

Summary
B lymphocytes undergo metabolic reprogramming upon activation to meet the bioenergetic demands for proliferation and differentiation. Yet, little is known if and how the fate of naive B cells is metabolically regulated. Here, we specifically delete von Hippel-Lindau protein (VHL) in B cells using CD19-Cre and demonstrate that metabolic balance is essential for naive B cell survival. Loss of VHL disturbs glycolytic and oxidative metabolic balance and causes severe reduction in mature B cells. Mechanistically, the metabolic imbalance in VHL-deficient B cells, arising from over-stabilization of hypoxia-inducible factor-1α (HIF-1α), triggers reductive glutamine metabolism leading to increased Fas palmitoylation and caspase-8-mediated apoptosis. Blockade of reductive glutamine metabolic flux by lactate supplementation and ATP citrate lyase inhibition restores the metabolic balance and rectifies the impaired survival of VHL-deficient B cells. Hence, we unravel that the VHL/HIF-1α pathway is required to maintain the metabolic balance of naive B cells and ensure their survival.
Subject Areas: Biological Sciences, Immunology, Cell Biology
Graphical Abstract
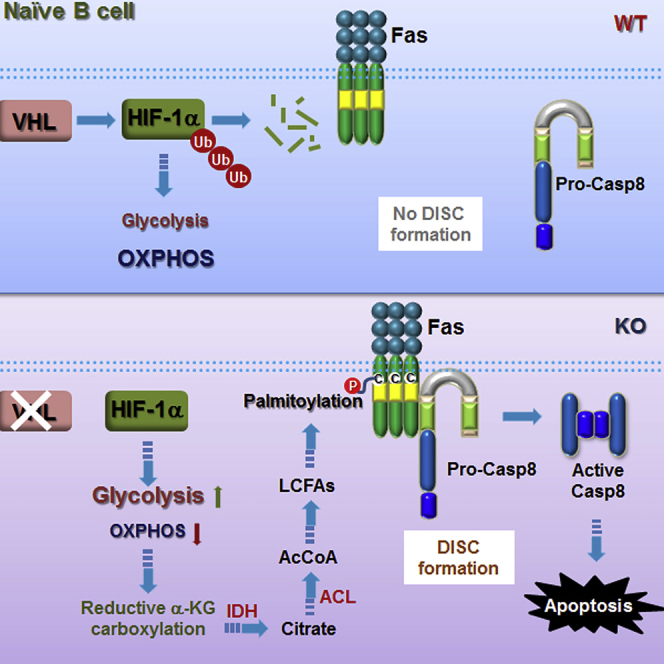
Highlights
-
•
vHL ablation in naive B cells leads to diminishment of mature B cell populations
-
•
B cells lacking vHL manifest perturbed metabolism and impaired survival
-
•
vHL deficiency in B cells triggers reductive carboxylation of α-KG
-
•
Metabolic rewiring in vHL-deficient naive B cells causes caspase-8 activation
Biological Sciences; Immunology; Cell Biology
Introduction
B lymphocytes develop from committed hematopoietic stem cells in the bone marrow (BM) and populate peripheral lymphoid organs as naive B cells. Upon activation by foreign antigens, naive B cells undergo intensive clonal expansion and differentiate into memory B and antibody-secreting plasma cells that provide an individual with lifelong humoral immunity (Rajewsky, 1996, Goodnow et al., 2010). Recent studies demonstrate that B cells undertake metabolic reprogramming upon activation to meet the bioenergetic demands for proliferation and differentiation (Boothby and Rickert, 2017, Olenchock et al., 2017). Upon B cell receptor cross-linking or stimulation with interleukin-4 or lipopolysaccharide (LPS) in vitro, glucose uptake and glycolysis are rapidly increased in B cells (Doughty et al., 2006, Dufort et al., 2007, Caro-Maldonado et al., 2014). Concurrently, glucose oxidation is increased in the activated B cells (Caro-Maldonado et al., 2014). When B cells are stimulated by proteinaceous antigens and differentiate into germinal center (GC) B cells in vivo, both glucose uptake and mitochondrial biogenesis are increased (Jellusova et al., 2017). This indicates that activated B cells undergo a balanced glycolytic and oxidative metabolic reprogramming, which differs from the disproportionally increased glycolytic metabolism in activated T cells (Wang et al., 2011). However, it remains unclear if and how metabolism affects the fate of quiescent naive B cells.
Hypoxia-inducible factor 1 (HIF-1) is a basic-helix-loop-helix transcription factor that regulates the expression of genes involved in metabolism and adaption to low oxygen stress. HIF-1 plays multifaceted roles in innate and adaptive immunity (Palazon et al., 2014). It is important for neutrophil survival and bactericidal activity of macrophages (Cramer et al., 2003) and essential for regulating glycolysis in CD4+ T cells and Th17 cell differentiation (Shi et al., 2011, Dang et al., 2011). HIF-1 is heterodimeric and comprises an inducibly expressed oxygen-sensitive α subunit and a constitutively expressed β subunit. Its activity is largely determined by protein stability of the α subunit. Under normoxia, HIF-1α is hydroxylated at specific proline residues and associates with von Hippel-Lindau tumor suppressor protein (VHL), which is the substrate recognition subunit of an E3 ubiquitin-protein ligase, and subsequently ubiquitinated and degraded (Semenza, 2001). Loss of VHL leads to constitutive stabilization of HIF-1α and upregulation of HIF-1's target genes even under normoxia. Mutations in vhl gene are found in patients with von Hippel-Lindau disease and in various spontaneous renal cell carcinomas (RCCs) (Gallou et al., 1999, Lemm et al., 1999).
Deletion of VHL disturbs cellular metabolism and affects activation and differentiation of various immune cells. For example, deletion of VHL in Treg cells results in their acquiring Th1-like inflammatory properties with excessive interferon-γ production due to augmented glycolysis (Lee et al., 2015). VHL deficiency in CD8+ T cells enhances their effector response against chronic infection and tumor growth by averting T cell exhaustion (Doedens et al., 2013). Recent studies showed that HIF-1 over-stabilization by VHL deletion inhibits activation-induced deaminase expression and mTORC1 activation and compromises B cell differentiation in GC (Abbott et al., 2016, Cho et al., 2016), suggesting that VHL is important for B cell activation.
Here, we investigate VHL's role in naive B cells, in which Vhl is specifically deleted in B cells using CD19-Cre. We show that VHL ablation causes imbalanced glycolytic and oxidative metabolism in quiescent naive B cells and leads to reduced mature B cell populations. Strikingly, VHL-deficient B cells manifest augmented caspase-8-dependent apoptosis. The metabolism and survival defects of VHL-deficient B cells are largely due to HIF-1α over-stabilization and could be rectified by its deletion. We further demonstrate that the metabolic imbalance in naive VHL-deficient B cells triggers reductive glutamine metabolism, causing constitutive Fas palmitoylation and caspase-8 activation. These data suggest that the VHL-HIF-1α pathway is important for the survival of naive B cells by maintaining metabolic balance and restraining caspase-8 activation.
