

Abstract
Congenital myopathies are early onset, slowly progressive neuromuscular disorders of variable severity. They are genetically and phenotypically heterogeneous and caused by pathogenic variants in several genes. Multi-minicore Disease, one of the more common congenital myopathies, is frequently caused by recessive variants in either SELENON, encoding the endoplasmic reticulum glycoprotein selenoprotein N or RYR1, encoding a protein involved in calcium homeostasis and excitation–contraction coupling. The mechanism by which recessive SELENON variants cause Multi-minicore Disease is unclear. Here, we extensively investigated muscle physiological, biochemical and epigenetic modifications, including DNA methylation, histone modification and non-coding RNA expression, to understand the pathomechanism of Multi-minicore Disease. We identified biochemical changes that are common in patients harbouring recessive RYR1 and SELENON variants, including depletion of transcripts encoding proteins involved in skeletal muscle calcium homeostasis, increased levels of class II HDACs and DNA methyltransferases. CpG methylation analysis of genomic DNA of patients with RYR1 and SELENON variants identified more than 3500 common aberrantly methylated genes, many of which are involved in calcium signalling. These results provide the proof of concept for the potential use of drugs targeting HDACs and DNA methyltransferases to treat patients with specific forms of congenital myopathies.
Keywords: Congenital myopathies, excitation-contraction coupling, ryanodine receptor, gene expression, epigenetics
INTRODUCTION
Multiminicore Disease (MmD; MIM# 255320) is an early onset congenital myopathy usually transmitted by autosomal recessive inheritance, characterized by the presence of areas of sarcomeric disorganization and multiple small cores running along the length of most fibers (Engel et al., 1971; Ferreiro et al., 2000). The clinical features and genetic backgrounds associated with multiple minicores on muscle biopsy are heterogeneous; the classic, most common form of MmD is characterized by onset during infancy, prominent axial involvement, scoliosis and severe respiratory impairment, and is allelic to Rigid Spine Muscular Dystrophy (RSMD1; MIM# 602771) (Moghadaszadeh et al., 2001; Okamoto et al., 2006). This phenotypic heterogeneity is mirrored by genetic heterogeneity. Patients with the classic form typically carry recessive variants in SELENON (previously known as SEPN1), the gene encoding Selenoprotein N (SelN), whereas those with MmD with extraocular muscle involvement typically carry recessive variants in RYR1, the gene encoding the ryanodine receptor 1 (RyR1) calcium channel of the sarcoplasmic reticulum (Jungbluth et al., 2011; Jungbluth et al., 2018) an important player of excitation contraction coupling (ECC) the process whereby depolarization of the plasma membrane brings about an increase in the myoplasmic calcium concentration, leading to muscle contraction (Rios & Pizarro 1991; Schneider & Chandler, 1972).
SelN is a ubiquitously expressed glycoprotein of the endoplasmic reticulum (Petit et al., 2003) but its absence causes a disease primarily affecting skeletal muscle. The proposed disease pathomechanisms include impaired muscle regenerative capacity due to defects in the satellite cell population (Castets et al., 2011) and oxidative stress leading to abnormal skeletal muscle calcium regulation and altered ECC (Arbogast et al., 2009; Marino et al, 2015).
An interesting feature associated with MmD caused by recessive RYR1 variants is the decrease of RyR1 protein content in affected muscles (Rokach et al., 2015; Zhou et al., 2007; Zhou et al., 2006). This is paralleled by the activation of epigenetic modifications including abnormal RYR1 methylation and high levels of expression of class II histone deacetylases (HDACs) (Rokach et al., 2015; Zhou et al., 2006). In the present study we show that similar changes occur in muscles of patients with SELENON variants, including depletion of RYR1, ATP2B2, STAC3 and ATP2A1 transcripts, abnormal gene methylation and increased levels of class II HDACs and DNA methyltransferases. Our findings provide the basis for the design of new molecularly based drug targeting strategies in patients with congenital myopathies characterized by reduced RyR1 protein content.
MATERIALS AND METHODS
Muscle biopsies and cell cultures:
Quadriceps muscle biopsies from patients with genetically confirmed SELENON and RYR1 variants and healthy non-affected individuals undergoing the in vitro contracture test were used. Human myoblasts from healthy controls and patients were cultured in skeletal muscle growth medium as previously described (Rokach et al., 2015; Bachmann et al., 2017).
Calcium measurements:
Muscle cells were cultured and differentiated on laminin-coated 0.17 mm glass cover slips as previously described (Bachmann et al., 2017). Calcium measurements were performed as previously described (Bachmann et al., 2017).
Brightfield and confocal microscopy:
Muscle cells were cultured and differentiated on laminin-coated Ibidi® glass bottom slides. Myotubes were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) and permeabilised with 1% Triton® X-100 in PBS. Labelling with primary antibodies (Supp. Table S1), Alexa Fluor conjugated secondary antibodies (Life technologies) and DAPI counterstaining were performed and cells were visualized on a Nikon® A1plus confocal microscope as previously described (Bachmann et al., 2017).
Electrophoresis and immunoblotting:
Proteins in muscle biopsy homogenates were separated on SDS PAGE and immunoblotted overnight as previously described (Bachmann et al., 2017). The primary antibodies that were used are listed in Supp. Table S1. Bands were visualized using the Advansta WesternBright ECL (Advansta #K-12045-D50) or BM Chemiluminescence kit (Roche; catalogue #11520709001). Densitometry of immunopositive bands was carried out by Epson Perfection V700 Photo instrument as previously described (Bachmann et al., 2017). For MyHC isoform separation, gels were prepared according as described (Talmadge & Roy, 1985) and stained with Coomassie Blue R250. Single fibres isolated from mouse FDB or soleus muscles were used to identify the different MyHC isoforms.
Quantitative PCR:
RNA was extracted from myotubes or muscle biopsies using TRIzol® reagent, transcribed into cDNA and amplified by quantitative real-time polymerase chain reaction (qPCR) using the comparative ΔΔCt method as previously described (Bachmann et al., 2017). The sequences of the primers used for qPCR and for miRNA quantification are listed in Suppl. Table 2 and Supp. Table 3. Each reaction was performed in duplicate and results are expressed as relative gene expression normalized to desmin (DES) for qPCR and to the human housekeeping gene RNU44 for miRNAs. Expression levels obtained from biopsies of healthy controls were set to 1.
Genome-wide DNA Methylation Analysis:
Genome-wide DNA methylation was measured by Infinium Epic BeadChip array (Illumina) according to manufacturer’s instructions. Illumina GenomeStudio software was used to extract the raw signal intensities of each CpG. Computational and statistical analyses were performed using R and Bioconductor. Preprocessing, correction and normalization steps were performed using minfi package. For probe-wise differential methylation analysis was performed using limma package (Wettenhall & Smyth, 2004). Statistical analyses were performed on logit transformation of β-values known as M-values (Du et al., 2010), whereas β-values were used for biologic interpretation. P-values were adjusted to control for the false discovery rate (FDR) using the Benjamini–Hochberg method. For the log2 fold change (logFC) calculation, the differences between the averages of groups were considered. Significantly differentially methylated CpGs (DMCs) in SELENON subgroups were defined as those having an adjusted P<0.05 and absolute methylation difference to control samples >10%. Functional correlation between methylation and biological pathway, KEGG pathway analysis was performed within hypermethylated and hypomethylated CpG associated genes. Shown are only those pathways that were enriched (P<0.01) exclusively. Differentially methylated CpGs in RYR1 subgroup were defined as those having absolute methylation difference to average of control samples >10% in all three samples. Data generated from the genome-wide DNA methylation is deposited at the NCBI Gene expression Omnibus (GEO) under the accession series number GSE121961.
