removed by PCR mutagenesis. The transcription factor reporter constructs were purchased from Promega (see Table S6). Two days before transfection, HEK293T cells were split to yield approximately 9×106 cells/15-cm plate in 2 days. On the day of transfection each well of a poly-L-lysine-coated 384-well white clear bottom cell culture plate received 30ng of DNA,15ng of one transcription factor reporter and 15ng of either GPCR or pCDNA3.1, pre-incubated with TransIT-2020. One plate was prepared for each time point, with 8 wells of GPCR and 8 wells of empty vector for each reporter construct. The prepared cells were then washed with PBS, dissociated using trypsin, centrifuged, and resuspended in DMEM supplemented with 1% dialyzed FBS. Cells were seeded in the DNA pre-loaded plates at 10,000 cells/well to a final volume of 40 μL and incubated for 24 hr. The following day single drug concentrations and vehicle (DMSO) were prepared in drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at 3 x and added to cells (20μl per well) at the appropriate time point for each plate. For each reporter 4 replicates of drug treated and 4 replicates of vehicle were added for each transfection condition at each time point. After incubation, media and drug solutions were removed from plates and 20μl per well of BrightGlo reagent (purchased from Promega, after 1:20 dilution) was added per well. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a function of time in GraphPad Prism 8.0.
SRE Reporter Assay
The GPCR constructs from the transcription factor reporter primary screen were used in these assays. Nanobody constructs were direct clones from Sindbis virus transgene isolation to pCDNA3.1. The day before transfection, HEK293T cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 15μg DNA per 15-cm dishes; 5μg of SRE-luc2P (Promega, pGL4.33), 5μg of GPCR, and an appropriate ratio of Nb and empty vector to 5ug. The next day, media and transfection reagents were removed, cells were washed with PBS, dissociated using versene, centrifuged and resuspended in DMEM supplemented with 0.1% dialyzed FBS. Transfected cells were then plated onto poly-L-lysine-coated 384- well white clear bottom cell culture plates at a density of 10,000 cells/well in a total of 40μl. The cells were incubated for 12 hr and then developed for untreated assays or received drug stimulation. Drug solutions were prepared in drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at 3 x and added to cells (20μl per well) for overnight incubation. After 6 hr incubation, media and drug solutions were removed from plates and 20 ml per well of BrightGlo reagent (purchased from Promega, after 1:20 dilution) was added per well. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a bar graphs or as a function of drug concentration and analyzed using “log(agonist) vs. response (three parameters)” in GraphPad Prism 8.0.
TANGO β;-arrestin recruitment assay
The MRGPRX2 Tango construct, which contains the TEV cleavage site and the tetracycline transactivator (tTA) fused to the C terminus of the receptor, were designed and assays were performed as previously described (Kroeze et al., 2015; Lansu et al., 2017). Each VEGAS-derived MRGPRX2 mutant was subcloned over the wildtype sequence via HiFi assembly. HTLA cells expressing TEV fused- β-Arrestin2 and a tetracycline transactivator-driven luciferase (kindly provided by Dr. Richard Axel, Columbia Univ.) were grown in HTLA media (10% FBS DMEM containing 5 mg/mL Puromycin and 100 mg/mL Hygromycin B). The day before transfection, HTLA cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 10μg DNA per 15-cm with MRGPRX2 Tango, or one of the VEGAS derived mutants, using TransIT-2020. The next day, media and transfection reagents were removed, cells were washed with PBS, dissociated using versene, centrifuged and resuspended in DMEM supplemented with 1% dialyzed FBS. Transfected cells were then plated onto poly-L-lysine-coated 384-well white clear bottom cell culture plates at a density of 10,000 cells/well in a total of 40μl. The cells were incubated for 12 hr before receiving drug stimulation to allow for recovery and adherence to the plate. Drug solutions were prepared in drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at 3 x and added to cells (20μl per well) for overnight incubation. Drug solutions used for the Tango assay were exactly the same as used for the SRE assay, which was conducted in parallel to the Tango assay. After 6 hr incubation, media and drug solutions were removed from plates and 20 ml per well of BrightGlo reagent (purchased from Promega, after 1:20 dilution) was added per well. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % wild-type stimulation, and analyzed using “log(agonist) vs. response (three parameters)” in GraphPad Prism 8.0.
Calcium flux assay
Assays were designed and performed as previously described (Wacker et al., 2017b), using the same 5-HT2AR stable cell line created with the Flp-In 293 T-Rex Tetracycline inducible system (Invitrogen). The day before transfection, 5-HT2AR cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 8μg DNA per 15-cm with adjusted ratios of empty vector (pCDNA3.1) or pCDNA3.1-VGS-Nb2 using TransIT-2020. Six hours after transfection, 1μM final concentration of doxycycline HCl was applied to induce receptor expression. The next day, media and transfection reagents were removed, cells were washed with PBS, dissociated using versene, centrifuged and resuspended in DMEM supplemented with 0.1% dialyzed FBS. cells were seeded in 384-well poly-L-lysine plates at a density of 10,000 cells/well at least 16–24 hr before the calcium flux assay. On the day of the assay, the cells were washed in FLIPR buffer (1 x HBSS, 2.5 mM probenecid, and 20 mM HEPES, pH 7.4), pre-treated with the 1μM alkylating agent phenoxybenzamine (Sigma-Aldrich, B019) for 30 min, washed again in FLIPR buffer and then incubated with 20μL/well Fluo-4 Direct dye (ThermoFisher, F10471) reconstituted in FLIPR buffer for 1 hr at 37°C. After dye loading, cells were placed in a FLIPRTETRA fluorescence imaging plate reader (Molecular Dynamics). Drug dilutions were prepared at 3 x final concentration in drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), aliquoted into 384-well plates. and placed in the FLIPRTETRA for drug stimulation. The fluidics module and plate reader of the FLIPRTETRA were programmed to read baseline fluorescence for 10 s (1 read/s), then 10μl of drug/well was added and read for 5 min (1 read/s). Fluorescence in each well was normalized to the average of the first 10 reads (i.e., baseline fluorescence). Then, the maximum-fold increase, which occurred within the first 60 s after drug addition, was determined and fold over baseline was plotted as a function of drug concentration. Data were analyzed using “log(agonist) vs. response (three parameters)” in Graphpad Prism 8.0.
Bioluminescence resonance energy transfer (BRET) association assay
Assays were designed and performed as previously described (Che et al., 2018), with additions. The day before transfection, HEK293T cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 12μg DNA per 15-cm dishes; 2μg of GPCR-RLuc DNA held constant and an adjusted ratio of empty vector (pCDNA3.1) or pCDNA3.1-mVenus-Nb using TransIT-2020. The next day, media and transfection reagents were removed, cells were washed with PBS, dissociated using versene, centrifuged and resuspended in DMEM supplemented with 1% dialyzed FBS. Transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% dialyzed FBS) at a density of 40–50,000 cells in 200μl per well and incubated overnight. The next day, media was aspirated and cells were washed twice with 60μL of drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), then 60μL of drug buffer was added per well, followed by 30μL of drug (3X) per well, and finally 10μL of the RLuc substrate, coelenterazine h (Promega, S2011, 5 mM final concentration). Plates were incubated for 5 min to allow for substrate diffusion, and then read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 s per well using a Mithras LB940 multimode microplate reader. The ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without mVenus-Nb present. The net BRET ratio was plotted as a function of nanobody concentration using Graphpad Prism 8 (Graphpad Software Inc., San Diego, CA).
Phosphoinositide hydrolysis assay
Phosphoinositide (PI) hydrolysis assays measuring inositol phosphates (IP) were performed using the scintillation proximity assay (Bourdon et al., 2006; Huang et al., 2009). The MRGPRX2 WT construct was developed from the TANGO system as described above and mutant constructs were directly subcloned as viral transgenes to pCDNA3.1. The day before transfection, HEK293T cells were split to yield approximately 9×106 cells/15-cm plate next day. The following day cells were transfected with 5μg DNA per 15-cm dish with TransIT-2020. On the day before the assay, transfected cells were seeded into 96-well poly-lysine coated plates at a density of 40–50,000 cells/well in 100μL inositol-free DMEM (Caisson Labs, DML13) containing 1% dialyzed FBS. After 6 hr, an additional 100μL of label media was added containing 1μCi/well (final concentration) of [3H]-myo-inositol (PerkinElmer, NET1177001MC) in inositol-free DMEM containing 1% dialyzed FBS and plates were incubated overnight for 16–18 hr. The next day, label media was removed and cells were washed twice with 60μL of drug buffer (1 x HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), then 60μL of drug buffer was added per well. Afterward, 30μL of drug (3X) was added per well and incubated at 37°C for 1 hr. To capture IP accumulation, lithium chloride (10μL/well, 15 mM final concentration) was added 30 min before lysis. The assay was terminated by replacement of the incubation medium with 40μL of 50 mM formic acid. After overnight incubation at 4°C, 10μL of lysates were added to 96-well flexible, clear microplates (PerkinElmer, 1450–401C) containing 75μL of 0.2 mg/well RNA binding yttrium silicate beads (PerkinElmer), and incubated for 1 hr on a shaker. Afterward, plates were centrifuged at 300 x g for 1 min, and radioactivity was measured using a Wallac MicroBeta Trilux plate reader (PerkinElmer). Data were plotted as counts per minute (CPM) as a function of drug concentration, and analyzed using “log(agonist) vs. response (three parameters)” in GraphPad Prism 8.0.
