

SUMMARY
Hcr1/eIF3j is a sub-stoichiometric subunit of eukaryotic initiation factor 3 (eIF3) that can dissociate the post-termination 40S ribosomal subunit from mRNA in vitro. We examine this ribosome recycling role in vivo by ribosome profiling and reporter assays and find that loss of Hcr1 leads to reinitiation of translation in 3′ UTRs, consistent with a defect in recycling. However, the defect appears to be in the recycling of the 60S subunit, rather than the 40S subunit, because reinitiation does not require an AUG codon and is suppressed by overexpression of the 60S dissociation factor Rli1/ABCE1. Consistent with a 60S recycling role, overexpression of Hcr1 cannot compensate for loss of 40S recycling factors Tma64/eIF2D and Tma20/MCT-1. Intriguingly, loss of Hcr1 triggers greater expression of RLI1 via an apparent feedback loop. These findings suggest Hcr1/eIF3j is recruited to ribosomes at stop codons and may coordinate the transition to a new round of translation.
Graphical Abstract
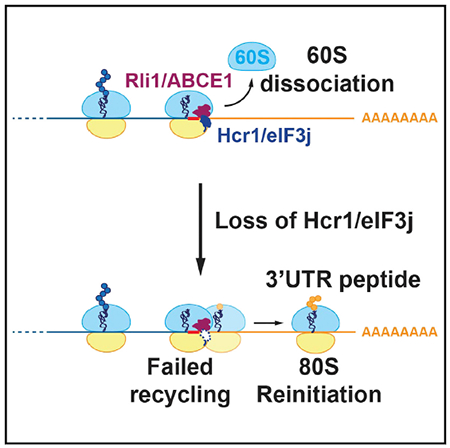
In Brief
Young and Guydosh show that Hcr1/eIF3j is an accessory factor for Rli1-catalyzed 60S subunit dissociation during ribosome recycling in vivo. The involvement of a translation initiation factor in ribosome recycling links the final step of translation with the next round of protein synthesis.
INTRODUCTION
Ribosome recycling is the critical cellular process that dissociates post-termination ribosomes into their constituent components, thereby freeing them up to initiate new rounds of translation. Ribosome recycling begins after eukaryotic release factor 1 (eRF1) recognizes the stop codon and releases the completed protein (Hellen, 2018). In the first step of recycling, the ATPase Rli1 (yeast)/ABCE1 (mammals) catalyzes splitting of the 60S ribosomal subunit from the tRNA- and mRNA-bound 40S subunit (Pisarev et al., 2010; Shoemaker and Green, 2011; Young et al., 2015). The second step of recycling, dissociation of tRNA and mRNA from the 40S subunit, has been reconstituted in vitro via two distinct pathways. In one pathway, recycling of the 40S subunit is promoted by the translation-machinery-associated factors, Tma64, Tma20, and Tma22 (yeast) or eIF2D, MCT-1, and DENR (mammals) (Skabkin et al., 2010). We have recently shown that Tma64, Tma20, sand Tma22 (Tma64/−20/−22) also promote recycling of post-termination 40S subunits in vivo and that double knockout of TMA64 with either TMA20 or TMA22 generates a nearly identical phenotype (Young et al., 2018).
In an alternative pathway, 40S recycling can be promoted in a reconstituted system with canonical initiation factors, including multiple subunits of eIF3. (Pisarev et al., 2007, 2010). In yeast, eIF3 contains five essential subunits, Tif32/eIF3a, Prt1/eIF3b, Nip1/eIF3c, Tif34/eIF3i, and Tif35/eIF3g, and one non-essential subunit, Hcr1/eIF3j (nomenclature for yeast or mammals, respectively) (Hinnebusch, 2017). Because Hcr1/eIF3j is not a stoichiometric subunit of eIF3, we hereafter referto eIF3 as a core complex without Hcr1/eIF3j. During ribosome recycling, it was observed that eIF1, in association with eIF1A, enhanced the dissociation of the deacylated tRNA from a tRNA- and mRNA-bound 40S subunit and that added eIF3j, in the presence of purified eIF3, could subsequently promote dissociation of the mRNA from the 40S subunit (Pisarev et al., 2007, 2010). The idea that Hcr1/eIF3j promotes removal of mRNA from a post-termination 40S complex is also supported by the finding that Hcr1/eIF3j and mRNA bind anti-cooperatively to the 40S subunit in both human cells and yeast (Fraser et al., 2007; Mitchell et al., 2010). Hcr1/eIF3j has been shown by hydroxyl radical probing, cryoelectron microscopy (cryo-EM), and crosslinking to bind the 40S in the region that forms the mRNA entry channel and ribosome A site (Aylett et al., 2015; Erzberger et al., 2014; Fraser et al., 2007), offering a potential explanation for how it interferes with mRNA binding.
Hcr1/eIF3j has also been shown to be important in multiple steps of translation initiation (Elantak et al., 2010; Nielsen et al., 2006), including the recruitment of core components of eIF3 to the 40S ribosomal subunit (Aylett et al., 2015; Fraser et al., 2004). Deletion of the HCR1 gene results in slow growth and a severe leaky scanning defect, a characteristic of poor start-codon recognition that can be partially suppressed by expression of the N-terminal domain (NTD) of Hcr1/eIF3j or by overexpression of eIF1A (Elantak etal., 2010). Structural data showing direct contact between eIF1A and Hcr1/eIF3j when bound to the 40S subunit is consistent with this observation (Aylett et al., 2015). Hcr1/eIF3j interacts with both the C terminus of Tif32/eIF3a and the N-terminal RNA recognition motif (RRM) domain of Prt1/eIF3b (Valásek et al., 2001). Interestingly, the allele of the Tif32/eIF3a gene that was used to identify Hcr1 (tif32-R7311) lies within that C-terminal region of Tif32/eIF3a (Valásek et al., 1998,1999). The tif32-R731I mutation and another nearby ts allele, tif32-H725P, significantly weaken binding between eIF3a and Hcr1/eIF3j, impair mRNA recruitment by 43S preinitiation complexes, and cause a leaky scanning defect (Chiu et al., 2010). A conserved tryptophan residue in the Hcr1/eIF3j N-terminal acidic (NTA) motif is held in a hydrophobic “pocket” of the eIF3b RRM. Mutating the tryptophan and NTA motif of Hcr1/eIF3j (NTA1 mutant) orthe corresponding pocket residues in Prt1/eIF3b eliminates Hcr1/eIF3j association with Prt1/eIF3b in vitro and in vivo and produces similar initiation defects to those observed for the eIF3a ts alleles (Elantak et al., 2010). Although these interactions are key to the role of Hcr1 in initiation, it remains unknown how they would affect any role of Hcr1 in recycling.
Other in vivo results suggest Hcr1/eIF3j is involved in translation termination. Deletion of the HCR1 gene results in apparent increased stop-codon readthrough in a dual-luciferase reporter assay and a shift of release factor 3 (eRF3) to heavy polysomes (Beznosková et al., 2013). Surprisingly, the effect could be suppressed by overexpression of Rli1/ABCE1, a factor known to catalyze recycling of the 60S subunit. Although overexpression of Rli1/ABCE1 fully suppresses the defects in apparent read-through and growth, it did not suppress the defect in leaky scanning (initiation). Interestingly, interactions have been detected between both Hcr1/eIF3j and eIF3 with terminating ribosomes, consistent with a role for these factors in termination and recycling (Beznosková et al., 2013, 2015).
The suppression of the hcr1Δ slow-growth phenotype by overexpression of Rli1/ABCE1 is consistent with biochemical studies linking the function of the two proteins. Hcr1 has been shown to physically interact with Rli1 by yeast two-hybrid analysis (Khoshnevis et al., 2010; Kispal et al., 2005). In addition, in vitro pull-down assays showed the second C-terminal ABC ATPase domain of Rli1 is sufficient to mediate the interaction (Khoshnevis et al., 2010). However, the region of Hcr1/eIF3j that interacts with Rli1/ABCE1 remains unknown. eIF3 has been shown to form a multi-initiation factor complex (MFC) by interacting directly with eIF1, eIF5, and the ternary complex (Met-tRNAMeti-eIF2-GTP) (Asano et al., 2000; Hinnebusch, 2017). Affinity-tagged Rli1/ABCE1 has been shown to pull down components of the MFC (Chen et al., 2006; Dong et al., 2004), presumably through an interaction with Hcr1/eIF3j. However, it is unknown whether any of these interactions are important for the function of Hcr1 in termination and recycling.
In this study, we sought to determine the role of Hcr1/eIF3j in termination and recycling in vivo. We employed ribosome profiling to show that hcr1Δ cells have increased ribosome foot-print occupancy in 3′ UTRs and confirmed with reporter genes that ribosomes engage in translation after a reinitiation event, two hallmarks of a ribosome recycling defect. The pattern of 3′-UTR footprint levels across the transcriptome and size of the 3′-UTR translation products was consistent with 3′-UTR translation occurring via an 80S-reinitiation mechanism, similar to what we have observed in cells depleted of the 60S subunit dissociation factor Rli1/ABCE1. In contrast, overexpression of Hcr1/eIF3j could not compensate for the simultaneous loss of the 40S recycling factors Tma64/eIF2D and Tma20/MCT-1. Moreover, loss of HCR1 did not lead to 40S reinitiation, as would be expected if it had a role in 40S recycling. We, therefore, conclude that Hcr1/eIF3j is an accessory factor that promotes recycling of 60S subunits by Rli1/ABCE1 at stop codons.
RESULTS
Deletion of HCR1 Results in Increased 3′ UTR Ribosome Occupancy, Consistent with a Defect in Translation Termination or Ribosome Recycling
We previously showed that defects in ribosome recycling in cells are evident in ribosome profiling data as an increase in ribosome footprints in 3′ UTRs due to unrecycled 80S ribosomes or 40S subunits reinitiating translation downstream of the stop codon (Young et al., 2015, 2018). Therefore, to determine whether Hcr1/eIF3j is involved in ribosome recycling in vivo, we performed ribosome profiling on a WT (BY4741) yeast strain and an hcr1Δ strain. Cells were grown and harvested, and ribosome-profiling libraries were constructed for two biological replicates, as previously described (Guydosh and Green, 2014; Young et al., 2015). Ribosome occupancy in open reading frames (ORFs) was highly correlated between the two biological replicates (Spearman’s R2 of 0.99), indicating consistency in the preparation methods. Analysis was generally performed on pooled data from the two replicates, with the exception of examples in some supplemental figures, as noted.
