

Abstract
Stabilized peptide therapeutics have the potential to hit currently undruggable targets, dramatically expanding the druggable genome. However, major obstacles to their development include poor intracellular delivery, rapid degradation, low target affinity, and membrane toxicity. With the emergence of multiple stabilization techniques and screening technologies, the high efficacy of various bioactive peptides has been demonstrated in vitro albeit with limited success in vivo. Here we discuss the chemical and pharmacokinetic barriers to achieving in vivo efficacy, analyze the characteristics of FDA-approved peptide drugs, and propose a developmental tool that considers the molecular properties of stabilized peptides in a comprehensive and quantitative manner in order to achieve the necessary rates for in vivo delivery to the target, efficacy, and ultimately, clinical translation.
Keywords: alpha helices, stabilized peptides, cyclic peptides, macrocycles, pharmacokinetics, lipophilicity
Opportunities and Challenges in the Peptide Drug Landscape
The field of peptide therapeutics has come a long way from utilizing unmodified naturally occurring peptides as in the case of insulin therapy discovered in the early 20th century. Today, over 60 peptide-based therapeutic agents have been approved by the FDA and many more are in the drug development pipeline. The majority of these peptide drugs are analogs of previously discovered endogenous peptides, many of which target G-protein coupled receptors or other cell surface receptors with endogenous protein ligands[1]. These receptors, however, make up only a fraction of molecular targets to treat disease. Targeting other protein-protein interactions (PPIs) has proven to be a much larger challenge since most of them occur inside the cell, out of the reach of traditional biologics such as monoclonal antibodies.
Currently, intracellular targeted therapeutics are dominated by lipophilic small molecule drugs that bind ‘druggable’ proteins that contain a small, hydrophobic binding pocket or enzyme active site. However, the majority of disease-associated proteins lack such features[2]. In contrast, 62% of PPIs have an alpha-helical motif in their binding interfaces[3], and alpha helix-based surrogate peptide structures are able to disrupt the much larger (1000–2000Å2) binding interfaces between proteins[4]. Therefore, alpha helices are being explored for targeting these PPIs. In principle, alpha helical peptide scaffolds are an attractive drug class for targeting PPIs due to their balanced size: large enough to specifically disrupt PPIs like biologics but compact enough to enter cells like small molecules. However, without modification, they suffer from rapid degradation, fast clearance, insufficient membrane permeability, and poor target affinity.
Over the past two decades and continuing with ongoing research, the barriers to intracellular delivery of alpha helix scaffolds are slowly being eroded with the emergence of helix stabilization chemistries, modification of peptide physicochemical properties, and screening technologies to select stand-alone high affinity agents, which is a challenge given the much larger chemical space of macromolecular drugs. Helix stabilization approaches (Fig. 1) help decrease degradation rates and potentially increase target affinities[5, 6]. Several groups have since adopted these techniques to engineer novel therapeutic and diagnostic constrained alpha helices with targets spanning a variety of human diseases. Combined with the development of various peptide library screening methods (phage, mRNA and bacterial display)[7, 8], these advances have propelled the field forward, and some agents have entered clinical trials[9]. Until one or more FDA approved drugs are developed, it remains unclear whether alpha helices are generally suitable for targeting intracellular PPIs. However, multiple approaches have been explored for overcoming the intrinsic challenges in developing these agents, and significant progress is being made[10]. Here, we focus on the quantitative pharmacology aspects of stabilized peptides, where multiple properties must be considered simultaneously for translational development.
Figure 1. Peptide Staple Examples.
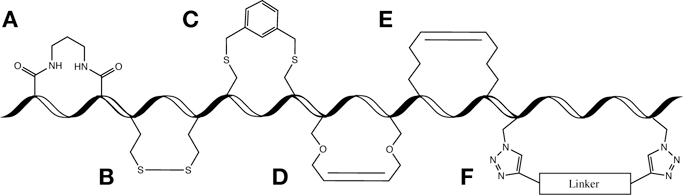
A (non-exhaustive) representation of several reported peptide staples is shown. All examples are drawn as i,i+4 staples except E which is shown as i,i+7, a more common stapling position for that staple type. A. Lactam bridge. B. Disulfide bridge. C. Bis-alkylation of cysteine residues. D. Ring-closing olefin metathesis of O-allylserine residues. E. All-hydrocarbon staple via ring-closing metathesis of α, α-disubstituted non-natural amino acids with all-hydrocarbon side chains. F. Double-click stabilization of azido non-natural amino acids.