Surface expression enzyme-linked immunosorbent assay (ELISA)
To confirm cell surface expression of MRGPRX2 and its mutants, immunohistochemistry was done using cells plated on 384-well plates, as described earlier, at 10,000 cells/well. Cells were fixed with 20μl/well 4% paraformaldehyde (Fisher, #AAJ19943K2) for 10 minutes at room temperature. After fixation, cells were washed twice with 40μl/well PBS. Blocking was performed with 20μl/well 5% normal goat serum (Vector Laboratories, #S-1000) in PBS for 30 minutes at room temperature. After blocking, 20μl/well monoclonal ANTI-FLAG M2-Peroxidase (HRP) antibody (Sigma-Aldrich, A8592) diluted 1/10,000 in PBS was added and incubated for 1 hour at room temperature. This was followed by two washes with 80μl/well PBS. Then, 20μl/well SuperSignal ELISA Pico Chemiluminescent Substrate (Sigma-Aldrich, #37069) was added, and luminescence was counted using a MicroBeta Trilux luminescence counter. Data were plotted as relative luminescent units (RLU) in GraphPad Prism 8.0.
Nanobody Production
Construct Preparation
5-HT2AR proteoliposomes for immunization were prepared using a receptor construct with truncated intracellular loop 3 (icl3) and C-terminus. The final construct lacking residues 278–304 and 404–471 was cloned into a modified pFastBac vector introducing a haemagglutinin (HA) signal sequence followed by a FLAG tag at the N-terminus, and a PreScission protease site followed by a 10×His tag at the C-terminus.
Expression and purification of 5-HT2AR
High-titer recombinant baculovirus (>109 viral particles per ml) was generated using the Bac-to-Bac Baculovirus Expression System (Invitrogen, 10359016). Recombinant baculovirus was obtained by transfecting ~5μg of recombinant bacmid into 5×105 settled Spodoptera frugiperda cells (Sf9, Expression Systems, 94–001S) in a 24 well plate (Sigma-Aldrich, CLS3527) using 3μl Cellfectin II Reagent (ThermoFisher, 10362100). After 5–12 hrs, media was exchanged for 1 ml Sf-900 II SFM media (ThermoFisher, 10902096) and incubated for 4–6 days at 27 °C. P0 v iral stock with ~109 virus particles per ml was harvested as the supernatant and used to generate high-titer baculovirus stock by infection of 40–1000 mls of Sf9 cells and incubation for several days. Expression of 5-HT2A was carried out by infection of Sf9 cells at a cell density of 2–3 × 106 cells/ml in ESF921 media (Expression Systems) with P1 virus at a MOI (multiplicity of infection) of 3–5. After 48 hrs of expression at 27 °C in the presence of 10 μM Methiothepin (Sigma-Aldrich, M149), cells were harvested by centrifugation, washed in PBS, and stored at −80 °C until use. Cells were dis rupted by thawing frozen cell pellets in a hypotonic buffer (10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and protease inhibitors 500μM AEBSF, 1μM E-64, 1μM Leupeptin, 150 nM Aprotinin). Membranes were purified by repeated centrifugation in a high osmolarity buffer containing 1.0 M NaCl, 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl, to remove soluble and membrane associated proteins. Purified membranes were directly flash-frozen in liquid nitrogen and stored at −80 °C.
Purified membranes were resuspended in buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl, 150 mM NaCl, 20μM LSD (synthesized in house, see Wackeret al., 2017b), and protease inhibitors before incubating at room temperature for 1 h. After 30 min incubation in the presence of 2 mg/ml iodoacetamide (Sigma, I6125), membranes were solubilized in 10 mM HEPES, pH 7.5, 150 mM NaCl, 1% (w/v) n- dodecyl-ȕ-D-maltopyranoside (DDM, Anatrace, D310), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma, C6512), 20μM LSD, and protease inhibitors for 2 h at 4 °C. Unsolubilized material was removed by centrifug ation at 150,000 × g for 30 min, and 15 mM imidazole (Sigma, I5513) was added to the supernatant. Proteins were bound to TALON metal affinity resin (Takara, 635653) overnight at 4 °C using approximately 750μl resin for protein purified from 1 L of cells. The resin was then washed with 10 column volumes (cv) of Wash Buffer I (50 mM HEPES, pH 7.5, 800 mM NaCl, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 20 mM imidazole, 10% (v/v) glycerol, and 20μM LSD, followed by 10 cv of Wash Buffer II (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% (v/v) glycerol, and 20μM LSD). Proteins were eluted in 2.5 cv of Wash Buffer II + 250 mM imidazole, concentrated in a 100 kDa molecular weight cut-off Vivaspin 20 concentrator (Cole-Parmer, VS2002) to 500μl, and imidazole was removed by desalting the protein over PD MiniTrap G-25 columns (GE Life Sciences, 28918007). The C-terminal 10× His-tag was removed by addition of His-tagged PreScission protease (GenScript) and incubation overnight at 4 °C. Protease, cleaved His-tag and uncleaved protein were removed by passing the suspension through equilibrated TALON IMAC resin (Clontech) and collecting the flow-through. 5-HT2A /LSD complexes were then concentrated to ~8.5 mg/ml with a 100 kDa molecular weight cut-off Vivaspin 500 centrifuge concentrator (Cole-Parmer, VS0141). Protein purity and monodispersity were tested by analytical size-exclusion chromatography.
100 μl of 5-HT2A concentrated to 8.5 mg/ml were mixed with 300 μl of 4 mg/ml 1,2- dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, Avanti Polar Lipids, 850355C) and cholesteryl hemi succinate (CHS) at a ratio of 9:1 (w/w) and 100 μl 5 mg/ml Lipid A (Sigma, L5399) both in 20 mM HEPES, pH 7.5, 100 mM NaCl, 1% (w/v) n-Octyl-ȕ-D- Glucopyranoside (OG, Anatrace, O311). Lipids, detergents, and protein were allowed to equilibrate on ice for 90 min, before rapid dilution with 1 ml of 20 mM HEPES, pH 7.5, 100 mM NaCl to bring OG below its critical micelle concentration. To remove detergent and form liposomes the sample was dialyzed against 20 mM HEPES, pH 7.5, 100 mM NaCl for 24 hrs, and subsequently incubated with Bio-Beads SM2 resin (BioRad, 1523920) for 4 hrs.
Llama immunization was done by Capralogics using a first injection of 200 μg, followed by 5 additional injections of 100μg of 5-HT2A proteoliposomes.
Library Preparation
A nanobody library was generated as described previously (Pardon et al., 2014). In brief, immunized llama blood was taken to isolate peripheral blood lymphocytes. RNA was purified from these lymphocytes and reverse transcribed by PCR to obtain cDNA. The resulting library was cloned into pTSin plasmid to a complexity of ~1 × 107 colonies.
Nanobody Purification
VGS-Nb2 was subcloned from its original pCDNA3.1 destination vector after VEGAS directed evolution to pMESy4 (kindly provided by Jan Steyaert, Vrije Universiteit, Brussels) and purified following steps 70–73 described in the previous protocol (Pardon et al., 2014). Nanobodies were concentrated, desalted (10 mM HEPES, 100 mM NaCl, and 10% Glycerol), and stored at 80°C for future use.
