

Abstract
Background:
Laboratory findings have suggested that systemic and vascular inflammation can impair the anti-atherogenic function of high density lipoproteins (HDL). However, evidence from population studies is sparse.
Objective:
To assess if blood inflammatory markers modify the risk of recurrent coronary events associated with apolipoprotein A-I (apoA-I) and HDL-C among post-infarction patients.
Methods:
ApoA-I, HDL-C and inflammatory markers (C-reactive protein [CRP], serum amyloid A [SAA], fibrinogen, von Willebrand Factor [vWF], and D-dimer) were measured from blood samples of 1028 patients drawn 2 months after an index myocardial infarction (MI). Patients were followed up for the composite coronary endpoint (non-fatal MI, coronary death, or unstable angina) for an average of 26 months. Cox proportional hazards models were used to assess effect modifications for the association of apoA-I and HDL-C with coronary risk by each inflammatory marker.
Results:
CRP significantly modified the risk of recurrent coronary events associated with apoA-I. Among the entire population, multivariable-adjusted hazard ratios (HRs) associated with each standard deviation (SD) increase in ApoA-I for those with low and high CRP levels were 0.89 and 1.35, respectively (P value for interaction = 0.008). VWF was a significant effect modifier of the apoA-I/coronary risk association only among diabetic patients (HRs were 0.56 and 1.43, for diabetic patients with low and high vWF levels, respectively; P value for interaction = 0.002). No effect modification was observed for the HDL-C/coronary risk association.
Conclusion:
Among stable post-MI patients, CRP modified the risk of recurrent coronary events associated with apoA-I. VWF modified this association only among the diabetic subgroup.
Keywords: high density lipoprotein cholesterol, apolipoprotein A-I, inflammation, myocardial infarction, unstable angina
Introduction
High density lipoprotein cholesterol (HDL-C) has been shown to be protective against atherosclerosis via diverse mechanisms including macrophage cholesterol efflux and reverse cholesterol transport, amelioration of endothelial function, anti-oxidative and anti-inflammatory activities. As the main protein constituent of HDL particles, apolipoprotein A-I (apoA-I) plays a major role in these anti-atherosclerosis effects. Although epidemiologic studies have generally found decreased risk of cardiovascular disease associated with high HDL-C and apoA-I 1–3, HDL particles have been shown to undergo a loss of function in certain pathophysiological states and may then demonstrate impaired anti-atherogenic or even pro-atherogenic effects 4. Recent biological studies have shown that systemic and vascular inflammation is one of these states that can convert HDL particle or apoA-I to a dysfunctional form through a variety of mechanisms. These mechanisms are primarily oxidative modification of apoA-I by the enzyme myeloperoxidase and changes in the HDL proteome and lipidome 4. Despite findings from biological studies, epidemiological studies that directly assess the influence of inflammation on the association of HDL-C or apoA-I with cardiovascular risk is sparse.
The aim of the present study is to assess whether blood inflammatory markers modify the risk of recurrent coronary events (unstable angina, nonfatal myocardial infarction, or coronary death) associated with apoA-I and HDL-C among post-MI (myocardial infarction) patients. Inflammatory markers examined were selected based on results of a principal component analysis published previously in which C-reactive protein (CRP), fibrinogen, von Willebrand factor (vWF) and D-dimer comprised a factor labeled as “vascular inflammatory” with factor loadings greater than 0.4 5. We also examined serum amyloid A (SAA), which is an acute phase protein and was re-analyzed from stored blood samples later. We examined the effect modification in the entire study population, as well as in those with and without diabetes separately, given that diabetic patients may have different HDL metabolism compared to non-diabetic patients 6. We hypothesized that the protective association of apoA-I and HDL-C with coronary risk would be attenuated for those with higher levels of inflammatory markers compared to those with lower levels of these markers.
Material and Methods
Study population
The study population comprised 1,028 patients of the THROMBO study (Thrombogenic Factors and Recurrent Coronary Events) who had complete laboratory data on apoA-I and HDL-C (98.4%). Details of the THROMBO study population have been reported previously 2. Briefly, patients were enrolled from 13 participating hospitals between October 1, 1994 and June 30, 1997 after an enzyme-confirmed MI (the index MI), and were followed up for recurrent coronary events including unstable angina, non-fatal MI, and cardiac death. Patients who had coronary bypass graft surgery during the hospitalization of the index MI, who were receiving heparin-type anticoagulant therapy, and who had significant comorbidity were excluded from enrollment. This study was approved by the Research Subject Review Board of all participating institutions. Written content was obtained from all patients who agreed to participate.
Measurements of blood markers
Fasting blood samples were drawn at baseline, which was defined as 2 months after the index MI. Patients were asked to refrain from smoking for 24 hours before blood drawn. HDL-C, apoA-I, fibrinogen, d-Dimer and vWF were quantified as previously described 2. CRP and SAA were measured using high-sensitivity assays (Dade Behring, Newark, Dlaware) 7. CRP was available for 945 subjects, and SAA was available for 896 subjects.