This review will focus on the need for a ‘systems’ approach to stabilized peptide scaffold development, where changes in one property impact the distribution of the agent across multiple length and time scales within a living organism. In early stages of development, beyond measuring target binging affinities, groups typically establish the efficacy of their peptides via in vitro cellular assays such as cell killing and gene reporter assays. Many also perform imaging of fluorophore-conjugated variants to demonstrate cellular uptake (for intracellular targets) or cell surface binding (for extracellular targets) as well as immunoprecipitation and western blotting to show up- or downregulation of native proteins. Much of the development work at this stage is carried out in cell culture, and this makes sense from the standpoint that the most formidable barrier to development of macromolecular agents that bind intracellular targets is accessing the cytosol. However, there are far fewer reports of in vivo efficacy studies, the precursor to clinical trials, and some of the strategies and mechanisms employed towards efficacy in cells may have deleterious effects in the context of an animal model or patient. Furthermore, various studies have revealed uncertainties in how effectively some of these recently developed peptides penetrate cells and bind their targets[11, 12]. Efficient cell membrane permeability is just one of many variables to consider for successful in vivo drug delivery. Ultimately, stabilized peptide design must be focused on reaching systemic circulation (following various routes of administration), remaining in circulation for extended periods of time (delayed renal and/or hepatic clearance), evading protease degradation (systemic and local degradation/’clearance’), reaching target organs and distributing in the tissue, penetrating cell membranes for intracellular targets, reaching the subcellular location (e.g. nuclear targets), and binding tightly to the target (Figure 2, Key Figure). In this review, we examine various classes of constrained peptides that have exhibited robust pharmacokinetics for clinical translation from a quantitative systems pharmacology view and examine them in comparison to stabilized alpha helical peptides specifically.
Figure 2. Key Figure: Multiscale Pharmacokinetic Challenges.
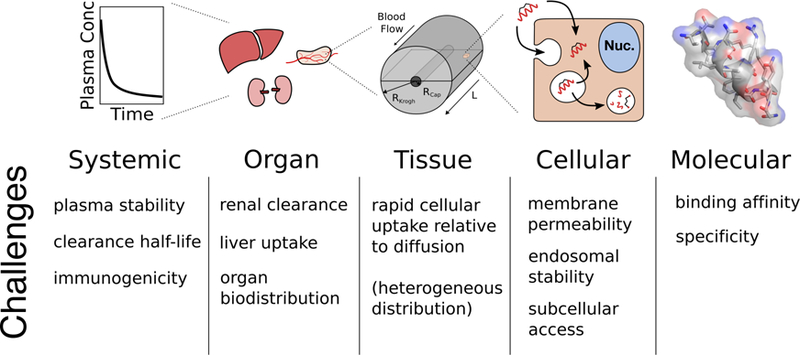
Determining the pharmacokinetic outcomes of a drug must be approached with careful consideration of the relevant physiological length scales associated with drug absorption, distribution, metabolism, excretion, and toxicity. These scales and their challenges are summarized in this figure ranging from the systemic level to the molecular level.