Compliance with Ethical standards:
All procedures were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This study was approved by the Ethikkommission Nordwest- und Zentralschweiz (permit N° EKNZ 2014-065); all subjects gave written informed consent to carry out this work.
Statistical analysis:
Statistical analysis was performed using the ‘R’ version 3.2.4 running on platform x86_64-apple-darwin13.4.0 (64-bit). For dose-response curves the package ‘drc’ version 2.5-12 was used (Ritz et al., 2015). Comparisons of two groups were performed using the Student’s t-test. Means were considered statistically significant when P values were < 0.05. All figures were created using Adobe Photoshop CS6 or R Studio (version 0.99.891 or newer).
RESULTS
We studied a cohort of 18 patients with genetically confirmed SELENON-associated congenital myopathy (Table 1). Muscle cells for calcium studies, qPCR and immunohistochemistry were only available from patient N° 17 and 18 (Table 1), control cells were obtained from 2 healthy individuals with no signs of neuromuscular disorders. Compared to controls, myotubes from both patients showed significant changes in calcium homeostasis (Fig.1 A, C and Table 2); in particular the peak Ca2+ response triggered by plasma membrane depolarization was significantly reduced (30.97±2.71 fluorescence units to 18.58±1.44 fluorescence units in controls and patients, respectively; Student’s t test P<0.001); the EC50 of KCl-induced calcium release was also reduced in patients compared to controls (11.12±2.33 and 23.69±3.79 mM KCl, respectively, p<0.0001 Student’s t test). Myotubes from the two patients were not identical in their Ca2+ response to direct RyR1 activation with 4-chloro-m-cresol (Zorzato et al., 1993), namely cells from patient 17 were similar to controls, while those from patient 18 who was more severely affected, showed a decrease in the peak Ca2+ transient elicited by 4-chloro-m-cresol (35.22±3.48 and 17.17±3.10 fluorescence units in controls and patient, respectively; P<0.0001 Student’s t test) (Fig.1 B, D and Table 2). Myotubes from both patients exhibited an increase in the EC50 to 4-chloro-m-cresol compared to controls (324.19±32.23 µM and 284.17±64.71 μM, respectively (P<0.0001 Student’s t test) (Table 2). Furthermore, analysis of the kinetics of the Ca2+ transient showed that the Full Width at Half Maximum (FWHM) and Half Relaxation Times (HRT) were increased in patients’ cells in response to 60 mM KCl, from 5.66±0.54 and 4.05±0.51 to 9.35±1.05 and 7.06±0.89 in controls and patients, respectively (P<0.01 Student’s t test); the kinetics in response to 600 µM 4-chloro-m-cresol were similar in myotubes from controls and patients (Table 2).
Table 1:
Genotypes and phenotypes of SELENON patients investigated in the present study
| # | Sample ID | Sample type | Origin | Age of biopsy | #SELENON variants | Phenotypic characteristics | †HGMD access N° | |
|---|---|---|---|---|---|---|---|---|
| Patient | 1 | UK 3358 | Muscle biopsy | U.K. | 2 years | c.[713dupA];[802C>T] p.[(N238fs)];[(R268C)] |
Normal early motor milestones, rigidity of the spine and rapidly progressive scoliosis from the age of 5 years, requiring scoliosis surgery aged 11. Respiratory insufficiency from the age of 10 years, requiring nocturnal respiratory BiPap |
CI022931 CM114823 |
| 2 | UK 3866 | Muscle biopsy | U.K. | 6 years | c.[1282-2A>C];[ 1282-2A>C] p.[?];[?] |
Motor delay with sitting at 9 months and walking holding at 1.5 years of age. Recurrent chest infections in the first few years and heart failure due to sleep hypoventilating at age 16 (he was initiated on cuirass jacket ventilation for 2 years then changed to BIPAP). Scoliosis first noticed at age 10 and surgery performed at age 15. Currently walking but limited distance. | CS114832 | |
| 3 | UK 7220 |
Muscle biopsy | U.K. | 6 years | c.[1A>G];[ 883G>A] p.[(M1V)];[(E295K)] |
Progressive muscle weakness. Lumbar lordosis and Rigidity of spine. Recurrent respiratory infections since the age of 9 months. Noctural BiPAP from age 5. |
CM022835 CM114829 |
|
| 4 | UK 5314 | Muscle biopsy | U.K. | 13 years | c.[1384T>G];[1384T>G] p.[(U462G)]; [(U462G)] |
Diagnosed at 14 months with axial hypotonia and forward flexed posture on sitting. Through childhood developed spinal rigidity with scoliosis. At the age of 12 referred due to symptoms of severe nocturnal hypoventilation, managed with nocturnal BIPAP. Underwent spinal fusion aged 17 years for progressive scoliosis. | CM022839 | |
| 5 | UK 7264 |
Muscle biopsy | U.K. | 2 years | c.[1282-2A>C];[ 1282-2A>C] p.[?];[?] (intron 9) |
First concern aged 9 months with axial and proximal muscle weakness, and dropped head. Early scoliosis initially noticed aged 2, and frequent falls since acquisition of deambulation at 19 months | CS114832 | |
| 6 | UK 6508 |
Muscle biopsy | U.K. | 12 years | c.[1315C>T];[1315C>T] p.[(R439*)];[(R439*)] |
Delayed motor milestones, right from the first 2 years of life with inability to jump, hop or run, but maintained ability to walk on level ground and going upstairs. Axial proximal facial weakness, contractures and some distal laxity present at age 15. Nocturnal ventilation since the age of 13 years. Obese phenotype. | CM050744 | |
| 7 | NL 00682 | Muscle biopsy | Netherlands | 4 years | c.[713dupA];[ 1332_1334del] p.[(N238fs)];[(N444del)] |
Delayed motor development, hypotonia and weakness, respiratory weakness for which non-invasive nocturnal ventilation since 10 years of age, permanent wheelchair use since at age 27. Scoliosis operation at age 10 |