Saturation and competitive radioligand binding assays
Radioligand assays were performed in parallel utilizing the same membrane preparations, binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), and purified VGS-Nb2 nanobody. For saturation assays, 5-HT2A-Gαq membrane (50 μL) treated with purified VGS-Nb2 (5μM final concentration) or vehicle were added to round-bottom 96-well plates. A range of [N-Methyl-3H]-Lysergic Acid Diethylamide ([3H]-LSD, PerkinElmer, #NET638250UC) concentrations up to 6.80nM was added (25 μL) along with either vehicle (25 μL) or unlabeled 10 mM 1-(4- iodo-2,5-dimethoxyphenyl)propan-2-amine HCl (25 μL, DOI HCl, Tocris, #2643) to determine non-specific binding. For competition assays, 5-HT2A and 5-HT2A-Gαq membranes (50μL) treated with purified VGS-Nb2 (7.5μM final concentration) or vehicle were added to round-bottom 96-well plates. 5-HT2A membranes were additionally treated with 50mM GppNHp (Abcam, ab146659) to uncouple all GĮ proteins. The antagonist radioligand [3H]Ketanserine (PerkinElmer, #NET791025) was added (25μL) to 1nM final concentration alongside a range of unlabeled DOI concentrations (25μL). Plates were incubated for 1.5 hrs at room temperature. Both saturation and competition binding plates were harvested immediately after incubation by vacuum filtration onto 0.3% polyethyleneimine pre-soaked 96-well filter mats (PerkinElmer, 1450–421) using a 96-well Filtermate harvester, followed by three washes of cold wash buffer (50 mM Tris, pH 7.4). Solid scintillant (PerkinElmerm 1450–441) was melted onto dried filters and radioactivity was counted using a Wallac Trilux MicroBeta counter (PerkinElmer). For analysis of saturation binding data non-specific counts were removed using “Remove Baseline and Column Math” and then analyzed using “One Site – Specific Binding” with asymmetrical 95% confidence intervals in Graphpad Prism 8.0. Statistics were performed as extra sum-of-squares F test (α, 0.05) for difference between best-fit values of each data set. Competitive binding data was analyzed using the “One Site – Fit logIC50” model and compared for best fit versus a “Two sites – Fit logIC50” model in Graphpad Prism 8.0 using an extra sum-of-squares F test (α, 0.05). Each data set was then normalized to the “Top” value of the best fit model and replotted with shared parameters. Within parameter comparisons of IC50 were performed with an extra sum- of-squares F test (α, 0.05).
Co-Immunoprecipitation Analysis
Co-Immunoprecipitation
The FLAG-5-HT2A and FLAG-DRD2 constructs were developed from their respective TANGO plasmids with the V2Tail-TEV-tTA C-terminal tag removed. The mVenus-VGS- Nb2 plasmid is the same described for use in BRET experiments. The day before transfection, HEK293T and HEK GĮq/11/sΔ cells were split to yield approximately 6×106 cells/15-cm plate next day. The following day cells were transfected with 10μg DNA per 15-cm; 8μg GPCR construct and 2μg nanobody using TransIT-2020. 6 hrs after transfection media and transfection reagents were removed, cells were washed with PBS, and incubated in DMEM supplemented with 0.1% dialyzed FBS. 48 hours after transfection the cells were washed, lysed, and FLAG-GPCR was FLAG- immunoprecipitated as described previously (Staus et al., 2014). In brief, cells were scraped in to ice-cold lysis buffer [20 mM HEPES, 100mM NaCl, 0.5% decyl maltose- neopentyl glycol (DMNG, Anatrace, NG322)] and incubated with gentle rotation at 4°C for 1 hr. Insoluble material was separated by centrifugation at 14,000 x g for 15 minutes at 4°C. The soluble lysate was immunoprecipitated with magnetic FLAG-M2 beads (Sigma-Aldrich, M8823), washed with TBS-M (50mM Tris HCl, 150mM NaCl, 0.5% MNG, pH 7.4), and eluted with 3μL of 5ug/μL 3x FLAG peptide (Sigma-Aldrich, #F4799) in TBS-M. Samples were then used immediately for downstream applications.
Western Blot
Co-immunoprecipitated fractions taken from solubilized cell lysate (L), unbound waste (U), bead wash (W), and final elution (E) were mixed with LDS gel loading buffer (Thermo-Fisher, #NP0007) containing 50mM fresh dithiothreitol and heated to 65°C for 5 minutes. Samples were then run on NuPage 4–12% Bis-Tris Protein Gels (Thermo-Fisher, #NP0322) according to manufacturer’s instructions. Samples were then transferred to Immobilon PVDF membranes for Western blotting (Sigma-Aldrich, #IPSN07852) according to manufacturer’s instructions. Protein-adhered membranes were blocked with TBS-T (50mM Tris, 150mM NaCl, 0.1% Tween 20) with 3% BSA for 1 hr followed by overnight incubation with primary antibodies to detect mVenus (Anti- GFP, rabbit, 1:1000 dilution, Novus Biologicals, #NB600–308) and FLAG (Anti-FLAG- M2, mouse, 1:1000, Sigma-Aldrich, F1804) at 4°C. The following day membranes were washed 4 × 15 min in TBST and probed for 1 hr with anti-rabbit IgG HRP (1:5000, Jackson ImmunoResearch, #711-035-152) for mVenus detection or anti-mouse IgG HRP (1:3000, Cell Signaling, #7076S) for FLAG detection. Blots were washed again 4× 15 min in TBST, mixed with Clarity Western ECL Substrate (BioRad, #1705061) and imaged on a ChemiDoc Touch Imaging System (BioRad, #1708370).
LC-MS/MS Analysis
Immunoprecipitated samples were run on SDS-PAGE in every other lane to avoid cross contamination and bands were excised from 150kDa to 15kDa as determined by Precision Plus Protein Dual Color Standard (BioRad, #1610374). The proteins were reduced, alkylated, and in-gel digested with trypsin overnight at 37°C. Peptides were extracted and dried via vacuum centrifugation. All peptide samples were stored at −80°C until further analysis.
The samples were analyzed by LC/MS/MS using an Easy nLC 1200 coupled to a QExactive HF mass spectrometer (ThermoScientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 45 min method. The gradient for separation consisted of 5–38% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% acetonitrile. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 350–2000) was set to 120,000 with a target value of 3 × 106 ions. MS/MS scans resolution was set to 15,000 with a target value of 1 × 105 ions, 100 ms max IT. The normalized collision energy was set to 27% for HCD. Dynamic exclusion was set to 30 s and precursors with unknown charge or a charge state of 1 and Ł 8 were excluded.
Data were searched against a Uniprot Human database (containing ~22,000 protein sequences), appended with the mVenus-VGS-Nb2 protein sequences, using Sequest within Proteome Discoverer 2.1 (Thermo Scientific). The following parameters were used to identify tryptic peptides for protein identification: 10 ppm precursor ion mass tolerance; 0.02 Da production mass tolerance; up to two missed trypsin cleavage sites. Carbamidomethylation of Cys was set as a fixed modification, and oxidation of Met and acetylation of N-terminus were set as variable modifications. Scaffold (version Scaffold_4.7.3) was used to validate MS/MS based peptide and protein identifications. Protein/peptide identifications were accepted if they could be established at greater than 95% probability by the Scaffold Local FDR algorithm.
Quantification and Statistical Analysis
All statistical analysis was performed using Prism 8 (GraphPad). The specific details of each analysis type are outlined in the experimental methods section, figures, and figure legends of the specific experiment. The number of biological experimental replicates per experiment (herein, N) is outlined in each figure legend and experimental methods section.
Database Analysis and Software
All graphs and data plots were generated using Prism 8 (GraphPad) unless noted otherwise herein. Alignment of tTA nucleotide sequences (Figure 2A) was performed and graphically rendered using Benchling (Benchling.com). Alignment of tTA peptide sequences (Figure 2C) as well as VEGAS cloned Nb sequences (Figure S4B) was performed using T-COFFEE (Di Tommaso et al., 2011) and graphically rendered using ESPript 3.0 (Robert and Gouet, 2014). Analysis of codon usage frequency (Figure S2D) was plotted using Morpheus (https://software.broadinstitute.org/morpheus). The datasets for human, hamster, and Sindbis virus codon usage frequency were obtained in 2018 from the Codon Usage Database (Nakamura et al., 2000). Protein structure renderings (Figure 2D and S2B) were created using PyMol 2.2 (Schrödinger) from PDB 4AC0 (Volkers, G. et al., unpublished) and PDB 1QPI (Orth et al., 2000). Alignment of GPCR switch regions (Figure S3A) was performed using GPCRdb (Isberg et al., 2015; van der Kant et al., 2014; Pándy-Szekeres et al., 2018) and graphically plotted using Morpheus. Snake plots (Figure S3D) were rendered using GPCRdb. Alignment of nanobody NGS reads was graphically rendered using the Integrative Genomics Viewer (Robinson et al., 2011; Thorvaldsdóttir et al., 2013).