Stabilized Alpha Helices as a Peptide Drug Scaffold
It has long been recognized that more rigid/constrained molecules have the potential for higher binding affinity (see Box 1 on helicity), and the alpha helix, one of the most prevalent secondary structures in proteins, has many attractive properties as a scaffold. The pursuit of structurally locking alpha helices in their conformation began several decades ago. Early techniques included the use of disulfide and lactam bridges (Fig. 1) as well as metal ion complexes. In 2000, Schafmeister and Verdine published an all-hydrocarbon stapling method adapted from a previously published ring-closing olefin metathesis stapling method by Blackwell and Grubbs[13], [14] (Fig. 1). By incorporating terminal alkene-functionalized non-natural amino acids with varying chain lengths, R and S stoichiometries to account for stereochemistry effects, and placing the reactive residues in i, i+4 and i, i+7 locations of the peptide, they optimized their stapling method to efficiently stabilize the scaffold[13]. This all-hydrocarbon stapling technique was first tested by Walensky and colleagues (Verdine and Korsmeyer groups), who designed peptides after the BH3 domain of a BCL-2-member protein involved in apoptosis pathways. They stabilized these peptides using their all-hydrocarbon stapling technique, and showed that the peptides were able to bind pro-apoptotic proteins and cause apoptosis of cancer cells in vitro and in vivo[5]. Similarly, Bernal and colleagues, also from the Verdine group, tested their stabilization technique on p53-based peptides. P53 is a transcription factor that regulates cell growth and apoptosis as a response to DNA damage and cellular stress. Its activity is regulated by MDM2, an E3 ubiquitin ligase that marks p53 for degradation when its functions are not necessary. Loss of p53 activity is the most common deficiency in human cancer, whether due to mutation of the gene or due to overexpression of MDM2[15]. Bernal et al. designed several p53-based peptides and showed that one of their variants, p53-SAH-8, had a higher binding affinity for MDM2 (Kd=55nM) than the wild-type p53 (Kd = 410nM). They also demonstrated in vitro cell permeability, which caused apparent upregulation of native p53—reactivating the apoptotic pathway[16].
Box 1. Helicity.
Alpha helices are coiled protein secondary structures held together by hydrogen bonding between amino acids that are four residues apart (i, i+4), which are commonly found in proteins and at interfaces[3]. Helicity is a widely measured parameter for stabilized peptides, but the multiple quantitative impacts of this property on binding have led to substantial debate. Increases in helicity result in a more ‘structured’ peptide, and a more rigid structure has long been known to enable selection of higher binding affinity molecules, whether this is from disulfide ‘constrained’ libraries in phage display[80], fixing the termini by displaying peptides as ‘loops’ in a protein[81], or disulfide bonds that are selected in CDR loops of antibodies[82]. However, this general finding is far from uniform and, at the molecular level, is complicated by the allowable ensemble of molecular conformations in the bound versus free states and the contribution of solvent effects[83].
The net impact on affinity can best be described by the thermodynamics of binding. The dissociation constant is related to the Gibbs free energy of binding, which in turn is comprised of the enthalpy of binding (a measure of the energy associated with charge interactions, van der Waals forces, and hydrogen bond contacts at the binding interface versus in free solution) and entropy (a measure of the loss in available conformations when it is bound to the target versus free in solution).
By ‘locking’ a peptide in one conformation (or more accurately, increasing the probability that the peptide in free solution is found in this conformation), the ‘entropic penalty’ for binding can be decreased significantly. This is demonstrated in a study by Sia et al. as a uniform decrease in the –TΔS term with increased helicity; the difference in entropy between bound and free goes down, increasing affinity[84]. However, unless the molecular conformation of the helix is perfectly aligned with the conformation maximizing all molecular contacts, a more rigid structure can lower the enthalpy of binding (e.g. slightly mis-aligning a hydrogen bond), resulting in an eventual decrease in affinity with increasing rigidity.
From a thermodynamic perspective, a more rigid structure, pre-organized in the optimal bound conformation, will always be higher affinity due to the reduced entropic penalty upon binding ceteris paribus. However, in practice, all other aspects of binding are not equal. Improvements in affinity by rigidifying the ligand are often less than (and sometimes opposite of) those predicted by the entropy of binding due to the ‘enthalpy/entropy compensation’ phenomenon. This effect, where changes in the entropy and enthalpy of binding oppose each other when engineering an affinity ligand, have their origins in a variety of molecular mechanisms including perturbations in a system with multiple closely spaced energy levels, higher ‘enthalpic’ interactions restricting ‘entropic’ motion, and importantly, the large thermodynamic contribution of structured waters before and after binding[83]. This makes helicity an imperfect measurement for changes in binding affinity. In summary, while increased structure generally has the potential for higher affinity, this is not guaranteed for any individual interaction.
