

Introduction
Endovascular drug-eluting stents (DES) and more recently drug-eluting balloons, have and continue to revolutionize the treatment of atherosclerosis in coronary and peripheral vasculature. The key has been to identify biological agents that can counter the hyperplastic tissue responses to device expansion/implantation and to develop effective local delivery strategies that can maintain efficacious drug levels across the artery wall over the course of device effects (Figure 1). In the current article we review the various local drug delivery strategies implemented in approved and emerging endovascular devices, explain the mechanisms they employ for drug release and provide a mechanistic basis for relating drug release mode to arterial drug distribution and effect.
Figure 1.

Endovascular drug delivery occurs in the context of tissue response to mechanical forces. A.i Stent implantation mechanically injures the arterial wall and induces strut-proximal (K-L) and strut-distal (M-N) recirculation zones. A.ii This triggers four phases of vascular repair: Platelet-rich thrombus accumulates at areas of deep strut injury, accounting for most early luminal loss. Coincident with thrombus deposition, inflammatory cells, predominantly surface-adherent monocytes (SAM), are recruited to the injury site, both at and between the struts, before migrating into the neointima as tissue-infiltrating monocytes (TIM). Proliferation of smooth muscle cells and monocyte/macrophages within the neointima peaks at 7 days after implantation and continues above baseline levels for weeks thereafter. Collagen deposition in the adventitia and throughout the tunica media and neointima leads to arterial shrinkage, or remodeling, causing compression of the artery on the stent struts from without. B. Model predicted drug distribution surrounding the square strut depicted in A.i. Maximal concentrations (red) occur immediately beneath the strut, minimal concentrations (blue) occur between struts. C. Depiction of the processes governing arterial drug distribution and effect: transmural drug convection along a pressure gradient, diffusion driven by concentration gradients, drug binding to non specific binding tissue proteins and intracellular receptors. A.i and B adapted with permission from(30), A.ii adapted with permission from(31) and C reproduced with permission from(15).
Tissue pharmacokinetics can limit drug efficacy
Restenosis was recognized early as a clinical syndrome and a range of systemic pharmacological therapies showed promise in vitro but failed in animals or humans. It became apparent (Figure 1) that the lesions to be combated were focal not diffuse and that systemic delivery not only exposed the great mass of unaffected tissues but diluted the desired target effects. Local therapy, once embraced, required a different mindset than other administration modes for issues of targeting, penetration and retention now dominated over dosing. Indeed, administered dose pales in comparison to consideration of these other forces, necessitating not simply a change in perspective but obviating qualitative, inferential approaches. The complexity of the issues required experimental and computational modeling analyses which created a quantitative framework by which to evaluate temporal and spatial extents of drug distribution in the arterial wall and correlate patterns with successful tissue effects (Figure 2).
Figure 2.

Local drug efficacy can be limited by drug penetration and retention. (A) Enface microscopy of stent-based delivery of fluorescent mirrors stet geometry. (B) Maximal drug deposition occurs immediately beneath the strut though distal recirculation zones can also deposit high drug concentrations as evidence by fibrin deposition 28 days post implantation of Paclitaxel-eluting stents. (C) Uniform endovascular drug delivery only saturates the artery wall after sufficiently prolonged exposures. Thus, while the a 15-min luminal exposure to paclitaxel results in an intimal drug concentration >10-fold higher than in the lunminal infusate, the arterial media is devoid of drug. (D) Fractional drug retention by pre-saturated arteries scales with the tissue’s binding capacity for the drug (D). (A) Reproduced with permission from(32), (B) from(33) and (C) from(34). (D) adapted with permission from(8).
The challenge of optimizing local delivery rises dramatically given the innovations and complexity in modern stent designs, as tissue distribution after stent delivery tends to mirror stent-coating geometry (Figure 2A). Different designs differentially affect luminal washout relative to drug diffusion in the tissue, and can result in peak drug concentrations and toxicity immediately adjacent to stent struts (Figure 2B). The disparity between peak and trough drug concentrations can be reduced by altering the rate of drug elution(1) or through engineering of strut shape to increase the spatial extent and drug delivery role of strut-distal recirculation zones(2) but require a sophisticated perspective (Figure 1B). As reviewed below, stent-based delivery can be rendered more uniform by intentionally varying drug loading along the device surface or through deployment of drug loaded coating or particles to inter-strut zones(3–4).
Notably, even uniform endovascular drug delivery does not ensure adequate transmural drug distribution unless the duration of delivery is sufficiently long (Figure 2C). The minimal duration for adequate arterial distribution increases with endothelial integrity and resistance to drug absorption, and tends to be higher for larger drugs that exhibit lower tissue diffusivities and higher steric retardation, though drug charge and lipophilicity are also important factors(5–6).