Results
Reduced Mature B Cell Populations in VHL-Deficient Mice
To investigate the role of VHL in naive B cells, we crossed mice bearing loxP-flanked (floxed) Vhl alleles with Cd19Cre/+ mice, which express Cre recombinase specifically in B cells, to generate Vhlfl/flCd19Cre/+ (VHL-deficient) mice. These mice were grossly normal and had no obvious phenotype when compared with wild-type Vhl+/+Cd19Cre/+ (WT) mice. Quantitative reverse-transcriptase PCR revealed that Vhl mRNA was significantly decreased in splenic B cells of VHL-deficient compared with control mice (Figure S1A). Immunoblotting further showed barely detectable VHL but markedly increased HIF-1α protein in VHL-deficient compared with WT B cells (Figure S1B). The level of HIF-2α, which can be targeted by VHL, was also elevated in VHL-deficient splenic B cells. These results suggest that VHL is efficiently deleted and HIF proteins are accumulated in mutant B cells.
Next, we assessed if VHL deficiency would perturb B cell development in the BM of VHL-deficient mice, as B cell development was impaired in the BM of HIF-1α-deficient RAG2 chimeric mice (Kojima et al., 2002). Flow cytometric analysis revealed that the total cellularity of BM and the frequency and absolute number of IgM+IgDlow/- immature B cells, which are c-kit- and correspond to Hardy fraction E (data not shown), are normal in VHL-deficient mice (Figures S1C and S1D left, Figures 1A and 1B left). In contrast, the population of IgM+IgDhigh mature B cells, which are c-kit- and correspond to Hardy fraction F (data not shown), was significantly reduced in VHL-deficient mice (Figure S1D right, Figures 1A and 1B right). However, VHL-deficient and WT mice had comparable IgM−IgD-B220+CD43+ pro-B and IgM−IgD- B220+CD43- pre-B cell populations (Figures S1E–S1G), which are c-kit+ and correspond to Hardy fractions A–C and fraction D, respectively (data not shown). Although it is not clear whether VHL is important for early B cell populations, due to apparently less efficient CD19-Cre-mediated Vhl deletion in these cells (50%–65%) compared with the deletion in mature B cells (>75%) (Figure S1H) and the considerable amount of VHL proteins present in these cells (Figure S1I), our results clearly suggest that VHL is required for mature B cells in the BM.
Figure 1.

Reduced Mature B Cell Populations in VHL-Deficient Mice
(A, C, and F) Flow cytometric analysis of B cells in various lymphoid organs of WT and VHL-deficient mice. Numbers adjacent to outline areas indicate the percentage of gated cells over total cells in the BM (A), spleen (Spl) (C, upper panel), inguinal lymph nodes (LN) (C, lower panel), and peritoneal cavities (F).
(B and D) Enumeration of B cells in various lymphoid organs of WT and VHL-deficient mice. Number of IgM+IgD− immature and IgM+IgD+ mature B cells in the BM (B) and total IgM+B220+ B cells in the spleen and lymph nodes (D) of WT (red) and VHL-deficient (blue) mice was calculated based on total cell count and their percentage as determined by flow cytometry.
(E) Microscopy of spleen sections of unchallenged WT and VHL-deficient mice. B cell follicles (red), T cell zones (green), and red pulps (blue) were stained with anti-IgD, anti-CD3, and F4/80 antibodies, respectively. Scale bar, 500 μm.
(G) Flow cytometric analysis of B cell survival in vitro. WT and VHL-deficient B cells were cultured for 16 h and analyzed by flow cytometry for Annexin V and propidium iodide (PI) staining.
(H) Percentage of PI− Annexin V− live B cells in different cultures. B cells were cultured in medium alone or in the presence of anti-IgM (20 μg/mL) or anti-CD40 (1 μg/mL) antibodies or LPS (100 ng/mL) for 16 h and examined by flow cytometry. Each symbol represents one mouse, and the height of bars indicates the mean.
Data shown are representative of more than five independent experiments (A, n = 9; C, n = 10 for spleen and n = 8 for lymph nodes; E, n = 5; F, n = 8; and G, n = 6). n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t test). See also Figure S1.
In the spleen and lymph nodes, the frequency and number of B220+IgM+ total B cells were significantly decreased in VHL-deficient compared with WT mice (Figures 1C and 1D). Moreover, the total cellularity of spleen was also reduced in VHL-deficient mice (Figure S1J). The defect was equally evident by histological analysis, as splenic B cell (IgD+) follicles were smaller in VHL-deficient than WT mice (Figure 1E and data not shown). However, the positioning of B cells, T cells, CD169+ marginal metallophilic macrophages (Figure 1E), and IgMhigh marginal zone B cells (Figure S2A) was unaffected in the spleen of VHL-deficient mice. The frequencies of B220+CD93−CD23+CD21lo follicular and B220+CD93−CD23−CD21+ marginal zone B cells within the B220+CD93- mature B cell compartment were also comparable between VHL-deficient and WT mice (Figure S2B), suggesting that VHL deficiency causes a general reduction in all B cell populations rather than in a specific B cell subset. Also, the frequency of B220+CD19+ B cells in peritoneal cavities were reduced by ∼3-fold in VHL-deficient compared with WT mice (Figure 1F), although the relative abundancies of conventional B2 and B1 B cell subsets within the total B cell population of peritoneal cavities were comparable between VHL-deficient and WT mice (data not shown). In addition, the percentage and total number of T cells in the spleen and lymph nodes were comparable between VHL-deficient and WT mice (data not shown). Together, these data suggest that VHL deficiency diminishes naive B cell populations.