Supplementary Material
Figure S4. VEGAS for Evolution of Active-State Nanobodies, Related to Figure 4. VEGAS was used to develop nanobodies that selectively activate diverse GPCR targets from a single cDNA library. A-F. Transcription factor reporter primary screen. Cells transfected with GPCRs (A,C,E) or pCDNA3.1 (B,D,F), and the indicated transcription response element (TRE) were stimulated. TRE-RLuc production equates to drug- mediated activation of the GPCR, quantified over time. Data are represented as mean ± SEM, N=1.
Figure S5. Positive Allosteric Modulation of GPCRs by VEGAS-Evolved Nanobodies, Related to Figure 5. VEGAS derived nanobodies were tested for direct association and allosteric modulation of their targets. A. Bioluminescence resonance energy transfer (BRET) association assay between GPCR-RLuc constructs and mVenus-VGS-Nb2 in the presence of increasing concentrations of 5HT. Data are represented as mean ± SEM, N=3. B. Co-immunoprecipitation determination from FLAG pull-down by Western blot analysis. Loaded (L), unbound (U), washed, (W), and eluted (E) fractions annotated. C. Bioluminescence resonance energy transfer (BRET) association assay between DRD2-RLuc or 5HT2A-RLuc and mVenus-VGS-Nb8 at increasing transfection ratios of nanobody. Data are represented as mean ± SEM, N=3 D. Bioluminescence resonance energy transfer (BRET) association assay between DRD2-RLuc or 5HT2A-RLuc and mVenus-VGS-Nb7 at increasing transfection ratios of nanobody. Data are represented as mean ± SEM, N=3. E. Co-immunoprecipitation determination from FLAG pull-down by LC-MS/MS analysis. 5HT2A, blue; mVenus, green; VGS-Nb2, orange; DRD2, red; other peptides, grey. Data are represented as total background subtracted spectral counts, N=2. See Table S5 for raw data. F. Calcium flux assay. Calcium mobilization equates to relative GPCR-mediated Gαq activity. Data are represented as mean ± SEM, N=3. G-I. Bioluminescence resonance energy transfer (BRET) association assay between 5HT2A-RLuc and mVenus- bArrestin-2 in the presence of increasing concentrations of DOI. H receptor only, I VGS-Nb2 co-transfected, G linear regression of area under the curve datasets for G, H, and additional nanobody concentrations. Data are represented as mean ± SEM, N=3. J. Serum response element (SRE) reporter assay. DRD2 alone or co-transfected with 5x VGS-Nb7 or VGS-Nb8. RLuc production equates to relative receptor activation. Data are represented as mean ± SEM, N=2.
Table S1. Evolution derived TETR sequences, Related to Figure 2. FASTA formatted list of TETR sequences obtained through evolution of tTA over multiple rounds with increasing doxycycline concentrations as outlined in Figure 2A. Names identify the sequence by round number (R#) and well location (_A-H,1–12).
Table S2. Previously reported TETR mutations from R7, Related to Figure 2. Mutations identified in the R7 consensus sequence are indexed. pIǻ, isoelectric point change from original to mutant sequence. @_, non-identical mutations published at the same position, published amino acids listed after the @. Δ, identical mutations published at the same position. D, TETR(B) to TETR(D) mutation.
Table S3. Alignment of MRGPRX2 to critical residues of canonical opiate receptors, Related to Figure 3. Alignment of μ opioid receptor (MOP), į opioid receptor (DOP), ț opioid receptor (KOP), nociceptin receptor (NOP), and MRGPRX2 amino acid sequences at key functional residues. Residues include conserved (Ł 3, gray highlight), ligand interacting (bold green text, adapted from Filizola and Devi, 2013), sodium pocket (Na+, adapted from Katritch et al., 2014), DRY, PIF, CWxP, and NPxxY domains. Mutations generated through VEGAS evolution are also included for reference.
Table S4. Deposited Llama glama amino acid sequences, Related to Figure 4. Sequences retrieved from GenBank and PDB databases and displayed in FASTA format.
Table S5. LC-MS/MS Raw Data, Related to Figure 5. Data from pull-down of FLAG-5HT2A, FLAG-DRD2, or FLAG-free cells provided as both spectral counts and total peptide counts. Target proteins (red font) are highlighted.
Table S6. Plasmids used in this study, Related to STAR Methods. List of plasmids used in this study, both pre-existing and generated in this work. Plasmid name, source, description, genbank (GBK) map, and primers used for construction are listed.
Data S1. VEGAS Resource Overview, a streamlined graphical outline of the VEGAS selection process, which is described in detail within Data S2 and related to Figures 1–5.
Data S2. VEGAS Resource Protocol, a complete protocol for the implementation of the VEGAS platform, as outlined in Data S1, related to Figures 1–5, and described in the STAR Methods Section “Sindbis Virus Production” and “Sustained Passage of Sindbis for Directed Evolution and Transgene Isolation”.
Figure S1. Sindbis Virus for Facile Directed Evolution in Mammalian Cell Culture, Related to Figure 1. A. Epifluorescence microscope images of BHK-21 cells in culture 0, 4, 12, and 24 hrs after application of pTSin-EGFP virus. B. Mutations observed per base measured via Illumina paired-end sequencing at each position in the EGFP transgene over time. Data are plotted as individual replicates. C. Read distribution from Illumina paired-end sequencing per position in the EGFP transgene over time. Data are plotted as individual replicates. D. Insert number and size and deletion number observed from sequencing of Sindbis packaged EGFP transgene over time. Data are plotted as individual replicates. E. Mutations observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band. F. Insertions observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band. E. Deletions observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band.
Figure S2. Directed Evolution of Transcription Factors with Sindbis, Related to Figure 2. A. qRT-PCR quantification of Sindbis virus production from cell culture. BHK- 21 cells transfected with TETO7-SSG (red) or TETO7-GFP (blue) were infected with pTSin-tTA and treated with 1μM Doxycycline (filled circles) or vehicle (open circles). Data are plotted as individual replications. B. Crystal structure PDB 1QPI of TETR in complex with TET operator DNA sequence. Residue interactions (yellow dash), waters (dot spheres), and DNA (orange) highlighted. R7 mutations modeled in purple and annotated with residue number and mutation. C. Codon mutation frequency of all acquired TETR sequences vs. initially packaged variants. Synonymous (blue) and nonsynonymous (red) mutations plotted at each position. D. Codon usage frequencies (%) for wildtype tTA, R7, Homo sapiens (human), Mesocricetus auratus (hamster), and Sindbis plotted as a heat map. The R7-tTA (%) column summarizes the % change in usage for a particular codon triplet between R7 and tTA. Arrows, >10% conversion of R7 codon usage to those more frequently used in humans and hamsters.