Demographic and medical data
At baseline, the study coordinator interviewed and examined the patients and recorded demographic data, smoking behavior, and medical history. Medical records from the index hospitalization were abstracted and results of medical examinations as well as cardiac medications were recorded. Routine clinical data were missing in <0.5% of patients. Ejection fraction and chest roentgenogram results were not obtained in 11.0% and 9.1% of patients (not ordered by the attending physician), respectively 2.
Measurements of end-points
The primary endpoint of the present study was the composite endpoint of non-fatal MI, unstable angina, or death due to coronary heart disease, whichever occurred first, during the follow-up period from baseline to Mar 31, 1998. As previously described 2, for fatal events the Hinkle-Thaler criteria were used to classify the cause of death based on information collected from witness, relatives, death certificates, autopsy reports (when available), and medical records. Nonfatal MI was defined as MB isoenzyme fraction >4% of the total creatine kinase with symptoms and/or ECG changes consistent with an acute infarction (Troponin was not available at the time of the study). These diagnosis criteria were the same as those applied to the index MI. Unstable angina was defined as chest pain with ECG changes indicating ischemia (without enzyme elevation) that requires hospitalization 8. Endpoint events were adjudicated by an independent Endpoint Event Committee based on a review of all information collected.
Statistical analysis
Comparisons of baseline characteristics between diabetic and non-diabetic subgroups were performed by Chi-square test for categorical variables and two-sample t test or Mann–Whitney U test (for skewed distribution) for continuous variables. Effect modification of the hazard of recurrent coronary events associated with apoA-I and HDL-C by inflammatory markers was examined using Cox proportional hazards model. The proportional hazards assumption was examined by interactions between log survival time and each covariate and no violation was found. Person-time at risk was calculated from baseline (2 months after the index MI) until the occurrence of the composite endpoint (for those who developed coronary events during follow-up), last contact (for those who were lost to follow-up), time of death (for those died of causes other than coronary events), or end of the study (Mar 31, 1998). A stepwise forward selection procedure was applied to select clinical risk predictors for the composite endpoint from 10 pre-specified covariates (age, gender, race, prior MI, prior stroke, history of diabetes mellitus, thrombolytic therapy, pulmonary congestion by chest roentgenogram, ejection fraction during the index MI, and smoking status at baseline), with a significance level of P < 0.10 for entry into the model. The enrolling hospital (n=13) was entered into all models as a stratification factor. Coding of each clinical variable has been described previously 2. In the present study, apoA-I and HDL-C were analyzed as continuous variables. Inflammatory markers were dichotomized into low vs. high using the third quartile as cut-off value.
We first assessed the hazard ratios of coronary events associated with each biomarker (apoA-I, HDL-C, and each of the five inflammatory markers) in separate models with adjustment for selected clinical variables. Among the 10 pre-specified clinical variables, diabetes mellitus, prior myocardial infarction, and prior stroke were entered into the model by the forward selection procedure and thus were adjusted for. To assess effect modification of the apoA-I/coronary risk association by CRP, we first fitted a model including apoA-I with adjustment for the selected clinical variables, and then added CRP and an interaction term between CRP and apoA-I to the model described above. From this model, we present the relative hazard of recurrent coronary events associated with each SD increase in apoA-I, separately among those with low and high levels of CRP. We then repeated this analysis for each inflammatory marker (SAA, fibrinogen, D-dimer, and vWF) in separate models. Effect modification of the HDL-C/coronary risk association by inflammatory markers was examined in the same way. Analyses were performed in the entire study population and among those with and without diabetes separately.
We performed sensitivity analyses to assess the influence of 7 cardiac medications (acetylsalicylic acid, ACE inhibitors, β-adrenergic blockers, calcium channel blockers, diuretics, oral anticoagulants, and lipid-lowering drugs) that the patients were receiving at baseline on results by adding these therapies into the model one at a time. We also assessed the influence of each lipid variable (total cholesterol, low density lipoprotein cholesterol, triglyceride, and apolipoprotein B) in the same way. All analyses were performed using SAS software (version 9.4; SAS institute Inc., Cary, NC) with p < 0.05 used to define statistical significance.
Results
The original THROMBO study population has been described elsewhere 2. Baseline clinical characteristics of the 1,028 patients included in the present study are shown in Table 1. The majority of patients were white (75%) and male (76%) with a mean age of 59 years (range: 23-92 years). Nineteen percent of the population had a history of diabetes, and 38% of the population was on lipid lowering treatment. Diabetic patients had lower percent of male and white race, higher prevalence of cardiovascular disease history, and higher prevalence of pulmonary congestion and lower ejection fraction during the index MI compared to non-diabetic patients. As for baseline medications, use of ACE inhibitors and diuretics was more prevalent while use of beta blockers was less prevalent among diabetic patients compared to non-diabetic patients. Diabetic patients also had higher levels of CRP, fibrinogen, SAA and vWF compared to non-diabetic patients.
Table 1.