CI022931 Not reported |
|
| 8 | NL 05244 | Muscle biopsy | Netherlands | 2 years | c.[943G>A];[ 943G>A] p.[(G315S)];[G315S)] |
Delayed motor development, hypotonia and weakness, respiratory weakness for which non-invasive nocturnal ventilation since 16 years of age, intermittent wheelchair use since age 19. Scoliosis operation | CM022836 | |
| 9 | NL 22413 | Muscle biopsy | Netherlands | 13 years | c.[943G>A];[ 1332_1334del] p.[(G315S)];[N444del] |
Delayed motor development, hypotonia and weakness, respiratory weakness for which no non-invasive nocturnal ventilation yet at 19 years of age, limited walking distance (100 m), scolioisis |
CM022836 Not reported |
|
| 10 | NL 3508 | Muscle biopsy | Netherlands | 1.5 years | c.[713dupA];[ 1332_1334del] p.[(N238fs)];[(N444del)] |
Delayed motor development, hypotonia and weakness, respiratory weakness for which non-invasive nocturnal ventilation since 10 years of age, intermittent wheelchair use |
CI022931 |
|
| 11 | Den 01 | Muscle biopsy | Denmark | 14 years | c.[446dupC];[943G>A] p.[(P149fs)];[(G315S)] |
24-hr ventilator, rigid spine, spine operation age 30 years. Wheelchair bound since age 25. | CI1111271 | |
| 12 | Den 02 | Muscle biopsy | Denmark | 7 years | c.[893T>C];[1396C>T] p.[(L298P)];[(R466W)] |
Nocturnal ventilator, rigid spine, scoliosis operation age 19, wheelchair user, but can walk small distances | CM174139 CM174140 |
|
| 13 | Den 03 | Muscle biopsy | Denmark | 10 years | c.[943G>A];[943G>A] p.[(G315S)];[(G315S)] |
Nocturnal ventilator, rigid spine, wheelchair user, but can walk small distances. | CM022836 | |
| 14 | Den 04 | Muscle biopsy | Denmark | 14 years | c.[713 dup];[943G>A] p.[(N238fs)];[(G315S)] |
Rigid spine, normal lung function, proximal weakness, reduced walking distance (1 km) | CI1111271 CM022836 |
|
| 15 | BOS1457-1 | Muscle biopsy | USA | 3 years | c.[1A>G];[943G>A], p.[(M1V)];[(G315S)] |
6 year female w/ moderate hypotonia at birth, delayed motor milestones, dysarthria. Currently ambulant for short distances, uses manual wheelchair for longer. On nocturnal ventilation by BiPAP |
CM022835 CM022836 |
|
| 16 | CH 01 | Muscle biopsy | Switzerland | c.[683_689dup];[ 683_689dup] p.[(M230fs)];[(M230fs)] |
CI147558 | |||
| 17 | UK 852A | Muscle cells | U.K. | 8 years | c.[827_829dup];[ 827_829dup] p.[(A276_C277insS)]:[(A276_C277insS)] |
Presented aged 8 with joint laxity and frequent falls.. At age 14 presents: Motor difficulties. Spinal rigidity. Neck muscle tightness. Short stature. Nocturnal hypoventilation - under BiPAP (started at age 11). |
CI160952 | |
| 18 | UK L1753 | Muscle cells | U.K | 2 years | c.[802C>T];[1406G>A] p.[(R268C)];[(R469Q)] |
At age 4 presents: early onset scoliosis, currently being treated in brace. Axial weakness, hypermobility, hypotonia, slow weight gain, and abnormal sleep study, with plans to initiate BiPap nocturnal ventilator support. | CM114823 CM091416 |
All variants listed relative to SELENON reference sequence NM_020451.2
All previously unreported variants have been submitted to the LOVD database (http://www.LOVD.nl/SEPN1 ; all variants: http://databases.lovd.nl/shared/variants/SEPN1/unique)
The HGMD column shows the ID accession numbers of all listed previously reported variants.
Figure 1: Calcium homeostasis and ECC are affected by the presence of SELENON mutations.
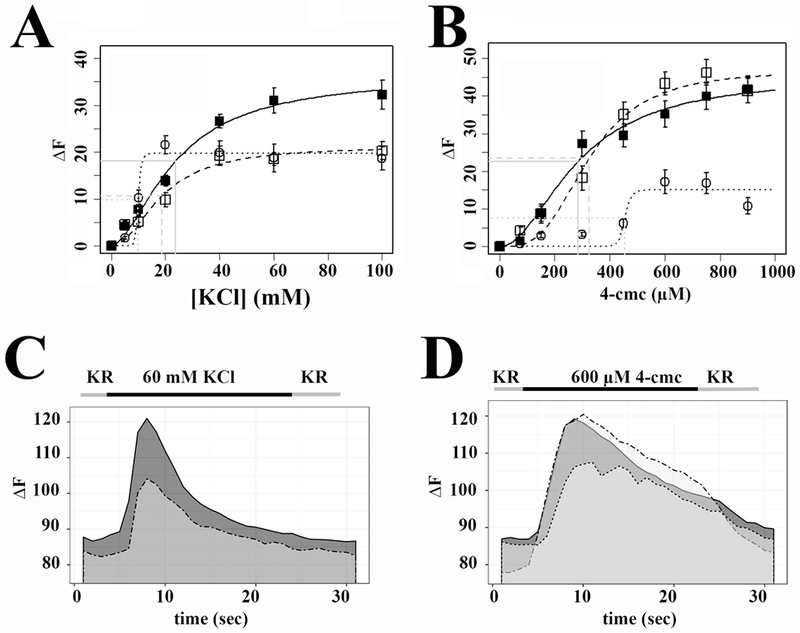
A. Depolarization (KCl) dose response curve; controls, black squares continuous line; patient N° 17, open squares dashed line; patient N°18, open circles, dotted line. B 4-chloro-m-cresol dose response curve; controls, black squares continuous line; patient N° 17, open squares dashed line; patient N°18, open circles dotted line C. Representative traces showing changes in [Ca2+]i induced by stimulation with 60 mM KCl. Continuous line, control; dashed line, patient N°17. D. Representative trace showing changes in [Ca2+]i induced by stimulation with 600 µM 4-chloro-m-cresol. Continuous line, control; dashed line patient N° 17; dotted line patient N° 18. In panels A and B, each symbol represents the mean (±SEM) peak calcium released by 5-10 myotubes.
Table 2:
Analysis of the parameters of Ca2+ homeostasis and of the kinetics of the Ca2+ transients in myotubes from patients and controls
| Controls | Pooled patients | Patient N° 17 | Patient N° 18 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Calcium homeostasis: | Mean±s.e.m. | N | Mean±s.e.m. | N | P value Controls vs patients | Mean±s.e.m | N | Mean±s.e.m | N | P value Patient 17 vs patient 18 |
| EC50 (KCl) | 23.69±3.79 | - | 11.12±2.33 | - | **** | 18.60±16.90 | - | 9.96±0.74 | - | **** |
| EC50 (4-CMC) | 284.17±64.17 | - | 324.19±32.23 | - | **** | 329.16±46.85 | - | 452.95±46.93 | - | **** |
| Peak (KCl) | 30.97±2.71 | 39 | 18.58±1.44 | 39 | *** | 18.48±1.42 | 23 | 18.72±2.93 | 16 | n.s. |
| Peak (4-CMC) | 35.22±3.48 | 38 | 29.92±2.95 | 43 | n.s. | 43.29±3.05 | 21 | 17.17±3.10 | 22 | **** |
| Resting Ca2+ (nM) | 87.39±0.36 | 445 | 84.55±0.37 | 482 | **** | 82.07±0.42 | 249 | 87.20±0.58 | 233 | **** |
| Calcium kinetics: | ||||||||||
| FWHM (4-CMC) | 15.01±1.10 | 28 | 16.56±0.35 | 25 | n.s. | 16.42±0.42 | 19 | 17.02±0.58 | 6 | n.s. |
| HRT (4-CMC) | 10.99±0.92 | 28 | 12.61±0.41 | 25 | n.s. | 12.55±0.46 | 19 | 12.80±0.96 | 6 | n.s. |
| FWHM (KCl) | 5.66±0.54 | 19 | 9.35±1.05 | 18 | ** | 7.42±0.98 | 12 | 13.19±1.58 | 6 | * |
| HRT (KCl) | 4.05±0.51 | 19 | 7.06±0.89 | 18 | ** | 5.35±0.65 | 12 | 10.48±1.65 | 6 | * |
P<0.05;
P<0.01;
P<0.001,
P<0.0001 Student’s t test.