Figure S3. VEGAS for the Evolution of GPCRs, Related to Figure 3. A. Residue identity alignment of all human Class A GPCRs across functional domains associated with transition from inactive to active receptor conformation. Ballesteros-Weinstein annotation of residue position was used for alignment. The receptor orphan classification (*) and conservation score (%) are listed. Red, conserved; blue, non- conserved. B. Transcription factor reporter primary screen for MRGPRX2 mediated (+)- Morphine response. RLuc production equates to drug-mediated activation. Left, MRGPRX2 transfected cells. Right, empty vector transfected cells. Data are represented as mean ± SEM. C. Snake plots of MRGPRX2 sequence and predicted secondary structure. Mutants from each round are marked in blue, purple, and orange. The locations of the Na+ pocket (red), ionic lock (green), PIF (yellow), and NPXXY (pink) domains are also highlighted. D. Surface expression enzyme-linked immunosorbent assay (ELISA) to quantify the total expression of MRGPRX2 (WT), untransfected cells (UT), and the MRGPRX2 mutants obtained from VEGAS. Data are represented as mean ± SEM, N=3.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Donkey polyclonal anti-rabbit IgG HRP | Jackson ImmunoResearch | 1711-035-152 |
| Horse polyclonal anti-mouse IgG HRP | Cell Signaling | 7076S |
| Mouse monoclonal anti-FLAG M2- Peroxidase (HRP) | Sigma-Aldrich | A8592 |
| Mouse polyclonal anti-FLAG-M2 | Sigma-Aldrich | F1804 |
| Rabbit polyclonal anti-GFP | Novus Biologicals | NB600–308 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Thermofisher | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (+)-Morphine base | NIDA Drug Supply | 9300–012 |
| [3H]Ketanserine | PerkinElmer | NET791025 |
| [3H]-myo-inositol | PerkinElmer | NET1177001MC |
| [N-Methyl-3H]-Lysergic Acid Diethylamide ([3H]-LSD) | PerkinElmer | NET638250UC |
| 1-(4-iodo-2,5-dimethoxyphenyl)propan-2- amine HCl (DOI) | Tocris | 2643 |
| 1,2-dipalmitoyl-sn-glycero-3- phosphocholine (DPPC) | Avanti Polar Lipids | 850355C |
| 10x Hank’s Buffered Saline Solution (HBSS) | Life Technologies | 14065–056 |
| 3x FLAG Peptide | Sigma-Aldrich | F4799 |
| 4% paraformaldehyde | Fisher | AAJ19943K2 |
| 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) | Sigma-Aldrich | A8456 |
| Aprotinin | Sigma-Aldrich | A1153 |
| Bovine Serum Albumin (BSA), fatty-acid free | Akron Biotech | AK8909 |
| Bright-Glo | Promega | E2620 |
| Carbenicillin | Gold Bio | C-103–25 |
| cholesteryl hemisuccinate (CHS) | Sigma-Aldrich | C6512 |
| Coelenterazine h | Promega | S2011 |
| Decyl Maltose Neopentyl Glycol (DMNG) | Anatrace | NG322 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | 276855 |
| Dopamine HCl | Tocris | 3584 |
| Doxycycline HCl (DOX) | Sigma-Aldrich | D3447 |
| D-Phosphate Buffered Saline (D-PBS), Ca2+/Mg2+ free | ThermoFisher | 14190144 |
| E-64 | Sigma-Aldrich | E3132 |
| GppNHp | Abcam | ab146659 |
| Hygromycin B | KSE | 98–923 |
| imidazole | Sigma-Aldrich | I5513 |
| iodoacetamide | Sigma-Aldrich | I6125 |
| Leupeptin | Sigma-Aldrich | L2884 |
| Lipid A | Sigma-Aldrich | L5399 |
| lysergic acid diethylamide | synthetic | See Wacker et al. 2017b |
| Methiothepin mesylate salt | Sigma-Aldrich | M149 |
| n-dodecyl-β-D-maltopyranoside (DDM) | Anatrace | D319 |
| n-Octyl-β-D-Glucopyranoside | Anatrace | O311 |
| nuclease-free water (H2O) | NEB | B1500 |
| Penicillin/Streptomycin | ThermoFisher | 15140122 |
| Phenoxybenzamine HCl | Sigma-Aldrich | B019 |
| poly-L-lysine | Sigma-Aldrich | P2636 |
| Puromycin | Gemini | 400–128P |
| Serotonin HCl (5-HT) | Sigma-Aldrich | H9523 |
| Trypsin | VWR | 45000–660 |
| Tryptose Phosphate Broth (TPB) | ThermoFisher | T8159 |
| UltraPure Phenol:Chloroform:Isoamyl Alcohol (25:25:1, w/v) | ThermoFisher | 15593031 |
| Versene | ThermoFisher | 15040066 |
| Critical Commercial Assays | ||
| Agencourt AMPure magnetic beads | Beckman Coulter | A63881 |
| Bac-to-Bac Baculovirus Expression System | Invitrogen | 10359016 |
| Bio-Beads SM2 Resin | BioRad | 1523920 |
| Bioruptor Pico sonication device | Diagenode | B01060010 |
| Cellfectin II reagent | ThermoFisher | 10362100 |
| Clarity Western ECL Substrate | BioRad | 1708370 |
| KAPA Hyperprep kit | Roche | KK8500 |
| LDS Gel Loading Buffer | ThermoFisher | NP0007 |
| MagMax Viral RNA Isolation Kit | ThermoFisher | AM1939 |
| Magnetic FLAG-M2 Beads | Sigma-Aldrich | M8823 |
| mMessage mMachine in vitro RNA transcription kit | ThermoFisher | AM1340 |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | E2621 |
| Neon Transfection Kit | ThermoFisher | MPK10096 |
| Neon Transfection System | ThermoFisher | MPK5000 |
| NuPage 4–12% Bis-Tris Protein Gels | ThermoFisher | NP0322 |
| PD MiniTrap G-25 Columns | GE Life Sciences | 28918007 |
| PowerPrep HP Plasmid Maxiprep system | Origene | NP100010 |
| Precision Plus Protein Dual Color Standard | BioRad | 1610374 |
| PreScission Protease | Genscript | Z02799 |
| PrimeSTAR Max DNA polymerase | Takara Bio | R045 |
| QIAprep spin miniprep kits | Qiagen | 27104 |
| QIAquick Gel Extraction Kit | Qiagen | 28115 |
| RNA binding beads (YSI) | PerkinElmer | RPNQ0013 |
| Sartorius Vivaspin 20 Centrifugal Concentrators 100kDa MWCO | Cole-Parmer | VS2002 |
| Sartorius Vivaspin 500 Centrifugal Concentrators 100kDa MWCO | Cole-Parmer | VS0141 |
| SuperScript IV One-Step RT-PCR System | Invitrogen | 12594025 |
| SuperSignal ELISA Pico Chemiluminescent Substrate | Sigma-Aldrich | 37069 |
| TALON Metal Affinity Resin | Takara | 635653 |
| TaqMan Fast Virus 1-Step Master Mix | ThermoFisher | 4444432 |
| TransIT-2020 Transfection Reagent | VWR | MIR5400 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE123269 |
| Experimental Models: Cell Lines | ||
| Hamster: BHK21 | ATCC | CCL-10 |
| Human: HEK293T | ATCC | CRL-3216 |
| Human: HEK-Gα11Δ/sΔ/olfΔ/12Δ/13Δ | gift | Asuka Inoue, Tohoku |
| Human: HEK-P | gift | Asuka Inoue, Tohoku University |
| Human: HTLA | gift | Richard Axel, Columbia University |
| Insect: Sf9 cells, suspension in ESF 921 1M cells/mL | Expression Systems | 94–001S |
| Oligonucleotides | ||
| See Table S6 | - | - |
| Recombinant DNA | ||
| See Table S6 | - | - |
| Other | ||
| 0.45uM media filters | EMD Millipore | SCGP00525 |
| 10 μg/mL Carbenicillin supplemented LB agar plates | Teknova | L1010 |
| 24-well plates | Sigma-Aldrich | CLS3527 |
| 384-well white plates | Black Dog | 781098 |
| 5% normal goat serum | Vector Laboratories | S-1000 |
| 96-well white plates | Black Dog | 655098 |
| BamHI-HF | NEB | R3136 |
| Canted neck culture flasks | Sigma-Aldrich | CLS430641U |
| ClaI | NEB | R0197 |
| CutSmart Buffer | NEB | B7204 |
| Dulbecco’s Modified Eagle Media (DMEM) | VWR | 45000 |
| Fetal Bovine Serum (FBS) | VWR | 89510–186 |
| Filtermat A, GF/C | PerkinElmer | 1450–421 |
| Immobilon PVDF membranes | Sigma-Aldrich | IPSN07852 |
| LB Broth | ThermoFisher | 10855001 |
| MeltiLex Solid Scintillant, for Microbeta | PerkinElmer | 1450–441 |
| MEM-α with nucleosides | ThermoFisher | 32571036 |
| myo-inositol-free DMEM | Caisson Labs | DML13 |
| NheI-HF | NEB | R3131 |
| NotI-HF | NEB | R3189 |
| PET-A, FLEX, 96-well clear sample plates | PerkinElmer | 1450–401C |
| RNAsin | Promega | N2111 |
| Sf-900 II SFM media | ThermoFisher | 10902096 |
| T7 DNA Ligase | NEB | M0318 |
| XbaI | NEB | R0145 |
Highlights.
One day per round of directed molecular evolution in mammalian cells
Mutation rates of 10−3 from each round, surpassing many in vitro systems
System does not require resetting or recycling of hits
Three unique campaigns are presented, each succeeding in less than a week
ACKNOWLEDGEMENTS
This work was supported by NIH grant 1U01MH105892–01, RO1MH112205, R37DA045657 and a grant from the University of North Carolina Lineberger Comprehensive Cancer Center to B.L.R. We thank our colleague Gary Johnson for providing the resources for our RNAseq analysis. We also thank our colleague Mark Heise for providing the initial Sindbis virus vectors used in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
EDITOR’S IN BRIEF:
VEGAS, a platform for directed evolution in mammalian cells, overcomes limitations of previous approaches to allow evolution of multiple classes of proteins, including GPCRs.
