

Abstract
Background
Despite the medical importance of the genus Culex, the mitochondrial genome (mt genome) characteristics of Culex spp. are not well understood. The phylogeny of the genus and particularly the generic status of the genus Lutzia and the subgenus Culiciomyia remain unclear.
Methods
The present study sequenced and analyzed the complete mt genomes of Lutzia halifaxia, Lutzia fuscanus and Cx. (Culiciomyia) pallidothorax and assessed the general characteristics and phylogenetics of all known 16 mt genome sequences for species in the genera Culex and Lutzia.
Results
The complete mt genomes of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax are 15,744, 15,803 and 15,578 bp long, respectively, including 13 PCGs, 22 tRNAs, two tRNAs and a control region (CR). Length variations in the Culex and Lutzia mt genomes involved mainly the CR, and gene arrangements are the same as in other mosquitoes. We identified four types of repeat units in the CR sequences, and the poly-T stretch exists in all of these mt genomes. The repeat units of CR are conserved to different extent and provide information on their evolution. Phylogenetic analyses demonstrated that the Coronator and Sitiens groups are each monophyletic, whereas the monophyletic status of the Pipiens Group was not supported; Cx. pallidothorax is more closely related to the Sitiens and Pipiens groups; and both phylogenetics analysis and repeat unit features in CR show that Lutzia is a characteristic monophyletic entity, which should be an independent genus.
Conclusions
To our knowledge, this is the first comprehensive review of the mt genome sequences and taxonomic discussion based on the mt genomes of Culex spp. and Lutzia spp. The research provides general information on the mt genome of these two genera, and the phylogenetic and taxonomic status of Lutzia and Culiciomyia.
Electronic supplementary material
The online version of this article (10.1186/s13071-019-3625-2) contains supplementary material, which is available to authorized users.
Keywords: Lutzia halifaxia, Lutzia fuscanus, Culex pallidothorax, Lutzia, Culiciomyia, Culex, Mitochondrial genome, Phylogeny
Background
The genus Culex is the largest genus in the Culicidae in terms of the number of species and is distributed worldwide [1]. Some Culex species are important vectors of infectious and arboviral diseases such as epidemic encephalitis and lymphatic filariasis [1]. The genus Lutzia was established by Theobald in 1903 [2] and was then reduced to a subgenus of the genus Culex by Edwards in 1932 [3]. Subsequent authors treated it as a subgenus until 2003, when Tanaka formally restored Lutzia to its original generic status [4]. However, the taxonomic level of Lutzia has remained controversial. For example, phylogenetic analysis based on larval and adult morphological characteristics placed Lutzia outside the clade that comprised the genus Culex (including representative species in all 26 subgenera) [5, 6]. In contrast, molecular phylogenetic analyses placed Lutzia among species of the genus Culex based on ITS1 and ITS2 sequences and cox1 [7–9]. The molecular phylogenetic analysis based on ITS1 and ITS2 sequences using neighbor-joining approach indicated that the genus Lutzia (one species included) formed the sister group to the subgenus Culex (11 species included) [7]. In contrast, the analysis based on 478 bp of cox1 using Bayesian method suggested that the genus Culex (17 species included) is paraphyletic relative to Lutzia (one species included) [8]. The analysis based on ITS2 using neighbor-joining approach showed that the genus Lutzia (one species included) was placed inside the genus Culex (16 species included) [9].
Lutzia is distributed in the Afrotropical, Oriental, southern Palaearctic, Australasian and Neotropical regions and has eight known species, with only two species (Lutzia halifaxia and Lutzia fuscanus) recorded in China. Subgenus Culiciomyia was established by Edwards in 1921 [10] and has 55 known species with a geographical distribution in Afrotropical, Oriental and Australasian regions [1]. Culex pallidothorax in the subgenus Culiciomyia was grouped into the subgenus Culex with a low bootstrap support of 11% based on the results of phylogenetic analysis of cox1 sequences [11]. The mitochondrial genome (mt genome) sequence of the subgenus has not yet been investigated.
Mitochondria are related to various biological processes, from power production to programmed cell death and ageing [12]. Mitochondrial DNA (mtDNA) sequences have been widely used as molecular markers for the identification of organisms and in research investigations on insect population genetics and phylogenetics [13–19]. As of 20 March 2018, a total of 13 different mt genome sequences have been reported in the genus Culex, and these sequences are all from nine species/subspecies within the subgenus Culex (Cx. camposi, Cx. coronator, Cx. gelidus, Cx. pipiens pallens, Cx. pipiens pipiens, Cx. quinquefasciatus, Cx. tritaeniorhynchus, Cx. usquatus and Cx. usquatissimus) [20–23]. To date, no mt genome sequence has been reported for Lutzia or Culiciomyia.
In the present study, we sequenced and analyzed the complete mt genomes of Lt. halifaxia and Lt. fuscanus in Lutzia and Culex (Culiciomyia) pallidothorax in the subgenus Culiciomyia, comprehensively analyzed the characteristics of all 16 mt genome sequences in the genus Culex available to date (including three mt genome sequences obtained in the present study), and conducted phylogenetic reconstruction using these 16 mt genomes. The study also generated insights into the taxonomic status and position of Lutzia and Culiciomyia.
Methods
Sample collection and total DNA extraction
Specimens of Lt. halifaxia and Cx. (Culiciomyia) pallidothorax were collected from Leishan County, Guizhou Province, China (26°29′27″N, 108°09′27″E) in July 2015. Specimens of Lt. fuscanus specimens were collected from Shuicheng County, Guizhou Province, China (26°35′40″N, 104°48′07″E) in August 2015. All collected samples were stored in 100% alcohol and stored at − 20 °C until use. These three species of mosquitoes were initially identified using morphological characteristics [24] and then confirmed by sequencing the cox1 and ITS2 loci as reported elsewhere [25]. Total DNA was separately extracted from a female adult of each species using a TIANamp Genomic DNA Kit (TianGen, Shanghai, China) following the manufacturerʼs instructions, and then total DNAs were preserved at − 80 °C for subsequent mt genome sequencing.
Mt genome sequencing, assembly and annotation
The mt genome fragments of these three species were amplified by the universal primers for Diptera [26]. Due to the amplification difficulty of the control region (CR) of Lt. halifaxia and Lt. fuscanus mt genomes, one additional pair of primers (F: 5′-TCA ATT TAC TAT TAT ATT TAT TGG AG-3′ and R: 5′-TAA TTT CAA TAG TTT GTC CAT GTA-3′) was designed with online Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) according to known Culicidae mt genomes and applied to fill the sequence gap of the CR. All PCR amplifications were performed in 25 μl reactions containing 4 μl of dNTPs, 1 μl of each primer, 2.5 μl of 10× LA PCR buffer I, 1–2 μl of DNA template, 0.25 μl of LA Taq polymerase (TaKaRa, Dalian, China) and 14.25–15.25 μl ddH2O. The PCR amplification conditions were as follows: an initial denaturation at 94 °C for 1 min; 35 cycles of 94 °C for 40 s (denaturation), 47–58 °C for 45 s (annealing) and 68 °C for 1 min (extension); followed by a final extension at 72 °C for 10 min. All PCR fragments were successfully amplified using the extracted DNA template, but the CR was cloned into the vector pMD-19T (TaKaRa) and then amplified due to extensive sequence variations. All PCR fragments were subsequently purified with a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and were sequenced using a DNA Sequencer (ABI3730) at Life Technologies™ Company (Shanghai, China) in both directions.