Impaired Survival of VHL-Deficient B Cells
It is possible that the reduced naive VHL-deficient B cell populations was due to defective proliferation caused by accumulation of HIF-1α, which was showed to influence cell-cycle arrest of hypoxic B cells (Goda et al., 2003). However, the percentage of Ki-67+ proliferating B cells in spleens of unchallenged VHL-deficient and WT mice was found to be similar (Figure S3A). The live carboxyfluorescein succinimidyl ester (CFSE)-labeled VHL-deficient B cells also formed blasts, as envisaged in the forward scatter/side scatter (FSC/SSC) profiles (data not shown), and underwent equivalent cell divisions compared with WT B cells upon stimulation with anti-IgM antibody or LPS (Figure S3B). Moreover, the levels of activation markers, such as CD25, CD69, CD86, and major histocompatibility complex II, in the ex vivo naive or in vitro stimulated VHL-deficient and WT B cells were comparable (Figures S3C and S3D). Conversely, the survival of VHL-deficient B cell was drastically impaired, as the percentage of Annexin V−PI- live VHL-deficient B cells was ∼3-fold lower than that of WT B cells (Figures 1G and 1H upper left), whereas the percentage of PI+Annexin V+ VHL B KO cells was significantly higher that of WT cells (83.8% ± 2.6% versus 55.2% ± 3.8%; p < 0.001) after overnight culture. Besides, the exacerbated cell death of VHL-deficient cells could not be rescued by treatment with anti-IgM antibodies, anti-CD40 antibodies, or LPS (Figure 1H). These data suggest that VHL is important for the survival but not the proliferation or activation of naive B cells.
Loss of VHL Perturbs Glycolytic and Oxidative Metabolism Balance in Naive B Cells
Next, we investigated how VHL deficiency impaired naive B cell homeostasis by comparing the gene expression profiles of VHL-deficient and WT B cells. Microarray study revealed 155 genes to be differentially expressed in the two cell populations (Figure 2A and Table S1), with 141 genes (90%) significantly upregulated in mutant B cells. Gene set enrichment analysis further revealed that 29 of the 50 HIF-1α target genes were significantly upregulated in VHL-deficient cells (Figures 2B and 2C), consistent with their augmented expression of HIF-1α protein. Expression of HIF-2α target genes were also increased in VHL-deficient cells (Figure 2C), correlating with increased HIF-2α protein in these cells (Figure S1B). Notably, genes encoding glycolysis enzymes were significantly upregulated in VHL-deficient cells, including hexokinase (HK) 1, HK2, phosphofructokinase liver type, phosphofructokinase platelet and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2, fructose bisphosphate aldolase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate mutase, enolase-1, pyruvate kinase M, and lactate dehydrogenase α (LDHα) (Figure 2D). Immunoblotting further confirmed the enhanced expression of these enzymes in naive VHL-deficient compared with WT B cells (Figures 2E and S7).
Figure 2.

Imbalanced Glycolytic and Oxidative Metabolisms in VHL-Deficient B Cells
(A) Scatterplot of gene expression of microarray analysis of VHL-deficient and WT B cells. Genes significantly upregulated (fold-change > 1.5, p < 0.05) and downregulated (fold-change < −1.5, p < 0.05) were shown in red and blue, respectively.
(B) Gene set enrichment analysis (GSEA) enrichment curve of HIF-1α target genes in VHL-deficient B cells (enrichment score [ES] = 0.927, normalized ES = 2.41, false discovery rate q value < 0.01). Vertical black lines indicate HIF-1α target genes detected by microarray analysis.
(C) Heatmap depicting log2 fold change of expression of HIF-1α and HIF-2α target genes in VHL-deficient versus WT B cells. Genes that were identified as differentially expressed with statistical significance were indicated with a black border.
(D) Schematic of glycolytic pathway.
(E) Immunoblotting of glycolytic enzymes in VHL-deficient and WT B cells. β-Actin was used as the loading control.
(F and G) Analysis of extracellular acidification rate (ECAR) in VHL-deficient (blue) and WT (red) B cells. Real-time changes in ECAR of B cells with the addition of glucose (Glc) (10 mM), oligomycin (Oligo) (1.0 μM), and 2-deoxy-d-glucose (2-DG) (50 mM) was measured (F). Basal glycolysis was quantified as the difference of ECAR before and after addition of glucose (G).
(H) Quantification of lactate produced by B cells. The amount of lactate in the supernatant of equal number of VHL-deficient and WT B cells cultured for 1 h was quantified.
(I) Oxygen consumption rate (OCR) in VHL-deficient and WT B cells. OCR of B cells with the addition of oligomycin (Oligo) (1.0 μM), FCCP (1.0 μM), and rotenone plus antimycin (Rot/AA) (0.5 μM for each) was measured in real time. Spare respiratory capacity (SRC) is indicated as the difference between basal OCR and maximum OCR after addition of FCCP.
(J) Quantification of basal OCR (leaf panel) and SRC (right panel) in VHL-deficient and WT B cells. Bar graphs represent mean ± SD (n = 5 for G and J; n = 3 for H).
Data shown are representative of more than 3 experiments (E, n = 3; F and I, n = 5). For F and I, each data point represents mean ± SD, n = 5. ***p < 0.001 (Student's t test). See also Figures S2 and S7.
The increased mRNA levels of glycolytic enzyme genes and protein levels of these enzymes in VHL-deficient B cells would suggest that the overall enzymatic activity was augmented due to the increased amount of enzymes, although we did not directly measure the enzymatic activities of these enzymes. Consistent with the enhanced levels of glycolytic enzymes, VHL-deficient B cells manifested increased extracellular acidification rate (ECAR), a surrogate for glycolysis, in response to glucose (Figures 2F and 2G), and produced much more lactate compared with WT cells (Figure 2H), indicating enhanced glycolysis in the mutant cells. This is similar to situations wherein much more enhanced glycolysis in Th17 cells than Treg cells and VHL-deficient CD8+ T cells than WT CD8+ T cells were demonstrated to be consistent with the increased HIF-1a protein level and the mRNA levels of genes encoding various glycolytic enzymes in Th17 and VHL-deficient CD8+ T cells (Shi et al., 2011, Doedens et al., 2013). However, VHL-deficient B cells exhibited reduced basal mitochondrial oxygen consumption rate (OCR), which is reflective of oxidative phosphorylation (OXPHOS), and spare respiratory capacity (SRC) as indicated by the difference between basal OCR and maximal OCR achieved after adding oligomycin and the ionophore FCCP (Figures 2I and 2J). These data suggest that VHL deficiency causes imbalanced glycolytic and oxidative metabolism in naive B cells.
Removal of HIF-1α Rectifies Metabolic and Survival Defects of VHL-Deficient B Cells
We next asked if the defects of VHL-deficient B cells were due to HIF-1α-accumulation. We generated Hif-1αfl/flCd19Cre/+ and Vhlfl/fl Hif-1αfl/flCd19Cre/+ mice (denoted as HIF-1α bKO and dbKO mice, respectively) and compared them with VHL-deficient and WT mice. Removal of HIf-1α did not affect naive B cells as the percentage and total number of B cells in the spleen and lymph nodes were equivalent between HIF-1α bKO and WT mice (Figures 3A and 3B). However, ablating HIF-1α almost completely restored the diminished B cell compartment in VHL-deficient B mice, as both the percentage and number of B cells were comparable between dbKO and WT mice. Immunoblotting confirmed that HIF-1α accumulation in VHL-deficient B cells was obliterated in dbKO B cells (Figure 3C). Interestingly, dbKO B cells survived much better than VHL-deficient cells and as normal as WT B cells (Figure 3D). Also, different from the VHL-deficient B cells, which had distorted glycolytic and oxidative metabolisms, the dbKO B cells exhibited normal ECAR and OCR profiles, same as WT cells (Figures 3E and 3F). These findings suggest that the survival and metabolic defects of VHL-deficient B cells are due to HIF-1α over-accumulation.