Patient characteristics.
| Characteristics | All patients (N=1028) | No diabetes (N=834) | Diabetes (N=194) |
|---|---|---|---|
| Age | |||
| Mean ±SD | 59±12 | 59±12 | 60±11 |
| ≥60y | 492 (48) | 390 (47) | 102 (53) |
| Male* | 780 (76) | 650 (78) | 130 (67) |
| Race, white* | 768 (75) | 653(78) | 115 (59) |
| Medical history | |||
| Prior Myocardial infarction | 193 (19) | 147 (18) | 46(24) |
| Treated hypertension* | 450 (44) | 331 (40) | 119 (62) |
| Diabetes mellitus | 194 (19) | 0 (0) | 194 (100) |
| Stroke* | 29 (3) | 16 (2) | 13 (7) |
| Smoking status | |||
| Never smoked | 347 (34) | 277 (33) | 70 (36) |
| Ex-smoker | 436 (42) | 357 (43) | 79 (41) |
| Current smoker | 245 (24) | 200 (24) | 45 (23) |
| Index myocardial infarction | |||
| Pulmonary congestion * | 176 (17) | 125 (15) | 51 (26) |
| Ejection fraction | |||
| Mean ± SD* | 0.48±0.12 | 0.48±0.12 | 0.45±0.12 |
| ≤0.30 | 109 (11) | 85 (10) | 24 (12) |
| Thrombolytic therapy | 353 (34) | 297(36) | 56 (29) |
| Medications at enrollment | |||
| Acetylsalicylic acid | 840 (82) | 686 (82) | 154 (79) |
| ACE inhibitors* | 379 (37) | 275 (33) | 104 (54) |
| Beta blockers* | 779 (76) | 646 (77) | 133 (69) |
| Calcium channel blockers | 208 (20) | 168 (20) | 40 (21) |
| Diuretics* | 169 (18) | 113 (14) | 56 (29) |
| Oral anticoagulants | 181 (18) | 149 (18) | 32 (16) |
| lipid lowering drugs | 395 (38) | 328 (39) | 67 (35) |
| Blood biomarkers | |||
| apoA-I, mg/dL | 118±25 | 118±25 | 120±25 |
| apoB, mg/dL | 123±28 | 123±28 | 123±29 |
| Cholesterol, mg/dL | 197±44 | 197±44 | 198±43 |
| HDL-C, mg/dL | 39±12 | 39±11 | 40±13 |
| LDL-C, mg/dL | 120±38 | 120±38 | 119±38 |
| Triglyceride, mg/dL | 168 (122-247) | 168 (121-247) | 170 (130-247) |
| CRP, mg/dL#* | 0.24 (0.10-0.59) | 0.22 (0.09-0.51) | 0.42 (0.15-0.84) |
| Fibrinogen, mg/dL* | 352±88 | 346±83 | 377±102 |
| SAA, mg/dL#* | 0.38 (0.18-0.70) | 0.34 (0.78-0.65) | 0.50 (0.29-0.93) |
| D-Dimer, ng/mL* | 376 (237-581) | 363 (232-553) | 452 (278-695) |
| Von Willebrand factor, U/dL* | 149±69 | 142±65 | 179±76 |
Data are N (%) for categorical variables and mean ± SD or median with interquartile range (if the distribution is highly skewed) for continuous variables.
CRP was available for 945, 174, and 771 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively. SAA was available for 896, 172, and 724 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively.
indicate P <0.05 for comparisons between patients with and without diabetes.
During an average follow-up of 26 months (1,731 person-years), 195 patients experienced a first recurrent coronary event (126 patients had unstable angina, 49 patients had non-fatal MI, and 20 patients died due to coronary heart disease). In the main effect analysis, none of the seven biomarkers (apoA-I, HDL-C, CRP, SAA, fibrinogen, d-Dimer, and vWF) was a significant predictor of coronary risk (Table 2).
Table 2.
Relative hazards of recurrent coronary events associated with apoA-I, HDL-C, and inflammatory markers
| Biomarkers | HR(95%CI) | HR(95%CI) | HR (95%CI) | |||
|---|---|---|---|---|---|---|
| All | P | Without diabetes | P | With diabetes | P | |
| Per SD increase | ||||||
| ApoA-I | 0.98 (0.84, 1.10) | 0.639 | 1.00 (0.84, 1.19) | 0.929 | 0.88 (0.65, 1.16) | 0.370 |
| HDL-C | 0.95 (0.81, 1.11) | 0.519 | 1.00 (0.82, 1.21) | 0.989 | 0.92 (0.69, 1.25) | 0.599 |
| High vs. low* | ||||||
| CRP | 1.31 (0.94, 1.83) | 0.106 | 1.43 (0.96, 2.15) | 0.077 | 1.19 (0.66, 2.14) | 0.564 |
| SAA | 1.19 (0.85, 1.68) | 0.306 | 1.27 (0.84, 1.92) | 0.260 | 1.08 (0.59, 1.95) | 0.813 |
| Fibrinogen | 1.09 (0.79, 1.51) | 0.607 | 1.25 (0.85, 1.83) | 0.256 | 0.89 (0.48, 1.64) | 0.700 |
| D-dimer | 1.15 (0.84, 1.58) | 0.375 | 1.24 (0.85, 1.81) | 0.271 | 1.20 (0.67, 2.15) | 0.544 |
| vWF | 1.08 (0.78, 1.49) | 0.648 | 1.12 (0.75, 1.68) | 0.586 | 1.07 (0.61, 1.86) | 0.819 |
Each biomarker was examined in separate models.