The Verdine group took some of the first steps toward the design of stapled peptides with intracellular therapeutic effects that overcame several barriers associated with delivery of peptides. However, particularly in these early days, the low throughput in synthesizing individual peptide sequences and testing for cell permeability combined with the gap in knowledge around the pharmacokinetic properties of molecules in this size range made progress arduous. Since this time, high throughput screening methods combined with novel chemistry have allowed isolation of high affinity sequences, aiding this step in development[17–19]. Recently, Lau and colleagues designed a p53-based peptide for therapeutic modulation of the p53-MDM2 interaction using two bioorthogonal reactions (an approach first reported by Torres and colleagues[20]). Instead of cross reacting two complementary residues, they placed two azido-functionalized residues in i, i+7 locations and stapled the peptide with a di-alkyne linker by copper-catalyzed azide-alkyne cycloaddition—a well characterized click chemistry reaction[21] (Fig. 1). This strategy, termed “double-click stabilization,”[22] conveniently allows the introduction of additional functionality on the linker (e.g. modification of charge, lipophilicity, imaging tag incorporation, etc.)[6, 23]. In addition to modifying the physicochemical properties of peptides, these polyfunctional linkers can contribute to protease stability and binding affinity through reducing the entropic penalty of binding and through direct (enthalpic) contributions - Box 1.
Stapling techniques allow for added complexity in peptide drug design, giving more nuanced control over the properties one can change to help achieve desired peptide outcomes. Despite these advances in peptide chemistry and molecular engineering, the field of constrained peptides still faces formidable challenges, particularly in the delivery of these agents to the site of action, which will need to be overcome to gain widespread clinical relevance. Now that more tools are becoming available to generate defined molecular properties, the major question remains: what properties are needed for clinically translatable drugs?
Stabilized peptide scaffolds sit at the interface of biologics and small molecule drugs, and guidance for development can be found in both fields. Several of the high throughput approaches have taken advantage of directed evolution from the field of biologics to screen a billion or more (109 to 1012) variants. (For comparison, a 12-mer peptide with only natural amino acids has a sequence space of 2012 ~ 4×1015 unique sequences.) To access the cytosol, where many relevant protein-protein interactions occur, inspiration can be found in the world of small molecules.
Non-Lipinski Drugs: Cyclic Peptides and Related Macrocycles
Most drugs against intracellular targets are small molecules—a class of compounds containing molecules sharing a specific set of physicochemical properties summed up by Lipinski’s Rule of Five (Ro5)[24]. Imaging studies show that this class of agents can enter cells and reach their subcellular target within seconds to minutes[25, 26]. Drugs that adhere to these guidelines tend to be well-absorbed making them good candidates for oral delivery. However, due to their small size (<500 Da), they lack large contact surface area for binding, making them prone to low specificity for their intended targets. Conversely, biologic drugs typically have molecular weights above 5000 Dalton (e.g. antibodies). Though they boast high target specificities due to large contact surface areas for binding, they often require intravenous delivery (see Box 2) and lack intracellular access[27]. Peptides and other non-traditional (non-Ro5) drug molecules falling in between small molecules and biologics have the potential to provide the best of both worlds. Though stabilized peptides have yet to be FDA-approved, other constrained peptide types (Fig. 3) have been approved or are in clinical development. Among them are cyclic peptides, which compared to all other peptide drugs have shown the highest abundance of orally bioavailable agents[28]. It is important to note that oral absorption is distinct from intracellular delivery/cytosolic access. Oral availability requires efficient membrane permeation prior to transit through the gut while stabilized peptides require membrane permeation prior to systemic clearance or local degradation (which can vary based on specific peptide properties). Also, the rate of transport through the intestinal epithelium for oral absorption may be different from cell membrane permeation of stabilized peptides in the tissue of interest. However, from a kinetic perspective, oral absorption and intracellular delivery are both controlled by the rate of membrane permeability relative to clearance/degradation (either in the gut or tissue of interest, respectively), and efficient membrane partitioning and chemical stability in various physiological conditions (discussed in Box 2) are useful characteristics for both orally and non-orally delivered peptides. Therefore, the properties that promote oral bioavailability in cyclic peptides may inform stabilized peptide development as well, regardless of the route of administration.