Figure 3.

Over-accumulation of HIF-1α Underlies Survival Defect and Distorted Metabolism in VHL-Deficient B Cells
(A) Flow cytometric analysis of B cells in the spleen and lymph nodes of WT, VHL-deficient, HIF-1α bKO, and VHL/HIF-1α bdKO mice. Numbers adjacent to the outline areas indicate percentage of gated cells over total cells.
(B) Enumeration of total splenic B cells in mice of various genotypes. Each symbol represents one mouse, and the height of bars indicates the mean.
(C) Immunoblotting of HIF-1α and VHL in B cells of different genotypes as indicated. A non-specific (ns) band served as loading control.
(D) Flow cytometric analysis of the survival VHL/HIF-1α bdKO B cells. Splenic B cells were cultured in medium for 16 h and analyzed for Annexin V and propidium iodide (PI) staining. Numbers adjacent to outline areas indicate the percentage of PI−Annexin V− live B cells (mean ± SD, n = 5).
(E and F) ECAR and OCR analyses of B cells. ECAR (E) and OCR (F) of splenic B cells from mice of various genotypes were measured in real time with the addition of different compounds as indicated.
Data are representative of 12 (A), 3 (C), or 5 (D, E, and F) experiments. For E and F, each data point represents mean ± SD, n = 5. n.s., not significant; ***p < 0.001 (Student's t test). See also Figure S3.
VHL-Deficient B Cells Manifest Caspase-8-Dependent Apoptosis
Albeit the survival and metabolic defects in VHL-deficient B cells were rescued by HIF-1α-deletion, the mechanism whereby HIF-1α over-accumulation impairs naive B cell survival was unclear. Apparently, VHL-deficient B cells had higher expression of bnip3, which encodes atypical BH3-only proteins BNIP3 (Figure S4A and Table S1), and its upregulation was HIF-1α-dependent as its protein level was markedly reduced in dbKO B cells (Figure S4B). BNIP3 has been known to be involved in cell death induced by hypoxic injury such as ischemia (Hamacher-Brady et al., 2007, Schmidt-Kastner et al., 2004) and regulate opening of mitochondrial permeability transition pore (mPTP) to induce necrosis or caspase-9-dependent apoptosis (Chinnadurai et al., 2008, Gustafsson, 2011). BNIP3 was also shown to promote survival and memory formation of natural killer cells (O'Sullivan et al., 2015). However, the mPTP opening was not affected in VHL-deficient B cells despite the upregulated BNIP3 (Figure S4C). Treatment of VHL-deficient B cells with mPTP inhibitors, bongkrekic acid or 2-aminoethoxydiphenyl borate, had no effect on their survival (Figure S4D). Also, no enhanced caspase-9 activation was found in VHL-deficient cells (Figure S4E). These data suggest that the impaired survival of VHL-deficient B cells is not likely due to BNIP3 upregulation. However, compared with WT cells, VHL-deficient B cells exhibited enhanced caspase-3 activation, which was evident after 2 h and became more pronounced after 4 h of normoxic culture (Figure 4A). Addition of pan-caspase inhibitor Z-VAD largely rescued the survival defect of mutant cells (Figure 4B). Interestingly, WT B cells cultured under hypoxia also had increased caspase-3 activation and apoptosis compared with cells under normoxia (Figure 4C). These data suggest that VHL deficiency or hypoxia can trigger caspase-3-dependent apoptosis of naive B cells.
Figure 4.

VHL-Deficient B Cells Exhibit Exacerbated Caspase-8 Activation
(A) Flow cytometric detection of caspase-3 activation in VHL-deficient B cells. VHL-deficient and WT B cells were cultured for 2 or 4 h and stained for activated caspase-3.
(B) Survival of VHL-deficient B cells after treatment with pan-caspase inhibitor Z-VAD. VHL-deficient B cells were cultured in medium alone or in the presence of Z-VAD (10μM) for 8 h followed by flow cytometry to detect Annexin V− live cells. Numbers indicate percentage of Annexin V− live cells (mean ± SD, n = 5).
(C) Survival and caspase-3 activation in B cells under normoxia and hypoxia. WT B cells were cultured under normoxia (21% O2) or hypoxia (1% O2) for 16 h followed by flow cytometry to detect Annexin V− live cells and caspase-3 activation. Numbers indicate percentage of Annexin V− live cells (upper panel, mean ± SD, n = 5) or activated caspase-3+ cells (lower panel, mean ± SD, n = 5).
(D) Immunoblotting of caspase-8 activation in VHL-deficient B cells. Whole-cell lysates of VHL-deficient and WT B cells were subjected to immunoblotting to detect pro- and cleaved-caspase-8. β-Actin was used as loading control.
(E) Survival of VHL-deficient B cells after treatment with pan-caspase inhibitor Z-VAD (10μM), caspase-8 inhibitor Z-IETD (10μM) or caspase-9 inhibitor Z-LEHD (10μM) for 8 h. Numbers indicate percentage of Annexin V− live cells (mean ± SD, n = 5).
(F) DISC formation in VHL-deficient B cells. VHL-deficient and WT B cells were lysed and subjected to immunoprecipitation with anti-Fas antibody. Fas-associated caspase-8 and FADD were detected by immunoblotting subsequently.
Data are representative of 4 (A) or 5 (D, F) experiments. See also Figures S4 and S7.
A previous study showed that Vhl deletion using Lck-Cre caused enhanced mRNA expression and activity of caspase-8 in thymocytes (Biju et al., 2004). Despite the comparable transcript and protein levels of caspase-8 in WT and VHL-deficient B cells, the amount of cleaved/activated forms of caspase-8 was significantly increased in mutant B cells (Table S1, Figures 4D and S7). In addition, caspase-8 specific inhibitor Z-IETD-fmk largely rescued the survival defect of VHL-deficient cells and to the same extent as the pan-caspase inhibitor Z-VAD (Figure 4E). In contrast, caspase-9 was not activated (Figure S4E) and caspase-9-specific inhibitor Z-LEHD-fmk had no effect on VHL-deficient B cell survival. These data suggest that enhanced caspase-8 activation is responsible for the caspase-3 activation and apoptosis of VHL-deficient B cells.