An average of ribosome-profiling footprints from the pooled dataset for all genes aligned at their stop codons in wild-type (WT) cells showed footprint density with a strong 3-nt periodicity in the ORF, corresponding to translation in a single reading frame (Figure 1A). We also noted an increase in the level of ribosome footprints in the 3′ UTR for the hcr1Δ strain, showing that loss of Hcr1 leads to a defect in ribosome recycling. The lack of 3-nt periodicity in the average 3′ UTR data implies that the ribosomes did not simply read past the stop codon in a single frame. Instead, the existence of footprints in all three reading frames implies the ribosomes reinitiate new translation in all three frames of the 3′ UTR after termination. These findings were consistent for both replicate experiments (Figure S1A). The magnitude of apparent 3′ UTR translation was similar to that observed in a tma64Δ/tma20Δ (tmaΔΔ) strain, known to exhibit a defect in 40S recycling, but less than that in a strain depleted of Rli1 (rli1-d), a known 60S recycling factor (Figure 1A, zoomed view). The tma64Δ/tma20Δ and rli1-d datasets were derived from pooled replicates that were previously published and shown to reproducibly exhibit elevated 3′ UTR read levels (Young et al., 2015, 2018).
Figure 1. HCR1 Deletion Results in Increased 3′ UTR Ribosome Occupancy.
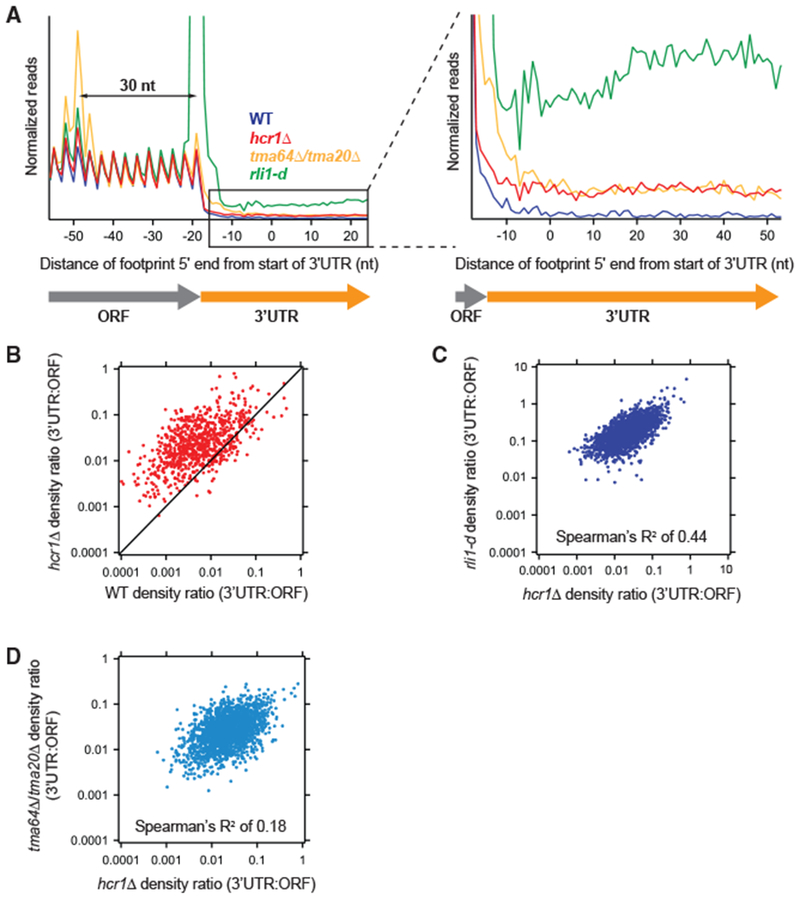
(A) Normalized average ribosome footprint occupancy (equally weighted by reads within the ORF) for all genes aligned at their stop codons. Footprints plotted by 5′ ends. (Inset) Magnified view of (A), showing increased 3′ UTR ribosome occupancy in the hcr1Δ strain relative to WT.
(B) Ratio of footprint densities in 3′ UTRs to their respective ORFs plotted for hcrlΔ. versus WT cells. Each point represents the data for one gene and shows increased 3′ UTR ribosome occupancy for most genes in the hcrlΔ. strain (most dots above the diagonal).
(C and D) Correlation analysis of ratio of footprint densities in 3′ UTRs to their respective ORFs plotted for hcrlΔ versus either rli1-d (C) or tma64Δ/20Δ. (D) cells, showing that transcripts with increased 3′ UTR ribosome occupancy are better correlated between hcr1Δ and rli1-d cells compared with the correlation between hcrlΔ. and tma64Δ/tma20Δ. cells.
Reads were pooled from the two hcrlΔ. replicates. Data for rli1-d and tma64Δ/20Δ. were taken from previous publications (described in Table S3). These datasets are created from pooled replicates that have each been shown previously to significantly differ from WT controls (Young et al., 2015, 2018). Footprint reads in (B)–(D) were quantitated by shifting them by 13 nt and then counting reads in ORFs and 3′ UTRs. Reads in the first and last 15 nt of ORFs were excluded and 3′ UTRs were extended by 25 nt to ensure collection of reads mapping partially in poly(A) tails.
We also examined the relative heights of ribosome footprint peaks at, or just upstream of, the stop codon for further insight into the mechanism of possible recycling failure. We have previously observed that failure to recycle the 60S subunit (as in a rli1-d strain) results in an enhanced peak at the stop codon, because of the presence of an unrecycled 80S ribosome, but that failure to recycle the 40S subunit (as in the tmaΔΔ strain) does not result in an increased 80S ribosome density peak because the method does not preserve any footprints generated by the 40S ribosome (Young et al., 2018). Instead, loss of 40S recycling results in a peak of enhanced ribosome density from a queued 80S ribosome positioned ~30 nt (approximately one footprint length) upstream of the stop codon (Young et al., 2018). In the hcr1Δ data, we did not observe strong evidence for either trend. We found the peak at the stop codon was no different from that in WT cells and that the queued ribosome peak was only slightly enhanced but still below that in the tmaΔΔ strain and even a small queued peak observable in the rli1-d strain. Therefore, it is not possible to determine, from this analysis, whether the recycling defect in the hcr1Δ cells was more likely the result of faulty 60S recycling or 40S recycling.
We, therefore, more closely quantitated the number of reads in individual 3′ UTRs in hcr1Δ cells to ask whether the distribution of footprints was more like that in rli1-d or tmaΔΔ cells. Given differences in the mechanism for 40S versus 80S reinitiation, it is likely that the distribution of footprints among 3′ UTRs should depend somewhat on the mechanism of recycling failure in a given strain. We first computed the ratio of 3′ UTR to ORF ribosome density for each gene and noted that this ratio was higher in the hcr1Δ strain versus WT (Figure 1B, most dots above the diagonal) and was observed for both replicates (Figure S1B). Analysis of these data with boxplots also showed that there was no statistical significance between replicates and that both differed significantly from WT (Figure S1C). As in the average analysis, the magnitude of the effect was less than that observed in the rli1-d cells (Figure S1D) but similar to that observed in the tmaΔΔ strains (Figure S1E). We then performed a correlation analysis to compare the 3′ UTR versus ORF ratio for each gene between different mutants (Figures 1C and 1D). The 3′ UTR:ORF ratios for hcr1Δ and rli1-d were correlated (Figure 1C; Spearman’s R2 of 0.44), showing that the gene-specific dependence of the recycling failure was similar in both strains. This result was consistent across two hcr1Δ. replicates (Figure S1F; Spearman’s R2 of 0.42 and 0.38, respectively). In contrast, the 3′ UTR:ORF ratio for hcr1Δ and tma64Δ/tma20Δ showed considerably less correlation (Figure 1D; Spearman’s R2 of 0.18) and was consistent for both hcr1Δ replicates (Figure S1G; Spearman’s R2 of 0.17 and 0.23, respectively). These data suggest that the variation in ribosome footprints among 3′ UTRs in hcr1Δ cells is more like that found in rli1-d cells versus tmaΔΔ cells. This implies the apparent recycling failure in the hcr1Δ strain is more consistent with a 60S recycling defect, rather than a 40S recycling defect.
Direct Detection of Epitope-Tagged 3′ UTR Translation Products in the hcr1Δ Strain Is Consistent with 80S Reinitiation, Not 40S Reinitiation
To confirm the 3′ UTR footprints resulted from translation reinitiation (as opposed to readthrough of stop codons) and to further characterize that mechanism, we assayed for peptide production from the 3′ UTR reporter gene constructs. In particular, we examined two previously described reporter genes, YDR524C-B and YMR122W-A, which were used to show 80S reinitiation in the rli1-d strain (Young et al., 2015) and two additional genes, RPL14A and RPS30B, which were used to assess 40S reinitiation in the tma64Δ/tma20Δ strain (Young et al., 2018).
We first examined the patterning of ribosome footprints across 3′ UTRs of the native genes that were chosen to create the reporters to test 80S reinitiation (YDR524C-B and YMR122W-A). Consistent with the evidence that Fieri promotes 60S recycling like Rli1, we found the pattern of footprint density in the hcr1Δ strain was remarkably similar to what was found in the rli1-d strain (Figure 2A), except that the overall magnitude was lower and the strong stop codon peaks previously observed in the rli1-d strain were absent (orange dots). Western analysis of these reporters in the hcr1Δ strain revealed 3′ UTR polypeptides of a similar size to those observed in the rli1-d strain (Figure 2B), consistent with the model in which 80S reinitiation occurs in both strains. In contrast, a strain depleted of the termination factor eRF1 (eRF1d; Wu etal., 2019) led to a longer protein product, consistent with the ribosome reading past the stop codon after shifting frames and adding a C-terminal extension to the protein encoded by the main ORF. These results show that loss of HCR1 leads to defects in ribosome recycling, rather than termination.
Figure 2. Detection of Epitope-Tagged 3′ UTR Translation Products Results from 80S Reinitiation in the hcr1Δ Strain.