Box 2. Routes of Administration.
The three routes of administration that span common delivery approaches for both small molecule drugs and biologics are oral (PO), subcutaneous (SC), and intravenous (IV) delivery. Oral delivery is the most challenging for stabilized peptides due to the harsh conditions found in the gastrointestinal tract, where stomach acid can destabilize the helical conformation by hydrogen bond disruption. Many enzymes including pepsin, trypsin and chymotrypsin can hydrolyze the peptide at specific sites along the backbone, and transit time in the gut provides a limited window for absorption. Studies of various peptide drugs have shown that cyclic peptides (including cyclosporin) and cyclotides are generally more stable than non-constrained peptides when exposed to gastric and intestinal fluids[85]. This is partly attributable to a folded backbone being less prone to acid and protease attack. Sequence modifications can further enhance stability by avoiding favored proteolytic sequences. In the case of stabilized peptides, the improved stability from side-chain crosslinking can enable some absorption, albeit low, following oral gavage as seen with a 36-residue double stapled enfuvirtide agent[28, 86] (Fig. 3A). A more clinical advanced example is the phase III trial of a formulated version of semaglutide[87]. Here, a permeability enhancer in the formulation aids in protection and absorption for this 4.1 kDa molecular weight peptide. Even with these advances, absolute absorption is still relatively low, but semaglutide is an ideal candidate for this approach given its safety at high doses, high potency, and slow clearance (so a relatively large oral dose and small absorbed fraction are tolerable).
Subcutaneous injection is a common delivery route for peptides since it enables self-administration and avoids degradation in the GI tract, though proteases also exist in subcutaneous tissue. The formulation and dose become important considerations given that a typical SC injection volume cannot exceed ~1 mL. Highly concentrated doses, depending on the physicochemical properties, could also cause local reactions. Recently, Zhang et al. demonstrated that peptide backbone stabilization can help increase the fraction of stabilized peptide absorbed after subcutaneous administration in mice[23].
Intravenous administration is a common delivery method for biologics, such as monoclonal antibodies, that have to be given at high (e.g. multiple mg/kg) doses. The stapled peptide currently in clinical trials, ALRN-6924, is also being administered intravenously and has a maximum tolerated dose of 3.1 mg/kg when given as a weekly intravenous infusion (three doses every 4 weeks)[9]. Proteolysis at the site of administration is largely avoided by intravenous delivery, however degradation of peptides can occur in the plasma, within clearance organs (e.g. Kupffer cells of the liver or proximal tubule of the kidney), in the extracellular matrix of tissues (e.g. matrix metalloproteinases in tumors), and within lysosomes of target tissues. Furthermore, one disadvantage for stabilized peptides delivered intravenously is that they have faster clearance than antibodies, necessitating more frequent delivery.
Figure 3. Constrained Peptide Types.