Caspase-8 activation is initiated when its proenzyme is recruited together with adaptor protein FADD to death receptors, such as Fas, to form death-inducing signaling complex (DISC) which causes its cleavage and activation (Kischkel et al., 1995). Interestingly, B cell-specific deletion of Fas leads to severe autoimmunity in mice, suggesting that Fas is important for the homeostasis of naive B cells despite its minimal expression (Hao et al., 2008). Hence, we examined if Fas-mediated DISC formation was affected in VHL-deficient B cells. Immunoprecipitation assay revealed increasing amount of caspase-8 and FADD associated with Fas in resting VHL-deficient compared with WT B cells (Figure 4F), suggesting a constitutive Fas-mediated DISC formation in the mutant cells.
VHL Deficiency Decouples Glycolysis from TCA and Induces Reductive Carboxylation of α-KG and Fas Palmitoylation in B Cells
Given the distorted metabolism and yet largely normal expression of cell survival genes in VHL-deficient B cells, we next investigated the metabolism of these cells in detail by performing a metabolomic profiling study. We obtained totally ∼100 metabolites with confirmed identities in B cells using liquid chromatography-mass spectrometry (LC-MS) (Table S2). Strikingly, VHL-deficient B cells exhibited high levels of various long-chain acylcarnitines (LCACs) (Figure 5A), which are metabolic intermediates of fatty acid (FA) oxidation (FAO). The high level of LCACs could be due to an impaired FAO in VHL-deficient cells. To test this, we treated B cells with etomoxir, an inhibitor of carnitine palmitoyltransferase-1 that is a critical enzyme for FAO, to totally block FAO. The reduction in basal OCR and SRC by etomoxir would reflect the FAO contribution to OCR. Etomoxir caused similar reduction in basal OCR and SRC levels in VHL-deficient and WT B cells (Figure S5A), and percent reduction was even slightly more in mutant B cells (Figure S5B), suggesting that LCACs' accumulation in mutant cells was not ascribed to an impaired FAO. It was also not due to enhanced uptake of exogenous triacylglycerol substrates via scavenger receptor CD36 and the subsequent lipolysis by lysosomal acid lipase (LAL) for releasing FAs, as VHL-deficient and WT B cells had comparable CD36 and LAL expression (Figures S5C and S5D) and equivalent amount of acylglycerol species (Table S2).
Figure 5.

Increased Reductive Carboxylation of α-KG and Constitutive Palmitoylation of Fas in VHL-Deficient B Cells
(A) Heatmap depicting lipid metabolite difference between WT and VHL-deficient (bKO) B cells. Lipid metabolites from bKO and WT B cells were subjected to ultra-performance LC-MS. Data were pre-processed and normalized to total protein concentration in each sample.
(B) Immunoblotting of PDK1 in B cells. Whole-cell lysates from WT and VHL-deficient B cells were subjected to immunoblotting with anti-PDK1 antibody. β-Actin was used as loading control.
(C) PDH activity in VHL-deficient and control B cells. The activity of PDH in VHL-deficient B cells (blue) was measured and presented relative to that in control B cells (red), set as 1.
(D) Real-time changes in OCR of VHL-deficient (blue) and control (red) B cells with the addition of Glc (10 mM), Oligo (1.0 μM), and 2-DG (1.0 μM).
(E) α-Ketoglutarate (α-KG)/citrate ratio in VHL-deficient and control B cells. The ratio in VHL-deficient cells (blue) was presented relative to that in control cells (red), set as 1.
(F) Carbon atom transition map depicting patterns of metabolites derived from [13C5,15N2] glutamine during oxidative (blue arrows) and reductive (red arrows) metabolism. Mass isotopomers for reductive glutamine metabolism detection include M5 citrate (Cit), M3 oxaloacetate (Oac), M3 aspartate (Asp), M3 malate (Mal), and M3 fumarate (Fum) from the labeled glutamine.
(G) Evidence for increased reductive carboxylation in VHL-deficient B cells. VHL-deficient (blue) and control (red) B cells were cultured in medium containing 2 mM [13C5,15N2] glutamine for 4 h and 13C enrichment in various intracellular metabolites was measured by LC-MS.
(H) Measurement of Fas palmitoylation. Whole-cell lysates from WT and VHL-deficient B cells were treated with (+) or without (−) hydroxylamine (HA) followed by biotin moiety replacement for pull down with streptavidin-conjugated agarose beads. The pulled down proteins were subjected to immunoblotting with Fas-specific antibody.
Bar graphs represent mean ± SD (n = 3 for C and E and n = 2 for G). Data are representative of 2 (A, G, and H) and 5 (B and D) experiments. For D, each data point represents mean ± SD, n = 5. *p < 0.05, **p < 0.01; ***p < 0.001 (Student's t test). See also Figures S5 and S7.
It is possible that the LCACs' accumulation in mutant B cells was indicative of increased de novo FA synthesis. In fact, recent studies showed that VHL-deficient RCC or hypoxic tumor cells had enhanced FA synthesis from glutamine (Mullen et al., 2011, Metallo et al., 2011, Wise et al., 2011, Filipp et al., 2012). In these cells, glucose was mainly converted to lactate and uncoupled from tricarboxylic acid (TCA) cycle, and isocitrate dehydrogenase-mediated reductive carboxylation of glutamine-derived α-ketoglutarate (α-KG) was triggered to fuel citrate production and lipogenesis for cell growth. We postulated that the distorted metabolism in VHL-deficient cells, arising from over-stabilization of HIF-1α, could also uncouple glycolysis from TCA cycle and trigger reductive carboxylic metabolism for lipogenesis. To test this, we first examined the activity of pyruvate dehydrogenase (PDH) that drives hypoxic glycolysis decoupling from TCA cycle and is negatively regulated by PDH kinase isoform 1 (PDK1) (Fendt et al., 2013, Kim et al., 2006), a direct target of HIF-1α. Consistent with the HIF-1α′s accumulation, PDK1 expression was enhanced (Figures 5B and S7) and PDH activity was decreased concurrently in VHL-deficient B cells (Figure 5C). Interestingly, whereas glucose slightly increased OCR in WT cells, as they fuel into TCA cycle through pyruvate, glucose addition reduced OCR in VHL-deficient B cells (Figure 5D), indicating that their abnormally enhanced glycolysis could further dampen OXPHOS. These data suggest that glycolysis is indeed decoupled from TCA cycle in the naive mutant B cells.