To establish if the changes in calcium homeostasis were caused by altered expression or mis-localization of the main proteins involved in ECC, we performed high-resolution immunohistochemistry and qPCR analysis. The subcellular distribution of RyR1, Cav1.1, SERCA1 and SERCA2 was similar in myotubes from the patients and controls (Fig. 2A and B). Notably, both in controls and patients myotubes, the two SERCA isoforms did not share subcellular localization, as SERCA2 staining was punctate, similar to that of RyR1 and Cav1.1, whereas SERCA1 staining was predominantly perinuclear (Fig.2B, arrows). SELENON mRNA was decreased in patients myotubes relative to controls (0.444±0.042 and 1.000±0.096 respectively; P<0.0001 Student’s t test), while genes involved in skeletal muscle ECC were up-regulated (Fig. 2C). Specifically, the relative expression of RYR1, CACNA1S and ATP2A2 transcripts was 2.675±0.333, 1.312±0.124 and 2.346±0.297 in patients and 1.000±0.125, 1.000±0.081 and 1.000±0.151 in control myotubes, respectively. Furthermore, we found mis-regulation of several epigenetic factors. In particular, class I HDACs (HDAC1 and HDAC3) were down-regulated (0.415±0.033 and 0.636±0.048 in patients and 1.000±0.087 and 1.000±0.059 in controls, respectively, P<0.0001 Student’s t test). As to the expression pattern of the class II HDACs, (i) HDAC9 was up-regulated (1.559±0.252 and 1.000±0.035 in patients and controls, respectively; P<0.05 Student’s t test), (ii) HDAC5 was down-regulated (0.664±0.056 and 1.000±0.068 in patients and controls, respectively; P<0.001 Student’s t test). No changes were detected in HDAC4 expression. Interestingly, the alterations in HDACs expression were paralleled (Fig. 2C) by down-regulation of the DNA methyltransferases DNMT1, DNMT2 (0.833±0.066 and 0.634±0.045 versus 1.000±0.018 and 1.000± 0.041, in patients and controls, respectively; P<0.05 and P<0.0001).
Figure 2: Cellular distribution of RyR1, Cav1.1, SERCA1 and SERCA2 and expression of selected transcripts, in differentiated myotubes from SELENON patients and healthy control.
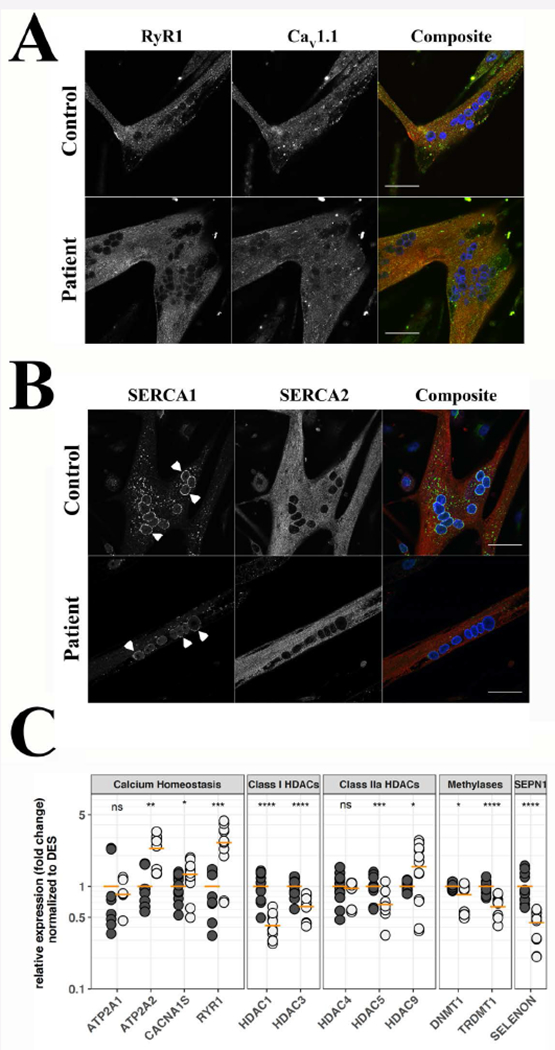
A. Subcellular distribution of RyR1 and Cav1.1. B. Subcellular distribution of SERCA1 and SERCA2. Myotubes were visualized with a Nikon A1 plus confocal microscope equipped with a Plan Apo 60× oil objective (NA 1.4). Top panels show myotubes from a healthy control, bottom panels show myotubes from patient N° 17. The images corresponding to RyR1 are pseudocoloured in red and those corresponding to Cav1.1 in green in the composite images (right); the images corresponding to SERCA1 are pseudocoloured in green and those corresponding to SERCA2 in red in the composite images (right); DAPI (nucleus) staining is blue; orange pixels show areas of co-localization. Arrowheads in B indicate perinuclear staining of SERCA1. Bar indicates 50 µm. C. Expression levels of the indicated transcripts, as assessed by qPCR. Black circles control cells, grey circles patient cells. qPCRs were repeated 4 times on separate myotube cultures from the two patients. Horizontal orange line indicates the mean expression level in pooled data from patient N° 17 and 18. Pooled levels from control myotubes were set as 1. *P<0.05; ** <0.01; *** P<0.001; **** P<0.0001; n.s. not significantly different. Student’s t test.
Since the phenotype of myotubes does not exactly recapitulate that of fully differentiated muscle fibres, we investigated the pattern of expression of the epigenetic enzymes in muscle biopsies (Fig. 3). Patients muscles exhibited decreased expression of class I HDACs: HDAC1 expression was 0.524±0.157 and 1.000±0.122, in patients and controls, respectively (P<0.05 Student’s t test), and HDAC3 expression was 0.340±0.087 and 1.000±0.103 in patients and controls, respectively (P<0.0001 Student’s t test). On the other hand, transcripts encoding class IIa HDACs were significantly increased: HDAC5 expression was 7.750±1.970 and 1.000±0.078 in patients and controls, respectively (P< 0.01 Student’s t test) and HDAC9 was 2.783±0.448 and 1.000±0.155 in patients and controls, respectively (P<0.05 Student’s t test). Similarly, the transcripts encoding DNA methylating enzymes were increased: DNMT1 expression levels were 8.010±2.080 and 1.000±0.126 in patients and controls, respectively (P< 0.05 Student’s t test) and TRDMT1 levels were 3.720±0.585 and 1.000±0.140 in patients and controls, respectively (P<0.01 Student’s t test). The transcripts encoding RYR1, CACNA1S and ATP2A1 were significantly reduced (0.464±0.081***, 0.483±0.059*** and 0.262±0.073* versus 1.000±0.085, 1.000±0.094 and 1.000±0.211, in patients and controls, respectively. *P<0.05, ***P<0.001, Student’s t test) (Fig.3A). Interestingly, the levels of SELENON transcripts in mature muscle biopsies from healthy controls were lower than their levels in myotubes, as assessed by qPCR. Indeed, the mean 2-(ΔΔCt) values were 1.063±0.171 for myotubes and 0.049±0.004 for muscle biopsies, indicating an approximate 20-fold difference between the two samples. These results are compatible with previous observation suggesting that SELENON is down-regulated in adult skeletal muscles (Petit et al., 2003).
Figure 3: Muscles from SELENON patients show significant changes in the expression levels of transcripts and proteins involved in ECC and in epigenetic regulation.