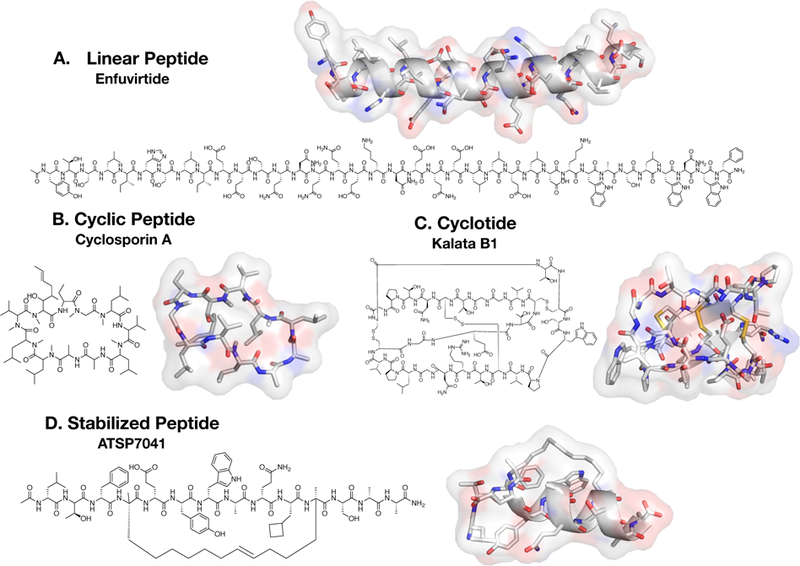
A. Linear peptides are those containing no backbone cyclization (between termini or residues). Enfuvirtide is a 36 amino acid linear peptide for HIV treatment. The complete 3D structure is not yet available; however a truncated structure lacking 8 C-terminal residues is shown (PDB 5Y14). B. Cyclosporin A is an immunosuppressant natural product cyclic peptide with a completely cyclic backbone, therefore containing no terminus. It has several N-methylations in its backbone (PDB 1QNG). C. Cyclotides are cyclic peptides with intramolecular disulfide bonds (3 shown in yellow), termed cysteine knots. Kalata B1 is a natural product cyclotide commonly used as a scaffold for its physical and chemical stability (PDB 1NB1). D. Stabilized peptides encompass peptides having residues chemically cross-linked by any one of various methods such as those outlined in Figure 1. ATSP-7041 is a clinical-lead all-hydrocarbon stapled peptide by Aileron Therapeutics for inhibiting MDM2 in relevant cancer types (PDB 4N5T).
Physicochemical Properties
What properties allow cyclic peptides to be well-absorbed? Nielsen and colleagues provide an exhaustive analysis of 125 orally bioavailable cyclic peptides and their properties, including molecular weight, predicted lipophilicity, and polar surface area. They profiled the properties of a variety of cyclic peptides with greater than four residues, including circular cysteine-knotted peptides, termed cyclotides (Fig. 3C), natural product cyclic peptide Cyclosporin A (Fig. 3B) and its derivatives, and, for comparison sake, the doubly stapled alpha helix SAH-gp41(626–662)—the only stabilized alpha helix with reported oral bioavailability thus far (though percent oral bioavailability was never reported for this peptide). Nielsen examined the correlation between multiple physicochemical properties and oral bioavailability. First, a high molecular weight (>500 Da) did not appear to preclude oral bioavailability as it does in the case of small molecules. They found that even in the higher molecular weight range for the peptides analyzed (960–1350 Da), 23 peptides had oral bioavailability greater than or equal to 10%, and lower molecular weight did not correlate with higher oral bioavailability[28]. This suggests that for peptides, other properties may be of greater importance when considering rates of drug absorption. Furthermore, upon comparing predicted octanol-water partition coefficients (logP, Box 3) as measured by Molinspiration cheminformatics or QikProp software, Nielsen et al. reports that the lipophilicity of orally bioavailable cyclic peptides tend to fall within Ro5 guidelines, 0≤LogP≤5, but that a slightly higher range of 0–8 (as calculated with Molinspiration) seemed permissible.
Box 3. Lipophilicity and LogP.
Lipophilicity is a key determinant of the pharmacokinetic behavior of small molecule drugs. More specifically, it is an important determinant of whether a drug is suitable for oral delivery. A drug must be lipophilic enough to partition into membranes for efficient absorption and distribution and for long circulation time (high plasma protein binding and kidney reabsorption). However, it must not be too lipophilic that it suffers from high first-pass metabolism or insufficient aqueous solubility making it unsuitable for delivery. Lipophilicity is traditionally quantified as LogP, the octanol-water partitioning coefficient, a measure of the extent to which a molecule partitions into octanol (a surrogate for the lipid bilayer) versus the aqueous phase. Molecules with a logP>0 favor lipids over aqueous dissolution. Lipinski’s rule of 5 states that a drug molecule should have a logP < 5 (generally between 0 and 5)[24]. This quantity can be calculated experimentally; however several computational programs exist allowing for high-throughput logP measurements for drug libraries[24]. Though computational programs are often accurate for small molecules, it has previously been shown that calculated logP values for peptides often deviate greatly from the experimentally measured values and that the discrepancy increases as peptide size increases[88], [89]. This is attributed to the failure of fragment-based approaches to adequately consider intramolecular interactions such as hydrogen bonding and the effects of cyclization[29]. One method to circumvent inaccuracies of computational peptide logP measurements is to measure HPLC retention times and compare it to the retention times of small molecules with known logPs using the same HPLC method. Bird and colleagues have recently used HPLC retention times to compare lipophilicities of several peptide variants to aid in optimization of stapled Bcl-2 targeting peptides and have correlated this parameter with improved cellular uptake[44]. Valko et al. have also recently developed HPLC retention time methods to assess the lipophilicities of various potential peptide therapeutics[90]. This approach could be useful for giving more accurate predictions of lipid partitioning given the challenges of predicting intramolecular hydrogen bonding.