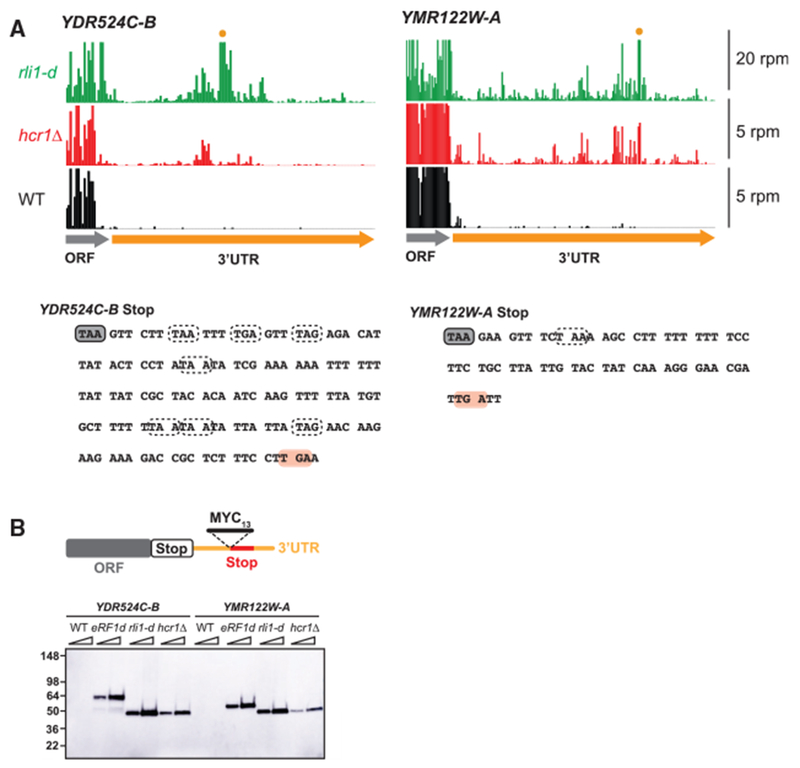
(A) Ribosome footprint profiles of the 3′ UTRs of genes YDR524C-B and YMR122W-A showing a similar pattern of footprint density between hcr1Δ and rli1-d cells (data from pooled replicates). The scale is zoomed in so reads in the ORF region are clipped. Two cases in which reads in the 3′ UTR for rli1-d exceed the scale because of a strong stop codon peaks are marked (orange dot). Note that reads from WT and hcr1Δ cells are scaled by 4x for better comparison. Sequences for each gene are shown below with stop codons in all frames indicated with dotted lines, except the stop codon corresponding to the schematic in (B) that the MYC13 tag is inserted in front of, which is indicated in pink.
(B) MYC13-tagged reporters that had previously been used to detect 80S reinitiation in the 3′ UTR of Rli1-depleted cells (Young et al., 2015) were inserted into the chromosome of hcr1Δ cells.
WT, eRF1d, rli1-d, and hcr1Δ cells carrying the MYC13- tagged reporters were grown to log-phase before reparation of whole cell extracts (WCEs). WCEs were prepared from five optical densities (ODs) of cells by trichloroacetic acid (TCA) extraction. WCEs were subjected to western analysis using an antibody against c-Myc. Two different amounts of protein were loaded for each sample (1 × and 2 ×). Migration of molecular weight (MW) standards (kDa) is indicated on the left. The 3′ UTR polypeptides in the hcr1Δ strain are of a similar size to those observed in the rli1-d strain and run at the expected weight (Young et al., 2015) for the tag plus a small amount of translated 3′ UTR sequence. In contrast, products from the eRF1-depleted strain run at a heavier weight because of inclusion of the entire protein encoded by the main ORF in the observed product.
The failure to recycle post-termination 40S subunits at the stop codon in the tmaΔΔ strains results, in some cases, in 40S subunits reinitiating translation by a process that shares some features with canonical 40S scanning in the 5′ UTR. One of these characteristics is increased 80S ribosome occupancy peaks at 3′ UTR AUG (start) codons, presumably because of a somewhat slow step between 60S subunit joining and subsequent elongation (Young et al., 2018). In contrast, such peaks are not observed if a 60S recycling defect is induced by depletion of Rli1 because reinitiation by the 80S ribosome does not depend on an AUG start codon (Young et al., 2015). If Hcr1/eIF3j promotes mRNA dissociation from the post-termination 40S subunit (i.e., 40S recycling), deletion of HCR1 should result in an increase in ribosome footprint peaks at AUG codons in the 3′ UTR. We, therefore, looked to see whether we could observe ribosome occupancy peaks at 3′ UTR AUG codons in the hcr1Δ deletion strain by averaging ribosome density around all 3′ UTR AUG codons. We found no peak when ribosome P sites were aligned at AUGs (Figure 3A, left) or at the near-cognate start codon UUG (Figure 3A, right) in the hcr1Δ strain, much like the rli1-d strain and in contrast to the tmaΔΔ strain. The lack of peaks at 3′ UTR AUGs and near-cognate UUG start codons was consistent across both hcr1Δ replicates (Figure S2). These results offer further evidence that Hcr1/eIF3j is not involved in dissociating mRNA from post-termination 40S subunits in vivo.
Figure 3. Footprint and Reporter Expression Data Do Not Support 3′ UTR Translation in the hcr1Δ Strain Resulting from an AUG-Dependent, 40S Reinitiation Mechanism.
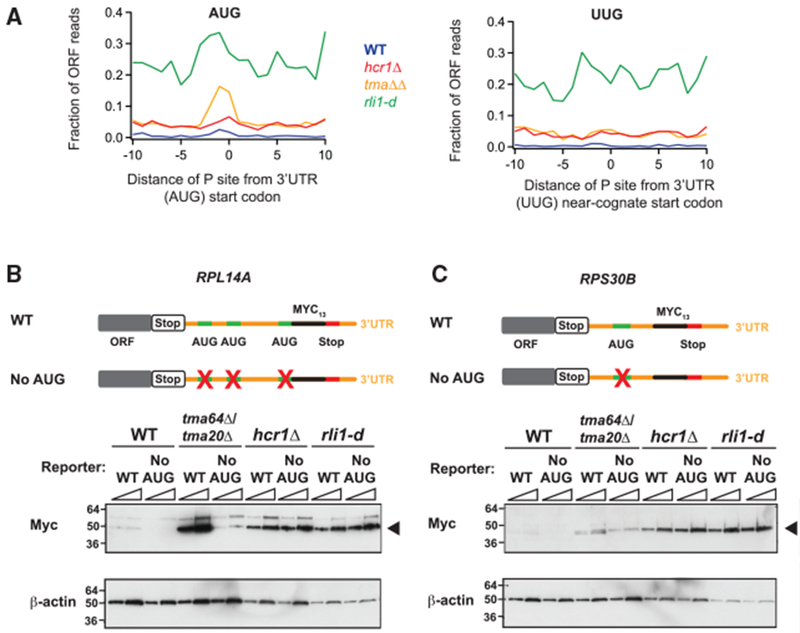
(A) Average fraction of ribosome occupancy (pooled replicate datasets) in a window surrounding 3′ UTR AUG codons (left) and 3′ UTR near-cognate UUG codons (right) normalized to ORF ribosome occupancy level (all frames included). Reads are shifted (12 nt), so peaks approximately correspond to ribosome P sites. A peak above background level is only evident for the tma64Δ/tma20Δ strain on AUG codons, as previously reported (Young et al., 2018), indicating 40S reinitiation.
(B and C) WCEs from WT, tma64Δ/tma20Δ, hcr1Δ, and rli1-d cells expressing reporters (RPL14A or RPS30B) were subjected to western analysis with antibodies against c-Myc (top blots) or β-actin (control, bottom blots). WCEs were prepared from five ODs of cells by TCA extraction. Two different amounts of protein were loaded for each sample (1× and 2×). Reporters include either a WT sequence or a “No AUG” version, to eliminate any 40S reinitiation. Products are observed (arrowheads) for the reinitiation product, in addition to a heavier readthrough product for RPL14A, attributable to high base-line readthrough for this gene, as reported previously (Young et al., 2018). Mutations of the AUG codons in the 3′ UTRs of the RPL14A (B) and RPS30B (C) reporters did not result in loss of the 3′ UTR reinitiation product in the hcr1Δ strain, supporting an 80S reinitiation model, in contrast to the tma64Δ/tma20Δ strain in which the product was AUG dependent.
See also Figure S2.
We then examined the western blots from reporter constructs (RPL14A and RPS30B genes) that we previously used to characterize 40S reinitiation (Young et al., 2018). We inserted a tag into the 3′ UTR sequences of these two genes at a position in-frame with an upstream AUG codon that could serve as a start codon for a 40S reinitiation event. We also examined versions of these reporters in which the putative AUG initiation codon had been mutated (“No AUG”) to an AAA codon to eliminate 40S reinitiation at that site (Figures 3B and 3C, schematics). The reporter that includes an AUG codon (WT sequence) would confirm 3′ UTR translation, whereas any loss of expression in the No AUG reporter would show that 3′ UTR translation originates from a 40S reinitiation event (i.e., requires a canonical start codon). The WT reporter sequences showed expression in all the mutant strains, confirming that footprints on these sequences are created by translating ribosomes that had reinitiated in the 3′ UTR (Figures 3B and 3C, westerns). In the tma64Δ/tma20Δ strain, mutation of the AUG codon to AAA reduced protein expression from the reporter, as previously observed and consistent with Tma20 and Tma64 acting as 40S recycling factors. In contrast, elimination of the AUG codon did not reduce protein expression in the rli1-d strain, consistent with loss of Rli1 leading to inhibition of 60S ribosome recycling and codon-independent 80S reinitiation, but not 40S reinitiation. In the hcr1Δ strain, the expression was similar from the WT and No AUG reporters, mirroring the result from the rli1-d strain, rather than the tmaΔΔ strain, and, therefore, is consistent with Hcr1 promoting 60S recycling, rather than 40S recycling.
Overexpression of RLI1 Suppresses the Recycling Defect Observed in the Absence of Hcr1/eIF3j
As a further test of whether Hcr1/eIF3j functions as a 60S recycling factor, we reasoned that overexpression of the known 60S recycling factor, Rli1, could compensate for the loss of Hcr1 activity. To test that hypothesis, we performed ribosome profiling in an hcr1Δ strain that expressed the RLI1 gene on a high-copy (hc) vector. Overexpression of Rli1 was confirmed by immunoblotting using C-terminally, FLAG-tagged versions of the RLI1 gene (RLI1-F) on a single copy and hc vectors (Figure S3). Compared with hcr1Δ cells expressing an empty control vector (YEplac195), we found that footprint reads in the 3′ UTR were sharply reduced in a strain expressing hc RLI1 (YEplac195-RLI1), thus showing that the phenotype could be suppressed (Figure 4A, zoomed view). As a positive control, we also found that hc HCR1 (Yep-j/HCR1-DS-U) could complement the knockout of HCR1 (Figure 4A, zoomed view). These effects were also apparent at the individual gene level, as shown for the example genes YDR524C-B and YMR122W-A (Figure 4B). These data, therefore, support the model that Rli1 and Hcr1 both promote dissociation of 60S subunits and are consistent with prior reports that Rli1/ABCE1 and Hcr1/eIF3j physically interact (Khoshnevis et al., 2010; Kispal et al., 2005). The fact that overexpression of Rli1 is capable of suppressing the hcr1Δ recycling defect also suggests that Rli1 acts genetically downstream of Hcr1 in the 60S recycling pathway.
Figure 4. Overexpression of RLI1 Suppresses the Recycling Defect Observed in the hcr1Δ Strain.