Moreover, even when drug saturates the artery wall, drug clearance can render the therapy inefficacious. Thus for example, though heparin pharmacology is well suited to countering the acute and sustained vascular responses to angioplasty, balloon based and catheter based delivery of this drug were plagued by high rates of restenosis. This was attributed to the fast tissue clearance of heparin (Figure 2D) owing to its aqueous solubility(7). Namely hydrophilic molecules such as heparin have a greater propensity for distributing into blood than into tissue, and within the tissue tend to reside in extracellular spaces. Consequently, tissue uptake and clearance rates of soluble drugs tend to scale with their diffusion coefficient and can be prolonged through the use of high molecular weight analogs or charged analogs.
Conversely, lipophilic drugs exhibit a significant preference for tissue over blood (Figure 2C) and can passively enter cells and bind to high affinity pharmacological targets. Indeed it was not until compounds like Paclitaxel and Sirolimus analogs were used that we appreciated that there was also a need for local tissue binding to ensure adequate uptake and retention. These drugs, smaller than heparin and less soluble than many compound are more sustainably retained by arterial tissue (Figure 2D) in a manner that correlates with the expression of drug bindings sites in the tissue(6,8); these properties underlie their emergence as the drugs of choice for stent and balloon based delivery in coronary and peripheral vascular beds. Importantly, drug distribution and retention are not solely determined by the mode of delivery and the properties of the drug, and are also dependent on tissue morphology as different ultrastructural tissue elements can exhibit disparate resistance and capacitance for the same drug(9–10).
Mechanisms controlling local drug release
As described above, duration of drug exposure impacts the extent of arterial distribution and retention and by that token, the resulting biological effects of endovascular drug delivery. Thus, strategies for controlling drug dose and release kinetics have played an important role in endovascular drug delivery. In lieu of additional controlling factors, drug that is coated onto device surfaces tends to elute in a burst fashion, potentially overdosing the local tissue environment without sustaining efficacious levels. While many DES exhibit some level of burst release, a significant fraction of the drug load is typically eluted in a sustained manner. The sustained elution component can vary between DES, and are broadly classified as zero order if the rate of release is near constant until depletion of the drug load, or first order if the release rate declines with time.
Despite the abundance of drug eluting stent and balloons designs developed over the last three decades, only several distinct physical mechanisms have been utilized to control drug release (Figure 3). These include diffusion of drug molecules along concentration gradients (usually through a rate limiting polymer material layer), dissolution of the drug particles within a coating or within the tissue, dissolution and hydrolytic degradation of rate limiting polymer layers, use of ion exchange for ionized drugs and reversible binding to immobilized antibodies for chemokines. Physical mechanisms have the advantage of depending in a predictable manner on the properties of the drug and carriers and their spatial distribution in the device. Although physical mechanisms forces are predictable and amenable to computational modeling, tightly controlling drug and carrier properties and spatial distribution is more difficult than realized(11–12).
Figure 3.

Popular mechanisms for controlling endovascular drug release
Chemical mechanisms can also control drug release and delivery through the breaking of covalent bonds that connect drug molecules to a delivery vehicle, such as polymer chains, by either chemical or enzymatic degradation(13). These mechanisms have been underutilized due to the need to chemically modify drugs for grafting to the delivery vehicle. Such modifications result in new chemical entities called prodrugs, adding regulatory scrutiny to previously approved drugs.
Device based endovascular drug delivery strategies
Durable adherent coatings
First and second generation DES employed durable polymer coatings to adhere therapeutic drug loads to the stent and release it in diffusion controlled manner. Such diffusion controlled devices are broadly classified as matrix type or reservoir type (Figure 4 A–B). In matrix type devices, such as the Paclitaxel eluting Taxus stent and the Zotarolimus eluting Endeavor stent, drug is released from the polymer matrix directly into the environment. For this reason matrix devices are also referred to as monolithic devices. By contrast, reservoir type devices such as the Sirolimus eluting Cypher stent and the Everolimus eluting Xience stent utilize at least two distinct layers, an internal drug reservoir and a thin external polymer layer, designed to limit the rate of drug elution from the internal drug reservoir. Hybrid multilayer coating designs (Figure 4 C) have also been developed as a means of providing greater control over the release of one or more drugs, while optimizing acute and sustained biocompatibility(13)
Figure 4.