The obtained sequences were assembled using DNAMANx software. All genes [13 protein-coding genes (PCGs) and two ribosomal RNA genes (rRNAs)] and the CR were identified by comparing with the corresponding sequences in other known Culex mt genomes with ClustalX [27], whereas transfer RNA genes (tRNAs) were identified using tRNAscan-SE Search Server v.1.21 (http://lowelab.ucsc.edu/tRNAscan-SE/) [28]. Some tRNAs that could not be identified by tRNAscan-SE were diagnosed by the multiple sequence alignment with the tRNA sequences of known Culex mt genomes. The base composition, relative synonymous codon usage (RSCU), and amino acid content were computed with MEGA v.5.0 software [29]. AT-skew [(A − T)/(A + T)] and GC-skew [(G − C)/(G + C)] were estimated in order to investigate nucleotide composition bias [30]. The graphical maps of the mt genomes were visualized with the CGView Comparison Tool [31]. The three-dimensional scatter plot of the AT-skew and GC-skew of these 16 mt genomes was drawn using Origin Pro v.9.0 [32]. The tandem repeats in the CRs were identified using the Tandem Repeats Finder program [33]. The secondary structures of tRNAs were predicted by tRNAscan-SE Search Server v.1.21.
Phylogenetic analysis
Phylogenetic analysis of the 16 Culex mt genomes (including three mt genomes produced in the present study and 13 Culex mt genomes deposited in GenBank; accession numbers are listed in Table 1) were performed using the Bayesian Inference (BI) analysis in MrBayes v.3.2.6 [34]. The amino acid sequence of each protein-coding gene was aligned individually based on codon-based multiple alignments using the MAFFT algorithm within the TranslatorX server (www.translatorx.co.uk) [35]. Poorly aligned sites were removed from the amino acid alignment before translating back to nucleotides using GBlocks in TranslatorX with default settings. The nucleotide sequences of the 13 PCGs were applied in the analysis because these are considered most suitable for inferring the phylogenetic relationships of known mt genome sequences of genus Culex [22]. The mt genome sequences of Anopheles gambiae (GenBank: NC002084) and Aedes aegypti (GenBank: NC010241) were used as outgroups. The best-fit model for each gene was chosen under the Akaike information criterion by Modeltest [36]. The concatenated matrix of the 13 PCGs was used to carry out the BI analysis. For the latter, two independent runs were performed, each with three hot chains and one cold chain, with posterior distributions estimated using Markov Chain Monte Carlo (MCMC) sampling. The MCMC chains were set for 5,000,000 generations, with tree sampling every 1000 steps and a relative ‛burn-inʼ of 25%. The convergence of the two runs was evaluated by average standard deviation of split frequencies (< 0.01). The phylogenetic tree was drawn in FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Table 1.
Detailed sequence information of 16 mt genomes of species in genera Culex and Lutzia
| Genus/Subgenus | Species | Total size (bp) | PCGs size (bp) | tRNA size (bp) | rRNA size (bp) | CR size (bp) | GenBank ID | Reference |
|---|---|---|---|---|---|---|---|---|
| Lutzia | Lt. halifaxia | 15,744 | 11,226 | 1484 | 2134 | 899 | MH316119 | This study |
| Lt. fuscanus | 15,803 | 11,218 | 1481 | 2126 | 920 | MH316118 | This study | |
| Culiciomyia | Cx. pallidothorax | 15,578 | 11,222 | 1482 | 2128 | 724 | KY400104 | This study |
| Culex | Cx. camposi | 15,570 | 11,228 | 1483 | 2124 | 719 | NC_036008.1 | [20] |
| Cx. coronator | 15,576 | 11,228 | 1482 | 2124 | 725 | NC_036006.1 | [20] | |
| Cx. gelidus | 15,600 | 11,230 | 1414 | 2143 | 721 | KX753344 | [22] | |
| Cx. pipiens pallens | 15,617 | 11,234 | 1482 | 2138 | 747 | KT851543.1 | [21] | |
| Cx. pipiens pipiens | 14,856 | 11,188 | 1475 | 2118 | a | NC_015079.1 | GenBankb | |
| Cx. pipiens TU | 14,856 | 11,216 | 1475 | 2118 | a | HQ724616.1 | GenBankb | |
| Cx. quinquefasciatus | 15,587 | 11,220 | 1467 | 2137 | 704 | NC_014574.1 | [19] | |
| Cx. quinquefasciatus USA | 14,856 | 11,216 | 1476 | 2118 | a | HQ724617.1 | GenBankb | |
| Cx. tritaeniorhynchus CQ | 14,844 | 11,219 | 1498 | 2143 | a | KT851544.1 | [21] | |
| Cx. tritaeniorhynchus JS | 14,861 | 11,222 | 1473 | 2128 | a | NC_028616.1 | GenBankb | |
| Cx. usquatissimus AC | 15,573 | 11,228 | 1482 | 2124 | 721 | MF040165.1 | [20] | |
| Cx. usquatissimus RO | 15,574 | 11,228 | 1483 | 2124 | 722 | NC_036007.1 | [20] | |
| Cx. usquatus | 15,573 | 11,228 | 1483 | 2124 | 719 | NC_036005.1 | [20] |
aDoes not harbor the CR
bReported only in GenBank
Results
Genome organization and nucleotide composition
The complete length of the mt genomes of Lt. halifaxia (GenBank: MH316119), Lt. fuscanus (GenBank: MH316118) and Cx. pallidothorax (GenBank: KY400104) was 15,744, 15,803 and 15,578 bp, respectively (Fig. 1). All mt genomes included 37 genes (13 PCGs, 22 tRNAs and 2 rRNAs) and a control region (CR), with 9 PCGs and 13 tRNAs encoded on the majority strand (J-strand) and 4 PCGs, 9 tRNAs and 2 rRNAs on the minority strand (N-strand). Comparison of the mt genomes of the two Lutzia spp. with nine Culex spp. which all have complete mt genome sequences indicated that those of Lt. halifaxia and Lt. fuscanus are 127–233 bp longer (Table 1). The PCGs, tRNAs and rRNAs are conservative in length, and the CRs are relatively variable in length, with the Lt. halifaxia and Lt. fuscanus CRs being much longer (898 and 920 bp, respectively) than the nine Culex mt genome CRs, which ranged from 704 bp in Cx. quinquefasciatus to 747 bp in Cx. p. pallens (Additional file 1: Table S1). Similarly to the 13 published Culex mt genomes, the nucleotide compositions of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax mt genomes are biased toward A and T, with A being the most favored nucleotide and C as the least favored. The observed adenine + thymine (AT) content of the mt genomes was high, accounting for 77.96% (A = 39.28%; T = 38.68%; G = 9.30%; C = 12.74%), 78.40% (A = 39.70%; T = 38.70%; G = 9.10%; C = 12.60%) and 78.50% (A = 39.70%; T = 38.80%; G = 9.10%; C = 12.70%) in Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax, respectively (Additional file 1: Table S1).