Low: the combined three lower quartiles (quartile 1, 2, and 3). High: the top quartile (quartile 4).
CRP was available for 945, 174, and 771 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively. SAA was available for 896, 172, and 724 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively.
For the entire population, analyses were adjusted for diabetes, prior MI, prior stroke, and enrolling sites. For each subgroup of diabetes, analyses were adjusted for prior MI, prior stroke, and enrolling sites.
In Table 3 we present the adjusted relative hazard of coronary events associated with each standard deviation increase in apoA-I, stratified by high and low levels of CRP, SAA, fibrinogen, d-Dimer, and vWF. The hazard ratios (HRs) are shown for the entire population and for those with and without diabetes separately. We observed significant effect modification of the association between apoA-I and coronary risk by CRP in all subjects (P value for interaction= 0.008). Among those with low CRP levels, apoA-I was not significantly associated with the risk of coronary events (HR = 0.88, 95% CI: 0.73 - 1.06), while among those with high CRP levels, each SD increase in apoA-I was significantly associated with a 33% increase in the risk (HR = 1.33, 95% CI: 1.04 - 1.71). This increased risk associated with apoA-I was primarily driven by the non-diabetic group (HR = 1.53 [95% CI: 1.15 - 2.04] for non-diabetic patients with high CRP levels; HR = 1.09 [95% CI: 0.68 - 1.75] for diabetic patients with high CRP levels). Nevertheless, the patterns of effect modification were consistent across subgroups by diabetes in that the protective effect estimates for apoA-I among those with low levels of CRP was attenuated or reversed in those with high levels of CRP.
Table 3.
Hazard ratios of recurrent coronary events associated with each SD increase in apoA-I in each stratum of inflammatory factors.
| Inflammatory and hemostatic factors | HR (95%CI) | HR(95%CI) | HR(95%CI) | |||
|---|---|---|---|---|---|---|
| All (n=1028) | P | No diabetes (n=834) | P | Diabetes (n=194) | P | |
| CRP*§ | ||||||
| Low | 0.88 (0.73, 1.06) | 0.170 | 0.94 (0.75, 1.16) | 0.550 | 0.67 (0.44, 1.01) | 0.058 |
| High | 1.33 (1.04, 1.71) | 0.022 | 1.53 (1.15, 2.04) | 0.004 | 1.09 (0.68, 1.75) | 0.725 |
| P for interaction | 0.008 | 0.007 | 0.128 | |||
| SAA*§ | ||||||
| Low | 0.98 (0.81, 1.18) | 0.833 | 1.05 (0.85, 1.31) | 0.634 | 0.70 (0.46, 1.08) | 0.110 |
| High | 1.06 (0.81, 1.39) | 0.663 | 1.25 (0.88, 1.78) | 0.217 | 0.93 (0.60, 1.46) | 0.766 |
| P for interaction | 0.633 | 0.418 | 0.367 | |||
| Fibrinogen* | ||||||
| Low | 0.94 (0.79, 1.11) | 0.440 | 0.96 (0.79, 1.17) | 0.708 | 0.83 (0.58, 1.17) | 0.280 |
| High | 1.09 (0.80, 1.49) | 0.567 | 1.25 (0.87, 1.81) | 0.226 | 0.99 (0.57, 1.73) | 0.972 |
| P for interaction | 0.383 | 0.214 | 0.587 | |||
| d-Dimer* | ||||||
| Low | 0.99 (0.84, 1.17) | 0.928 | 1.00 (0.82, 1.22) | 0.989 | 0.94 (0.67, 1.30) | 0.699 |
| High | 0.89 (0.66, 1.20) | 0.453 | 1.02 (0.72, 1.45) | 0.895 | 0.76 (0.43, 1.32) | 0.329 |
| P for interaction | 0.540 | 0.913 | 0.514 | |||
| vWF* | ||||||
| Low | 0.88 (0.74, 1.06) | 0.174 | 0.98 (0.80, 1.19) | 0.808 | 0.60 (0.38, 0.93) | 0.022 |
| High | 1.17 (0.91, 1.50) | 0.234 | 1.10 (0.79, 1.53) | 0.577 | 1.42 (0.92, 2.20) | 0.110 |
| P for interaction | 0.077 | 0.545 | 0.006 | |||
Low: the combined three lower quartiles (quartile 1, 2, and 3). High: the top quartile (quartile 4).
CRP was available for 945, 174, and 771 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively. SAA was available for 896, 172, and 724 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively.
For the entire population, analyses were adjusted for diabetes, prior MI, prior stroke, and enrolling sites. For each subgroup of diabetes, analyses were adjusted for prior MI, prior stroke, and enrolling sites.