Schematic side views of adherent coatings: monolithic (A), reservoir (B) and hybrid reservoir/monolith (C). The Local drug efficacy can be limited by drug penetration and retention
In theory, drug release from durable coated DES designs can be predictably controlled based on the designed thicknesses of the polymer and drug layers and the concepts of diffusion and dissolution. In reality though, multiple rounds of spray coating can lead to considerable mixing between sequential layers(12) and to enrichment of drug near the surface(14). Thus, while the Cypher stent was designed as a reservoir type DES(13), imaging reveals drug presence in the external rate limiting polymer layer, resulting in monolithic type diffusion controlled release(15). As spray coating is traditionally applied to stents that are mounted to a spinning mandrel, this technique also limits control over spatial drug coating design. Thus, it is difficult to optimize inter-strut drug delivery via enrichment of luminal and distal coating, or to coat different drugs on the abluminal and luminal aspects of stents for inhibiting restenosis while promoting endothelialization. More sophisticated inkjet and microdrop injection technologies can provide better controlled spatial coating design at the micro and macro levels(16). In addition, layer-by-layer assembly of polyelectrolytes is a promising strategy for high fidelity engineering of device surfaces and ion-exchange controlled release of charged biological agents such as DNA and small interfering RNA(13).
Remarkably, application of the spray iterations to static flat metal sheets can overcome the limitations seen with spray application to spinning stents. Such a procedure allows for shorter spray run times but longer dry times and therefore minimal mixing between successive layers. Moreover, en face spraying onto a static sheet is ideally suited for high fidelity abluminal coating and can be programmed to provide controlled heterogeneous coatings. We recently showed that en face spray coating of flat cobalt-chromium sheets with tightly controlled concentrations of the drug Ridaforolimus and two polymer types provided monolithic and hybrid reservoir/monolithic type coatings. Drug release from these DES was well predicted by a diffusion-based model that accounts for the predetermined layer thicknesses and compositions and a composition-dependent diffusion coefficient(17). Such a combination of tightly controlled spray coating and computational modeling provides for truly customizable release kinetics.
Biodegradable adherent coatings
Concern for persistent adverse responses to durable polymeric coatings prompted the development of more biocompatible durable polymeric materials or bioerodible coatings that are absorbed over the course of stent implantation(18). Some designs sought to minimize polymer-tissue contact by incorporating polymer/drug formulations into sculpted surface inlays in the form of grooves and holes available on the struts using microdispensers(13,18). The grooves and holes vary in size and shape and location relative to the struts and can incorporate the polymer in monolithic, reservoir, or hybrid reservoir/monolithic design. On the contrary, others attributed the persistence of safety concerns with durable coated metallic stents to the persistence of the metallic scaffold long after acute responses and drug delivery have ceased, providing an impetus for the use of biodegradable polymeric scaffolds as vascular mechanical supports and drug delivery platforms(18).
Despite the abundance of biodegradable DES and scaffold designs, the optimum temporal balancing of erosion and drug release still awaits full definition as most erodible scaffolds and stents with erodible coatings release their entire drug load prior to erosion(Figure 5), maintaining diffusion-limited release kinetics similar to durable coated DES(15) but also necessarily prolonging the duration of any adverse polymer effects. Indeed, due to the notion that restenosis inhibition by sirolimus analogs requires sustained delivery, the duration of drug release from biodegradable coatings and scaffolds is typically comparable to that of first generation Cypher stents (Figure 5). This misguided focus on release kinetics as a driver of effect, rather than tissue retention has restricted the choice of DES coating materials. In particular, natural bioerodible polymers that permeabilize quickly during hydrolysis and absorption have been shunned in favor of synthetic polymers that are absorbed over the course of several months(18), e.g., poly lactic acid, poly glycolic acid, or copolymer or other variations thereof (Figure 5). These synthetic materials may produce local irritation due to the release of acidic degradation products and can delay healing and transiently place the artery at increased risk of adverse reaction.(19)
Figure 5.

Durations of drug release and coating absorption for a range of Sirolmus analog eluting stents. Abbreviations: BIO9, Biolimus A9; CL, Caprolactone; COR, Corolimus; EVR, Everolimus; NOV, novolimus; SIR, Sirolimus; ZOT, Zotarolimus. Data compiled from(4,18,20,35–36).
We hypothesized that naturally derived coating compositions which degrade rapidly can deliver controlled volumes of drug without loss of biological effect and at reduced periods of tissue vulnerability. To examine this hypothesis, cobalt chromium stents were conformally coated with a novel cross-linked omega-3 fatty acid (O3FA) based coating(20) that is 85% absorbed (Figure 6A) and elutes 97% of its Sirolimus analog (Corolimus) load within 8 days of implantation (Figure 6B). Evaluation in pig coronary arteries revealed sustained efficacious drug levels that were similar to those achieved by slow eluting durable coated Sirolimus-eluting stents (Figure 6C) and resulted in superior efficacy and more benign tissue response. Computational modeling confirmed that Corolimus distributes in arterial tissue with the same diffusion and binding constants as Sirolimus and explained that its sustained retention after fast elution from the O3FA DES was facilitated by high affinity binding of drug to intracellular FKBP12(Figure 6C). The same computational model predicts that Corolimus saturates>65% up to 8 days post implantation and that the dissociation of the drug-FKBP12 complex linearly tracks with coating absorption (Figure 6D). Thus, late coating absorption and drug inhibitory effects decline in a linearly parallel manner, suggesting that the former drives the latter, and representing a new paradigm in stent based drug delivery. Clinical data suggest that late lumen loss stabilizes already after 6 months in the presence of O3FA(Cinatra™ DES) and continues to rise with the Cypher stent(20), supporting the clinical validity of the O3FA DES product design approach.