Fig. 1.

Mt genome structure of Lt. halifaxia (a), Lt. fuscanus (b) and Cx. pallidothorax (c). The blue-, pink-, yellow- and gray-filled blocks indicate PCGs, tRNAs, rRNAs and CR, respectively. The genes on the outer circle are located on the J-strand, whereas the genes on the inner circle are located on the N-strand. L, L2, S1 and S2 represent the tRNAs trnL1, trnL2, trnS1 and trnS2, respectively. Arrows indicate the transcriptional direction of mitochondrial genes
The three-dimensional scatter plot of the AT content, AT-skew and GC-skew in the 14 Culex spp. and 2 Lutzia spp. mt genomes is shown in Fig. 2. The AT-skew of Lt. halifaxia (0.0078) and Cx. pallidothorax mt genome (0.0078) are lower than the average AT-skew of all investigated mt genomes (0.0107), whereas the AT-skew of Lt. fuscanus mt genome (0.0128) is higher than the average AT-skew value. The GC-skew in Lt. halifaxia (-0.1613) and Cx. pallidothorax (-0.1651) are a bit lower than the average investigated GC-skew value (-0.1572), whereas the GC-skew of Lt. fuscanus mt genome (-0.1559) is slightly higher than the average GC-skew value. In general, the AT-skew and GC-skew are highly variable in the investigated mt genomes. For example, species of the Coronator group [Cx. camposi, Cx. coronator, Cx. usquatissimus AC (geographical name as published), Cx. usquatissimus RO and Cx. usquatus)] have similar AT content and AT/GC-skew, which are closely distributed in the three-dimensional scatter plot, whereas the species of the Pipiens group (Cx. p. pallens, Cx. p. pipiens, Cx. pipiens TU, Cx. quinquefasciatus and Cx. quinquefasciatus USA) are widely distributed in the plot for AT content, AT-skew and GC-skew (Fig. 2).
Fig. 2.
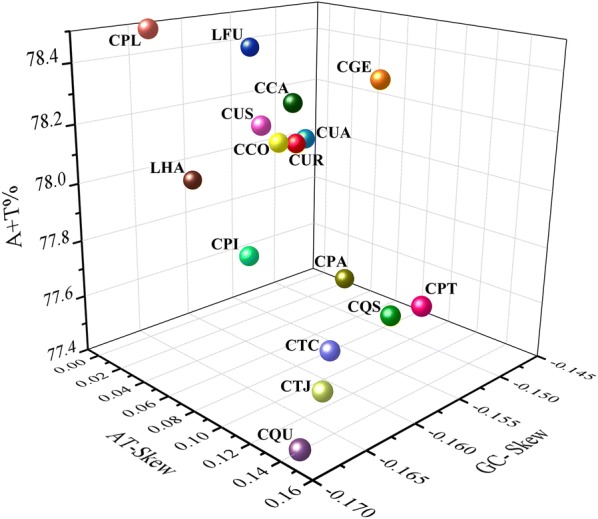
Three-dimensional scatterplot of the AT-Skew, GC-Skew and AT% of 16 Culex and Lutzia mt genome sequences. Abbreviations: CCA, Cx. camposi; CCO, Cx. coronator; LFU, Lt. fuscanus; CGE, Cx. gelidus; LHA, Lt. halifaxia; CPA, Cx. p. pallens; CPI, Cx. p. pipiens; CPL, Cx. pallidothorax; CPT, Cx. pipiens TU; CQS, Cx. quinquefasciatus USA; CQU, Cx. quinquefasciatus; CTC, Cx. tritaeniorhynchus CQ; CTJ, Cx. tritaeniorhynchus JS; CUA, Cx. usquatissimus AC; CUR, Cx. usquatissimus RO; CUS, Cx. usquatus
Protein-coding genes
The total nucleotide length of the 13 PCGs of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax was 11,226, 11,218 and 11,222 bp, respectively, falling within the range of total nucleotide length variations of the 13 PCGs in the 16 Culex spp. mt genomes (from 11,188 bp in Cx. p. pipiens to 11,234 bp in Cx. p. pallens) (Table 1). ATN is used as the start codon of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax PCGs, except for cox1, which uses TCG as a start codon (Tables 2, 3, 4). Specifically, six PCGs (cox2, cox3, atp6, nad4, nad4L and cob) begin with ATG, four PCGs (atp8, nad1, nad3 and nad6) with ATA and two PCGs (nad2 and nad5) with ATC. The most frequently used codon among the PCGs is TAA, followed by TA and T. Among the 16 investigated mt genomes, ATN is the most frequently used start codon, followed by TCG, and TAA is the most frequently used stop codon, followed by TA and T.
Table 2.