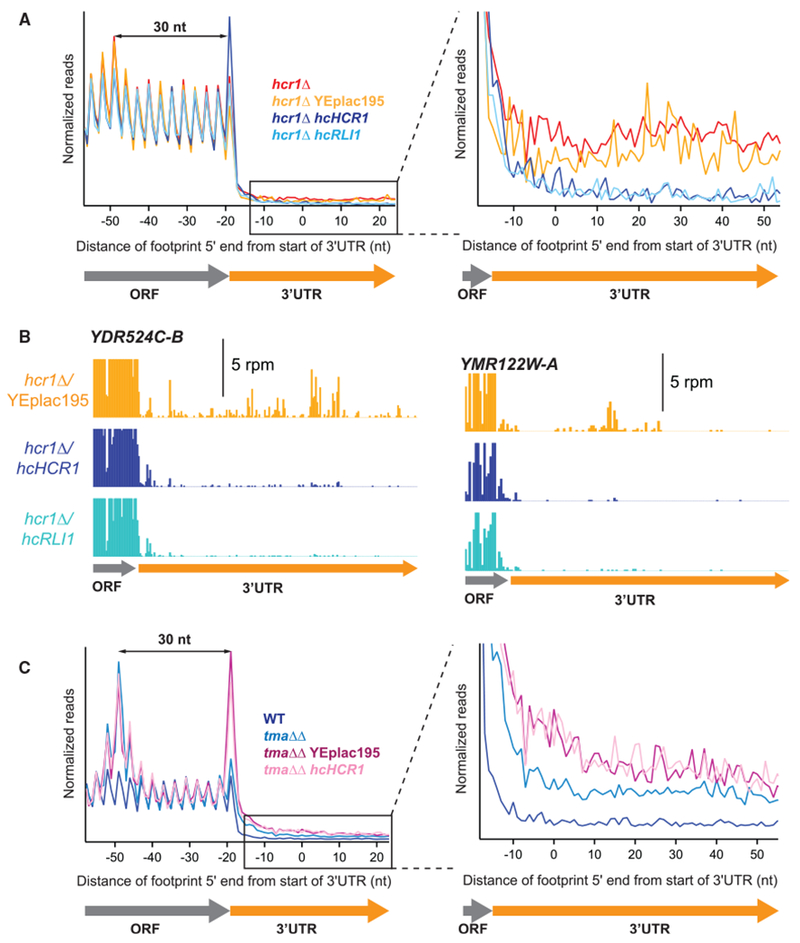
(A) Normalized average ribosome footprint occupancy (equally weighted by reads within the ORF) for all genes aligned at their stop codons. Footprints plotted by 5′ ends. (Inset) Magnified view of (A), showing complementation and suppression of 3′ UTR ribosome occupancy in the hcr1Δ hcHCR1 and hcr1Δ hcRLI1 strains, respectively.
(B) Individual examples of the elimination of 3′ UTR reads shown in the average in (A). Ribosome footprint profiles for the genes YDR524C-B and YMR122W-A showing loss of 3′ UTR footprint density in the hcr1Δ hcHCR1 and hcr1Δ hcRLI1 strains relative to hcr1Δ with an empty vector.
(C) Normalized average ribosome footprint occupancy as for (A) but showing the effect of overexpression of HCR1 on tma64Δ/tma20Δ. (Inset) Magnified view of (C), showing that 3′ UTR ribosome occupancy in the tma64Δ/tma20Δ strain is not suppressed by hcHCR1.
See also Figure S3.
In the alternative model, where Hcr1/eIF3j can promote recycling of 40S ribosomal subunits from mRNA in the cell, we might expect that hc HCR1 could compensate for loss of the known 40S recycling factors, Tma64, Tma22, and Tma20 (Young et al., 2018). We, therefore, performed ribosome profiling on the tma64Δ/tma20Δ strain transformed with the hc HCR1 plasmid or an empty-vector control. In the ribosome profiling data, we found that expression of our empty vector construct slightly increased 3′ UTR ribosome occupancy in the tma64Δ/tma20Δ strain above that of WT, an effect that is likely related to growing these cells in synthetic medium instead of YPD medium. Importantly, overexpression of HCR1 in this strain under these conditions did not reduce the level of 3′ UTR ribosome occupancy observed in the empty vector control (Figure 4C). Overexpression of Hcr1 cannot, therefore, complement loss of known 40S recycling factors, consistent with its in vivo role being a 60S recycling factor.
The N-Terminal Half of Hcr1 Is Essential for Its Function in Ribosome Recycling
Prior studies showed that the slow-growth phenotype of the hcr1Δ strain can be suppressed by expression of the NTD but not the C-terminal domain (CTD) of Hcr1/eIF3j (Elantak et al., 2010). In addition, the NTD is thought to be critical for its interaction with other subunits of eIF3 (Elantak et al., 2010; Nielsen et al., 2006), whereas the CTD has been proposed to be critical for 40S binding (Fraser etal., 2007). To gain insight into which regions are critical for the 60S recycling activity of Hcr1/eIF3j, we tested each half of the protein for its role in promoting ribosome recycling of 60S subunits. In particular, we performed ribosome profiling on hcr1Δ strains separately expressing the NTD and CTD of HCR1 from hc vectors (Figure 5A). The truncated proteins were expressed from high-copy vectors to overcome any defects in the stability that could arise from the shortened ORF. Expression levels have previously been shown to be approximately 3-fold greater than the physiological level of Hcr1 (Elantak etal., 2010).
Figure 5. The N-Terminal Half of Hcr1 Is Required for Its Function in Ribosome Recycling.
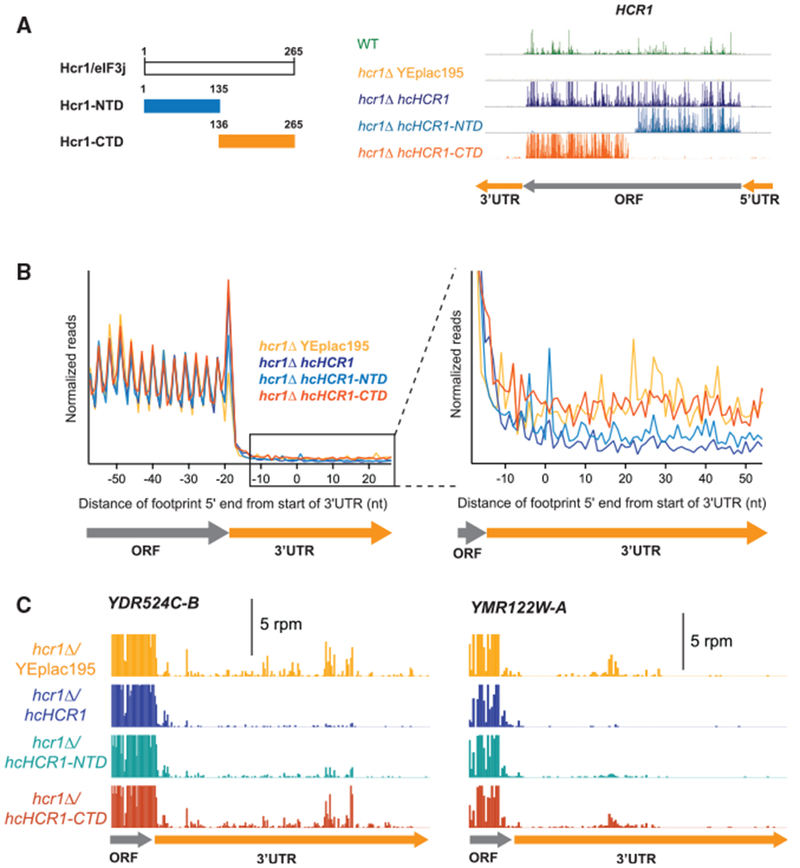
(A) Schematic of the Hcr1 protein showing the lengths (aa) of the HCR1-NTD and HCR1-CTD gene fragments (left), and the ribosome footprint profile of HCR1 confirming expression of HCR1-NTD and HCR1-CTD in the hcr1Δ strain (right).
(B) Normalized average ribosome footprint occupancy (equally weighted by reads within the ORF) for all genes aligned at their stop codons. Footprints plotted by 5′ ends. (Inset) Magnified view of (B) showing complementation of 3′ UTR ribosome occupancy in the hcr1Δ strain by HCR1-NTD but not HCR1-CTD.
(C) Individual examples of the elimination of 3′ UTR reads shown in the average in (B). Ribosome footprint profiles of the 3′ UTRs of genes YDR524C-B and YMR122W-A showing a loss of 3′ UTR footprint density in the hcr1Δ hcHCR1-NTD strain relative to the hcr1Δ YEplac195.
Based on clear changes in average 3′ UTR ribosome density in the strain expressing HCR1-NTD, we found the NTD was sufficient to partially suppress the recycling defect of hcr1Δ cells (Figure 5B), whereas the CTD had no apparent effect on the hcr1Δ recycling defect. It, how ever, remains a possibility that the CTD could rescue if the expression level were higher. Figure 5C shows examples of two individual genes, YDR524C-B and YMR122W-A, with partial suppression of 3′ UTR ribosome occupancy in the hcr1Δ strain when the HCR1-NTD is expressed or no suppression when the HCR1-CTD is expressed. These results indicate that the N-terminal 135 aa are important for 60S subunit dissociation.
eIF3 May Not Be Required for Hcr1/eIF3j Function in Ribosome Recycling
The above results support a role for Hcr1/eIF3j in 60S subunit dissociation during ribosome recycling. One possible mechanism by which Hcr1/eIF3j performs this role is by serving as a bridge between eIF3 and Rli1 to recruit Rli1 to the post-termination ribosome or, alternatively, Hcr1/eIF3j may activate the ATPase activity of Rli1 (Figure 6A). These two models are not necessarily mutually exclusive. To determine whether other subunits of eIF3 are required for Hcr1/eIF3j to promote 60S ribosome recycling, we performed ribosome profiling on yeast strains in which the interaction between Hcr1/eIF3j and the remainder of the eIF3 is weakened. We tested whether temperature-sensitive mutants of Tif32/eIF3a known to decrease Hcr1/eIF3j association with eIF3 at their non-permissive temperature (Chiu et al., 2010), tif32-H725P and tif32-R731I, and a hcr1Δ strain carrying a single-copy plasmid expressing the hcr1-NTA1 mutant that disrupts the interaction between Hcr1/eIF3j and Prt1/eIF3b (Elantak et al., 2010), produced a phenotype similar to the hcr1Δ strain (Figure 6A). The average footprint data for the eIF3a temperature-sensitive mutants grown at the non-permissive temperature of 36°C for 6 h did not show an increase in 3′ UTR ribosome occupancy relative to a control strain with WT TIF32 (Figure 6B). The hcr1Δ strain expressing the hcrl-NTA1 mutant also did not show an increase in 3′ UTR ribosome occupancy relative to a control strain expressing WT HCR1 (Figure 6B). The absence of an increase in 3′ UTR ribosome occupancy in these mutants suggests that the core of eIF3 may not be required for Hcr1/eIF3j to promote 60S subunit recycling. However, it cannot be ruled out that other interactions between Hcr1/eIF3j or Rli1 and the core of eIF3 exist.