Compared with WT cells, mutant B cells also had higher α-KG to citrate ratio (Figure 5E), the trigger for reductive carboxylation of α-KG (Fendt et al., 2013). To assess directly the reductive α-KG carboxylation, we performed isotopic 13C-labeling assay by culturing B cells in [13C5,15N2] glutamine-containing medium for 4 h and measuring 13C enrichment in various metabolites by LC-MS. Reductive carboxylation of α-KG would generate mass isotopomers, such as M5 citrate, M3 oxaloacetate, M3 aspartate, M3 malate, and M3 fumarate, from the labeled glutamine (Figure 5F). It was found that the amounts of M5 citrate, M3 malate, M3 fumarate, and M3 aspartate were increased in VHL-deficient compared with WT B cells (Figure 5G), suggesting that mutant B cells have enhanced reductive carboxylation of α-KG.
However, it was still elusive how enhanced reductive α-KG carboxylation was linked to compromised survival of VHL-deficient B cells. Lipogenesis via reductive α-KG carboxylation was engaged to support the growth of tumor cells lacking VHL or under hypoxia, but it is obviously unnecessary for naive B cells as they do not have such metabolic needs. Other than contributing to membrane synthesis, LCFAs can also modify proteins at their cysteine residues (Nadolski and Linder, 2007, James and Olson, 1990). For instance, palmitate, the most abundant FA in the cell, preferentially modifies proteins via palmitoylation (Resh, 2012, Salaun et al., 2010). Interestingly, Fas palmitoylation at membrane-proximal cysteine residues of its cytoplasmic region is known to facilitate DISC formation (Chakrabandhu et al., 2007, Feig et al., 2007). The high level of LCACs including palmitoyl carnitine (Figure 5A) and constitutive DICS formation in VHL-deficient B cells (Figure 4F) prompted us to ask if Fas was constitutively palmitoylated in the mutant cells. We examined Fas palmitoylation by employing a non-radioactive acyl-biotinyl exchange (ABE) assay, in which the S-palmitoyl thioester link was cleaved by hydroxylamine (HA) and subsequently replaced by a biotin moiety for protein pull-down. Fas protein was hardly detectable in both HA-treated and HA-untreated WT samples after ABE reaction, suggesting that Fas is not palmitoylated in these cells (Figure 5H). By contrast, a considerable amount of Fas was detected in HA-treated cell lysates from mutant B cells, indicating constitutive palmitoylation in these cells. Together, these results suggest that the distorted metabolism in VHL-deficient B cells activates reductive α-KG carboxylation for lipogenesis, leading to constitutive Fas palmitoylation.
Blockade of Reductive Carboxylation of α-KG Rescues Survival Defect of VHL-Deficient B Cells
As the distorted metabolism in VHL-deficient B cells triggers reductive α-KG carboxylation causing Fas palmitoylation, we next asked if interference with this metabolic pathway could rectify the survival defect of the mutant cells. Lactate supplementation was shown to reverse reductive glutamine metabolism in cells under hypoxia or with impaired respiration (Fendt et al., 2013), as it shifts the reaction direction of LDH from net synthesis to net consumption of lactate to generate pyruvate (Huckabee, 1956) and increases carbon entry into the TCA cycle via acetyl-CoA (Mintun et al., 2004). Thus, we assessed the effect of lactate supplementation on the metabolism and survival of VHL-deficient B cells. We found that lactate indeed reduced LCAC levels, especially the levels of palmitoyl carnitine (C16:0) and linoelaidyl carnitine (C18:2) (Figure 6A), and rectified the elevated ECAR and decreased OCR in VHL-deficient B cells (Figure S6). More interestingly, lactate greatly improved VHL-deficient B cell survival (Figures 6B and 6D). Consistently, caspase-3 activation was significantly reduced by lactate (Figures 6C and 6D), suggesting that blockade of reductive carboxylation of α-KG corrects the metabolic and survival defects in VHL-deficient B cells.
Figure 6.

Effect of Lactate Supplementation and ACLY Inhibition on VHL-Deficient B Cells
(A) Abundance of major acylcarnitine species in B cells. WT and VHL-deficient B cells were cultured for 2 h with or without lactate (Lac) (25 mM) as indicated and subjected to lipid metabolite extraction and ultra-performance LC-MS analysis. The relative abundance of acylcarnitine was normalized to total protein in each sample.
(B and C) Flow cytometric analysis of survival and caspase-3 activation of VHL-deficient B cells after lactate supplementation and ACLY inhibition. VHL-deficient B cells were cultured with lactate (25 mM), ACLY inhibitor SB 204990 (50 μM), or both for 6 h. Annexin V− live cells (B) and cells with active caspase-3 (C) were examined by flow cytometry.
(D) Quantification of live cells and cells with active caspase-3 in different cultures. Percentage of Annexin V− live (upper panel) and caspase-3+ (lower panel) cells was quantified based on flow cytometric analysis.
(E) Immunoblotting of caspase-8 activation and DISC formation in VHL-deficient B cells. Whole-cell lysate from VHL-deficient B cells cultured for 2 h under different conditions as indicated was subjected to immunoblotting for the detection of cleaved caspase-8 (upper panel). β-Actin was used as loading control. For the detection of DISC formation, whole-cell lysate was subjected to immunoprecipitation with anti-Fas antibody followed by the detection of Fas-associated caspase-8 by immunoblotting.
Bar graphs represent mean ± SD (A, n = 3; D, n = 5). Data are representative of 3 (E) or 5 (B and C) experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t test). See also Figure S6.
We also evaluated the effect of inhibiting ATP-citrate lyase (ACLY), another critical metabolic enzyme along the reductive glutamine metabolism pathway, on VHL-deficient B cell survival. We found that SB204990, a specific ACLY inhibitor, significantly rescued the survival defect and dampened caspase-3 activation in mutant B cells (Figures 6B–6D). Notably, combinatory treatment with lactate and SB204990 had more profound rescuing effect on VHL-deficient cells than individual treatment (Figures 6B–6D). The constitutive DISC formation and caspase-8 activation in VHL-deficient B cells were also significantly reduced by the combinatory treatment (Figure 6E). These data suggest that enhanced caspase-8-dependent apoptosis of VHL-deficient B cells is largely due to increased reductive carboxylation of α-KG arising from the distorted metabolism.
Discussion
In this study, we uncover an important mechanism whereby the survival of naive B cells is metabolically regulated. We demonstrate that VHL deletion in B cells causes over-stabilization of HIF-1α, which leads to imbalanced glycolytic and oxidative metabolism and diminishment of mature B cell. The distorted metabolism decouples glucose-derived pyruvate from entering the TCA cycle and triggered reductive glutamine metabolism in the quiescent mutant B cells. The abnormally activated reductive glutamine metabolism triggers increased lipogenesis that causes Fas palmitoylation and constitutive activation of caspase-8, leading to increased apoptosis of VHL-deficient B cells.