Figure 6.

Receptor mediated sustained tissue retention. In vivo coating absorption (A) and local tissue pharmacokinetics (B–D) of a fast eluting O3FA DES. The O3FA coating quickly elutes off the stent with low accumulation in arterial tissue (A). This results in faster release of Corolimus from O3FA DES (red) compared to Sirolimus release from Cypher Select Plus DES (green) in the same cohort (B). Yet in vivo tissue contents (symbols) for both DES are comparable tissue levels up to 56 days post implantation and closely predicted by a computational model that accounts for high affinity drug binding to FKBP12 (lines). The inset illustrates that modeling that does not account for high affinity drug binding to FKBP12 (dashes) predicts unrealistically fast tissue clearance.(D) Model predicted FKBP12 saturation by Corolimus (red line) correlates linearly with in vivo coating absorption (insert). Panels A–C reproduced with permission from(20).
Thus, tissue retention of Sirolimus analogs can be achieved even with fast elution kinetics following rapid coating erosion, with computational modeling identifying binding to tissue receptors as the mechanism of prolonged retention(20). The composition of drug-eluting coatings can then be designed for optimal biocompatibility and bioabsorption rather than predominantly for sustained drug elution kinetics. This paradigm shift creates an opportunity for the use of a range of biocompatible materials that may have not otherwise been considered.
Deployable coatings
The above highlighted DES designs employ durable and erodible coatings as stent adherent conduits for drug elution. By contrast, the absorbable PLGA coating of the MiStent Sirolimus-eluting stent is designed to soften and spread into the neointima by tissue remodeling forces (Figure 7A) while carrying along its microcrystalline Sirolimus load(3). These intra-tissue polymer encapsulated microcrystalline drug particles act as sustained drug delivery micro-depots (micro-reservoirs) that ensure high tissue contents long after the stent has reverted to the bare metal state (Figure 7B). By the same token, during neointimal growth, the area of drug delivery increases dynamically beyond the immediate vicinity of the struts. Computational modeling demonstrated that spread of coating to interstrut regions improves drug delivery to these areas and decreases gradients in drug distribution (Figure 7C). At small migration distances, drug deposition near the struts may actually increase as the greater surface area of elution can compensate for the decline in strut adherent coating. However, beyond a threshold distance (~100 µum) migration can appreciably decrease near strut drug deposition (Figure 7C). Peak-trough levels 150 µm into the media were predicted to decline at a near constant rate of 1.7 ng/mg per 100 µm coating migration – reaching unity as the coating migrates ~0.5mm distal to the strut(21).
Figure 7.

Enhanced drug delivery capabilities from stents coated with absorbable polymer and crystalline drug. (A) Histopathology at 30d identified coating as the ‘negative image’ of space occupying mass (arrows) between struts (S). (B) Drug release is complete within 45–60 days after stent implantation, yet tissue levels remain at near peak levels long after.(C,D) Computational modeling predicted drug (C) and receptor binding (D) distribution patterns around a strut pair that is fully coated (conformal) or where the bottom coating deployed 35, 100, or 400 µm into interstrut zones. Figures A–D reproduced with permission from(3).
Whether or not the more homogeneous drug delivery provided by tissue deployed coatings comes at the expense of suboptimal dosing cannot be determined from total drug levels and requires insight into the concentration of therapeutically active drug that is bound to its intracellular target. Whereas labeling of a drug may enable tracking of total drug concentration gradients, resolution of the concentration of Sirolimus bound to FKBP12 was only possible through computational modeling (Figure 7D). Such simulations predicted ≥80% saturation of FKBP12 by drug, even in arteries in which coating remains fully conformal to the stent. However, whereas conformal stents bind 15% more receptors near than between struts, deployed coatings saturate 94–96% of receptors throughout the neointima (Figure 7D).
The novel insights provided by these studies are of relevance to the expanding class of endovascular delivery devices that deliver drug in microcrystalline form, including drug coated balloons, and nanopolymer coated(4) stents and balloons. Quantitative experimental and computational analyses of these devices have yet to be published.