Figure 6. eIF3 Does Not Appear to Be Required for Hcr1 Function in Ribosome Recycling.
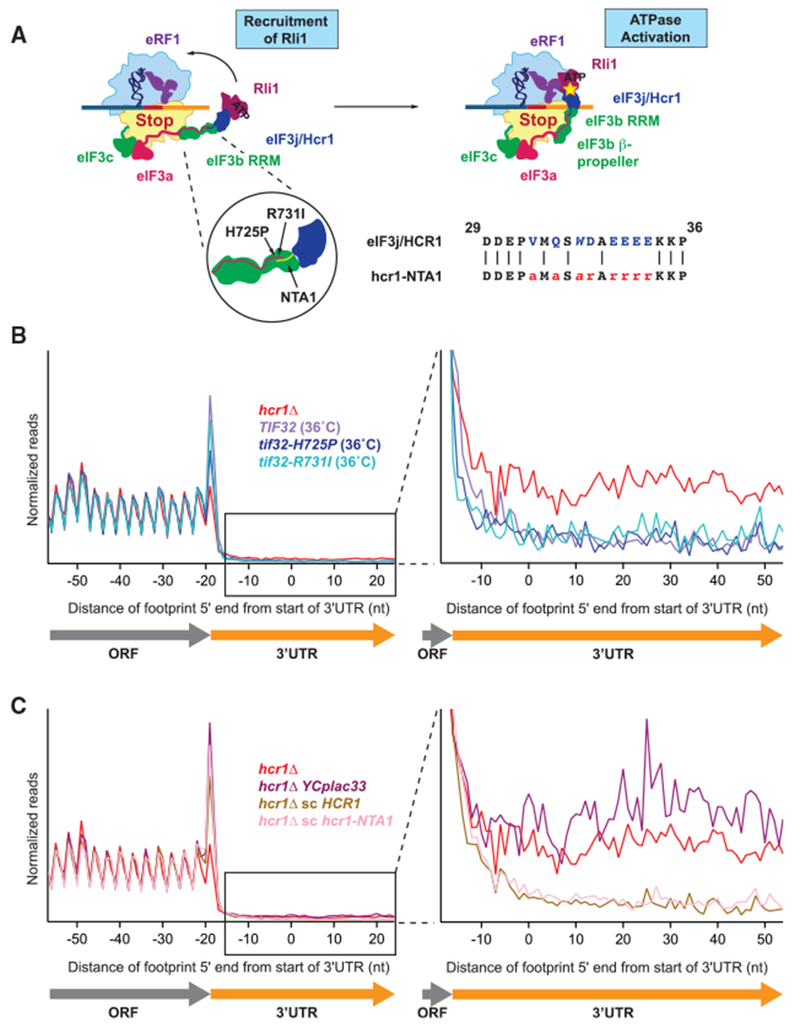
(A) Schematic diagram showing two potential models for Hcr1 function in ribosome recycling: by recruiting it to the ribosome (left), or by activating the ATPase activity (indicated by the star) of Rli1 (right). Tif32/eIF3a (shown in pink) has an extended C-terminal tail that interacts with both Prt1/eIF3b and Hcr1/eIF3j. The inset shows the location of the two tif32 ts alleles (tif32-H725P and -R731I) and the hcr1-NTA1 mutation that disrupts the interaction between Hcr1 and eIF3a or eIF3b, respectively. To the right of the insert is a short alignment showing the amino acids mutated in the hcr1-NTA1 mutant. We note that data shown in (B) and (C) suggest that these proposed interactions may not be strictly required for the ribosome recycling role of Rli1.
(B and C) Normalized average ribosome footprint occupancy (equally weighted by reads within the ORF) for all genes aligned at their stop codons. Footprints plotted by 5′ ends and replicate data pooled where available. (Inset) Magnified view of the 3′ UTR. There is no increase in 3′ UTR ribosome density in the two tif32 ts alleles grown at the non-permissive temperature of 36°C for 6 h (B), or for the hcr1-NTA1 mutant grown at 30°C (C), suggesting that the interaction between Hcr1 and the rest of the eIF3 complex may not be not required for ribosome recycling.
RLI1 Expression Is Increased in the Absence of HCR1
To gain insight on the biological pathways that rely on the apparent 60S recycling function of Hcr1/eIF3j, we performed mRNA sequencing (mRNA-seq) on two replicates of the hcr1Δ strain and identified significant changes in gene expression (Table S1) using DESEQ2 (Love et al., 2014). Taking genes where changes were highly significant (padj < 0.01), gene ontology analysis using PANTHER (Thomas et al., 2003) revealed several significant (pval < false discovery rate [FDR]) categories of enrichment, including sulfur metabolism, amino acid pathways, and translation (Figure 7A). Consistent with some of those categories, one of the top three most significantly (genes with padj < 10−14) upregulated genes in the cell was that encoding Rli1 itself, a known iron sulfer (Fe-S) protein (~2-fold increase). Similarly, we found the gene encoding Lto1, a protein known to specifically facilitate Fe-S maturation on Rli1, to be upregulated approximately 2-fold (padj < 10−5) (Paul et al., 2015; Zhai et al., 2014). Further analysis revealed significant upregulation of genes encoding key enzymes in the production of Fe-S sulfur clusters that are required for cell viability and Rli1 function, including: Isu2 (padj < 10−8), Dre2 (padj < 0.02), and Cfd1 (padj < 0.06) (Barthelme et al., 2011; Gerber et al., 2004; Stehling and Lill, 2013). We compared these data with that for depletion of Rli1 and found the pattern of gene expression changes was correlated (Spearman’s R2 = 0.27; Figure 7B, top plot) and that the same four Fe-S maturation factors noted above were upregulated in both strains (red dots in upper right quadrant). These trends were reflected in the ribosome footprinting data as well, as is generally expected for changes in underlying transcript levels, which rules out additional changes in translation efficiency. This finding suggests that the expression of RLI1 and genes required for the production of Fe-S clusters are regulated by a homeostatic feedback loop in response to defects in ribosome recycling (Figure 7C). One possible mechanism by which this could occur is nonstop decay, a process by which translation of poly(A) tails by ribosomes that reach the end of 3′ UTRs leads to mRNA degradation (Simms et al., 2017). However, the changes in gene expression noted above persisted in mRNA-seq data when HCR1 was knocked out in a ski2Δ background in which this pathway is less active (Spearman’s R2 = 0.45; Figure 7B, bottom plot), suggesting the existence of a novel sensor of 3′ UTR translation in the cell. To rule out the possibility that the upregulation of RLI1 results from a generalized stress response triggered by loss of HCR1, we examined the levels of RLI1 in previously published mRNA-seq datasets for many mutations and stress conditions (Table S2). The level of RLI1 is generally lower, and those for which it increased did not reach the level observed in the hcr1Δ strain, implying the increased expression was a specific response to translation of 3′ UTRs. Consistent with that, we noted a similar increase in RLI1 expression in the tmaΔΔ strain, which exhibited a similar level of 3′ UTR translation to that of the hcr1Δ strain (Figure S1E).
Figure 7. RLI1 Expression Is Increased in the hcr1 Deletion Strain.
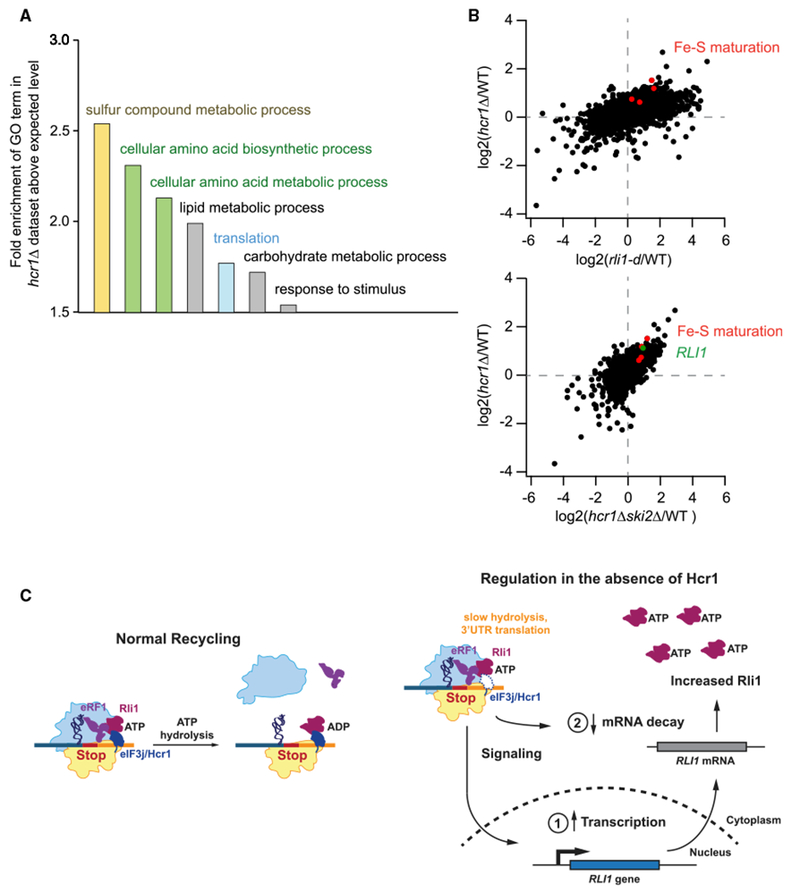
(A) Analysis of Gene Ontology (GO) terms in the dataset of genes that significantly change expression (assayed by mRNA-seq) in the hcr1Δ strain. GO terms that are most significantly enriched above the expected background level are shown.
(B) Correlation analysis of fold changes in mRNA-seq between hcr1Δ and rli1-d strains (top, Spearman’s R2 = 0.27) or between hcr1Δ strains in WT and ski2Δ backgrounds (bottom, Spearman’s R2 = 0.45) to assess the effect of inhibition of nonstop decay. Genes involved in iron sulfur (Fe-S) maturation that are significantly upregulated are identified in red (LTO1, ISU2, DRE2, and CFD1).
(C) (Left) Model for Hcr1 function in the cell. (Right) Schematic diagram showing that impairment of ribosome recycling results in increased expression of RLI1 either through increased transcription (1) or a decrease in decay of the RLI1 mRNA (2).
See also Table S2.
DISCUSSION
In this study, we employed a number of complementary approaches to ask how Hcr1/eIF3j is involved in the process of translation termination and ribosome recycling in the cell. We tested several proposed roles and established that Hcr1/eIF3j is an accessory factor for Rli1 that promotes 60S ribosomal subunit dissociation at stop codons in vivo (Figure 7C, left).