Organization of the Lt. halifaxia mt genome
| Gene | Strand | Position | Size (bp) | Space (+)/Overlap (−) | Anticodon | Codon | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| trnI | J | 1–69 | 69 | GAT | |||
| trnQ | N | 70–139 | 70 | 0 | TTG | ||
| trnM | J | 150–218 | 69 | 10 | CAT | ||
| nad2 | J | 219–1241 | 1023 | 0 | ATC | TAA | |
| trnW | J | 1242–1309 | 68 | 0 | TCA | ||
| trnC | N | 1309–1374 | 66 | − 1 | GCA | ||
| trnY | N | 1375–1440 | 66 | 0 | GTA | ||
| cox1 | J | 1439–2975 | 1537 | − 2 | TCG | T | |
| trnL2 | J | 2969–3034 | 66 | − 7 | TAA | ||
| cox2 | J | 3042–3726 | 685 | 7 | ATG | T | |
| trnK | J | 3727–3797 | 71 | 0 | CTT | ||
| trnD | J | 3798–3865 | 68 | 0 | GTC | ||
| atp8 | J | 3 875–4027 | 153 | 9 | ATA | TAA | |
| atp6 | J | 4021–4701 | 681 | − 7 | ATG | TAA | |
| cox3 | J | 4701–5489 | 789 | − 1 | ATG | TAA | |
| trnG | J | 5489–5555 | 67 | − 1 | TCC | ||
| nad3 | J | 5553–5909 | 357 | − 3 | ATA | TAA | |
| trnA | J | 5908–5971 | 64 | − 2 | TCG | ||
| trnR | J | 5972–6037 | 66 | 0 | TGC | ||
| trnN | J | 6038–6 104 | 67 | 0 | GTT | ||
| trnS1 | N | 6107–6173 | 67 | 2 | GCT | ||
| trnE | J | 6175–6240 | 66 | 1 | TTC | ||
| trnF | N | 6239–6305 | 67 | − 2 | GAA | ||
| nad5 | N | 6280–8025 | 1746 | − 26 | ATC | TAA | |
| trnH | N | 8023–8090 | 68 | − 3 | GTG | ||
| nad4 | N | 8090–9433 | 1344 | − 1 | ATG | TAA | |
| nad4L | N | 9427–9723 | 297 | − 7 | ATG | TAA | |
| trnT | J | 9729–9794 | 66 | 5 | TGT | ||
| trnP | N | 9795–9860 | 66 | 0 | TGG | ||
| nad6 | J | 9866–10,384 | 519 | 5 | ATA | TA | |
| cob | J | 10,400–11,536 | 1137 | 15 | ATG | TAA | |
| trnS2 | J | 11,536–11,601 | 66 | − 1 | TGA | ||
| nad1 | N | 11,620–12,576 | 957 | 18 | ATA | TAA | |
| trnL1 | N | 12,571–12,638 | 68 | − 6 | TAG | ||
| rrnL | N | 12,639–13,975 | 1337 | 0 | |||
| trnV | N | 13,976–14,047 | 72 | 0 | TAC | ||
| rrnS | N | 14,048–14,844 | 797 | 0 | |||
| CR | 14,845–15,744 | 899 | 0 | ||||
Table 3.
Organization of the Lt. fuscanus mt genome
| Gene | Strand | Position | Size (bp) | Space (+)/Overlap (−) | Anticodon | Codon | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| trnI | J | 1–69 | 69 | GAT | |||
| trnQ | N | 70–139 | 70 | 0 | TTG | ||
| trnM | J | 150–218 | 69 | 10 | CAT | ||
| nad2 | J | 219–1241 | 1023 | 0 | ATC | TAA | |
| trnW | J | 1242–1310 | 69 | 0 | TCA | ||
| trnC | N | 1311–1376 | 66 | 0 | GCA | ||
| trnY | N | 1377–1442 | 67 | 0 | GTA | ||
| cox1 | J | 1441–2977 | 1537 | − 2 | TCG | T | |
| trnL2 | J | 2978–3044 | 67 | 0 | TAA | ||
| cox2 | J | 3053–3737 | 685 | 8 | ATG | T | |
| trnK | J | 3738–3808 | 71 | 0 | CTT | ||
| trnD | J | 3817–3883 | 67 | 8 | GTC | ||
| atp8 | J | 3893–4045 | 153 | 9 | ATA | TAA | |
| atp6 | J | 4039–4719 | 681 | − 7 | ATG | TAA | |
| cox3 | J | 4719–5507 | 789 | − 1 | ATG | TAA | |
| trnG | J | 5507–5573 | 67 | − 1 | TCC | ||
| nad3 | J | 5571–5927 | 357 | − 3 | ATA | TAA | |
| trnA | J | 5926–5989 | 64 | − 2 | TCG | ||
| trnR | J | 5990–6055 | 66 | 0 | TGC | ||
| trnN | J | 6056–6122 | 67 | 0 | GTT | ||
| trnS1 | N | 6125–6191 | 67 | 2 | GCT | ||
| trnE | J | 6193–6258 | 66 | 1 | TTC | ||
| trnF | N | 6257–6323 | 67 | − 2 | GAA | ||
| nad5 | N | 6324–8068 | 1745 | 0 | ATC | TAA | |
| trnH | N | 8066–8131 | 66 | − 3 | GTG | ||
| nad4 | N | 8131–9471 | 1344 | − 1 | ATG | TAA | |
| nad4L | N | 9468–9764 | 297 | − 4 | ATG | TAA | |
| trnT | J | 9770–9834 | 65 | 5 | TGT | ||
| trnP | N | 9860–9925 | 66 | 25 | TGG | ||
| nad6 | J | 9931–10,446 | 515 | 5 | ATA | TAA | |
| cob | J | 10,446–11,582 | 1137 | − 1 | ATG | TAA | |
| trnS2 | J | 11,582–11,647 | 65 | − 1 | TGA | ||
| nad1 | N | 11,666–12,621 | 957 | 18 | ATA | TAA | |
| trnL1 | N | 12,617–12,683 | 67 | − 6 | TAG | ||
| rrnL | N | 12,687–14,019 | 1333 | 3 | |||
| trnV | N | 14,021–14,092 | 72 | 1 | TAC | ||
| rrnS | N | 14,093–14,885 | 793 | 0 | |||
| CR | 14,886–15,803 | 920 | 0 | ||||
Table 4.