A recent study shows that glucose fluxing via the glycolytic pathway does not end up as lactate or go into TCA in B cells stimulated in vitro. Instead, pyruvate and other glycolytic intermediates mainly branch into pentose phosphate pathway for DNA and RNA synthesis to support B cell proliferation. Interestingly, glutamine is shown to be oxidized for fueling TCA and enhancing OXPHOS in these activated B cells (Waters et al., 2018). Here, we show that in the quiescent naive B cells, HIF-1α is accumulated when VHL is ablated and glycolysis is enhanced, as evidenced by their enhanced ECAR and elevated lactate level. Similar to the stimulated B cells, our metabolomics study reveals that the VHL-deficient quiescent B cells also exhibit higher levels of some metabolites of nucleic acid species, such as adenosine monophosphate, uridine 5′-monophosphate, guanosine monophosphate, guanine, and guanosine (Table S2), suggesting that the carbon source from the enhanced glucose flux through glycolysis also deviates into the pentose phosphate pathway to certain extent. Together with the finding that the reductive glutamine metabolism is abnormally triggered in the VHL-deficient quiescent B cells, which eventually leads to their increased cell death, these results suggest that the fates of quiescent and stimulated B cells are differentially regulated by different metabolic pathways.
The activation of reductive glutamine metabolic pathway in VHL-deficient B cells is reminiscent of the metabolic re-programming in tumor cells under hypoxia or with VHL mutations, where reductive carboxylation of α-KG is also triggered (Metallo et al., 2011, Gameiro et al., 2013). However, the similar metabolic re-programming leads to vastly different outcomes in the two scenarios. In the tumor cells, which have high demands for lipids, reductive carboxylation of α-KG is necessary for de novo lipogenesis to support the growth of tumor cells under either bona fide or “pseudo” hypoxic conditions (Mullen et al., 2011, Metallo et al., 2011, Wise et al., 2011, Filipp et al., 2012). On the contrary, in the quiescent naive B cells, the activation of reductive carboxylation of α-KG is rather disastrous as it causes constitutive caspase-8 activation and apoptosis. Our data reveal for the first time a link between metabolic imbalance and activation of programmed cell death machinery in naive B cells.
Previous studies showed that VHL deletion in thymocytes caused dramatic reduction in thymic cellularity. The defect was apparently not due to enhanced intrinsic apoptotic pathway and could not be rescued by transgenic expression of Bcl-2 or Bcl-xL or inhibiting caspase-9. Although the activation of caspase-8 was shown to be significantly increased in VHL-deficient thymocytes (Biju et al., 2004), the detailed underlying mechanism remained unresolved. Our data suggest that the enhanced caspase-8 activation in VHL-deficient B cells is not through complexed transcriptional regulation of cell survival genes. Instead, post-translational palmitoylation of death receptor Fas, arising from distorted metabolism, causes enhanced DISC assembly and caspase-8 activation.
Equally fascinating is that pharmacological interference with the metabolic imbalance could reverse the survival defect of VHL-deficient B cells. Lactate supplementation partially reduces ECAR and increases OCR and more importantly, alleviates the increased LCAC levels in VHL-deficient B cells by blocking reductive carboxylation of α-KG. Interestingly, it significantly reduces caspase-8 activation and rescues the impaired survival of VHL-deficient B cells. More strikingly, blockade of reductive carboxylation of α-KG by combinatory lactate supplementation and ACLY inhibition could efficiently inhibit constitutive DISC formation and caspase-8 activation and largely rescue the survival defect of VHL-deficient cells, highlighting the importance of balanced metabolism for naive B cell survival.
In summary, our findings discover a previously unappreciated mechanism by which VHL/HIF-1 pathway metabolically regulates the survival of naive B cells. By balancing glycolytic and oxidative metabolisms, this pathway is able to control carbon entry into TCA cycle and the contribution of reductive glutamine metabolism to lipogenesis, thereby regulating the survival of naive B cells. Our study potentially identifies a pathway that could be metabolically targeted for B cell-driven ailments such as systemic lupus erythematosis.
Limitations of the Study
As VHL also regulates the stability of HIF2α, it cannot be ruled out that the accumulation of HIF2α in VHL-deficient B cells might contribute to the metabolic and survival defects of VHL-mutant B cells to certain extent. More complete studies of B cell-specific HIF1α/HIF2α, HIF2α/VHL DKO, and HIF1α/HIF2α/VHL TKO mice will be important addition to the current study for understanding exactly the role of these molecules in regulating the metabolism and survival of naive B cells. Moreover, it would be interesting to know if other hematopoietic lineages are affected indirectly in VHL-deficient mice.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Nicholas Seah for maintenance of mouse colonies and Wee-Lee Lim for help with histology studies. This work was funded by Singapore Agency for Science Technology and Research.
Authors Contributions
X.S. and L.K.P. designed experiments. X.S., H.J., H.Y., A.M., O.X., C.S., M.S., Y.L.Y., H.Y.S, N.S.W., T.A., and L.A. performed experiments. X.S. and L.K.P. wrote the manuscript. The authors declare no competing financial interests.
Declaration of Interests
The authors declare no competing interests.
Published: July 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.002.
Contributor Information
Shengli Xu, Email: xu_shengli@bti.a-star.edu.sg.
Kong-Peng Lam, Email: lam_kong_peng@bti.a-star.edu.sg.