As expected for loss of a recycling factor, we observed translation of 3′ UTRs in the hcr1Δ strain by both an accumulation of ribosome footprints protecting 3′ UTR sequences and detection of tagged 3′ UTR reporter peptides. By investigating the nature of those footprints and tagged peptides, we found that translation of 3′ UTRs occurred because of an unrecycled 80S ribosome reinitiating translation in the 3′ UTR, rather than an unrecycled 40S reinitiating at a 3′ UTR AUG codon or an 80S ribosome failing to terminate and reading past the stop codon. In particular, we found that the gene specificity of 3′ UTR translation in the hcr1Δ strain was better correlated with that observed in a strain deficient in Rli1, in which 80S reinitiation is known to occur, rather than a tmaΔΔ strain in which 40S reinitiation occurs (Young et al., 2015, 2018). Consistent with that, we did not observe accumulation of ribosome footprints on AUG codons in 3′ UTRs, as was observed in the tmaΔΔ strain. In further support, epitope-tagged reporters showed that the hcr1Δ strain produced 3′ UTR translation products of the same size as those observed in an Rli1-depleted strain, but not longer than C-terminally extended proteins produced in an eRF1-depleted strain in which stop codon readthrough occurs. Moreover, reporter sets designed to distinguish 40S versus 80S reinitiation showed that reinitiation in the hcr1Δ strain did not require an AUG codon as it did in the tmaΔΔ strain, consistent with reinitiation by an 80S mechanism. In summary, the data point to a model in which Hcr1/eIF3j is an accessory factor that promotes recycling of the 60S subunit at stop codons (Figure 7C, left). Loss of Hcr1/eIF3j permits unrecycled 80S ribosomes to then reinitiate translation in 3′ UTRs, generating a similar (though less severe) phenotype to that observed in cells depleted of Rli1/ABCE1. Consistent with this interpretation, the recycling defect observed in hcr1Δ cells could be suppressed by overexpression of Rli1/ABCE1.
Our evidence does not support a role for Hcr1/eIF3j in dissociation of mRNA from the post-termination 40S subunit in the cell. Although such activity has been observed in reconstituted systems, it appears other processes in the cell prevent that from occurring in vivo, and the activity is instead provided by the known 40S recycling factors Tma64, Tma20, and Tma22. Given the importance of Hcr1/eIF3j in maintaining the fidelity of the start codon selection during canonical initiation in the 5′ UTR (Elantak et al., 2010; Nielsen et al., 2006), a process that occurs with mRNA bound to the 40S, it stands to reason that the presence of other ribosome-bound factors may place limits on the conformation adopted by Hcr1 (Sokabe and Fraser, 2014), preventing it from dissociating the mRNA in the cell.
Given prior evidence that Hcr1/eIF3j and Rli1/ABCE1 interact (Khoshnevis et al., 2010; Kispal et al., 2005) and our finding that overexpression of Rli1 can suppress the recycling defect in hcr1Δ cells, we favor a model in which Hcr1/eIF3j promotes the 60S recycling activity of Rli1/ABCE1. In one version of this model, Hcr1 could stimulate the ATPase activity of Rli1/ABCE1 or, in an alternative version, recruit Rli1/ABCE1 to the terminating ribosome. In either case, our data showing the N-terminal half of Hcr1/eIF3j is sufficient to prevent a 60S recycling defect suggests the N terminus of Hcr1 interacts with Rli1. It is conceivable that the Hcr1-Rli1 interaction requires that Hcr1 be simultaneously bound to the core subunits of eIF3 (Figure 6A). This view is consistent with data showing eIF3 can associate with the terminating ribosome or Rli1/ABCE1 (Beznosková et al., 2015; Dong et al., 2004), and data from reconstituted systems showing eIF3 can have 80S recycling activity (Pisarev et al., 2007, 2010). However, our finding that disruption of the interaction between Hcr1 and eIF3a and eIF3b did not reduce the ability of Hcr1 to stimulate 60S recycling suggests that an interaction with ribosome-bound eIF3 may not be critical. Alternatively, it is possible that the converse is true and that the interaction of Hcr1/eIF3j and Rli1/ABCE1 is important for recruiting eIF3 to the post-termination ribosome, perhaps to coordinate new rounds of translation in the 5′ UTR.
In addition, it has been suggested that Hcr1/eIF3j could have a role in termination, upstream of the 80S recycling step in the translation cycle. Although our data appear inconsistent with a direct role in regulating the termination factor eRF1, it is conceivable that Hcr1/eIF3j could promote recognition of a nonsense suppressor tRNA, a process that was shown to be enhanced in the absence of Hcr1 (Beznosková et al., 2013). One model to explain these data is that loss of Hcr1 decreases recruitment of Rli1 to the terminating ribosome. Rli1 has also been shown to increase the rate of termination (Shoemaker and Green, 2011). In the absence of Rli1, eRF1 may dissociate from the ribosome, thus increasing the likelihood of a suppressor tRNA decoding the stop codon.
Finally, it’s intriguing that loss of Hcr1 appears to increase mRNA levels of a number of genes involved in iron-sulfur cluster production and the gene for Rli1 itself, an Fe-S protein, along with Lto1, a protein known to be required for Fe-S maturation on Rli1 and having a human ortholog, ORAOV1, known to be upregulated in many human tumors (Paul et al., 2015; Zhai et al., 2014). This finding strongly hints at a homeostatic pathway whereby the cell regulates ribosome recycling activity via tuning mRNA levels of these key factors in response to a novel 3′ UTR translation sensor (Figure 7C, right). In this way, the hcr1Δ phenotype is likely lessened by the natural upregulation of RLI1. Such a sensor may be important for regulating controlled recycling failure in cases in which the translation of the non-coding 3′ UTR is advantageous to the cell or responding to oxidative stress, known to lessen the available Rli1/ ABCE1 in the cell. As increased reactive oxygen species are a well-known characteristic of cancer cells and have also been proposed to inhibit ribosome recycling in aged neurons (Alhebshi et al., 2012; Sudmant et al., 2018), a sensor for 3′ UTR translation could have an important role in human health.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicholas Guydosh (nicholas.guydosh@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Saccharomyces cerevisiae strains used in this study are derived from the BY4741 and W303 backgrounds. They were maintained on either YPD plates or SC-Ura plates for transformants. Cultures were grown at 30°C. Escherichia coli DH5α transformants were maintained on LB + 100 μg/ml Ampicillin plates. Cultures were grown at 37°C.
METHOD DETAILS
Yeast Strain Constructions
Yeast strains used in this study are listed in Table S4. The primers used for strain construction and verification are listed in Table S5.
The eRF1d and hcr1Δ MYC13-tagged strains were constructed by transformation of eRF1d and 6704 (hcr1Δ) with fragments containing the 3′UTR MYC13 insertions and HIS3 coding sequences PCR-amplified from the appropriate WT 3′UTR-MYC13 tagged strains and selecting transformants on SC-His. In all cases, PCR amplification from chromosomal DNA and DNA sequencing of the amplified fragment was used to confirm the correct integration events.
The hcr1Δ/ski2Δ double deletion strain (YDY572) was constructed in two steps. First, the hcr1Δ::kanMX4 deletion allele in 6704 was converted to hcr1Δ::natMX4, by transformation with BamHI/SpeI digested pAG25 (carrying natMX4) followed by selection on YPD containing 100 μg/mL ClonNat to produce strain YDY526. SKI2 was then deleted in strain YDY526 by transformation with the ski2Δ::kanMX4 allele PCR-amplified from yeast strain 5307 (ski2Δ), and selecting transformants on YPD containing 200 μg/mL G418, to produce strain YDY572. PCR amplification from chromosomal DNA was used to confirm correct integrations.
Plasmid Constructions
Plasmids used in this study are listed in Table S6. The primers used for plasmid construction are listed in Table S5. Plasmid pDY122 (sc HCR1) was constructed by PCR amplifying the HCR1 gene from yeast genomic DNA using HCR1-YXpf and HCR1-YXpr, digesting with SphI and XbaI and inserting the resulting restriction fragment between the SphI and XbaI sites of YCplac33. Plasmid pDY139 (sc hcr1-NTA1) was constructed using the Q5 Site-directed mutagenesis kit (NEB; E0554S) and primers HCR1-NTA1_SDMf and HCR1-NTA1_SDMr.
Ribosome footprint profiling
Ribosome profiling of BY4741, hcr1Δ, hcr1Δ YEplac195, hcr1Δ hc HCR1, hcr1Δ hc RLI1, hcr1Δ hc HCR1-NTD, and hcr1Δ hc HCR1-CTD was performed as previously described in (Young et al., 2018) and is based on the method originally described in (Ingolia et al., 2012). Ribosome profiling of TIF32, tif32-H725P, tif32-R731I, hcr1Δ YCplac33, hcr1Δ sc HCR1, and hcr1Δ sc hcr1-NTA1 was performed using a modified protocol based on the updated ribosome profiling method described in (McGlincy and Ingolia, 2017).
The ribosome profiling datasets generated for this paper, including number of reads and alignment statistics, are described in Table S7. Previously published ribosome profiling datasets used in this paper are listed in Table S3.
Growth of yeast for ribosome profiling
BY4741 and 6704 (hcr1Δ) were grown in YPD. 6704 (hcr1Δ) transformed with YEplac195 (empty vector), YEp-j/HCR1-DS-U (hc HCR1), pDY35 (hc RLI1), YEp-j/hcr1-NTD-U (Hcr1-NTD), YEp-j/hcr1-CTD-U (Hcr1-CTD), YCplac33 (empty vector), pDY122 (sc HCR1), and pDY139 (sc hcr1-NTA1), and YDY10 (tma64Δ/tma20Δ) transformed with YEplac195 (empty vector) and YEp-j/HCR1-DS-U (hc HCR1) were grown in SC-Ura. WLCY01 (TIF32), WLCY02 (tif32-H725P), and WLCY03 (tif32-R731I) were grown overnight in YPD at 30°C inoculated into fresh YPD media in the morning to an OD600 of 0.075 and grown at 30°C to an OD600 of between 0.2 and 0.4 before being grown at the non-permissive temperature of 36°C for 6 h. All samples were grown to a final OD600 of 0.6, fast filtered, and frozen in liquid nitrogen.
Preparation of ribosome footprint libraries
Cells were lysed with a Retsch Cryomill (Retsch 20.749.0001) in the presence of frozen lysis buffer (20 mM Tris [pH8], 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100) containing 0.1 mg/ml cycloheximide (Sigma; C7698). The milled cells were transferred to a 50 mL conical tube, thawed, and spun at 3000 g for 5 min at 4°C. The supernatant was transferred to 1.5 mL Eppendorf tubes and spun at full speed for 10 min in a refrigerated benchtop centrifuge at 4°C. The clarified supernatant was divided into aliquots before being snap frozen in liquid nitrogen and stored at −80°C.