REFERENCES
- Agapov EV, Frolov I, Lindenbach BD, Prágai BM, Schlesinger S, and Rice CM (1998). Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc. Natl. Acad. Sci. U. S. A 95, 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TJ, Dailidiene D, Dailide G, Norton JE, Kalia A, Richmond TA, Molla M, Singh J, Green RD, and Berg DE (2005). Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat. Methods 2, 951. [DOI] [PubMed] [Google Scholar]
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, and Milligan G (2016). Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J. Biol. Chem 291, 27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, et al. (2007). A module of negative feedback regulators defines growth factor signaling. Nat. Genet 39, 503–512. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold FH (1998). Design by Directed Evolution. Acc. Chem. Res 31, 125–131. [Google Scholar]
- Badran AH, and Liu DR (2015). In Vivo Continuous Directed Evolution. Curr. Opin. Chem. Biol 0, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, and Kishony R (2016). Spatiotemporal microbial evolution on antibiotic landscapes. Science 353, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar M, Hao N, Dohlman HG, and Elston TC (2007). Mathematical and Computational Analysis of Adaptation via Feedback Inhibition in Signal Transduction Pathways. Biophys. J 93, 806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens C, Altschmied L, and Hillen W (1992). The role of the N terminus in Tet repressor for tet operator binding determined by a mutational analysis. J. Biol. Chem 267, 1945–1952. [PubMed] [Google Scholar]
- Bourdon DM, Wing MR, Edwards EB, Sondek J, and Harden TK (2006). Quantification of Isozyme-Specific Activation of Phospholipase C-β2 by Rac GTPases and Phospholipase CVε by Rho GTPases in an Intact Cell Assay System. In Methods in Enzymology, (Academic Press), pp. 489–499. [DOI] [PubMed] [Google Scholar]
- Bredenbeek PJ, Frolov I, Rice CM, and Schlesinger S (1993). Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J. Virol 67, 6439–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F, Angrand P-O, and Stewart AF (1998). Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol 16, 657–662. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, and Tsien RY (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A 99, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JC, Badran AH, Guggiana-Nilo DA, and Liu DR (2014). Negative selection and stringency modulation in phage-assisted continuous evolution. Nat. Chem. Biol 10, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu W-L, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, et al. (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, et al. (2018). Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172, 55–67.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, and Arnold FH (1993). Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc. Natl. Acad. Sci 90, 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crameri A, Whitehorn EA, Tate E, and Stemmer WPC (1996). Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol 14, 315–x1. [DOI] [PubMed] [Google Scholar]
- Darwin C, and Bynum WF (2009). The origin of species by means of natural selection: or, the preservation of favored races in the struggle for life (AL Burt).
- Das AT, Zhou X, Vink M, Klaver B, Verhoef K, Marzio G, and Berkhout B (2004). Viral Evolution as a Tool to Improve the Tetracycline-regulated Gene Expression System. J. Biol. Chem 279, 18776–18782. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, and Lefkowitz RJ (1980). A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem 255, 7108–7117. [PubMed] [Google Scholar]
- Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang J-M, Taly J-F, and Notredame C (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J (2002). Evolution, consequences and future of plant and animal domestication. Nat. Lond 418, 700–707. [DOI] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Drake JW, and Holland JJ (1999). Mutation rates among RNA viruses. Proc. Natl. Acad. Sci 96, 13910–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, and Crow JF (1998). Rates of Spontaneous Mutation. Genetics 148, 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll CA, Macdonald DW, and O’Brien SJ (2009). From Wild Animals to Domestic Pets, an Evolutionary View of Domestication. Proc. Natl. Acad. Sci. U. S. A 106, 9971–9978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobizhev M, Makarov NS, Tillo SE, Hughes TE, and Rebane A (2011). Two-photon absorption properties of fluorescent proteins. Nat. Methods 8, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011). Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic Reprogramming of the Kinome In Response to Targeted MEK Inhibition In Triple Negative Breast Cancer. Cell 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JG, Shellhammer JP, Malahe M, McCarter PC, Elston TC, and Dohlman HG (2015). MAPK feedback encodes a switch and timer for tunable stress adaptation in yeast. Sci Signal 8, ra5–ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Carlson JC, and Liu DR (2011). A system for the continuous directed evolution of biomolecules. Nature 472, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, and Sali A (2009). Molecular Docking Screens Using Comparative Models of Proteins. J. Chem. Inf. Model 49, 2512–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE (2002). Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol. 14, 140–148. [DOI] [PubMed] [Google Scholar]
- Filizola M, and Devi LA (2013). Grand opening of structure-guided design for novel opioids. Trends Pharmacol. Sci 34, 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin L-G, and Schiöth HB (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- Frolov I, Hardy R, and Rice CM (2001). Cis-acting RNA elements at the 5’ end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA N. Y. N 7, 1638–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GarciãMoreno M, Sanz MA, Pelletier J, and Carrasco L (2013). Requirements for eIF4A and eIF2 during translation of Sindbis virus subgenomic mRNA in vertebrate and invertebrate host cells. Cell. Microbiol 15, 823–840. [DOI] [PubMed] [Google Scholar]
- Garrington TP, and Johnson GL (1999). Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol 11, 211–218. [DOI] [PubMed] [Google Scholar]
- Gilman AG (1987). G Proteins: Transducers of Receptor-Generated Signals. Annu. Rev. Biochem 56, 615–649. [DOI] [PubMed] [Google Scholar]
- Gossen M, and Bujard H (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U. S. A 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, and Bujard H (1995). Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- Hammer SC, Kubik G, Watkins E, Huang S, Minges H, and Arnold FH (2017). Anti-Markovnikov alkene oxidation by metal-oxo-mediated enzyme catalysis. Science 358, 215–218. [DOI] [PubMed] [Google Scholar]
- Hanes J, and Plückthun A (1997). In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci 94, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, and Reid B (1970). Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. U. S. A 66, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht B, Müller G, and Hillen W (1993). Noninducible Tet repressor mutations map from the operator binding motif to the C terminus. J. Bacteriol 175, 1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GT, Frésard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, and Bassik MC (2016). Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods N. Y 13, 1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell AS, Jin M, Wu C-F, Zyla TR, Elston TC, and Lew DJ (2012). Negative Feedback Enhances Robustness in the Yeast Polarity Establishment Circuit. Cell 149, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MJ, and Summers J (1991). Infection initiated by the RNA pregenome of a DNA virus. J. Virol 65, 5435–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling R, et al. (2015a). Structural insights into μ-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X-P, Setola V, Yadav PN, Allen JA, Rogan SC, Hanson BJ, Revankar C, Robers M, Doucette C, and Roth BL (2009). Parallel Functional Activity Profiling Reveals Valvulopathogens Are Potent 5-Hydroxytryptamine2B Receptor Agonists: Implications for Drug Safety Assessment. Mol. Pharmacol 76, 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X-P, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, et al. (2015b). Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston M (1979). A General Hypothesis of Species Diversity. Am. Nat 113, 81–101. [Google Scholar]
- Isberg V, Graaf C. de, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin J-P, Stevens RC, Vriend G, et al. (2015). Generic GPCR residue numbers – aligning topology maps while minding the gaps. Trends Pharmacol. Sci 36, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan SBJ, Lewis RD, Chen K, and Arnold FH (2016). Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life. Science 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Vriend G, van der Kant R, and Vriend G (2014). Alpha-Bulges in G Protein-Coupled Receptors. Int. J. Mol. Sci 15, 7841–7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, and Stevens RC (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci 39, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisker C, Hinrichs W, Tovar K, Hillen W, and Saenger W (1995). The complex formed between Tet repressor and tetracycline-Mg2+ reveals mechanism of antibiotic resistance. J. Mol. Biol 247, 260–280. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, et al. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, and Deupi X (2007). Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci 28, 397–406. [DOI] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang X-P, Lansu K, McCorvy JD, Giguère PM, Sciaky N, and Roth BL (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchner O, and Arnold FH (1997). Directed evolution of enzyme catalysts. Trends Biotechnol. 15, 523–530. [DOI] [PubMed] [Google Scholar]
- Lansu K, Karpiak J, Liu J, Huang X-P, McCorvy JD, Kroeze WK, Che T, Nagase H, Carroll FI, Jin J, et al. (2017). In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol 13, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Venkatakrishnan AJ, and Dror RO (2017). GPCR Dynamics: Structures in Motion. Chem. Rev 117, 139–155. [DOI] [PubMed] [Google Scholar]
- Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim Y, Lee K, Jung I, Kim D, Kim S, et al. (2018). Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun 9, 3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles P-Y, Gauthier C, Lee M-H, Pani B, et al. (2018). Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 11, eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshri N, Koerber JT, Kaspar BK, and Schaffer DV (2006). Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol 24, 198–204. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. (2015). Structural insights into the dynamic process of ȕ2-adrenergic receptor signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Chen R, Anikeeva P, and Jasanoff A (2015). Engineering intracellular biomineralization and biosensing by a magnetic protein. Nat. Commun 6, 8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, and Roth BL (2015). Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther 150, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C, Baier AS, Pascolutti R, Wegrecki M, Zheng S, Ong JX, Erlandson SC, Hilger D, Rasmussen SGF, Ring AM, et al. (2018). Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol 25, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, and Shi L (2015). What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev 67, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley VJ, and Turner PE (2017). Dynamics of molecular evolution in RNA virus populations depend on sudden versus gradual environmental change: RNA VIRUS EVOLUTIONARY DYNAMICS IN CHANGING ENVIRONMENTS. Evolution 71, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller G, Hecht B, Helbl V, Hinrichs W, Saenger W, and Hillen W (1995). Characterization of non-inducible Tet repressor mutants suggests conformational changes necessary for induction. Nat. Struct. Biol 2, 693–703. [DOI] [PubMed] [Google Scholar]
- Muyldermans S, Cambillau C, and Wyns L (2001). Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem. Sci 26, 230–235. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, and Ikemura T (2000). Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR (2003). International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev 55, 597–606. [DOI] [PubMed] [Google Scholar]
- Onaran HO, and Costa T (2009). Allosteric coupling and conformational fluctuations in proteins. Curr. Protein Pept. Sci 10, 110–115. [DOI] [PubMed] [Google Scholar]
- Orth P, Cordes F, Schnappinger D, Hillen W, Saenger W, and Hinrichs W (1998). Conformational changes of the Tet repressor induced by tetracycline trapping11Edited by R. Huber. J. Mol. Biol 279, 439–447. [DOI] [PubMed] [Google Scholar]
- Orth P, Schnappinger D, Sum PE, Ellestad GA, Hillen W, Saenger W, and Hinrichs W (1999a). Crystal structure of the tet repressor in complex with a novel tetracycline, 9-(N,N-dimethylglycylamido)-6-demethyl-6-deoxy-tetracycline. J. Mol. Biol 285, 455–461. [DOI] [PubMed] [Google Scholar]
- Orth P, Saenger W, and Hinrichs W (1999b). Tetracycline-chelated Mg2+ ion initiates helix unwinding in Tet repressor induction. Biochemistry 38, 191–198. [DOI] [PubMed] [Google Scholar]
- Orth P, Schnappinger D, Hillen W, Saenger W, and Hinrichs W (2000). Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat. Struct. Biol. N. Y 7, 215–219. [DOI] [PubMed] [Google Scholar]
- Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsøe K, Hauser AS, Bojarski AJ, and Gloriam DE (2018). GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlkönig A, Ruf A, Muyldermans S, Hol WGJ, Kobilka BK, and Steyaert J (2014). A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc 9, 674–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T, and Scott JD (1997). Signaling Through Scaffold, Anchoring, and Adaptor Proteins. Science 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Peng Y, McCorvy JD, Harpsøe K, Lansu K, Yuan S, Popov P, Qu L, Pu M, Che T, Nikolajsen LF, et al. (2018). 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 172, 719–730.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Suk H-J, Kodandaramaiah SB, Yoshida F, DeGennaro EM, Drobizhev M, Hughes TE, Desimone R, Boyden ES, and Verkhusha VV (2017). Near-Infrared Fluorescent Proteins Engineered from Bacterial Phytochromes in Neuroimaging. Biophys. J 113, 2299–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, and Lefkowitz RJ (2002). Signalling: Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol 3, 639–650. [DOI] [PubMed] [Google Scholar]
- Purvis JE, and Lahav G (2013). Encoding and Decoding Cellular Information through Signaling Dynamics. Cell 152, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SGF, Choi H-J, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, et al. (2007). Crystal structure of the human ȕ2 adrenergic G-protein-coupled receptor. Nature 450, 383–387. [DOI] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero PA, and Arnold FH (2009). Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL (2016). DREADDs for Neuroscientists. Neuron 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Irwin JJ, and Shoichet BK (2017). Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol 13, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sane J, Kurkela S, Levanov L, Nikkari S, Vaheri A, and Vapalahti O (2012). Development and evaluation of a real-time RT-PCR assay for Sindbis virus detection. J. Virol. Methods 179, 185–188. [DOI] [PubMed] [Google Scholar]
- Sanjuan R, Nebot MR, Chirico N, Mansky LM, and Belshaw R (2010). Viral Mutation Rates. J. Virol 84, 9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, and Plückthun A (2008). Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc. Natl. Acad. Sci 105, 14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger S (1993). Alphaviruses — vectors for the expression of heterologous genes. Trends Biotechnol. 11, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell MJ, Buonocore L, Whitt MA, and Rose JK (1996). The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol 70, 2318–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz O, Henßler E-M, Bail J, Schubert P, BogdanskãUrbaniak J, Sopp S, Reich M, Wisshak S, Köstner M, Bertram R, et al. (2004). Activity reversal of Tet repressor caused by single amino acid exchanges. Mol. Microbiol 53, 777–789. [DOI] [PubMed] [Google Scholar]
- Schubert P, Schnappinger D, Pfleiderer K, and Hillen W (2001). Identification of a Stability Determinant on the Edge of the Tet Repressor Four-Helix Bundle Dimerization Motif †. Biochemistry 40, 3257–3263. [DOI] [PubMed] [Google Scholar]
- Schütz M, Schöppe J, Sedlák E, Hillenbrand M, Nagy-Davidescu G, Ehrenmann J, Klenk C, Egloff P, Kummer L, and Plückthun A (2016). Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts. Sci. Rep 6, 21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, and Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, and Tsien RY (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MG, Westmeyer GG, Romero PA, Szablowski JO, Küster B, Shah A, Otey CR, Langer R, Arnold FH, and Jasanoff A (2010). Directed evolution of a magnetic resonance imaging contrast agent for noninvasive imaging of dopamine. Nat. Biotechnol 28, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LD, and Bertrand KP (1988). Mutations in the Tn10 tet represser that interfere with induction: Location of the tetracycline-binding domain. J. Mol. Biol 203, 949–959. [DOI] [PubMed] [Google Scholar]
- Smith TF, and Waterman MS (1981). Identification of common molecular subsequences. J. Mol. Biol 147, 195–197. [DOI] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Strachan RT, Rasmussen SGF, Pardon E, Ahn S, Steyaert J, Kobilka BK, and Lefkowitz RJ (2014). Regulation of ȕ2-Adrenergic Receptor Function by Conformationally Selective Single-Domain Intrabodies. Mol. Pharmacol 85, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, et al. (2016). Allosteric Nanobodies Reveal the Dynamic Range and Diverse Mechanisms of GPCR Activation. Nature 535, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan RT, Sun J, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, and Lefkowitz RJ (2014). Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem 289, 14211–14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, and Strauss EG (1994). The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev 58, 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss EG, Rice CM, and Strauss JH (1984). Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 133, 92–110. [DOI] [PubMed] [Google Scholar]
- Subramaniam M, Schmidt LJ, Crutchfield Iii CE, and Getz MJ (1989). Negative regulation of serum-responsive enhancer elements. Nature 340, 64–66. [DOI] [PubMed] [Google Scholar]
- Tang J, Jose J, Chipman P, Zhang W, Kuhn RJ, and Baker TS (2011). Molecular Links between the E2 Envelope Glycoprotein and Nucleocapsid Core in Sindbis Virus. J. Mol. Biol 414, 442–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, and Kishony R (2012). Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet 44, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracewell CA, and Arnold FH (2009). Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol 13, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, and Hillen W (2000). Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci 97, 7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, et al. (2016). Distinct cortical and striatal actions of a β-arrestin–biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnaitơ R, and MacNeill SA (2016). Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. PROTEOMICS 16, 2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang X-P, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013). Structural features for functional selectivity at serotonin receptors. Science 340, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, and Roth BL (2017a). How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, et al. (2017b). Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AR (1855). XVIII.—On the law which has regulated the introduction of new species. Ann. Mag. Nat. Hist 16, 184–196. [Google Scholar]
- Wallace AR (1871). Contributions to the theory of natural selection (Macmillan; ). [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang X-P, Dror RO, Shoichet BK, et al. (2017). D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissmann A, Baumeister R, Müller G, Hecht B, Helbl V, Pfleiderer K, and Hillen W (1991). Amino acids determining operator binding specificity in the helix-turn-helix motif of Tn10 Tet repressor. EMBO J. 10, 4145–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, and Gaut BS (2005). The Effects of Artificial Selection on the Maize Genome. Science 308, 1310–1314. [DOI] [PubMed] [Google Scholar]
- Xiong C, Levis R, Shen P, Schlesinger S, Rice CM, and Huang HV (1989). Sindbis virus: an efficient, broad host range vector for gene expression in animal cells. Science 243, 1188–1191. [DOI] [PubMed] [Google Scholar]
- Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T, Lim AC, Liu H, Lohse PA, Sun L, et al. (2002). Directed Evolution of High-Affinity Antibody Mimics Using mRNA Display. Chem. Biol 9, 933–942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S4. VEGAS for Evolution of Active-State Nanobodies, Related to Figure 4. VEGAS was used to develop nanobodies that selectively activate diverse GPCR targets from a single cDNA library. A-F. Transcription factor reporter primary screen. Cells transfected with GPCRs (A,C,E) or pCDNA3.1 (B,D,F), and the indicated transcription response element (TRE) were stimulated. TRE-RLuc production equates to drug- mediated activation of the GPCR, quantified over time. Data are represented as mean ± SEM, N=1.