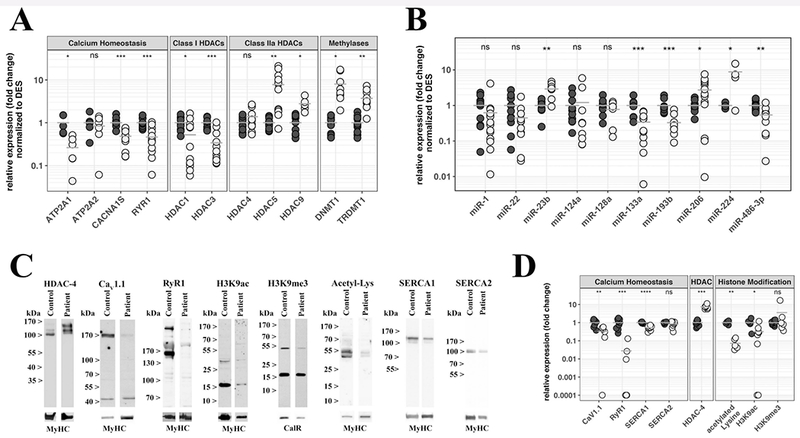
A. Expression levels of the indicated transcripts were determined by qPCR and normalized to the expression of DES. B. Expression levels of the indicated miRs were determined by qPCR and normalized to RNU44. Each reaction was performed in duplicate on muscle biopsies from healthy controls (black circles) and SELENON patients (empty circles). C. Western blot analysis and protein quantification of selected proteins. Top lanes, representative immunoblots probed with the indicated antibodies. Lower lanes show immunoreactivity to an antibody recognizing all MyHC isoform or the ER protein calreticulin used to normalize for protein loading. D. Comparison of the relative expression of the indicated proteins in muscle extracts from healthy controls (black circles), and SELENON patients (empty circles). Results were prepared using Excel’s “Conditional Formatting Plugin”. The relative protein content in patients was compared to that found in healthy controls that was set to 1. The horizontal grey bar represents the mean content levels in patient muscles. *P<0.05; **P<0.01; ***P<0.001; ****P< 0.0001 Student’s t test.
Gene expression is regulated at different levels, including regulation by transcription factors and by microRNAs (miRs); the latter epigenetic modifiers are not ubiquitously expressed but are differentially expressed in specific tissues (Bartel, 2009). In skeletal muscle miR-1, miR-133, miR-206 and miR-486 are under the control of myoD and MEF-2 and target a number of transcripts including HDAC4 (Ge & Chen, 2011; Eisenberg et al., 2008). We investigated muscle biopsies for the expression of muscle specific or muscle associated miRs (myomiRs) as well as other relevant miRs. Muscles from SELENON patients showed a significant decrease in miR-133a (0.340±0.068 and 1.000±0.150 in patients and controls, respectively; P<0.001 Student’s t test) and miR-486 (0.541±0.089 and 1.000±0.094 in patients and controls, respectively; P<0.01 Student’s t test), an increase in miR-206 (2.723±0.653 and 1.000±0.115 in patients and controls, respectively, P<0.05 Student’s t test) and in miR-23b (2.911±0.501 and 1.000±0.154 in patients and controls, respectively, P<0.01 Student’s t test), which according to its sequence, is predicted to bind in silico to SELENON. miR-22 which is predicted to bind to 3’ UTR of the RYR1, was decreased in most patients whereas miR-124 which is also predicted to bind to the 3’UTR of the RYR1 was decreased in the muscles of 5 patients and increased in the muscles of 2 patients. miR-128 levels were unchanged (Fig. 3B). The level of miR-193b, which is predicted to bind to several targets including SELENON, was decreased (0.328±0.071 and 1.000±0.134 in patients and controls, respectively, P<0.001 Student’s t test).
We next investigated by quantitative immunoblotting, the content of proteins in muscle homogenates. Figure 3C shows representative Western blots and figure 3D shows the protein content in muscles from patients (white circles) versus controls (black circles); protein levels were normalized to myosin heavy chain (MyHC) content or the endoplasmic/sarcoplasmic reticulum protein calreticulin and control levels were set to 1. The anti-MyHC antibody that was used recognizes all isoforms. Suppl. Table S4 shows the normalized content of each protein in each muscle sample. Quantification of all proteins could not be performed on all patient samples due to the lack of sufficient biological material but the results presented in figure 3D and Supp. Table S4 confirm that the protein content of RyR1, Cav1.1 and SERCA1 were reduced in muscles from all patients carrying SELENON-variants (normalized protein content was 0.033±0.025***, 0.331±0.090** and 0.527±0.046**** in patients versus 1.000±0.185, 1.000±0.169 and 1.000±0.043 in controls, for RyR1***, Cav1.1** and SERCA1****, respectively.**P<0.01, ***P<0.001 and ***P<0.0001), whereas SERCA2 content was unchanged. Since SERCA1 is expressed in fast (type 2) glycolytic fibres (Brandl et al., 1986) and patients with congenital myopathies often show atrophy of these fibres (Romero & Clarke, 2013) the decrease in SERCA1 could simply reflect a decrease in type 2 fibres. This was investigated by performing high-resolution gel electrophoresis (Talmadge & Roy, 1985). As indicated in Supp. Figure S1A and B, the decrease in SERCA1 in SELENON patients is associated with a larger fraction of fibres expressing the slow MyHC isoform.
Histone acetylation and methylation also influence gene expression: H3K9 acetylation is considered a mark for gene transcription whereas H3K9me3 is considered a mark for transcriptional repression (Barski et al., 2007). The protein content of the histone de-acetylating enzyme HDAC-4 was significantly increased in muscles of patients (7.403±0.747 and 1.000±0.107 in patients and controls, respectively, P<0.001 Student’s t test) and this was paralleled by a decrease in the overall content of acetylated proteins (0.072±0.019 and 1.000±0.114 in patients and controls, respectively, P<0.01 Student’s t test) and a decrease of H3K9 acetylation in patient muscles (0.320±0.138 and 1.000±0.244 in patients and controls, respectively, P<0.05 Student’s t test) (Fig. 3D and Supp. Table S4). The mean content of tri-methylated H3K9 was not changed. Histone de-acetylases bind to and function in association with DNA methylating enzymes to bring about chromatin methylation, and gene silencing (Kimura & Shiota, 2003). Based on these results and on previous studies (Rokach et al., 2015; Zhou et al., 2006, 14) we hypothesized that changes of gene expression, are caused by alterations in gene methylation.
To investigate DNA methylation in SELENON patients in comparison to healthy controls, we performed genome-wide DNA methylation using Infinium methylation EPIC Beadchip arrays in muscle biopsies from 5 SELENON patients and 4 healthy controls. Multidimensional scaling (MDS) analysis of the most variably methylated CpGs showed clear separation of SELENON patients from controls (Fig.4A). By using differentially methylation analysis, we identified 31623 differentially methylated CpGs, among them 22376 (71%) were hypermethylated and 9247 (29%) were hypomethylated (Fig. 4B) in SELENON patients. KEGG pathway analyses of hypermethylated-CpGs associated genes revealed a specific functional association with calcium signalling, focal adhesion and axonal guidance, while hypomethylated-CpGs associated genes were enriched in endocytosis, cancer as well as calcium signalling (Fig. 4C). Interestingly, methylation profiles of patients with recessive RYR1 variants (Suppl. Table S5) grouped closer to SELENON patients than to the control group (Fig. 4E), indicating common methylation targets between SELENON and RYR1 patients. Indeed, among hypermethylated sites, 3163 CpGs were common and among hypomethylated sites, 324 CpGs were common in both SELENON and RYR1 patients (Fig. 4E). We checked the expression level of transcripts encoding four genes that were hypermethylated and whose abnormal expression is relevant to skeletal muscle ECC, namely ATP2B2 (the gene encoding the plasma membrane Ca2+ ATPase), STAC3 (encoding the SH3 and cysteine rich domain 3 protein, which is mutated in Native American Myopathy patients)(Horstick et al., 2013), RYR1 and ATP2A1 (encoding SERCA1). In muscles from all patients bearing RYR1 and SELENON variants the transcript levels were reduced by more than 50% (Fig. 4F).