We also observed a pattern of effect modification by vWF in all subjects with borderline significance (P value for interaction = 0.077). Subgroup analysis by diabetes showed that the effect modification was only present in diabetic patients (P values for interaction were 0.006 and 0.545, for diabetic group and non-diabetic group, respectively). In the diabetic group, each SD increase in apoA-I was significantly associated with a 40% decrease in the risk of recurrent coronary events among those with low levels of vWF (HR = 0.60, 95% CI: 0.38 - 0.93), while among those with high levels of vWF, apoA-I was not significantly associated with the risk (HR = 1.42, 95% CI: 0.92 - 2.20). We did not observe clear patterns of effect modification by SAA, fibrinogen or d-Dimer of the association between apoA-I and the risk of coronary events. Nor did we observe significant effect modification of coronary risk associated with HDL-C by any of these inflammatory biomarkers (Table 4). None of the 7 baseline cardiac medications and 5 lipid variables had a substantial impact on the pattern of effect modification observed in the main analyses.
Table 4.
Hazard ratios of recurrent coronary events associated with each SD increase in HDL-C in each stratum of inflammatory factors.
| Inflammatory and hemostatic factors | HR (95%CI) | HR (95%CI) | HR (95%CI) | |||
|---|---|---|---|---|---|---|
| All (n=1028) | P | No diabetes (n=834) | P | Diabetes (n=194) | P | |
| CRP*§ | ||||||
| Low | 0.90 (0.73, 1.11) | 0.332 | 0.96 (0.75, 1.24) | 0.750 | 0.81 (0.52, 1.24) | 0.332 |
| High | 1.14 (0.88, 1.48) | 0.307 | 1.29 (0.93, 1.80) | 0.128 | 1.08 (0.70, 1.68) | 0.717 |
| P for interaction | 0.156 | 0.158 | 0.324 | |||
| SAA*§ | ||||||
| Low | 0.94 (0.76, 1.16) | 0.558 | 1.06 (0.82, 1.36) | 0.659 | 0.71 (0.43, 1.17) | 0.180 |
| High | 0.91 (0.68, 1.21) | 0.506 | 1.01 (0.68, 1.48) | 0.976 | 0.89 (0.56, 1.42) | 0.620 |
| P for interaction | 0.849 | 0.828 | 0.511 | |||
| Fibrinogen* | ||||||
| Low | 0.94 (0.79, 1.12) | 0.492 | 0.97 (0.78, 1.21) | 0.806 | 0.94 (0.67, 1.32) | 0.712 |
| High | 1.00 (0.73, 1.39) | 0.976 | 1.19 (0.78, 1.81) | 0.412 | 0.86 (0.47, 1.54) | 0.603 |
| P for interaction | 0.721 | 0.398 | 0.782 | |||
| d-Dimer* | ||||||
| Low | 0.93 (0.78, 1.12) | 0.452 | 0.94 (0.75, 1.18) | 0.614 | 0.98 (0.70, 1.37) | 0.904 |
| High | 1.02 (0.76, 1.38) | 0.882 | 1.18 (0.82, 1.68) | 0.376 | 0.82 (0.46, 1.46) | 0.496 |
| P for interaction | 0.605 | 0.307 | 0.578 | |||
| vWF* | ||||||
| Low | 0.94 (0.78, 1.12) | 0.483 | 0.94 (0.75, 1.18) | 0.599 | 0.99 (0.70, 1.38) | 0.935 |
| High | 0.98 (0.73, 1.32) | 0.912 | 1.19 (0.81, 1.74) | 0.379 | 0.79 (0.46, 1.33) | 0.375 |
| P for interaction | 0.786 | 0.304 | 0.466 | |||
Low: the combined three lower quartiles (quartile 1, 2, and 3). High: the top quartile (quartile 4).
CRP was available for 945, 174, and 771 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively. SAA was available for 896, 172, and 724 subjects in the entire population, in the diabetic group, and in the non-diabetic group, respectively.
For the entire population, analyses were adjusted for diabetes, prior MI, prior stroke, and enrolling sites. For each subgroup of diabetes, analyses were adjusted for prior MI, prior stroke, and enrolling sites.
We further fitted a model with three-way interactions among CRP, vWF and apoA-I, and present the relative hazards of recurrent coronary events associated with each SD increase in apoA-I for each of the four subgroups stratified by low or high levels of CRP and vWF (low CRP/low vWF, high CRP/low vWF, low CRP/high vWF, and high CRP/high wWF) in Figure 1. The group with both low CRP and vWF showed significant protective association between apoA-I and coronary risk with a hazard ratio of 0.81 (95% CI: 0.66 - 1.00). The group with both high CRP and vWF showed significant increased risk associated with higher apoA-I with a HR of 1.62 (95% CI: 1.16 - 2.25).
Figure 1.