Polymer free coated stents
Some metallic DES designs dispense with polymer coatings altogether, offering the potential advantages of avoiding long-term polymer material-induced hypersensitivity and potential thrombogenicity that necessitate long term dual antiplatelet therapy, and alleviating concerns for coating peeling and cracking(4,18,22). First generation polymer free stents (PFS) were dip coated in ethanolic Paclitaxel and did not meet the predetermined primary clinical end point of target vessel failure and the secondary end point of binary restenosis(13). Preclinical evaluation of these dip coated stents showed that most of the drug loss occurred before stent expansion and deployment(13), and prompted the development of several different techniques for controlled drug elution from stents in the absence of a polymer(23) (Figure 8A):
Direct attachment of drug to the stent surface using covalent bonding, or crystallization–chemical precipitation on the stent surface.
Dissolving of the drug in a nonpolymeric biodegradable carrier on the stent surface.
Impregnation of the drug into surface micropores or nanopores formed by mechanical or electrochemical treatment of metallic surfaces, or on specialized inorganic surface coating.
Microinjection of the pure drug or formulated with nonpolymeric excipients into sculpted surface inlays or slots
Figure 8.
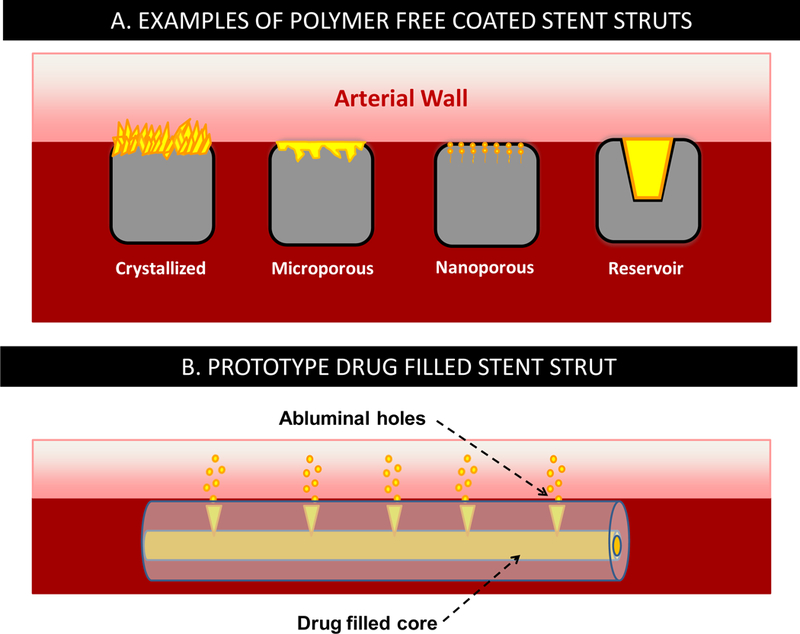
Schematics of various polymer free drug-eluting stent types.
Similar to drug coated balloons, second generation PFS rely on non-diffusive mechanisms, most commonly slow dissolution of sparsely soluble crystalline drug forms, to compensate for the absence of the tempering effects of a polymeric layer. This has again focused attention to hydrophobic drugs, mostly on Paclitaxel and Sirolimus analogs due to their well characterized antirestenotic pharmacological effects, favorable tissue distribution and retention properties Clinical experience with second generation PFS has been promising, with several coronary PFS receiving CE mark and the Paclitaxel-eluting peripheral PFS receiving FDA approval. Due to the differences in the eluted drugs, stent geometries, strut thickness and surface morphologies, as well as the paucity of in vivo drug release data, the optimal kinetic of drug elution from PFS has yet to be defined.
Preliminary insight into the dependence of PFS efficacy on Sirolimus analog release kinetics can be gained by contrasting the available tissue delivery profiles of these devices with efficacious bioresorbable coated and durable coated Sirolimus DES (Figure 9). For example, the Yukon PFS elutes 66.4% and 85.5% of Sirolimus loaded into its microporous surface within 7 and 21 days of in vitro deployment in a buffer solution(24–25). During the first week of implantation, tissue concentrations provided by Yukon PFS exceed those provide by slow eluting Cypher stents, but this is reversed by 10 days following a >95% decline in tissue levels for the PFS (Figure 9). Incorporation of a biodegradable polymer along with Sirolimus into the microporous surface results in the Yukon Choice stent, which releases Sirolimus at a near constant rate during 28 day deployments in PBS, and sustains high tissue levels up to 20 days post implantation (Figure 9). Clinical late lumen loss in denovo coronary lesions scaled with the duration of tissue retention, as Yukon PFS but not Yukon Choice was inferior to durable coated Cypher stents(26).