Organization of the Cx. pallidothorax mt genome
| Gene | Strand | Position | Size (bp) | Space (+)/Overlap (−) | Anticodon | Codon | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| trnI | J | 1–69 | 69 | GAT | |||
| trnQ | N | 67–135 | 69 | − 3 | TTG | ||
| trnM | J | 140–208 | 69 | 4 | CAT | ||
| nad2 | J | 209–1230 | 1022 | 0 | ATC | TA | |
| trnW | J | 1233–1301 | 69 | 2 | TCA | ||
| trnC | N | 1302–1367 | 66 | 0 | GCA | ||
| trnY | N | 1367–1432 | 66 | − 1 | GTA | ||
| cox1 | J | 1431–2967 | 1537 | − 2 | TCG | T | |
| trnL2 | J | 2968–3032 | 65 | 0 | TAA | ||
| cox2 | J | 3040–3724 | 685 | 7 | ATG | T | |
| trnK | J | 3725–3795 | 71 | 0 | CTT | ||
| trnD | J | 3806–3873 | 68 | 10 | GTC | ||
| atp8 | J | 3883–4036 | 154 | 9 | ATA | TAA | |
| atp6 | J | 4029–4709 | 681 | − 8 | ATG | TAA | |
| cox3 | J | 4709–5 498 | 790 | − 1 | ATG | TAA | |
| trnG | J | 5497–5563 | 67 | − 2 | TCC | ||
| nad3 | J | 5561–5917 | 357 | − 3 | ATA | TAA | |
| trnA | J | 5916–5979 | 64 | − 2 | TCG | ||
| trnR | J | 5980–6045 | 66 | 0 | TGC | ||
| trnN | J | 6046–6112 | 67 | 0 | GTT | ||
| trnS1 | N | 6129–6196 | 68 | 18 | GCT | ||
| trnE | J | 6184–6251 | 68 | − 13 | TTC | ||
| trnF | N | 6250–6316 | 67 | − 2 | GAA | ||
| nad5 | N | 6317–8062 | 1746 | 0 | ATC | TAA | |
| trnH | N | 8060–8125 | 66 | − 3 | GTG | ||
| nad4 | N | 8125–9469 | 1345 | − 1 | ATG | TAA | |
| nad4L | N | 9463–9759 | 297 | − 7 | ATG | TAA | |
| trnT | J | 9765–9830 | 66 | 5 | TGT | ||
| trnP | N | 9831–9896 | 66 | 0 | TGG | ||
| nad6 | J | 9902–10,417 | 516 | 5 | ATA | TAA | |
| cob | J | 10,417–11,551 | 1135 | − 1 | ATG | TA | |
| trnS2 | J | 11,552–11,617 | 66 | 0 | TGA | ||
| nad1 | N | 11,635–12,591 | 957 | 17 | ATA | TAA | |
| trnL1 | N | 12,586–12,652 | 67 | − 6 | TAG | ||
| rrnL | N | 12,654–13,989 | 1335 | 1 | |||
| trnV | N | 13,990–14,061 | 72 | 0 | TAC | ||
| rrnS | N | 14,062–14,854 | 793 | 0 | |||
| CR | 14,855–15,578 | 724 | 0 | ||||
The RSCU values of the 16 investigated mt genomes are presented in Additional file 2: Table S2. In Lt. fuscanus and Lt. halifaxia, UUA is the most frequently used codon, followed by CGA, GGA and UCU, whereas CCG and ACG are rarely used, and CGC is not used. In Cx. pallidothorax, UUA is the most frequently used codon, followed by CGA, UCU and GGA, whereas CCG, ACG and CGC are not used. Among the 16 investigated mt genomes, UUA is the most frequently used codon, followed by CGA, GGA and UCU, whereas CGC, CCG and ACG are rarely used. Among the 16 investigated mt genomes, a total of 20 different amino acids are encoded, and the amino acid Leu has the highest frequency (16.33%), whereas Cys has the lowest (1.05%) (Fig. 3).
Fig. 3.

Frequency percentage of each of the 20 amino acids coded in the 16 Culex and Lutzia mt genomes. Abbreviations: CCA, Cx. camposi; CCO, Cx. coronator; LFU, Lt. fuscanus; CGE, Cx. gelidus; LHA, Lt. halifaxia; CPA, Cx. p. pallens; CPI, Cx. p. pipiens; CPL, Cx. pallidothorax; CPT, Cx. pipiens TU; CQS, Cx. quinquefasciatus USA; CQU, Cx. quinquefasciatus; CTC, Cx. tritaeniorhynchus CQ; CTJ, Cx. tritaeniorhynchus JS; CUA, Cx. usquatissimus AC; CUR, Cx. usquatissimus RO; CUS, Cx. usquatus
Transfer RNAs, ribosomal RNAs and the CR
Twenty-two tRNAs were identified in the Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax mt genomes; their secondary structures are presented in Additional file 3: Figure S1. The length of the tRNAs varies from 64 (trnA) to 74 bp (trnN) among the three mt genomes (Tables 2, 3, 4). Most of the tRNAs can be folded as a typical cloverleaf structure, except for trnS2, whose DHU arm simply forms an 11-nucleotide loop (Additional file 3: Figure S1). A total of 27 mismatched base pairs were detected in Lt. halifaxia tRNAs, 18 of which are UG pairs, and the remaining nine pairs include three AC pairs, three UU pairs, two AA pairs and one GA pair. Twenty-one mismatched base pairs were observed in Lt. fuscanus tRNAs, including 17 UG pairs, two AA pairs and two UU pairs. There are 23 mismatched base pairs in Cx. pallidothorax tRNAs, including 18 UG pairs, three AG pairs, one UU pair and one UG pair.
In the three newly sequenced mt genomes, two rRNAs (rrnL and rrnS) are located between trnL2 and trnV, and between trnV and CR, respectively. The length of the rRNAs is 2134 bp, with an AT content of 82.61% in Lt. halifaxia; 2126 bp, with an AT content of 82.78% in Lt. fuscanus; and 2128 bp, with an AT content of 82.08% in Cx. pallidothorax.
The CR is located between rrnS and trnI and shows the highest AT content (88.88% in Lt. halifaxia, followed by 89.78% in Lt. fuscanus and 87.11% in Cx. pallidothorax) (Additional file 1: Table S1). The length of the CRs of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax are 899, 921 and 724 bp, respectively. For the nine Culex and two Lutzia mt genomes with known CRs, the CR lengths vary from 704 bp in Cx. quinquefasciatus to 920 bp in Lt. fuscanus, and their AT content ranges from 87.11% in Cx. pallidothorax to 90.58% in Cx. gelidus. The length of the CR in Lt. halifaxia and Lt. fuscanus is 174 to 216 bp greater than the other nine CRs (Table 1). We also detected a 49-bp repeat unit, a poly-T stretch of 17 bp and a 50-bp repeat unit in Lt. halifaxia; a 90-bp repeat unit, a poly-T stretch of 18 bp and a 47-bp repeat unit in Lt. fuscanus; and a 41-bp repeat unit, a poly-T stretch of 18 bp and a 320-bp microsatellite-like dinucleotide repeat region [(TA)n stretch] in Cx. pallidothorax (Fig. 4).
Fig. 4.

Conservative stretches of CRs in 11 Culex and Lutzia mt genomes with complete CR sequences. The ellipses filled with different colors indicate regions with different tandem repeat units (repeat number and unit bp length showing beneath the corresponding ellipses), the pentagrams denote regions with 17–20 repeats of nucleotide T, and the green-filled boxes demonstrate the non-repeat regions with the sequence length marked inside
Assessment of the CR of nine Culex and two Lutzia mt genomes identified four types of repeat units, and the structure of four types of repeat units are conservative along the taxonomic taxa (Fig. 4). The first repeat unit is 17–20 bp of poly-T tract, which is located in the central part of the CR and exists in all these mt genomes. The second is a 30–90 bp sequence with 2–3 repeats; these sequences are all situated nearby rrnS, vary among species and occur in all species but Cx. p. pallens, in which the repeat unit might have been lost during evolution. The third is a 47–50 bp sequence with two repeats; these two sequences are situated proximal to trnI and vary and exist only in two species in the genus Lutzia. The fourth is a microsatellite-like TA sequence ([TA(A)]n stretch) consisting of 97–179 repeats, which is also situated close to trnI and exists in all nine species in the subgenera Culiciomyia and Culex.