Figure 4: Muscles of patients with congenital myopathies leading to reduced RyR1 content show common changes in CpG methylation.
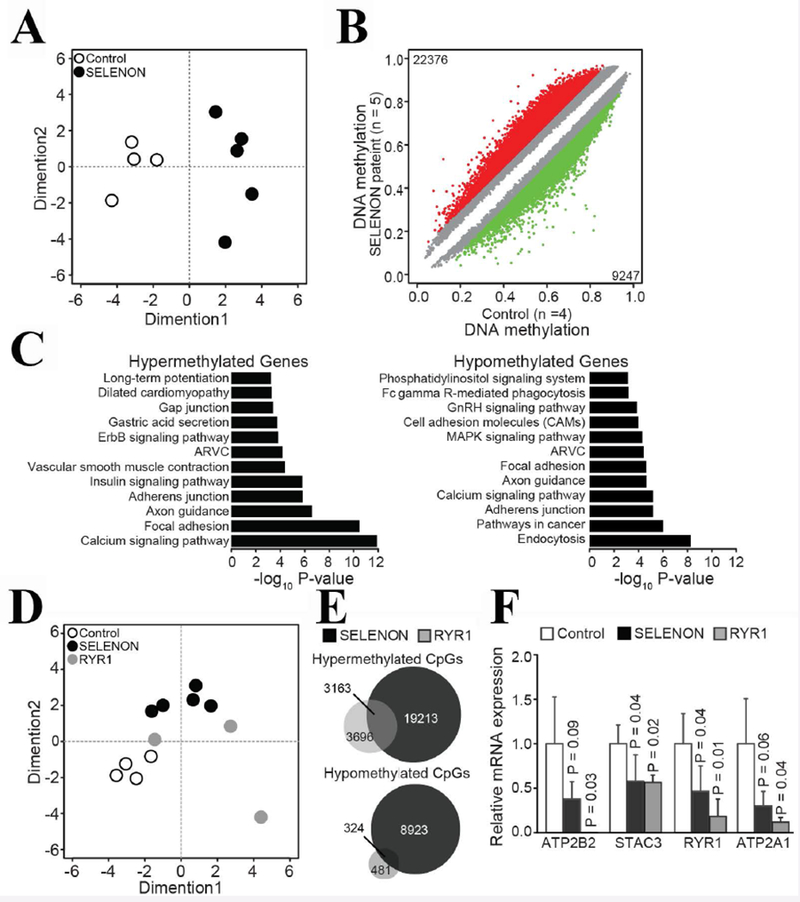
A. Multidimensional Scaling (MDS) plot analysis of genome-wide methylation profiles separated healthy control © muscle sample from patients with SELENON myopathy. MDS plot includes all probes on the array. B. Scatter plot shows differential methylated CpGs in patients with SELENON myopathy. Colored dots represent significant hypomethylated (green) or hypermethylated (red) CpGs together with numbers. Number of samples are indicated with n. C. Enriched KEGG pathways (P<0.001) within hypermethylated or hypomethylated CpGs associated genes. D MDS plot including controls, SELENON and RYR1 samples. E. Venn diagram shows overlap of CpGs hypermethylated in SELENON and RYR1 samples. F. qPCR analysis of selected transcripts. The ATP2B2 expression level in patients with recessive RYR1 mutations was below the detection level. The genes encoding these transcripts were hypermethylated in patients’ muscles. Results are presented as relative expression compared to control. P-values were calculated with Welch two sample t-test and error bars denote S.D.
DISCUSSION
A novel finding of the present investigation is that muscles of patients with SELENON variants contain lower amounts of proteins (as well as the transcripts encoding for proteins) playing a key role in calcium regulation and skeletal muscle ECC, namely RyR1, Cav1.1 (the alpha 1 subunit of the DHPR) and SERCA1. During ECC, depolarization of the sarcolemma is sensed by DHPRs leading to Ca2+ release from SR stores via RyR1 (Rios & Pizarro, 1991; Schneider & Chandler, 1972). Calcium release activates muscle contraction, while muscle relaxation is brought about by SERCA pumps which transport calcium back into the SR (MacLennan et al., 1997). Thus, a decrease in proteins involved in ECC is expected to result in muscle weakness and hypotonia since less Ca2+ would be released from the SR to activate the contractile proteins. Alterations of Ca2+ homeostasis in myotubes from patients carrying recessive SELENON variants have been previously reported (Arbogast et al., 2009) and those results were partially confirmed in the present study. Importantly however, 4-chloro-m-cresol induced calcium release as well as the kinetics of KCl-induced calcium release were affected only in myotubes from patient N°18 who displayed a more severe phenotype and early onset, while myotubes from patient N°17 (who is less severely affected), displayed parameters that were more similar to those of control myotubes. Furthermore, myotubes from both patients displayed a significant decrease in Ca2+ release triggered by sarcolemmal depolarization. This “uncoupling” was not caused by reduced RyR1 and DHPR content in myotubes, nor by the presence of smaller intracellular calcium stores caused by leaky RyR1 channels, nor was it associated with any apparent change in the subcellular localization of the main players involved in ECC. Furthermore, the absence of Selenoprotein N did not impact SERCA pump activity as recently suggested (Marino et al., 2015) since the kinetics of 4-chloro-m-cresol-induced Ca2+ transients (FWHM and HRT) were similar in control and patient myotubes. Such discrepancies could be due to different experimental approaches and/or to the fact that our experiments were conducted on myotubes that predominantly express SERCA1 and SERCA2a (Toral-Ojeda et al., 2016; Töth et al., 2015) while Marino et al. used non-muscle cells predominantly expressing SERCA3a/b/c and SERCA2b (Marino et al., 2015).
Nevertheless, our results demonstrate that recessive variants in SELENON, do result in severely compromised ECC. Of importance, the decrease in RYR1, CACNA1S and ATP2A1 gene expression only occurs in fully differentiated adult muscle fibres and not in myotubes. These observations together with the fact that the available animal models do not reproduce the human disease (Rederstorff et al., 2011) suggest the involvement of additional factors in the pathomechanism of SELENON-related disease. We postulate that the primary genetic defect results in changes in transcriptional regulation brought about by epigenetic enzymes that are only activated in fully developed and differentiated skeletal muscles.
The observed up-regulation of HDAC-4/5 in skeletal muscle can be caused by a variety of stimuli including: (i) ER stress where it may have a protective effect against apoptosis (Zhang et al., 2014); (ii) immobilization and nutrient deprivation induced-atrophy (Beharry & Judge, 2015) and (iii) denervation induced-atrophy, which induces nuclear accumulation of HDAC-4 with a concomitant reduction of transcription of MEF-2 dependent genes (Lu et al., 2000; Wang et al., 2014). On the other hand, stimuli that lead to increased expression of class I HDACs include muscle disuse caused by cast immobilization and denervation (Beharry et al., 2014). Furthermore HDAC-1 and HDAC-2 control autophagy in skeletal muscle (Moresi et al., 2012). Since the content of class I HDACs was decreased in the muscles of SELENON patients, neither denervation- nor immobilization-induced atrophy, or changes in autophagy are likely the primary cause of the increased expression of class II HDACs.