Figure 9.
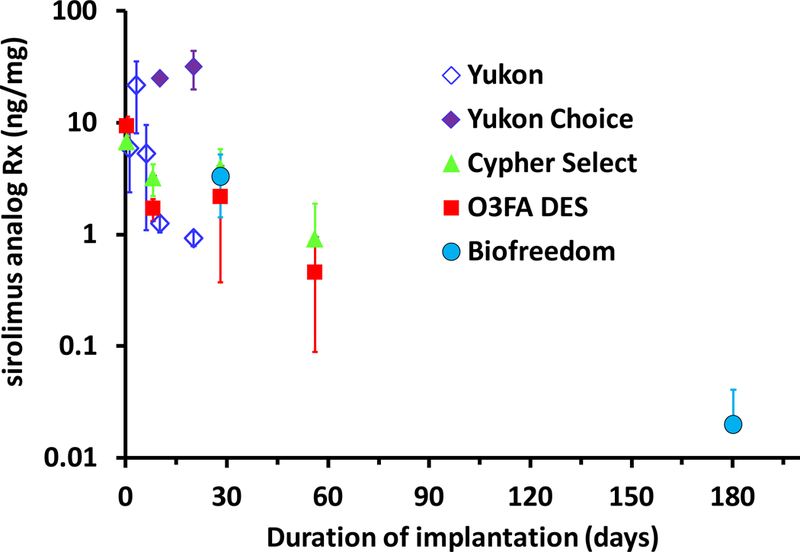
Tissue concentrations achieved by Sirolimus analog-eluting PFS, durable and bioerodible coated DES(20,24–25,27). O3FA DES and Cypher Select data are from Figure 6C
While these clinical findings also scale with the relative duration of Sirolimus release from the two microporous stents, the predictive power of local tissue concentrations over released dose becomes apparent for the Biofreedom PFS that elute the extremely lipophilic Sirolimus analog Biolimus A9. Thus, although Biofreedom PFS have been reported to release ~90% and ~99.9% by 2 and 28 days(23,27), the associated tissue concentration in porcine arteries at 28 days(27) is comparable to that sustained by slower eluting durable coated DES (Figure 9). Similar to our findings with the fast eluting O3FA coated DES(20), Biofreedom PFS exhibited reduced inflammation and wall thickening compared to the slower eluting durable coated Cypher stents at 28 and 180 days(27). Moreover, randomized clinical comparisons of Biofreedom PFS with Taxus stents found non-inferior late lumen loss at 1 year (0.17 vs 0.35 mm), significantly lower major cardiac event rates (6.8 vs 10.0%), and target lesion revascularization (3.4 vs 6.7%) at 2 years(22). In light of the sustained tissue concentration achieved by Biofreedom PFS, such positive preclinical and clinical results for a stent that releases ~90% of its drug load in a matter of 2 days are likely attributable to the pharmacokinetic profile of the eluted drug. In particular, Tada et al have highlighted that the extreme lipophilicity of Biolimus A9 relative to other Sirolimus analogs endows it with more favorable tissue absorption and cell uptake(27). This plausible assertion should be investigated further using animal and computational model.
Drug filled stents
Drug-Filled Stent (DFS) are a new class of a polymer free DES technology currently being developed (Medtronic). In this design, the stent struts are a tubular configuration, with a hollow core; small access holes connect the inner core to the abluminal surface. Drug is loaded in the inner core and diffuses out through the holes into the vessel wall (Figure 8B). Initial 90 day pig studies with a prototype Sirolimus-eluting DFS exhibited comparable drug release rates and tissue concentrations to the slow eluting durable coated Resolute stent and effective suppression of neointimal hyperplasia at 28 days compared to bare metal stents, with minimal inflammation through 90 days(28). Follow up analysis of the data revealed that Sirolimus release kinetics were biexponential, speaking to the existence of an easily available and releasable pool of drug that elutes within the first day (first time point) following arterial implantation, plus a pool of sustained-eluting drug with a half-life of 28–32 days(29). The pool of sustained-eluting drug and its half-life of release both decreased with increasing hole-size in a manner that cannot be explained by Fickian diffusion(29). These results suggest that drug release kinetics from DFS is predominantly dissolution controlled and can be modulating by altering hole-size. Modeling analysis of this system is underway.
Summary/Discussion
Though endovascular drug delivery is now a mature field, the last few years have heralded a resurgence of innovation aimed at improving performance and reducing cost. Every tenet of first generation DES has been reexamined and questioned, from the need for a persistent metallic scaffold, through the need for an adherent polymer coating as a drug release reservoir, to the need of sustaining drug release. While some call for the abolition of the polymer coating, others have expanded its role into a deployable carrier of crystalline drug. We attempted to review the mechanistic basis underlying these technological innovations and to illustrate the critical role that quantitative experiments and computational modeling have played so far. These techniques are critical for understanding the interplay between device design, drug release, tissue distribution and effect and offer powerful framework for further waves of innovation.