Phylogenetic relationships
The best-fit model chosen for each gene and the resulting phylogenetic tree from the BI analysis are provided in Additional file 4: Table S3 and Fig. 5, respectively. The Bayesian topology shows Lutzia as the sister taxon of Culex spp. with a maximum posterior probability (pp = 1.0). Inside the clade composed by Culex spp., the monophyly of the Sitiens and Coronator groups was strongly supported (pp = 1.0), whereas the monophyletic status of the Pipiens Group was not supported (pp = 0.81). Culex pallidothorax was resolved sister to the clade compounded of Sitiens Group + Pipiens Group but the support was poor (pp = 0.79). Within the Sitiens group, Culex gelidus was a placed as sister species to Culex tritaeniorhynchus with high posterior probability. On the other hand, internal relationships of the Coronator Group were poorly resolved: one individual of Culex usquatissimus (AC) was placed as sister to Culex camposi (pp = 0.8) and the other (Culex usquatissimus RO) was sister to Culex coronator. The placement of Culex usquatus was weakly supported (0.62 pp). Similarly, in the Pipiens Group, clustering of one individual Culex quinquefasciatus within the clade of Culex pipiens specimens was strongly supported (1.0 pp).
Fig. 5.

Phylogenetic relationships of 16 mt genomes based on nucleotide sequences of 13 protein-coding genes. The tree was constructed using BI method, and numbers at the nodes are Bayesian posterior probabilities. The newly sequenced mt genomes of three species are indicated by triangles. The GenBank accession numbers of mt genome sequences of the species are listed in Table 1
Discussion
General characteristics of 16 Culex spp. mt genome sequences
Among the 16 mt genome sequences (including the three newly sequenced) of species in the genera Culex and Lutzia, 11 complete sequences were 15,573 to 15,803 bp in length, with variations mainly occurring in the CR, similar to that earlier reported in insects [15, 37, 38]. These 16 Culex mt genomes include 37 genes (13 PCGs, 22 tRNAs and two rRNAs) with a similar gene arrangement as those reported in other mosquito genera [19]. The nucleotide composition is biased toward AT (77.40–78.50%), with A being the most favored nucleotide (39.11–39.70%) and G as the least favored (9.10–9.44%), and with an average AT-skew value of 0.0107 and an average GC-skew value of -0.1572, features also similar to those previously reported in insects [13, 14, 19, 20, 37, 38].
Among the 16 Culex mt genomes, 13 PCGs showed variations in total nucleotide length ranging from 11,188 to 11,234 bp, with ATN being the most frequently used start codon, followed by TCG, and TAA being the most frequently used stop codon, followed by TA and T. The PCGs of most other mosquito species are also predicted to mainly use ATN as the start codon and TAA as the stop codon [19]. The incomplete stop codons are common in insect mt genomes [13, 14, 17, 19, 20, 37, 38] and the complete termination codon is thought to be created by post-transcriptional polyadenylation [39]. UUA is the most frequently used codon, followed by CGA, GGA and UCU, whereas CGC, CCG and ACG are rarely used, which is consistent with the observed higher AT content in these mt genomes. This phenomenon has also been observed in the mt genome of another mosquito species, Anopheles minimus [40]. The amino acids encoded by the codons ending with U or A are overused, with Leu being the most frequently encoded amino acid (16.33%) and Cys as the least frequently used amino acid (1.05%), which is also similar to that reported in some Anopheles mt genomes [19, 40, 41]. In terms of the trnS2 of three newly sequenced mt genomes, the DHU arm is not formed, which is similar to other Culex species [20–23] and metazoans [42]. The location of the two rRNAs is the same as in other Dipteran mt genomes [43].
We identified four types of repeat units in the nine Culex and two Lutzia mt genomes with complete CR sequences. Among the four types, the poly-T stretch and [TA(A)]n stretches have also been found in other insect species [44–47]. The poly-T stretch is highly conserved in insects, and it is thought to contain several regulatory elements, including the origin of replication and transcription [45]. The [TA(A)]n stretch exists in all nine mt genomes in the subgenera Culiciomyia and Culex investigated in the present study and in some other insect species [43], and it does not in two species in the genus Lutzia as determined in this study and in some other insect species, which suggest multiple evolutionary origins. Two other types of repeat units have not been found in other species; however, three other types of repeat units were identified in other insects, namely a highly conserved stem-and-loop structure, a G(A)nT structure and a G+A-rich stretch, which were not detected in the genus Culex. The repeat units are relatively conserved and thus may be utilized in phylogenetic reconstruction.
Evolutionary relationships and taxonomy
The Coronator and Sitiens groups each form a unique clade with a posterior probability of 1, whereas the Pipiens group is poorly supported with a posterior probability of 0.82. These results support the earlier results of phylogenetic studies using mt genome sequences [21–23]. Morphologically, the Cx. pallidothorax has been classified within subgenus Culiciomyia [1, 24], whereas phylogenetic analysis based on cox1 sequences has shown that this species belongs to the subgenus Culex, with a low bootstrap support of 11% [11]. The phylogenetic analysis conducted in the present study indicates that Cx. pallidothorax belongs to the subgenus Culex and is sister of the groups Sitiens and Pipiens albeit with poor support. Additionally, the species has three types of repeat units, which is similar to that observed in species of the subgenus Culex. It appears that the taxonomic status of Cx. pallidothorax is doubtful and needs to be elucidated. In order to enlighten the position of Culiciomyia as subgenus, further analyses will be necessary using additional species.
Whether Lutzia should be considered as a genus or subgenus has long remained controversial. Morphological taxonomy identifies it as a genus [2, 4] or subgenus [3], whereas a phylogenetic analysis based on the morphological characteristics of larvae and adults has placed it outside the clade comprising the genus Culex. Molecular phylogenetic analysis using ITS1 and ITS2, including 14 species in the four subgenera of the genus Culex, showed that Lutzia tigripes was placed at the base of subgenus Culex (including three species in the Pipiens group and one species in the Sitiens group) [7]. Another analysis that also used ITS2, including 17 Neotropical species from five subgenera of genus Culex, classified Lutzia under subgenus Culex (including one species in the Pipiens group and two species in the Coronator group) [9]. The cox1-based analysis of 17 species from five subgenera of genus Culex (including one species in Lutzia, one species in the Pipiens group, and two species in the Coronator group in subgenus Culex) showed Lutzia as the sister taxon of the clade composed by the subgenera Culex + Phenacomyia [8]. Our phylogenetic analysis that included two Lutzia spp. (Lt. halifaxia and Lt. fuscanus) indicated Lutzia as a monophyletic entity and supports its original generic status. In the present study, the two Lutzia species have a 47–50-bp sequence with two repeats in the CR, which was not detected in other species. In addition, the two repeats lack the [TA(A)]n stretch, which is present in all other Culex species investigated. The assessment of features of the repeat units in the CR also supports the monophyly of this taxon.