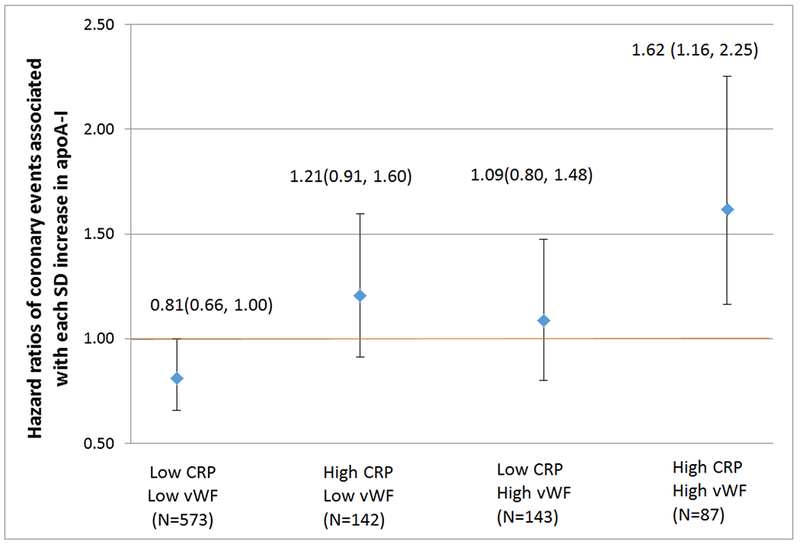
Hazard ratios of coronary events associated with each SD increase in apoA-I, stratified by high or low CRP and vWF status. Low: the combined three lower quartiles (quartile 1, 2, and 3). High: the top quartile (quartile 4). Analyses were adjusted for diabetes, prior MI, prior stroke, and enrolling sites.
Discussion
The present study assessed if blood inflammatory markers (CRP, SAA, fibrinogen, D-dimer, and vWF) modify the risk of recurrent coronary events associated with apoA-I and HDL-C. We identified CRP as a strong effect modifier of the association between apoA-I and coronary risk, with a muted protective association among patients with high CRP compared to patients with low CRP, and this pattern was consistent across subgroups by diabetes. We also observed a similar pattern of effect modification by vWF. However, this pattern was only observed in the diabetic subgroup.
HDL-C is a well-known protective factor of cardiovascular disease. However, in randomized controlled trials, high-dose niacin or inhibitors of cholesteryl ester transfer protein failed to show clinical benefit on cardiovascular events despite significantly increasing HDL cholesterol levels 9–11, which shifts the focus from HDL quantity to HDL function. A number of studies have demonstrated that HDL particles can undergo structural and functional changes during inflammatory responses. Increased oxidative stress accompanied with inflammation can lead to damaging oxidative modifications of apoA-I 12, 13. Additionally, inflammation was discovered to adversely affect HDL function via changing the HDL proteome and lipidome 4. Displacement of apoA-I by the acute phase protein SAA, loss of anti-oxidative proteins paraoxonase and platelet-activing factor acetylhydrolase, and decreased activity of lecithin: cholesterol acyltransferase that occur during inflammatory responses all contribute to HDL dysfunction 14, 15. Moreover, acute-phase HDL is enriched in triglycerides, which might lead to impaired HDL stability and enhanced apoA-I catabolism 4, 16. These inflammation-induced changes result in impaired anti-atherogenic function of HDL particles regarding its role in reverse cholesterol transport, anti-oxidative and anti-inflammatory activity, and amelioration of endothelial function.
Despite the well-established mechanisms of inflammation-induced HDL-dysfunction, only a few population studies to date examined how inflammation influences cardiovascular risk associated with apoA-I or HDL-C. A study by Tehrani et al. assessed if inflammatory markers CRP, interleukin-6 (IL-6), and lipoprotein-associated phospholipase A2 (Lp-PLA2) modify the association between HDL-C and incident coronary heart disease in 3,888 elder adults from the Cardiovascular Health Study 17. This study did not observe significant interactions between HDL-C and any inflammatory markers, although the protective relation of high HDL-C with incident coronary heart disease seemed to be attenuated by greater inflammation. Compared with this study of a general population without cardiovascular disease at baseline, our post-MI population had a different CRP distribution with a higher mean and tertiles and a larger standard deviation. The relative high levels of CRP with more variability make it more likely to detect the effect modification in this post-MI population.
Our previous study of non-diabetic patients of the THROMBO study demonstrated that elevated HDL-C was associated with increased risk of recurrent coronary events in a high-risk subgroup of patients characterized by simultaneous elevations in serum CRP and total cholesterol 18. In the present study, we did not observe significant effect modification of coronary risk associated with HDL-C by CRP among non-diabetic patients. The high-risk subgroup in the previous study identified by an exploratory three-dimensional graphical screening technique was very different from the overall non-diabetic patients with not only significantly higher CRP but also significantly higher levels of other risk factors (e.g. total cholesterol, triglycerides, fibrinogen et al.). Thus the null finding for HDL-C in the present study compared to our previous study might be explained by differences in the risk profile of the two populations. Nevertheless, our finding of the interaction between apoA-I and CRP supports the biological mechanism underlying the previous study that HDL particle can lose its protective association with coronary risk under certain states such as inflammation.