Lysates were digested with 15U of RNase I (Ambion; AM2294) per OD for 1h at room temperature (25°C), loaded onto 10%–50% sucrose gradients, and spun at 40,000 rpm for 3h at 4°C in an ultracentrifuge. The sucrose gradients were fractionated using a Brandel Density Gradient Fractionation System and the isolated 80S peaks were snap frozen in liquid nitrogen and stored at −80°C. RNA was purified from the 80S fractions using hot phenol chloroform, and 25–34 nt ribosome footprints were size selected (see Table S8 for size standards) from a 15% TBE-Urea gel.
Original protocol (based on Ingolia et al., 2012)
The purified RNA fragments were dephosphorylated using PNK (NEB; M0201L) and ligated to universal miRNA cloning linker (NEB; S1315S) using truncated T4 RNA ligase 2 (NEB; M0242L). The ligated RNA footprints were size selected on a 10% TBE-Urea gel, and reverse transcribed using the RT primer NI-NI-9 (Table S8) and Superscript III (Invitrogen; 18080044). The reverse transcribed footprints were separated from unutilized RT primer on a 10% TBE-Urea gel and circularized using CircLigase ssDNA Ligase (Biosearch Technologies; CL4115K). Ribosomal RNA footprints were removed from the circularized libraries by oligonucleotide subtraction hybridization using Dynabeads MyOne Streptavidin C1 (Invitrogen; 65001) and a pool of DNA oligonucleotides that are the reverse complement of common rRNA contaminants (Table S8). The subtracted libraries were amplified by PCR using Phusion DNA Polymerase (ThermoFisher Scientific; F530L) to add unique 6 nt indexes for each library and common Illumina primer and flow cell binding regions. Library quality and concentration was assessed by BioAnalyzer using the High Sensitivity DNA Kit (Agilent; 5067-4626). Libraries were pooled, and sequencing was performed on an Illumina HiSeq2500 machine at the NIDDK Genomics Core at NIH (Bethesda, MD).
Modified protocol (based on McGlincy and Ingolia 2017)
The purified RNA fragments were dephosphorylated using PNK (NEB; M0201L) and ligated to pre-adenylated linkers (Table S8) containing a randomized 5 nt Unique Molecular Index (UMI) and a 5 nt sample barcode unique for each sample using truncated T4 RNA ligase 2 (K227Q) (NEB; M0351L). The linkers were pre-adenylated using a 5′ DNAadenylation kit (NEB; E2610L). Unligated linker was removed from the ligation reaction by addition of 5′ deadenylase (NEB; M0331S) and RecJ exonuclease (Biosearch Technologies; RJ411250). Ligated samples were pooled and purified using an oligo clean & concentrator kit (Zymo Research; D4060). The ligated RNA footprints were reverse transcribed using the RT primer NI-802 (Table S8) containing a randomized 2 nt UMI, and Superscript III (Invitrogen; 18080044). The reverse transcribed footprints were separated from unutilized RT primer on a 10% TBE-Urea gel and circularized using CircLigase ssDNA Ligase (Biosearch Technologies; CL4115K). Ribosomal RNA footprints were removed, and PCR was carried out as described above, adding a 6-nt (“Illumina”) barcode to be read during the indexing step of the sequencing run. Libraries were assessed and quantified by BioAnalyzer. Sequencing was performed on an Illumina HiSeq3000 machine at the NHLBI DNA Sequencing and Genomics Core at NIH (Bethesda, MD).
RNA-seq
Libraries for mRNA-Seq were prepared by the NHLBI DNA Sequencing and Genomics Core at NIH (Bethesda, MD) using Illumina TruSeq stranded ribozero kits (with 50-nt sequencing) to prepare libraries from total RNA prepared using SDS and hot acid phenol and chloroform.
Computational analysis
Read analysis and sequence alignment were performed as previously described (Young et al., 2018) for footprint samples prepared using the protocol based on (Ingolia et al., 2012). Briefly, fastq files (debarcoded by the core facility) were trimmed of their linkers using CUTADAPT. Contaminating tRNA and rRNA were removed with a BOWTIE alignment to an index of noncoding RNAs (https://downloads.yeastgenome.org/sequence/S288C_reference/rna/archive/rna_coding_R64-1-1_20110203.fasta.gz). The resulting fastq files were then aligned to the genome and splice junctions using BOWTIE, allowing up to 2 mismatches but no multimapping reads. Reads that failed to align were trimmed of poly(A) on their 3′ ends and remapped to ensure that ribosomes that had partially read into poly(A) tails were not eliminated.
A slightly modified protocol was used for libraries generated using the method of (McGlincy and Ingolia, 2017). Fastq files (debarcoded at the core facility according to their 6-nt Illumina barcodes) were trimmed of their linkers and separated according to their 5-nt internal sample barcode by using CUTADAPT. Contaminating tRNA and rRNA were removed with a BOWTIE alignment to the index of noncoding RNAs. Then, all PCR duplicates were removed using a simple python script to compare the UMIs. The resulting fastq files were then aligned to the genome and splice junctions using BOWTIE, allowing up to 2 mismatches but no multimapping reads. Reads that failed to align were trimmed of poly(A) on their 3′ ends and remapped to ensure that ribosomes that had partially read into poly(A) tails were not eliminated.
For mRNA-Seq data, all reads that failed to map to noncoding RNA were mapped to the genome and a list of splice junctions.
We used BOWTIE version 1.1.2 or 1.01 (Langmead et al., 2009) and included the parameters: - y -a -m 1 -best -strata. Other analysis software used Biopython 1.58 or 1.63. Unless noted otherwise, replicate samples were pooled for all analysis. In general, ORFs marked dubious or those that overlapped with other transcripts were ignored in the analysis. Annotations for 3′ UTRs (Nagalakshmi et al., 2008) that used coordinates from the R64-1-1 genome assembly were downloaded from Saccharomyces Genome Database Project. Python code for the three basic analyses (Gene average, gene quantitation, and position average plots) has been published previously (Young et al., 2018).
Quantitation of gene-wide occupancy (i.e., counting reads mapping to genes) was performed by normalizing density in units of reads per kilobase per million mapped reads (rpkm) by taking reads mapping to an annotated sequence and dividing by the gene length in kilobases. These analyses were used to generate Figures 1B–1D and 7B, and S1B–G. For footprint gene quantitation analyses only, 3′ UTRs were extended 25 nt downstream of their annotated endpoints in order to ensure all ribosomes that partially protected poly(A) sequences were accounted for. Reads that mapped in the first or last 15 nt of ORFs were left out to eliminate artifacts associated with initiating and terminating ribosomes. To be included in the analysis, a threshold of 5 rpkm was required of ORF reads and 0.5 rpkm for 3′UTR reads, unless noted otherwise. Footprint reads were shifted by 13 nt to correspond to the center nucleotide of the P site. mRNA-Seq reads were unshifted. Correlation analysis of 3′UTR:ORF ratios was done with a required threshold of 20 rpkm in ORFs and 0.5 rpkm in 3′UTRs.
To compute significance of gene expression changes in the mRNA-Seq data, the number of raw reads per gene were quantitated for two replicates each for WT and hcr1Δ cells and then compared with DESEQ2 (Key Resources Table) within Rstudio to obtain Padj values (Table S1). Data in plots for new or previously published mRNA-Seq data (Table S1) show, where applicable, the average of both replicates. GO analysis was performed by using the PANTHER (Key Resources Table) Overrepresentation Test with Fisher’s Exact test and a calculated FDR on the PANTHER GO-Slim Biological Process annotation dataset.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal antibody c-Myc (9E10) | Roche | 1167203001 |
| Mouse monoclonal antibody ANTI-FLAG® M2 | Sigma | F1804 |
| Mouse monoclonal antibody Anti-beta actin | Abcam | ab8224 |
| Bacterial and Virus Strains | ||
| DH5alpha | NEB | C2987H |
| Biological Samples | ||
| N/A | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | C7698 |
| RNase I | Ambion | AM2294 |
| PNK | NEB | M0201L |
| Universal miRNA cloning linker | NEB | S1315S |
| T4 RNA ligase 2, truncated | NEB | M0242L |
| Superscript III Reverse Transcriptase | Invitrogen | 18080044 |
| Dynabeads MyOne streptavidin C1 | Invitrogen | 65001 |
| Phusion DNA polymerase | ThermoFisher Scientific | F530L |
| T4 RNA ligase 2, truncated K227Q | NEB | M0351L |
| 5′ deadenylase | NEB | M0331S |
| RecJ exonuclease | Biosearch Technologies | RJ411250 |
| Critical Commercial Assays | ||
| Q5 Site-directed mutagenesis kit | NEB | E0554S |
| CircLigase ssDNA ligase kit | Biosearch Technologies | CL4115K |
| Agilent high sensitivity DNA kit | Agilent | 5067-4626 |
| 5′ DNA adenylation kit | NEB | E2610L |
| Oligo clean & concentrator kit | Zymo Research | D4060 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE124204 |
| Experimental Models: Cell Lines | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| A full list of yeast strains used in this study is provided in Table S4. | This paper | N/A |
| Oligonucleotides | ||
| A full list of oligonucleotides used in this study is provided in Tables S5 and S8. | This paper | N/A |
| Recombinant DNA | ||
| A full list of plasmids used in this study are provided in Tables S6. | This paper | N/A |
| Software and Algorithms | ||
| Igor Pro Version 7.00 | Wavemetrics | 15-500 |
| Position average and metagene and makegenelist | Young et al., 2018 | makeposavg makeavggene makegenelist |
| BOWTIE | Github | Langmead et al., 2009 |
| DESEQ2 | Bioconductor | Love et al., 2014 |
| PANTHER 13.1 | http://www.pantherdb.org/ | Thomas et al., 2003 |
| RStudio 1.1.456 | RStudio | 1.1.456 |
Gene-average plots (“metagene”) were constructed by excluding genes with features that were smaller than the window size (300 nt upstream of the stop codon and 100 nt downstream). In all cases, a minimal threshold of 5 rpkm was required from the ORF reads and genes were equally weighted according to reads in the coding sequence.
Position-average plots (“metacodon”) were similarly created by averaging together (with equal weight) reads in a window about every occurrence of a particular motif in a 3′UTR. Genes with no reads in the 3′ UTR were excluded from these analyses. Reads were normalized to the total reads present in the ORF and only genes exceeding a threshold of 5 rpkm were used. Reads were shifted by 12 nt so that the traces correspond to the first nucleotide in the P site.