Key Points.
As the effects of combination drug-eluting devices are multi-factorial, designs abound and there is still room for innovation
Drug concentrations in tissues are predictive of effect and are not synonymous with delivered dose
Thus, while promising drug pharmacology is requisite, achieving adequate drug distribution and retention is key
Understanding and computationally modeling the determinants of drug release kinetics and tissue distribution can help further drive innovation at reduced cost
Acknowledgments
Funding: This study was supported in part by grants from the NIH (RO1 GM-49039) to ERE.
Footnotes
Disclosure: ART is an employee of CBSET, a nonforprofit contract research organization. ERE is a paid consultant to Atrium/Maquet, MiCell Medical Technologies and 480 Medical, and has sponsored research agreements with Boston Scientific and Medtronic.
References
- 1.Balakrishnan B, Dooley JF, Kopia G, Edelman ER. Intravascular drug release kinetics dictate arterial drug deposition, retention, and distribution. J Control Release 2007;123:100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolachalama VB, Tzafriri AR, Arifin DY, Edelman ER. Luminal flow patterns dictate arterial drug deposition in stent-based delivery. J Control Release 2009;133:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlyle WC, McClain JB, Tzafriri AR et al. Enhanced drug delivery capabilities from stents coated with absorbable polymer and crystalline drug. J Control Release 2012;162:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg S, Bourantas C, Serruys PW. New concepts in the design of drug-eluting coronary stents. Nat Rev Cardiol 2013;10:248–60. [DOI] [PubMed] [Google Scholar]
- 5.Hwang CW, Wu D, Edelman ER. Impact of transport and drug properties on the local pharmacology of drug-eluting stents. Int J Cardiovasc Intervent 2003;5:7–12. [DOI] [PubMed] [Google Scholar]
- 6.Tzafriri AR, Levin AD, Edelman ER. Diffusion-limited binding explains binary dose response for local arterial and tumour drug delivery. Cell Prolif 2009;42:348–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lovich MA, Edelman ER. Computational simulations of local vascular heparin deposition and distribution. Am J Physiol 1996;271:H2014–24. [DOI] [PubMed] [Google Scholar]
- 8.Levin AD, Vukmirovic N, Hwang CW, Edelman ER. Specific binding to intracellular proteins determines arterial transport properties for rapamycin and paclitaxel. Proc Natl Acad Sci U S A 2004;101:9463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang CW, Edelman ER. Arterial ultrastructure influences transport of locally delivered drugs. Circ Res 2002;90:826–32. [DOI] [PubMed] [Google Scholar]
- 10.Hwang CW, Levin AD, Jonas M, Li PH, Edelman ER. Thrombosis modulates arterial drug distribution for drug-eluting stents. Circulation 2005;111:1619–26. [DOI] [PubMed] [Google Scholar]
- 11.Saylor DM, Guyer JE, Wheeler D, Warren JA. Predicting microstructure development during casting of drug-eluting coatings. Acta Biomater 2011;7:604–13. [DOI] [PubMed] [Google Scholar]
- 12.Balss KM, Llanos G, Papandreou G, Maryanoff CA. Quantitative spatial distribution of sirolimus and polymers in drug-eluting stents using confocal Raman microscopy. J Biomed Mater Res A 2008;85:258–70. [DOI] [PubMed] [Google Scholar]
- 13.Acharya G, Park K. Mechanisms of controlled drug release from drug-eluting stents. Adv Drug Deliv Rev 2006;58:387–401. [DOI] [PubMed] [Google Scholar]
- 14.Belu A, Mahoney C, Wormuth K. Chemical imaging of drug eluting coatings: combining surface analysis and confocal Raman microscopy. J Control Release 2008;126:111–21. [DOI] [PubMed] [Google Scholar]
- 15.Tzafriri AR, Groothuis A, Price GS, Edelman ER. Stent elution rate determines drug deposition and receptor-mediated effects. J Control Release 2012;161:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarcha PJ, Verlee D, Hui HW et al. The application of ink-jet technology for the coating and loading of drug-eluting stents. Ann Biomed Eng 2007;35:1791–9. [DOI] [PubMed] [Google Scholar]