It is also of note that although apoA-I and HDL-C are closely related and are both considered cardioprotective, they are biologically distinct. ApoA-I is the main structural and functional protein of HDL particles that plays a pivotal role in HDL’s diverse anti-atherogenic effect, whereas HDL-C reflects the concentration of cholesterol transported by HDL particles. In our study, HDL-C and apoA-I concentrations were only moderately correlated (r = 0.44, P < 0.001). One of the major anti-atherogenic functions of ApoA-I is to mediate macrophage cholesterol efflux via the binding of Apo-AI to ATP-binding cassette transporter A1 (ABCA1). A previous study showed that ApoA-I is a selective target for oxidative modification via the myeloperoxidase pathway 19. Oxidation of methionine, tyrosine, or tryptophan residues of apoA-I by myeloperoxidase dramatically reduced the ability of apoA-I to promote cholesterol efflux through the ABCA1 pathway. Therefore, our inconsistent findings of effect modification by inflammation for HDL-C and apoA-I might be explained by the fact that apoA-I is a direct functional target in HDL particles adversely affected by high inflammation, and thus is more sensitive to effect modification by inflammation.
We also observed vWF as a strong effect modifier of the association between apoA-I and risk of recurrent coronary events among diabetic patients, but not among non-diabetic patients. VWF is released into the circulation by secretion from endothelial cells and is generally considered as a marker of endothelial dysfunction. Circulating vWF levels can be affected by a number of factors. Impaired endothelial nitric oxide production, inflammation, insulin resistance, and diabetes have all been associated with elevated vWF plasma levels 20, 21. Our previous study of the THROMBO compared diabetic versus non-diabetic patients regarding several thrombogenic factors, and vWF was the only factor found to be independently associated with diabetes, suggesting substantial endothelial damage in diabetic post-MI patients 22. In addition to endothelial dysfunction, diabetes can also contribute to HDL dysfunction. This is mediated by hyperglycemia and glycation of apoA-I and other HDL-associated proteins, as well as chronic inflammation and enhanced oxidative stress in diabetes 6. Furthermore, dysfunctional HDL is deficient in its ability to stimulate endothelial NO production and to protect endothelial cells from apoptosis, thus contributing to endothelial dysfunction 4. Therefore, diabetes, inflammation, HDL dysfunction, and endothelial dysfunction characterized by high vWF are likely to interfere with each other and together have complex influences on the association between apoA-I and coronary risk. Our study showed that among diabetic patients, the protective association between apoA-I and coronary risk only existed in patients with low vWF. However, it is unclear whether the presence of endothelial dysfunction diminished the protective effect of apoA-I among diabetic patients or whether high vWF levels are merely consequence of other states that produce endothelial damage such as inflammation and HDL dysfunction. We did not observe effect modification by vWF among non-diabetic patients. The significantly lower vWF levels among non-diabetic patients compared to diabetic patients may contribute to the null finding.
We did not observe clear patterns of effect modification by SAA, d-Dimer and fibrinogen of the association between apoA-I and risk of coronary events. Previously studies suggested that the apoA-I content of HDL particles dramatically decreased during an acute phase response and was replaced mostly by the acute phase protein SAA. SAA-enriched HDL particles have decreased cholesterol efflux capacity 14. However, in this stable post-MI population with blood drawn 2 months after the index MI, SAA levels were similar to that of the general population 23–25. Therefore, our null finding might be explained by the overall low levels of SAA in this population. Although fibrinogen and d-Dimer can be considered as markers of generalized inflammatory responses, these factors did not modify the association between apoA-I and coronary risk as suggested by the present study.
We previously demonstrated in the original report of the THROMBO study that higher apoA-I was significantly associated with lower risk of coronary events defined as non-fatal MI or death due to coronary heart disease 2. In the present study, neither apoA-I nor HDL-C was a significant predictor of coronary risk in the main effect analysis. It should be noted that a different composite endpoint that included unstable angina was used in the present study (unstable angina, non-fatal MI, or death due to coronary heart disease). Unstable angina is a less severe form of coronary events compared to non-fatal MI and death due to coronary heart disease. Therefore, the null finding for apoA-I in this study may be explained by the relative mild and heterogeneous endpoint. The null finding for HDL cholesterol in the present study is consistent with our previous report of the THROMBO study.
Our study has several limitations that should be considered when making inferences. First, some caution is needed in the interpretation of the positive findings due to multiple testing. However, given the significance of the results for interaction by CRP in the entire population and by vWF in the diabetic group, the chance of false positives should be small. Second, although we adjusted for multiple clinical covariates in our model, there might be residual confounders such as physical activity, diet and psychological factors that affect the main effect of apoA-I and HDL-C on coronary risk. Third, patients in this study were not under the clinical care considered optimal based on today’s practice guidelines (e.g. use of lipid lowering drugs was only around 40%). Therefore, findings may not be generalizable to post-infarction patients under today’s standard clinical care. However, adjustment for medications including lipid lowering drugs did not change the pattern of effect modification observed in the main analysis.