Other original Ro5 properties include the number of hydrogen bond donors and acceptors, limited to less than 5 and 10, respectively[24]. Nielsen’s study found that cyclic peptides with greater than 6 hydrogen bond donors had oral bioavailability of less than 10%. However, the number of hydrogen bond acceptors, defined as simply the total number of nitrogen and oxygen atoms in the molecule, was greater than 10 for all cyclic peptides in the study, therefore not meeting the Ro5 criteria. A number of studies conclude that few rotatable bonds (no more than 10–13) corresponds to good oral bioavailability, likely due to a decrease in solvent interactions and lower susceptibility to degradation because of structural rigidity[29], [30]. Cyclic peptides are advantaged in this area due to the fact that they are inherently constrained[28].
Backbone Modifications
Many peptide macrocycles and other ‘Ro5 violators’ that have had success in the clinic are derived from natural products[4]. These peptides have been widely studied as models for synthetic derivatives or de novo cyclic peptides. Cyclosporin A, for example, is a naturally occurring orally bioavailable (oral bioavailability = 29%) 11-residue peptide macrocycle used as an immunosuppressant drug[31]. Despite definitively falling outside of Lipinksi’s standards for drug-likeness, Cyclosporin A and other natural product macrocycles have the uncanny ability to traverse membranes, avoid degradation, and bind efficiently to their targets in their necessary conformations[4, 28, 31, 32]. One of the common characteristics of natural product macrocycles like Cyclosporin A is amide N-methylation[31, 33, 34]. In a study of 39 peptide macrocycles by Ahlbach and colleagues, all compounds with measurable passive permeability except one had amide N-methylation[31]. N-methylation has been widely shown to improve membrane permeability of natural product peptide macrocycle derivatives, a phenomenon attributed to a decrease in solvent-exposed polar groups (polar surface area), therefore reducing the desolvation energy required for crossing membranes[32–34]. In addition, designing molecules to enable the formation of intramolecular hydrogen bonds and introducing sterically shielding hydrophobic groups also adds to these permeability enhancing effects[31, 35]. This may be particularly important for imparting ‘chameleon’-like properties to maintain solubility: solvent hydrogen bonding in aqueous environments and intramolecular hydrogen bonding while crossing membranes[4]. These are all useful insights in the design of stabilized alpha helices since hydrogen bonding via staple choice and sequence optimization could prove useful for achieving enhanced permeability and in vivo efficacy.
Stabilized Alpha Helices: Properties and Challenges
Access to the Cytosol
Membrane permeability of stabilized peptide structures is the most challenging step in delivery to the site of action (the target). After all, membrane partitioning is a basic element of a functioning cell. In recent years, elucidating the nuances of how peptides enter cells has been an increasingly active area of research in the field of stabilized peptides. Early methods of verifying cellular uptake included conjugating peptides to fluorophores, treating cells with the fluorophore-conjugated peptides, and using microscopy or flow cytometry to detect intracellular fluorescence[5, 6, 16, 36, 37]. This is a challenging task, since care must be taken to distinguish cellular uptake in endosomes/lysosomes (which do not have access to cytosolic targets) versus localization in the cytosol itself. These methods however, do not directly measure target engagement. Even cell viability assays without the use of proper controls are at risk of false positives. This became clear with the emergence of methods directly assessing whether peptides have off-target toxicity[38]. Adopted from the viral and gene delivery fields, positive charge is one method of conferring membrane permeability of peptides[6, 39, 40]. However, methods such as the recombinase enhanced bimolecular luciferase complementation platform (ReBiL) and LDH release quantification highlighted some of the toxicity hurdles inherent in cationic agents and motivated other strategies to achieve membrane permeability[12, 38]. Just a few years since the first hydrocarbon stapled peptides were introduced, researchers in the field are developing new membrane permeable peptides with more robust methods of demonstrating on-target efficacy using in vitro assays and giving special attention to physicochemical properties outside of charge that can help improve cytosolic delivery[41–44]. However, careful consideration of drug delivery issues outside of the cellular challenges, including systemic clearance, organ uptake, and tissue distribution, is necessary for in vivo efficacy.