Figure S5. Positive Allosteric Modulation of GPCRs by VEGAS-Evolved Nanobodies, Related to Figure 5. VEGAS derived nanobodies were tested for direct association and allosteric modulation of their targets. A. Bioluminescence resonance energy transfer (BRET) association assay between GPCR-RLuc constructs and mVenus-VGS-Nb2 in the presence of increasing concentrations of 5HT. Data are represented as mean ± SEM, N=3. B. Co-immunoprecipitation determination from FLAG pull-down by Western blot analysis. Loaded (L), unbound (U), washed, (W), and eluted (E) fractions annotated. C. Bioluminescence resonance energy transfer (BRET) association assay between DRD2-RLuc or 5HT2A-RLuc and mVenus-VGS-Nb8 at increasing transfection ratios of nanobody. Data are represented as mean ± SEM, N=3 D. Bioluminescence resonance energy transfer (BRET) association assay between DRD2-RLuc or 5HT2A-RLuc and mVenus-VGS-Nb7 at increasing transfection ratios of nanobody. Data are represented as mean ± SEM, N=3. E. Co-immunoprecipitation determination from FLAG pull-down by LC-MS/MS analysis. 5HT2A, blue; mVenus, green; VGS-Nb2, orange; DRD2, red; other peptides, grey. Data are represented as total background subtracted spectral counts, N=2. See Table S5 for raw data. F. Calcium flux assay. Calcium mobilization equates to relative GPCR-mediated Gαq activity. Data are represented as mean ± SEM, N=3. G-I. Bioluminescence resonance energy transfer (BRET) association assay between 5HT2A-RLuc and mVenus- bArrestin-2 in the presence of increasing concentrations of DOI. H receptor only, I VGS-Nb2 co-transfected, G linear regression of area under the curve datasets for G, H, and additional nanobody concentrations. Data are represented as mean ± SEM, N=3. J. Serum response element (SRE) reporter assay. DRD2 alone or co-transfected with 5x VGS-Nb7 or VGS-Nb8. RLuc production equates to relative receptor activation. Data are represented as mean ± SEM, N=2.
Table S1. Evolution derived TETR sequences, Related to Figure 2. FASTA formatted list of TETR sequences obtained through evolution of tTA over multiple rounds with increasing doxycycline concentrations as outlined in Figure 2A. Names identify the sequence by round number (R#) and well location (_A-H,1–12).
Table S2. Previously reported TETR mutations from R7, Related to Figure 2. Mutations identified in the R7 consensus sequence are indexed. pIǻ, isoelectric point change from original to mutant sequence. @_, non-identical mutations published at the same position, published amino acids listed after the @. Δ, identical mutations published at the same position. D, TETR(B) to TETR(D) mutation.
Table S3. Alignment of MRGPRX2 to critical residues of canonical opiate receptors, Related to Figure 3. Alignment of μ opioid receptor (MOP), į opioid receptor (DOP), ț opioid receptor (KOP), nociceptin receptor (NOP), and MRGPRX2 amino acid sequences at key functional residues. Residues include conserved (Ł 3, gray highlight), ligand interacting (bold green text, adapted from Filizola and Devi, 2013), sodium pocket (Na+, adapted from Katritch et al., 2014), DRY, PIF, CWxP, and NPxxY domains. Mutations generated through VEGAS evolution are also included for reference.
Table S4. Deposited Llama glama amino acid sequences, Related to Figure 4. Sequences retrieved from GenBank and PDB databases and displayed in FASTA format.
Table S5. LC-MS/MS Raw Data, Related to Figure 5. Data from pull-down of FLAG-5HT2A, FLAG-DRD2, or FLAG-free cells provided as both spectral counts and total peptide counts. Target proteins (red font) are highlighted.
Table S6. Plasmids used in this study, Related to STAR Methods. List of plasmids used in this study, both pre-existing and generated in this work. Plasmid name, source, description, genbank (GBK) map, and primers used for construction are listed.
Data S1. VEGAS Resource Overview, a streamlined graphical outline of the VEGAS selection process, which is described in detail within Data S2 and related to Figures 1–5.
Data S2. VEGAS Resource Protocol, a complete protocol for the implementation of the VEGAS platform, as outlined in Data S1, related to Figures 1–5, and described in the STAR Methods Section “Sindbis Virus Production” and “Sustained Passage of Sindbis for Directed Evolution and Transgene Isolation”.
Figure S1. Sindbis Virus for Facile Directed Evolution in Mammalian Cell Culture, Related to Figure 1. A. Epifluorescence microscope images of BHK-21 cells in culture 0, 4, 12, and 24 hrs after application of pTSin-EGFP virus. B. Mutations observed per base measured via Illumina paired-end sequencing at each position in the EGFP transgene over time. Data are plotted as individual replicates. C. Read distribution from Illumina paired-end sequencing per position in the EGFP transgene over time. Data are plotted as individual replicates. D. Insert number and size and deletion number observed from sequencing of Sindbis packaged EGFP transgene over time. Data are plotted as individual replicates. E. Mutations observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band. F. Insertions observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band. E. Deletions observed per read length from sequencing of Sindbis packaged EGFP transgene over time. Data are represented individually and as linear regression, shading represents 95% confidence band.
Figure S2. Directed Evolution of Transcription Factors with Sindbis, Related to Figure 2. A. qRT-PCR quantification of Sindbis virus production from cell culture. BHK- 21 cells transfected with TETO7-SSG (red) or TETO7-GFP (blue) were infected with pTSin-tTA and treated with 1μM Doxycycline (filled circles) or vehicle (open circles). Data are plotted as individual replications. B. Crystal structure PDB 1QPI of TETR in complex with TET operator DNA sequence. Residue interactions (yellow dash), waters (dot spheres), and DNA (orange) highlighted. R7 mutations modeled in purple and annotated with residue number and mutation. C. Codon mutation frequency of all acquired TETR sequences vs. initially packaged variants. Synonymous (blue) and nonsynonymous (red) mutations plotted at each position. D. Codon usage frequencies (%) for wildtype tTA, R7, Homo sapiens (human), Mesocricetus auratus (hamster), and Sindbis plotted as a heat map. The R7-tTA (%) column summarizes the % change in usage for a particular codon triplet between R7 and tTA. Arrows, >10% conversion of R7 codon usage to those more frequently used in humans and hamsters.
Figure S3. VEGAS for the Evolution of GPCRs, Related to Figure 3. A. Residue identity alignment of all human Class A GPCRs across functional domains associated with transition from inactive to active receptor conformation. Ballesteros-Weinstein annotation of residue position was used for alignment. The receptor orphan classification (*) and conservation score (%) are listed. Red, conserved; blue, non- conserved. B. Transcription factor reporter primary screen for MRGPRX2 mediated (+)- Morphine response. RLuc production equates to drug-mediated activation. Left, MRGPRX2 transfected cells. Right, empty vector transfected cells. Data are represented as mean ± SEM. C. Snake plots of MRGPRX2 sequence and predicted secondary structure. Mutants from each round are marked in blue, purple, and orange. The locations of the Na+ pocket (red), ionic lock (green), PIF (yellow), and NPXXY (pink) domains are also highlighted. D. Surface expression enzyme-linked immunosorbent assay (ELISA) to quantify the total expression of MRGPRX2 (WT), untransfected cells (UT), and the MRGPRX2 mutants obtained from VEGAS. Data are represented as mean ± SEM, N=3.