- 17.Tzafriri AR, Markham PM, LaRochelle AW et al. CRT-500.10 Ridaforolimus Eluting Stents with Customizable Diffusion Controlled Release Kinetics and Tissue Uptake. JACC: Cardiovascular Interventions 2016;9:S56–S56. [Google Scholar]
- 18.Garg S, Serruys PW. Coronary stents: looking forward. J Am Coll Cardiol 2010;56:S43–78. [DOI] [PubMed] [Google Scholar]
- 19.Shazly T, Kolachalama VB, Ferdous J, Oberhauser JP, Hossainy S, Edelman ER. Assessment of material by-product fate from bioresorbable vascular scaffolds. Ann Biomed Eng 2012;40:955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artzi N, Tzafriri AR, Faucher KM et al. Sustained Efficacy and Arterial Drug Retention by a Fast Drug Eluting Cross-Linked Fatty Acid Coronary Stent Coating. Ann Biomed Eng 2015. [DOI] [PMC free article] [PubMed]
- 21.Tzafriri AR, Bailey L, Stanley J et al. TCT-570 Stents With Absorbable Tissue-Deployable Coatings Can Distribute Drug More Uniformly Between Struts. JACC 2012;60:B166. [Google Scholar]
- 22.Urban P, Abizaid A, Chevalier B et al. Rationale and design of the LEADERS FREE trial: A randomized double-blind comparison of the BioFreedom drug-coated stent vs the Gazelle bare metal stent in patients at high bleeding risk using a short (1 month) course of dual antiplatelet therapy. Am Heart J 2013;165:704–9. [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Habraken TC, Hennink WE, Kok RJ. Polymer-Free Drug-Eluting Stents: An Overview of Coating Strategies and Comparison with Polymer-Coated Drug-Eluting Stents. Bioconjug Chem 2015;26:1277–88. [DOI] [PubMed] [Google Scholar]
- 24.Steigerwald K, Merl S, Kastrati A et al. The pre-clinical assessment of rapamycin-eluting, durable polymer-free stent coating concepts. Biomaterials 2009;30:632–7. [DOI] [PubMed] [Google Scholar]
- 25.Wessely R, Hausleiter J, Michaelis C et al. Inhibition of neointima formation by a novel drug-eluting stent system that allows for dose-adjustable, multiple, and on-site stent coating. Arterioscler Thromb Vasc Biol 2005;25:748–53. [DOI] [PubMed] [Google Scholar]
- 26.Mehilli J, Byrne RA, Wieczorek A et al. Randomized trial of three rapamycin-eluting stents with different coating strategies for the reduction of coronary restenosis. Eur Heart J 2008;29:1975–82. [DOI] [PubMed] [Google Scholar]
- 27.Tada N, Virmani R, Grant G et al. Polymer-free biolimus a9-coated stent demonstrates more sustained intimal inhibition, improved healing, and reduced inflammation compared with a polymer-coated sirolimus-eluting cypher stent in a porcine model. Circ Cardiovasc Interv 2010;3:174–83. [DOI] [PubMed] [Google Scholar]
- 28.Stone G, Kirtane A, Abizaid A et al. Preclinical Results with a Novel Internally Loaded Drug -Filled Coronary Stent. JACC 2015;65:A1762. [Google Scholar]
- 29.Tzafriri AR, Markham PM, Goshgarian J et al. TCT-554 Titratable drug delivery from drug filled stents. Journal of the American College of Cardiology 2015;66.
- 30.Kolachalama VB, Levine EG, Edelman ER. Luminal flow amplifies stent-based drug deposition in arterial bifurcations. PLoS One 2009;4:e8105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edelman ER, Rogers C. Pathobiologic responses to stenting. Am J Cardiol 1998;81:4E–6E. [DOI] [PubMed] [Google Scholar]
- 32.Hwang CW, Wu D, Edelman ER. Physiological transport forces govern drug distribution for stent-based delivery. Circulation 2001;104:600–605. [DOI] [PubMed] [Google Scholar]
- 33.Balakrishnan B, Tzafriri AR, Seifert P, Groothuis A, Rogers C, Edelman ER. Strut position, blood flow, and drug deposition: implications for single and overlapping drug-eluting stents. Circulation 2005;111:2958–65. [DOI] [PubMed] [Google Scholar]
- 34.Creel CJ, Lovich MA, Edelman ER. Arterial paclitaxel distribution and deposition. Circ Res 2000;86:879–84. [DOI] [PubMed] [Google Scholar]
- 35.Parker T, Dave V, Falotico R. Polymers for drug eluting stents. Curr Pharm Des 2010;16:3978–88. [DOI] [PubMed] [Google Scholar]
- 36.Gao RL, Xu B, Lansky AJ et al. A randomised comparison of a novel abluminal groove-filled biodegradable polymer sirolimus-eluting stent with a durable polymer everolimus-eluting stent: clinical and angiographic follow-up of the TARGET I trial. EuroIntervention 2013;9:75–83. [DOI] [PubMed] [Google Scholar]