Conclusions
Among stable post-MI patients, CRP was a strong effect modifier of the association between apoA-I and the risk of recurrent coronary events. VWF was an effect modifier of the association between apoA-I and risk only among diabetic subgroup. These findings provide population evidence for the biological mechanism of inflammation-induced HDL dysfunction. Our study suggests that HDL quantitative measures may not be reliable predictors of cardiovascular risk and more focus on HDL function is needed. Further studies of different populations are needed to confirm or refute our findings.
Acknowledgments
Funding
This study was supported by research grant HL-48259 from the National Institutes of Health, Bethesda, MD.
Footnotes
Conflict of Interest: None
References
- 1.D’Agostino RB Sr., Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–753. [DOI] [PubMed] [Google Scholar]
- 2.Moss AJ, Goldstein RE, Marder VJ, et al. Thrombogenic factors and recurrent coronary events. Circulation. 1999;99:2517–2522. [DOI] [PubMed] [Google Scholar]
- 3.Ingelsson E, Schaefer EJ, Contois JH, et al. Clinical utility of different lipid measures for prediction of coronary heart disease in men and women. Jama. 2007;298:776–785. [DOI] [PubMed] [Google Scholar]
- 4.Rosenson RS, Brewer HB Jr., Ansell BJ, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nature reviews. Cardiology. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corsetti JP, Zareba W, Moss AJ, et al. Metabolic syndrome best defines the multivariate distribution of blood variables in postinfarction patients. Atherosclerosis. 2003;171:351–358. [DOI] [PubMed] [Google Scholar]
- 6.Farbstein D, Levy AP. HDL dysfunction in diabetes: causes and possible treatments. Expert review of cardiovascular therapy. 2012;10:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harb TS, Zareba W, Moss AJ, et al. Association between inflammatory markers, hemostatic, and lipid factors in postinfarction patients. The American journal of cardiology. 2003;91:1120–1123. [DOI] [PubMed] [Google Scholar]
- 8.Mieszczanska H, Pietrasik G, Piotrowicz K, McNitt S, Moss AJ, Zareba W. Gender-related differences in electrocardiographic parameters and their association with cardiac events in patients after myocardial infarction. The American journal of cardiology. 2008;101:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. The New England journal of medicine. 2007;357:2109–2122. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. The New England journal of medicine. 2012;367:2089–2099. [DOI] [PubMed] [Google Scholar]
- 11.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. The New England journal of medicine. 2011;365:2255–2267. [DOI] [PubMed] [Google Scholar]
- 12.Shao B, Bergt C, Fu X, et al. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. The Journal of biological chemistry. 2005;280:5983–5993. [DOI] [PubMed] [Google Scholar]
- 13.Bergt C, Pennathur S, Fu X, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13032–13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Lenten BJ, Hama SY, de Beer FC, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. The Journal of clinical investigation. 1995;96:2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. The Journal of biological chemistry. 1986;261:9644–9651. [PubMed] [Google Scholar]
- 16.Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. Journal of lipid research. 1996;37:2662–2674. [PubMed] [Google Scholar]
- 17.Tehrani DM, Gardin JM, Yanez D, et al. Impact of inflammatory biomarkers on relation of high density lipoprotein-cholesterol with incident coronary heart disease: cardiovascular Health Study. Atherosclerosis. 2013;231:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corsetti JP, Zareba W, Moss AJ, Rainwater DL, Sparks CE. Elevated HDL is a risk factor for recurrent coronary events in a subgroup of non-diabetic postinfarction patients with hypercholesterolemia and inflammation. Atherosclerosis. 2006;187:191–197. [DOI] [PubMed] [Google Scholar]
- 19.Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. The Journal of clinical investigation. 2004;114:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. Journal of thrombosis and haemostasis : JTH. 2006;4:1186–1193. [DOI] [PubMed] [Google Scholar]
- 21.Tousoulis D, Papageorgiou N, Androulakis E, et al. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. Journal of the American College of Cardiology. 2013;62:667–676. [DOI] [PubMed] [Google Scholar]
- 22.Zareba W, Pancio G, Moss AJ, et al. Increased level of von Willebrand factor is significantly and independently associated with diabetes in postinfarction patients. THROMBO Investigators. Thrombosis and haemostasis. 2001;86:791–799. [PubMed] [Google Scholar]
- 23.d’Eril GM, Anesi A, Maggiore M, Leoni V. Biological variation of serum amyloid A in healthy subjects. Clinical chemistry. 2001;47:1498–1499. [PubMed] [Google Scholar]
- 24.Hijmans W, Sipe JD. Levels of the serum amyloid A protein (SAA) in normal persons of different age groups. Clin Exp Immunol 1979;35:96–100. [PMC free article] [PubMed] [Google Scholar]
- 25.Fyfe AI, Rothenberg LS, DeBeer FC, Cantor RM, Rotter JI, Lusis AJ. Association between serum amyloid A proteins and coronary artery disease: evidence from two distinct arteriosclerotic processes. Circulation. 1997;96:2914–2919. [DOI] [PubMed] [Google Scholar]