Supplemental Information
References
- Abbott R.K., Thayer M., Labuda J., Silva M., Philbrook P., Cain D.W., Kojima H., Hatfield S., Sethumadhavan S., Ohta A. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J. Immunol. 2016;197:4014–4020. doi: 10.4049/jimmunol.1601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biju M.P., Neumann A.K., Bensinger S.J., Johnson R.S., Turka L.A., Haase V.H. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol. Cell. Biol. 2004;24:9038–9047. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothby M., Rickert R.C. Metabolic regulation of the immune humoral response. Immunity. 2017;46:743–755. doi: 10.1016/j.immuni.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A., Wang R., Nichols A.G., Kuraoka M., Milasta S., Sun L.D., Gavin A.L., Abel E.D., Kelsoe G., Green D.R., Rathmell J.C. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014;192:3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabandhu K., Herincs Z., Huault S., Dost B., Peng L., Conchonaud F., Marguet D., He H.T., Hueber A.O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 2007;26:209–220. doi: 10.1038/sj.emboj.7601456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai G., Vijayalingam S., Gibson S.B. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–S127. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.H., Raybuck A.L., Stengel K., Wei M., Beck T.C., Volanakis E., Thomas J.W., Hiebert S., Haase V.H., Boothby M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537:234–238. doi: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T., Yamanishi Y., Clausen B.E., Forster I., Pawlinski R., Mackman N., Haase V.H., Jaenisch R., Corr M., Nizet V. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang E.V., Barbi J., Yang H.Y., Jinasena D., Yu H., Zheng Y., Bordman Z., Fu J., Kim Y., Yen H.R. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens A.L., Phan A.T., Stradner M.H., Fujimoto J.K., Nguyen J.V., Yang E., Johnson R.S., Goldrath A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty C.A., Bleiman B.F., Wagner D.J., Dufort F.J., Mataraza J.M., Roberts M.F., Chiles T.C. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood. 2006;107:4458–4465. doi: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufort F.J., Bleiman B.F., Gumina M.R., Blair D., Wagner D.J., Roberts M.F., Abu-Amer Y., Chiles T.C. Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J. Immunol. 2007;179:4953–4957. doi: 10.4049/jimmunol.179.8.4953. [DOI] [PubMed] [Google Scholar]
- Feig C., Tchikov V., Schutze S., Peter M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2007;26:221–231. doi: 10.1038/sj.emboj.7601460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S.M., Bell E.L., Keibler M.A., Olenchock B.A., Mayers J.R., Wasylenko T.M., Vokes N.I., Guarente L., Vander Heiden M.G., Stephanopoulos G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013;4:2236. doi: 10.1038/ncomms3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp F.V., Scott D.A., Ronai Z.A., Osterman A.L., Smith J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012;25:375–383. doi: 10.1111/j.1755-148X.2012.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallou C., Joly D., Mejean A., Staroz F., Martin N., Tarlet G., Orfanelli M.T., Bouvier R., Droz D., Chretien Y. Mutations of the VHL gene in sporadic renal cell carcinoma: definition of a risk factor for VHL patients to develop an RCC. Hum. Mutat. 1999;13:464–475. doi: 10.1002/(SICI)1098-1004(1999)13:6<464::AID-HUMU6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Gameiro P.A., Yang J., Metelo A.M., Perez-Carro R., Baker R., Wang Z., Arreola A., Rathmell W.K., Olumi A., Lopez-Larrubia P. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–385. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda N., Ryan H.E., Khadivi B., McNulty W., Rickert R.C., Johnson R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow C.C., Vinuesa C.G., Randall K.L., Mackay F., Brink R. Control systems and decision making for antibody production. Nat. Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- Gustafsson A.B. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 2011;32:267–274. doi: 10.1007/s00246-010-9876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A., Brady N.R., Logue S.E., Sayen M.R., Jinno M., Kirshenbaum L.A., Gottlieb R.A., Gustafsson A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- Hao Z., Duncan G.S., Seagal J., Su Y.W., Hong C., Haight J., Chen N.J., Elia A., Wakeham A., Li W.Y. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29:615–627. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckabee W.E. Control of concentration gradients of pyruvate and lactate across cell membranes in blood. J. Appl. Physiol. 1956;9:163–170. doi: 10.1152/jappl.1956.9.2.163. [DOI] [PubMed] [Google Scholar]
- James G., Olson E.N. Fatty acylated proteins as components of intracellular signaling pathways. Biochemistry. 1990;29:2623–2634. doi: 10.1021/bi00463a001. [DOI] [PubMed] [Google Scholar]
- Jellusova J., Cato M.H., Apgar J.R., Ramezani-Rad P., Leung C.R., Chen C., Richardson A.D., Conner E.M., Benschop R.J., Woodgett J.R., Rickert R.C. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017;18:303–312. doi: 10.1038/ni.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.W., Tchernyshyov I., Semenza G.L., Dang C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt S., Behrmann I., Germer M., Pawlita M., Krammer P.H., Peter M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H., Gu H., Nomura S., Caldwell C.C., Kobata T., Carmeliet P., Semenza G.L., Sitkovsky M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc. Natl. Acad. Sci. U S A. 2002;99:2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Elly C., Park Y., Liu Y.C. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity. 2015;42:1062–1074. doi: 10.1016/j.immuni.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm I., Lingott A., Strandmann E., Zoidl C., Bulman M.P., Hattersley A.T., Schulz W.A., Ebert T., Ryffel G.U. Loss of HNF1alpha function in human renal cell carcinoma: frequent mutations in the VHL gene but not the HNF1alpha gene. Mol. Carcinog. 1999;24:305–314. [PubMed] [Google Scholar]
- Metallo C.M., Gameiro P.A., Bell E.L., Mattaini K.R., Yang J., Hiller K., Jewell C.M., Johnson Z.R., Irvine D.J., Guarente L. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun M.A., Vlassenko A.G., Rundle M.M., Raichle M.E. Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proc. Natl. Acad. Sci. U S A. 2004;101:659–664. doi: 10.1073/pnas.0307457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen A.R., Wheaton W.W., Jin E.S., Chen P.H., Sullivan L.B., Cheng T., Yang Y., Linehan W.M., Chandel N.S., DeBerardinis R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadolski M.J., Linder M.E. Protein lipidation. FEBS J. 2007;274:5202–5210. doi: 10.1111/j.1742-4658.2007.06056.x. [DOI] [PubMed] [Google Scholar]
- O'Sullivan T.E., Johnson L.R., Kang H.H., Sun J.C. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015;43:331–342. doi: 10.1016/j.immuni.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenchock B.A., Rathmell J.C., Vander Heiden M.G. Biochemicphenotypesal underpinnings of immune cell metabolic. Immunity. 2017;46:703–713. doi: 10.1016/j.immuni.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A., Goldrath A.W., Nizet V., Johnson R.S. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41:518–528. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- Resh M.D. Targeting protein lipidation in disease. Trends Mol. Med. 2012;18:206–214. doi: 10.1016/j.molmed.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun C., Greaves J., Chamberlain L.H. The intracellular dynamic of protein palmitoylation. J. Cell Biol. 2010;191:1229–1238. doi: 10.1083/jcb.201008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R., Aguirre-Chen C., Kietzmann T., Saul I., Busto R., Ginsberg M.D. Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res. 2004;1001:133–142. doi: 10.1016/j.brainres.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- Shi L.Z., Wang R., Huang G., Vogel P., Neale G., Green D.R., Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Dillon C.P., Shi L.Z., Milasta S., Carter R., Finkelstein D., McCormick L.L., Fitzgerald P., Chi H., Munger J., Green D.R. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L.R., Ahsan F.M., Wolf D.M., Shirihai O., Teitell M.A. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience. 2018;5:99–109. doi: 10.1016/j.isci.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise D.R., Ward P.S., Shay J.E., Cross J.R., Gruber J.J., Sachdeva U.M., Platt J.M., DeMatteo R.G., Simon M.C., Thompson C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.