Conclusions
The present study sequenced and analyzed the complete mt genomes of Lt. halifaxia, Lt. fuscanus and Cx. pallidothorax and assessed the general characteristics and phylogenetic relationships of all known 16 mt genome sequences in the genera Culex and Lutzia. Culex spp. mt genomes share the same gene arrangement as other mosquito species, and variations in length mainly involve the CR. The repeat units of the CR are relatively conserved and provide information that may be utilized in establishing the phylogeny of Culex and Lutzia. The Coronator and Sitiens groups are each monophyletic, whereas the monophyletic status of the Pipiens Group was not supported. The taxonomic status of subgenus Culiciomyia has yet to be elucidated using additional species. Both phylogenetic analysis and repeat unit features of the CR show that Lutzia is a characteristic monophyletic group at the generic level. To our knowledge, this is first comprehensive review of the mt genome sequences and taxonomic assessment based on mt genome sequences of species in the genera Culex and Lutzia.
Additional files
Additional file 1: Table S1. Composition and skewness of 16 Culex mt genomes.
Additional file 2: Table S2. Relative synonymous codon usage (RSCU) in the 16 Culex mt genomes.
Additional file 3: Figure S1. Predicted secondary structures of 22 tRNAs in the mt genomes of Lt. fuscanus (a), Lt. halifaxia (b) and Cx. pallidothorax (c).
Additional file 4: Table S3. Best-fit models chosen under Akaike information criterion by Modeltest for each of the 13 PCGs.
Acknowledgements
Not applicable.
Abbreviations
- mt genome
mitochondrial genome
- PCGs
protein-coding genes
- rRNAs
ribosomal RNA genes
- tRNAs
transfer RNA genes
- CR
control region
- RSCU
relative synonymous codon usage
- ML
maximum likelihood
- BI
Bayesian inference
- TR
tandem repeats
Authors’ contributions
BC and LS conceived and designed the study. LS, BC and DSB performed the experiments and data analysis, and drafted the manuscript. TJL, WBF, ZTY, FLS, YJZ and QMM participated in specimen collection and experiments. All authors read and approved the final manuscript.
Funding
This research was supported by the following: The National Natural Science Foundation of China (31872262, 31672363), Coordinated Research Project of the International Atomic Energy Agency (18268), National Key Program of Science and Technology Foundation Work of China (2015FY210300), Science and Technology Major Special Project of Guangxi (GKAA17129002), Chongqing Natural Science Foundation and Frontier Research Planning Project (CSTC2018JCYJA2487), the Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJ1600304) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP No. 2016/08551-4).
Availability of data and materials
All data are available as tables and figures in the main document and its additional files. The GenBank accession numbers for the three mt genomes produced in the present study are MH316119 (Lt. halifaxia), MH316118 (Lt. fuscanus) and KY400104 (Cx. pallidothorax).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ling Sun, Email: 1540313460@qq.com.
Ting-Jing Li, Email: 18929550@qq.com.
Wen-Bo Fu, Email: 297043713@qq.com.
Zhen-Tian Yan, Email: 525201877@qq.com.
Feng-Ling Si, Email: 30431057@qq.com.
Yu-Juan Zhang, Email: 41437909@qq.com.
Qi-Meng Mao, Email: 874561386@qq.com.
Bruna Demari-Silva, Email: bruna-demary@usp.br.
Bin Chen, Email: bin.chen@cqnu.edu.cn.
References
- 1.Harbach RE. Classification within the cosmopolitan genus Culex (Diptera: Culicidae): The foundation for molecular systematics and phylogenetic research. Acta Trop. 2011;120:1–14. doi: 10.1016/j.actatropica.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Theobald FV. A monograph of Culicidae or mosqutoes. London: British Museum (Natural History); 1903. [Google Scholar]
- 3.Edwards FW. Genera insectorum. Diptera, Fam. Culicidae. Fasc. 194. Brussels: Desmet-Verteneuil; 1932.
- 4.Tanaka K. Studies on the pupal mosquitoes of Japan Genus Lutzia, with establishment of two new subgenera, Metalutzia and Insulalutzia (Diptera: Culicidae) Jpn J Syst Entomol. 2003;9:159–169. [Google Scholar]
- 5.Navarro JC, Liria J. Phylogenetic relationships among eighteen neotropical Culicini species. J Am Mosq Control Assoc. 2000;16:75–85. [PubMed] [Google Scholar]
- 6.Harbach RE, Kitching IJ, Culverwell CL, Culverwell CL, Dubois J, Linton YM. Phylogeny of mosquitoes of tribe Culicini (Diptera: Culicidae) based on morphological diversity. Zool Scr. 2012;41:499–514. doi: 10.1111/j.1463-6409.2012.00546.x. [DOI] [Google Scholar]
- 7.Miller BR, Crabtree MB, Savage HM. Phylogeny of fourteen Culex mosquito species, including the Culex pipiens complex, inferred from the internal transcribed spacers of ribosomal DNA. Insect Mol Biol. 1996;5:93–107. doi: 10.1111/j.1365-2583.1996.tb00044.x. [DOI] [PubMed] [Google Scholar]
- 8.Demari-Silva B, Vesgueiro FT, Sallum MAM, Marrelli MT. Taxonomic and phylogenetic relationships between species of the genus Culex (Diptera: Culicidae) from Brazil inferred from the cytochrome c oxidase I mitochondrial gene. J Med Entomol. 2011;48:272–279. doi: 10.1603/ME09293. [DOI] [PubMed] [Google Scholar]
- 9.Vesgueiro FT, Demari-Silva B, Malafronte RS, Sallum MA, Marrelli MT. Intragenomic variation in the second internal transcribed spacer of the ribosomal DNA of species of the genera Culex and Lutzia (Diptera: Culicidae) Mem Inst Oswaldo Cruz. 2011;106:1–8. doi: 10.1590/S0074-02762011000100001. [DOI] [PubMed] [Google Scholar]
- 10.Edwards FW. A revision of the mosquitos of the Palaearctic Region Bull Entomol Res. 1921;12:263–351. [Google Scholar]
- 11.Wang G, Li CX, Guo XX, Xing D, Dong YD, Wang ZM, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS ONE. 2012;7:e47051. doi: 10.1371/journal.pone.0047051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae) BMC Genomics. 2008;9:331. doi: 10.1186/1471-2164-9-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai LS, Qian C, Zhang CF, Wang L, Wei GQ, Li J, et al. Characterization of the complete mitochondrial genome of Cerura menciana and comparison with other Lepidopteran insects. PLoS ONE. 2015;10:e0132951. doi: 10.1371/journal.pone.0132951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YY, Liu XY, Garzón-Ordunac IJ, Winterton SL, Yan Y, Aspöck U, et al. Mitochondrial phylogenomics illuminates the evolutionary history of Neuropterida. Cladistics. 2016;33:617–637. doi: 10.1111/cla.12186. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Wei SJ, Tang P, Wu Q, Shi M, Sharkey MJ, et al. Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae. Genome Biol Evol. 2016;8:2651. doi: 10.1093/gbe/evw184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breeschoten T, Doorenweerd C, Tarasov S, Vogler AP. Phylogenetics and biogeography of the dung beetle genus Onthophagus, inferred from mitochondrial genomes. Mol Phylogenet Evol. 2016;105:86–95. doi: 10.1016/j.ympev.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 18.Song N, An SH, Yin XM, Cai WZ, Li H. Application of RNA-seq for mitogenome reconstruction, and reconsideration of long-branch artifacts in Hemiptera phylogeny. Sci Rep. 2016;6:33465. doi: 10.1038/srep33465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hao YJ, Zou YL, Ding YR, Xu WY, Yan ZT, Li XD, et al. Complete mitochondrial genomes of Anopheles stephensi and An dirus and comparative evolutionary mitochondriomics of 50 mosquitoes. Sci Rep. 2017;7:7666. doi: 10.1038/s41598-017-07977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behura SK, Lobo NF, Haas B, Debruyn B, Lovin DD, Shumway MF, et al. Complete sequences of mitochondria genomes of Aedes aegypti and Culex quinquefasciatus and comparative analysis of mitochondrial DNA fragments inserted in the nuclear genomes. Insect Biochem Mol. 2011;41:770. doi: 10.1016/j.ibmb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demari-Silva B, Foster PG, Oliveira TMPD, Bergo ES, Sanabani SS, Pessoa R, et al. Mitochondrial genomes and comparative analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera: Culicidae), members of the Coronator group. BMC Genomics. 2015;16:831. doi: 10.1186/s12864-015-1951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo QC, Hao YJ, Meng FX, Li TJ, Ding YR, Hua YQ, Chen B. The mitochondrial genomes of Culex pipiens pallens and Culex tritaeniorhynchus (Diptera: Culicidae) and comparison analysis with two other Culex species. Parasit Vectors. 2016;9:406. doi: 10.1186/s13071-016-1694-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun L, Fu WB, Yan ZT, Li TJ, Ding YR, Chen B. Sequencing and analysis of the complete mitochondrial genome of Culex gelidus (Diptera: Culicidae) Mit DNA Part B. 2017;2:477–479. doi: 10.1080/23802359.2017.1361350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu BL. Fauna Sinica. Insecta. Diptera: Culicidae 1. Vol. 8. Beijing, China: Science Press; 1997.
- 25.Sun L, Fu WB, Yan ZT, Chen B. Molecular phylogenetic relationships among 40 species (subspecies) in the genus Culex from China (Diptera: Culicidae) Acta Entomol Sinica. 2018;61:94–102. [Google Scholar]
- 26.Zhang NX, Zhang YJ, Yu G, Chen B. Structure characteristics of the mitochondrial genomes of Diptera and design and application of universal primers for their sequencing. Acta Entomol Sinica. 2013;56:398–407. [Google Scholar]
- 27.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Peterson D, Peterson N, Stecher G, Nei M. MEGA5: molecular evolutionary genetics analysisusing maximum likelihood, evolutionary distance, and maxi-mum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353–358. doi: 10.1007/BF01215182. [DOI] [PubMed] [Google Scholar]
- 31.Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36:W181–W184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikrajuddin A, Khairurrijal A. A simple method for determining surface porosity based on SEM images using Origin Pro software. Indonesian J Phys. 2009;20:37–41. [Google Scholar]
- 33.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronquist F, Teslenko M, van derMark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucl Acids Res. 2010;38:W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 37.Chen YH, Huang DY, Wang YL, Zhu CD, Hao JS. The complete mitochondrial genome of the endangered Apollo butterfly, Parnassius apollo, (Lepidoptera: Papilionidae) and its comparison to other Papilionidae species. J Asia-Pac Entomol. 2014;17:663–671. doi: 10.1016/j.aspen.2014.06.002. [DOI] [Google Scholar]
- 38.Wang Y, Liu X, Yang D. The first mitochondrial genome for caddisfly (Insecta: Trichoptera) with phylogenetic implications. Int J Biol Sci. 2014;10:53–63. doi: 10.7150/ijbs.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- 40.Hua YQ, Ding YR, Yan ZT, Si FL, Luo QC, Chen B. The complete mitochondrial genome of Anopheles minimus (Diptera: Culicidae) and the phylogenetics of known Anopheles mitogenomes. Insect Sci. 2016;23:353–365. doi: 10.1111/1744-7917.12326. [DOI] [PubMed] [Google Scholar]
- 41.Hua YQ, Yan ZT, Fu WB, He QY, Zhou Y, Chen B. Sequencing and analysis of the complete mitogenome in Anopheles culicifacies species B (Diptera: Culicidae) Mit DNA Part A. 2015;27:2909–2910. doi: 10.3109/19401736.2015.1060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negrisolo E, Babbucci M, Patarnello T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaroniusand comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics. 2011;12:221. doi: 10.1186/1471-2164-12-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang NX, Yu G, Li TJ, He QY, Zhou Y, Si FL, et al. The complete mitochondrial genome of Delia antiqua and its implications in dipteran phylogenetics. PLoS ONE. 2015;10:e0139736. doi: 10.1371/journal.pone.0139736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang DX, Hewitt GM. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997;25:99–120. doi: 10.1016/S0305-1978(96)00042-7. [DOI] [Google Scholar]
- 45.Sun WY, Xu DL, Chen HX, Shi W, Sundberg P, Strand M, Sun SC. Complete mitochondrial genome sequences of two parasitic/commensal nemerteans, Gononemertes parasite and Nemertopsis tetraclitophila (Nemertea: Hoplonemertea) Parasit Vectors. 2014;19:273. doi: 10.1186/1756-3305-7-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spanos L, Koutroumbas G, Kotsyfakis M, Louis C. The mitogenome of the Mediterranean fruit fly. Ceratitis capitata. Insect Mol Biol. 2000;9:139–144. doi: 10.1046/j.1365-2583.2000.00165.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Z, Huang Y, Shi F, Ye H. The complete mitogenome of Deracantha onos (Orthoptera: Bradyporidae) Mol Biol Rep. 2009;36:7–12. doi: 10.1007/s11033-007-9145-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Composition and skewness of 16 Culex mt genomes.
Additional file 2: Table S2. Relative synonymous codon usage (RSCU) in the 16 Culex mt genomes.
Additional file 3: Figure S1. Predicted secondary structures of 22 tRNAs in the mt genomes of Lt. fuscanus (a), Lt. halifaxia (b) and Cx. pallidothorax (c).
Additional file 4: Table S3. Best-fit models chosen under Akaike information criterion by Modeltest for each of the 13 PCGs.
Data Availability Statement
All data are available as tables and figures in the main document and its additional files. The GenBank accession numbers for the three mt genomes produced in the present study are MH316119 (Lt. halifaxia), MH316118 (Lt. fuscanus) and KY400104 (Cx. pallidothorax).