Biochemical Techniques
Yeast strains were grown to log-phase before preparation of WCEs from 5 ODs of cells. Protein was prepared from WCEs by TCA extraction. Western analysis was conducted using mouse monoclonal antibodies against c-myc (Roche; 1167203001) FLAG (Sigma; F1804), and beta-actin (Abcam; ab8224). All Westerns were repeated at least twice on two independently grown cultures for each condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
For western blots, at least two individual replicates were performed to verify the result was reproducible. Graphs were made with Igor Pro 7 (Wavemetrics). Ribosome footprint data were processed with custom Python 2.7 scripts. To assess similarity in quantitated footprint data across genes, Spearman’s R2 statistic was used in Igor Pro. Boxplot analysis was performed and created by Rstudio ver. 1.1 to show statistical significance of the key result that 3′UTR translation increases in the hcr1Δ strain. To estimate significance in changes in mRNA-Seq data, DESEQ2 was used. For most analysis, replicate ribosome profiling runs were generally pooled. However, replicates are displayed separately to support key conclusions. GO analysis was performed with PANTHER.
DATA AND CODE AVAILABILITY
Raw and analyzed data have been deposited in the NCBI GEO database under the accession number GEO: GSE124204.
Supplementary Material
Highlights.
Hcr1/eIF3j is an in vivo accessory factor for Rlil-catalyzed 60S dissociation
Unrecycled 80S ribosomes in hcr1Δ cells can reinitiate translation in 3′ UTRs
The recycling defect in hcr1Δ can be suppressed by overexpression of Rli1/ABCE1
Perturbations in recycling result in higher levels of Rli1 via a feedback loop
ACKNOWLEDGMENTS
We thank Alan Hinnebusch, Jon Lorsch, Tom Dever, Petra Beznosková, and Leos Valásek for valuable feedback during this study. We thank Sezen Meydan Marks for helpful comments on the manuscript. We thank Theresa Marlin for help with submitting samples for mRNA sequencing. We thank Rachel Green for the eRF1d yeast strain and Alan Hinnebusch for yeast plasmids and strains. The work was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; DK075132 to N.R.G.).
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.111.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Celik et al. (2017); Cheng et al. (2019); Gerashchenko et al. (2012); Gietz and Sugino (1988); Goldstein and McCusker (1999); Solis et al. (2016); Thompson et al. (2016).
REFERENCES
- Alhebshi A, Sideri TC, Holland SL, and Avery SV (2012). The essential iron-sulfur protein Rli1 is an important target accounting for inhibition of cell growth by reactive oxygen species. Mol. Biol. Cell 23, 3582–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano K, Clayton J, Shalev A, and Hinnebusch AG (2000). A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 14, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylett CH, Boehringer D, Erzberger JP, Schaefer T, and Ban N (2015). Structure of a yeast 40S-eIF1-eIF1A-eIF3-eIF3j initiation complex. Nat. Struct. Mol. Biol. 22, 269–271. [DOI] [PubMed] [Google Scholar]
- Barthelme D, Dinkelaker S, Albers SV, Londei P, Ermler U, and Tampé R (2011). Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc. Natl. Acad. Sci. USA 108, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznosková P, Cuchalová L, Wagner S, Shoemaker CJ, Gunišová S, von der Haar T, and Valášek LS (2013). Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS Genet. 9, e1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznosková P, Wagner S, Jansen ME, von der Haar T, and áš LS (2015). Translation initiation factor eIF3 promotes programmed stop codon readthrough. Nucleic Acids Res. 43, 5099–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik A, He F, and Jacobson A (2017). NMD monitors translational fidelity 24/7. Curr. Genet. 63, 1007–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZQ, Dong J, Ishimura A, Daar I, Hinnebusch AG, and Dean M (2006). The essential vertebrate ABCE1 protein interacts with eukaryotic initiation factors. J. Biol. Chem. 281, 7452–7457. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Mugler CF, Keskin A, Hodapp S, Chan LY, Weis K, Mertins P, Regev A, Jovanovic M, and Brar GA (2019). Small and large ribosomal subunit deficiencies lead to distinct gene expression signatures that reflect cellular growth rate. Mol. Cell 73, 36–47. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WL, Wagner S, Herrmannová A, Burela L, Zhang F, Saini AK, Valásek L, and Hinnebusch AG (2010). The C-terminal region of eukaryotic translation initiation factor 3a (eIF3a) promotes mRNA recruitment, scanning, and, together with eIF3j and the eIF3b RNA recognition motif, selection of AUG start codons. Mol. Cell. Biol. 30, 4415–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Lai R, Nielsen K, Fekete CA, Qiu H, and Hinnebusch AG (2004). The essential ATP-binding cassette protein RLI1 functions in translation by promoting preinitiation complex assembly. J. Biol. Chem. 279, 4215742168. [DOI] [PubMed] [Google Scholar]
- Elantak L, Wagner S, Herrmannová A, Karásková M, Rutkai E, Lukavsky PJ, and Valásek L (2010). The indispensable N-terminal half of eIF3j/HCR1 cooperates with its structurally conserved binding partner eIF3b/PRT1-RRM and with eIF1A in stringent AUG selection. J. Mol. Biol. 396, 1097–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Stengel F, Pellarin R, Zhang S, Schaefer T, Aylett CHS, Cimermančič P, Boehringer D, Sali A, Aebersold R, and Ban N (2014). Molecular architecture of the 40S·eIF1·eIF3 translation initiation complex. Cell 158, 1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Lee JY, Mayeur GL, Bushell M, Doudna JA, and Hershey JW (2004). The j-subunit of human translation initiation factor eIF3 is required for the stable binding of eIF3 and its subcomplexesto 40 S ribosomal subunits in vitro. J. Biol. Chem. 279, 8946–8956. [DOI] [PubMed] [Google Scholar]
- Fraser CS, Berry KE, Hershey JW, and Doudna JA (2007). eIF3j is located in the decoding center of the human 40S ribosomal subunit. Mol. Cell 26,811–819. [DOI] [PubMed] [Google Scholar]
- Gerashchenko MV, Lobanov AV, and Gladyshev VN (2012). Genome wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 109, 17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber J, Neumann K, Prohl C, Mühlenhoff U, and Lill R (2004). The yeast scaffold proteins Isu1p and Isu2p are required inside mitochondria for maturation of cytosolic Fe/S proteins. Mol. Cell. Biol. 24, 4848–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Sugino A (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, and McCusker JH (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553. [DOI] [PubMed] [Google Scholar]
- Guydosh NR, and Green R (2014). Dom34 rescues ribosomes in 3′ untranslated regions. Cell 156, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CUT (2018). Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb. Perspect. Biol. 10, a032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG (2017). Structural Insights into the Mechanism of Scanning and Start Codon Recognition in Eukaryotic Translation Initiation. Trends Biochem. Sci. 42, 589–611. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnevis S, Gross T, Rotte C, Baierlein C, Ficner R, and Krebber H (2010). The iron-sulphur protein RNase L inhibitor functions in translation termination. EMBO Rep. 11, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispal G, Sipos K, Lange H, Fekete Z, Bedekovics T, Janáky T, Bassler J, Aguilar Netz DJ, Balk J, Rotte C, and Lill R (2005). Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J. 24, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT. (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Walker SE, Algire MA, Park EH, Hinnebusch AG, and Lorsch JR (2010). The 5′-7-methylguanosine cap on eukaryotic mRNAs serves bothto stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell 39, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, and Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KH, Valásek L, Sykes C, Jivotovskaya A, and Hinnebusch AG (2006).Interaction of the RNP1 motif in PRT1 with HCR1 promotes 40S binding of eukaryotic initiation factor 3 in yeast. Mol. Cell. Biol. 26, 2984–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul VD, Mühlenhoff U, Stümpfig M, Seebacher J, Kugler KG, Renicke C, Taxis C, Gavin AC, Pierik AJ, and Lill R (2015). The deca-GX3 proteins Yae1-Lto1 function as adaptors recruiting the ABC protein Rli1 for iron-sulfur cluster insertion. eLife 4, e08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Hellen CU, and Pestova TV (2007). Recycling of eukaryotic posttermination ribosomal complexes. Cell 131, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, and Pestova TV (2010). The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell 37, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, and Green R (2011). Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. USA 108, E1392–E1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Thomas EN, and Zaher HS (2017). Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip. Rev. RNA 8, e1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skabkin MA, Skabkina OV, Dhote V, Komar AA, Hellen CU, and Pestova TV (2010). Activities of Ligatin and MCT-1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev. 24, 1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe M, and Fraser CS (2014). Human eukaryotic initiation factor 2 (eIF2)-GTP-Met-tRNAi ternary complex and eIF3 stabilizethe 43 S preinitiation complex. J. Biol. Chem. 289, 31827–31836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, and Denic V (2016). Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol. Cell 63, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehling O, and Lill R (2013). The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect. Biol. 5, a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Lee H, Dominguez D, Heiman M, and Burge CB (2018). Widespread accumulation of ribosome-associated isolated 3′ UTRs in neuronal cell populations of the aging brain. Cell Rep. 25, 2447–2456. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, and Narechania A (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MK, Rojas-Duran MF, Gangaramani P, and Gilbert WV (2016). The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs. eLife 5, e11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valásek L, Trachsel H, Hasek J, and Ruis H (1998). Rpg1, the Saccharo myces cerevisiae homologue of the largest subunit of mammalian translation initiation factor 3, is required for translational activity. J. Biol. Chem. 273, 21253–21260. [DOI] [PubMed] [Google Scholar]
- Valásek L, Hasek J, Trachsel H, Imre EM, and Ruis H (1999). The Saccharomyces cerevisiae HCR1 gene encoding a homologue of the p35 subunit of human translation initiation factor 3 (eIF3) is a high copy suppressor of a temperature-sensitive mutation in the Rpg1p subunit of yeast eIF3. J. Biol. Chem. 274, 27567–27572. [DOI] [PubMed] [Google Scholar]
- Valásek L, Phan L, Schoenfeld LW, Valásková V, and Hinnebusch AG (2001). Related eIF3 subunits TIF32 and HCR1 interact with an RNA recognition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J. 20, 891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Zinshteyn B, Wehner KA, and Green R (2019). High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol. Cell 73, 959–970. e955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, and Green R (2015). Rli1/ABCE1 recycles terminating ribosomes and controls translation reinitiation in 3′UTRs in vivo. Cell 162, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DJ, Makeeva DS, Zhang F, Anisimova AS, Stolboushkina EA, Ghobakhlou F, Shatsky IN, Dmitriev SE, Hinnebusch AG, and Guydosh NR (2018). Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR Recycle posttermination 40s subunits in vivo. Mol. Cell 71, 761–774. e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai C, Li Y, Mascarenhas C, Lin Q, Li K, Vyrides I, Grant CM, and Panaretou B (2014). The function of ORAOV1/LTO1, a gene that is overexpressed frequently in cancer: essential roles in the function and biogenesis of the ribosome. Oncogene 33, 484–494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and analyzed data have been deposited in the NCBI GEO database under the accession number GEO: GSE124204.