What is the mechanism leading to the increased expression of class II HDACs? And does this mechanism also lead to the increased expression of DNA methylating enzymes? DNA methylating enzymes directly bind HDACs and it is the combined activity of both classes of epigenetic enzymes that results in transcriptional repression (Fuks et al., 2000). One mechanism that may be responsible for the increased expression of both classes of chromatin modifying enzymes may relate to muscle development. HDAC4, 5 and 9, DNMT1 and TRDMT1 (DNMT2) contain binding sites for the transcription factors SP1 and SP3 (Kishikawa et al., 2002; Liu et al., 2008) that are involved in the regulation of many cellular processes including cell-cycle progression, growth control and response to oxidative and DNA damage (Watanabe et al., 1998; Lee et al., 2006). Thus it is plausible that the reduced content of the mutated protein and/or the altered calcium homeostasis in developing muscles affect the muscle’s capacity to grow and differentiate leading to enhanced SP1 and SP3–dependent gene transcription. Simultaneously over-expression of class II HDACs and of DNA methyltransferases leads to a decreased transcription of MEF-2 dependent genes and miRs and to abnormal gene methylation. The combination of these events causes profound modifications in the transcription and translation of genes which are characteristics of mature skeletal muscle. Our observation that muscle samples from patients with recessive RYR1 and SELENON-related congenital myopathies, two muscle disorders which share as a common feature reduced RyR1 protein content, also share >3000 abnormally methylated CpG sites, is both intriguing and relevant to disease pathomechanism since four of the hypermethylated genes namely RYR1, STAC3, ATP2A1 and ATP2B2 were significantly down-regulated in patients’ muscles and the top hyper-methylated CpG sites were contained within genes involved in calcium signalling pathways.
In conclusion, we show that common epigenetic signatures including abnormally methylated genes and increased levels of chromatin modifying enzymes occur in muscles of patients with MmD as a consequence of different primary genetic variants. Epigenetic modifications, including gene hyper-methylation lead to a decrease in the expression of proteins directly involved in ECC and calcium regulation (in fully developed and differentiated skeletal muscles). Targeting these enzymes may constitute a valid pharmacologic approach to improve muscle function in this group of patients.
Supplementary Material
Acknowledgements
We would like to thank Pierpaolo Ala for his expert technical support and C.C. Verschuuren-Bemelmans for helping collect the clinical data This work was supported by a grant from the Swiss National Science Foundation (SNF N° 31003A-169316), a grant from the OPO-Stiftung, from the Swiss muscle foundation (FSRMM) and a grant from NeRAB and M.U.R.S.T Italy 2015. The MRC Centre for Neuromuscular Diseases Biobank and the UMCG Pathology department are greatly appreciated for providing patients’ samples. The Muscular Dystrophy UK support to the Dubowitz Neuromuscular Centre is also gratefully acknowledged. The support of the Department of Anesthesia Basel University Hospital is gratefully acknowledged. S.A.M. and K.D.M. are partially funded by NIH grant U54, NS053672 for the University of Iowa Wellstone Muscular Dystrophy Cooperative Research Center. E.M. is supported by a Wellcome Clinical Research Career Development Fellowship. A.H.B. and C.A.G. were supported by grants NIH R01 AR044345 and MDA383249 from the Muscular Dystrophy Association, U.S.A, and by a generous gift from the Lee and Penny Anderson Family Foundation. G.M. and R.C. were supported by Fondazione Malattie Miotoniche-Italy (FMM).
Grant numbers: SNF 31003A-169316, NIH R01 AR044345, MDA383249
ABBREVIATIONS:
- Cav1.1
alfa 1 subunit of the dihydropyridine receptor
- DHPR
dihydropyridine receptor
- ECC
excitation-contraction coupling
- HDAC
histone de-acetylase
- miRs
micro RNAs
- MyHC
myosin heavy chain
- MmD
multiminicore Disease
- RyR1
ryanodine receptor 1
- SR
sarcoplasmic reticulum
- SelN
Selenoprotein N
Footnotes
Conflict of interest: The Authors have declared that no conflict of interest exists.
References
- Arbogast S, Beuvin M, Fraysse B, Zhou H, Muntoni F & Ferreiro A (2009). Oxidative stress in SEPN1-related myopathy: from pathophysiology to treatment. Annals of Neurolology, 65, 677–686. doi: 10.1002/ana.21644. [DOI] [PubMed] [Google Scholar]
- Bachmann C, Jungbluth H, Muntoni F, Manzur AY, Zorzato F & Treves S (2017). Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Human Molecular Genetics, 26, 320–332. doi: 10.1093/hmg/ddw388. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Ron TY, Schones DE, Wang Z, Wei G, Chepelev I & Zhao K (2007). High resolution profiling of histone methylation in the human genome. Cell, 129, 823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell, 136, 215–233. doi: 0.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry AW & Judge AR (2015). Differential expression of HDAC and HAT genes in atrophying skeletal muscle. Muscle and Nerve, 52, 1098–1101. doi: 10.1002/mus.24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM & Judge AR (2014). HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. Journal of Cell Science, 12, 1441–1453. doi: 10.1242/jcs.136390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl CJ, Green NM, Korczak B & MacLennan DH (1986). Two Ca2+ ATPase genes: homologies and mechanistic implications of deduced amino acid sequences. Cell, 44, 597–607. [DOI] [PubMed] [Google Scholar]
- Castets P, Bertrand AT, Beuvin M, Ferry A, Le Grand F, Castets M, Chazot G, Rederstorff M, Krol A, Lescure A, Romero NB, Guicheney P & Allamand V (2011). Satellite cell loss and impaired muscle regeneration in selenoprotein N deficiency. Human Molecular Genetics, 20, 694–704. doi: 10.1093/hmg/ddq515 [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L & Lin SM (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics, 11, 587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg I, Alexander MS & Kunkel LM (2008). miRNAs in normal and diseased skeletal muscle. Journal of Cellular and Molecular Medicine 13, 2–11. doi: 10.1111/j.1582-4934.2008.00524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG, Gomez MR & Groover RV (1971). Multicore disease. A recently recognized congenital myopathy associated with multifocal degeneration of muscle fibers. Mayo Clinic Proceedings, 46, 666–681. [PubMed] [Google Scholar]
- Ferreiro A, Estournet B, Chateau D, Romero NB, Laroche C, Odent S, Toutain A, Cabello A, Fontan D, dos Santos HG, Haenggeli CA, Bertini E, Urtizberea JA, Guicheney P & Fardeau M (2000). Multi-minicore disease-searching for boundries: phenotype analysis of 38 cases. Annals of Neurolology, 48, 745–757. [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Brehm A, Hughes-Davies L & Kouzarides T (2000). DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genetics, 1, 88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- Ge Y & Chen J (2011). MicroRNAs in skeletal myogenesis. Cell Cycle 10, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstick EJ Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini-Armstrong C, Hirata H & Kuwada JY (2013). Stac3 is a component of the excitation–contraction coupling machinery and mutated in Native American myopathy. Nature Communications, 4, 1952 doi: 10.1038/ncomms2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H, Sewry CA & Muntoni F (2011). Core myopathies. Seminars in Pediatric Neurology, 18, 239–249. doi: 10.1010/.spen.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry CA, Phadke R, Gautel M & Muntoni F (2018). Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nature Reviews Neurology 14, 151–167. doi: 10.1038/nrneurol.2017.191. [DOI] [PubMed] [Google Scholar]
- Kimura H & Shiota K (2003). Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methlytransferase, Dnmt1. Journal of Biological Chemistry 278, 4806–4812. [DOI] [PubMed] [Google Scholar]
- Kishikawa S, Murata T, Kimura H, Shiota K & Yokoyama KK (2002). Regulation of transcription of the Dnmt1 gene by Sp1 and Sp3 zinc finger proteins. European Journal of Biochemistry, 269, 2961–2970. [DOI] [PubMed] [Google Scholar]
- Lee J, Kosaras B, Aleyasin H, Han JA, Park DS, Ratan RR, Kowall NW, Ferrante RJ, Lee SW & Ryu H (2006) Role of cyclooxygenase-2 induction by transcription factor Sp1 and Sp3 in neuronal oxidative and DNA damage response. FASEB Journal, 20, 2375–2377. doi: 10.1096/fj.06-5957fje [DOI] [PubMed] [Google Scholar]
- Liu F, Pore N, Kim M, Voong KR, Dowling M, Maity A & Kao GD (2006). Regulation of histone deacetylase 4 expression by the SP family of transcription factors. Molecular Biology of the Cell, 17, 585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL & Olson EN (2000). Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Molecular Cell, 6, 233–244. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Rice WJ & Green NM (1997). The mechanism of Ca2+ transport by serco(endo)plasmic reticulum Ca2+-ATPase. Journal of Biological Chemistry, 272, 28815–28828. [DOI] [PubMed] [Google Scholar]
- Marino M, Stoilova T, Giorgi C, Bachi A, Cattaneo A, Auricchio A, Pinton P & Zito E (2015). SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal pathology counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Human Molecula Genetics, 24, 1843–1855. doi: 10.1093/hmg/ddu602. [DOI] [PubMed] [Google Scholar]
- Moghadaszadeh B, Petit N, Jaillard C, Brockington M, Quijano- Roy S, Merlini L, Romero N, Estournet B, Desguerre I, Chaigne D, Muntoni F, Topaloglu H & Guicheney P (2001). Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nature Genetics, 29, 17–18. doi: 10.1038/ng713. [DOI] [PubMed] [Google Scholar]
- Moresi V, Carrer M, Grueter CE, Rifky OF, Shelton JM, Richardson JA, Bassel-Duby R & Olson E (2012). Histone deacetylase 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proceedings of the National Academy of Sciences of the United States of America, 109, 1649–1654. doi: 10.1073/pnas.1121159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Takashima H, Higuchi I, Matsuyama W, Suehara M, Nishihira Y, Hashiguchi A, Hirano R, Ng AR, Nakagawa M, Izumo S, Osame M & Arimura K (2006). Molecular mechanism of rigid spine with muscular dystrophy type 1 caused by novel mutations of selenoprotein N gene. Neurogenetics, 7, 175–183. [DOI] [PubMed] [Google Scholar]
- Petit N, Lescure A, Rederstorff M, Krol A, Moghadaszadeh B, Wewer UM & Guicheney P (2003). Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Human Molecual Genetics, 12, 1045–1053. [DOI] [PubMed] [Google Scholar]
- Rederstorff M, Castets P, Arbogast S, Lainé J, Vassilopoulos S, Beuvin M, Dubourg O, Vignaud A, Ferry A, Krol A, Allamand V, Guicheney P, Ferreiro A & Lescure,. A. (2011). Increased muscle stress-sensitivity induced by selenoprotein N inactivation in mouse: a mammalian model for SEPN1-related myopathy. PloS One 6(8),e23094. doi: 10.1371/journal.pone.0023094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC & Gerhard D (2015). Dose response analysis using R. PloS One, 10, e0146021 doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero NB & Clarke NF (2013). Congenital myopathies. Handbook of Clinical Neurology, 113, 1321–36. doi:10.1016B978-0-444-59565-2.00004-6. [DOI] [PubMed] [Google Scholar]
- Rios E & Pizarro G (1991). Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiological Reviews 71, 849–908. [DOI] [PubMed] [Google Scholar]
- Rokach O, Sekulic-Jablanovic M, Voermans N, Wilmhurst J, Pillay K, Heytens L, Zhou H, Muntoni F, Gautel M, Nevo Y, Mitrani-Rosenbaum S, Attali R, Finotti A, Gambari R, Mosca B, Jungbluth H & Treves S (2015). Epigenetic changes as a common trigger of muscle weakness in congenital myopathies. Human Molecular Genetics, 15, 4636–4647. doi: 10.1093/hmg/ddv195. [DOI] [PubMed] [Google Scholar]
- Schneider MF & Chandler WK (1972). Voltage dependent charge measurement of skeletal muscle: a possible step in excitation-contraction coupling. Nature 242, 244–246. [DOI] [PubMed] [Google Scholar]
- Talmadge RJ & Roy RR (1985). Electrophoretic separation of rat skeletal muscle myosin heavy chain isoforms. Journal of Applied Physiology 75, 2337–2340. [DOI] [PubMed] [Google Scholar]
- Töth A, Fodor J, Vincze J, Olah T, Juhasz T, Zakany R, Csernoch L & Zador E (2015). The effect of SERCA1b silencing on the differentiation and calcium homeostasis of C2C12 skeletal muscle cells. PloS One, 10, e0123583, doi 10.1371/journal.pone.0123583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toral-Ojeda I, Aldanondo G, Lasa-Elgarresta J, Lasa-Fernandez H, Fernandez-Torron R, Lopez de Munain A & Vallejo-Illarramendi A (2016). Calpain deficiency affects SERCA expression and function in the skeletal muscle. Expert. Rev. Mo.l Med, 18, e7, doi. 10.107/erm.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Qin G & Zhao TC (2014). Histone deacetylase 4 (HDCA4): mechanism of regulations and biological functions. Epigenomics, 6, 139–150. doi: 10.2217/epi.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein B & Pestell RG (1998). Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Molecular and Cellular Biology, 18, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall JM & Smyth GK (2004). limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics, 20, 3795–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]
- Zhang P, Sun Q, Zhao C, Ling S, Li Q & Chang YZ (2014). HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell Signaling, 26, 556–563. doi: 10.1016/j.cellsig.2013.11.026. [DOI] [PubMed] [Google Scholar]
- Zhou H, Brockington M, Jungbluth H, Monk D, Stainer P, Sewry CA, Moore GE & Muntoni F (2006). Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. American Journal of Human Genetics, 79, 859–868. doi: 10.1086/508500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H Jungbluth H, Sewry CA, Feng L, Bertini E, Bushby K, Straub V, Roper H, Rose MR, Brockington M, Kinali M, Manzur A, Robb S, Appleton R, Messina S, D’Amico A, Quinlivan R, Swash M, Müller CR, Brown S, Treves S & Muntoni F (2007). Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain, 130, 2024–2036. [DOI] [PubMed] [Google Scholar]
- Zorzato F, Scutari E, Tegazzin V, Clementi E & Treves S (1993). Chlorocresol: an activator of ryanodine receptor-mediated Ca2+ release. Molecular Pharmacology 44, 1192–1201. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.