Pharmacokinetics Beyond the Cell
Two of the main mechanisms for getting stabilized helices across membranes are charge and lipophilicity. Both are inspired by nature. Many natural compounds like cyclic peptides and cyclotides are highly stable and lipophilic. In contrast, viral approaches, such as the cationic TAT peptide from HIV, use charge to help penetrate membranes. As a cautionary note, cationic charge, particularly in combination with lipophilicity, is also found in another class of molecules: antimicrobial peptides. These membrane-disrupting agents have the potential for high toxicity and may have narrow concentration windows for efficacy without toxicity. Likewise, viruses (and related gene-delivery payloads) benefit from amplification and integration within the cell, requiring relatively low doses, whereas peptide-scaffold delivery requires target-saturating amounts. Examples of both approaches are prevalent in the literature; how do these strategies, designed to allow access to the cytosol, impact the quantitative systems pharmacology of these agents?
The number of clinical examples of intracellular therapeutics outside Lipinski’s Ro5 dwindles dramatically when approaching 1 kDa and above, and so too does the detailed data on tissue, organ, and systemic distribution. However, specific examples (both within and outside the class of stabilized peptides) can highlight some of the challenges and strategies to improve intracellular delivery in a clinically translatable manner. Although these agents do not currently have FDA approval, macromolecules against intracellular targets with significant published clinical results include imetelstat (a non-peptidic 4.6 kDa telomerase inhibitor) and the clinical test compound ALRN-6924[9] (the clinical variant of stapled peptide ATSP-7041). Typically molecules that have made it further in the clinical trial process have improved pharmacokinetic properties[45]. Looking at these molecules in conjunction with the cyclic peptides discussed previously, some trends are clear. Lipophilicity is much more prevalent than cationic charge for cytosolic access. This is also true for BCL-2 inhibitors (e.g. venetoclax and navitoclax), rapamycin, and a host of other ‘beyond Lipinski’ molecules (500–1500 Da)[46, 47]. Even at the other extreme of the ‘non-Lipinski’ space, close to 5 kDa, molecules with high lipophilicity tend to prevail. Cyclotides, as natural products, are isolated based on their stability and long HPLC retention time (i.e. lipophilicity)[48]. By integrating a target-binding helix into a cyclotide’s backbone, Ji et al. were able to target MDM2 inside cells with a 5.3 kDa cyclotide. While the doses are high (40 mg/kg in 5% dextrose which can enhance intestinal permeability) likely due to slow transport, they have shown remarkable evidence of activity after oral administration against this intracellular target[49]. The use of lipophilicity does not preclude other mechanisms of cytosolic access from being developed (and may be necessary for the intracellular delivery of larger cargos, such as DNA/RNA). However, it is worth highlighting the strengths of augmenting lipophilicity as an approach from a systems pharmacology perspective.
Focusing first on systemic delivery (plasma concentrations), lipophilicity can help slow down clearance in the blood to enable more efficient uptake in tissue. These peptide scaffolds are well below the roughly 60 kDa molecular weight filtration limit of the kidney, so hydrophilic scaffolds are rapidly excreted by the kidneys[50]. Lipophilic peptides (and small molecule drugs) typically bind albumin and other proteins to reduce renal filtration. With careful design, lipophilicity can be used to impart long half-lives to peptides, such as the 1 week half-life of semaglutide obtained by fatty acid conjugation[51]. Highly charged molecules can also stick to plasma proteins, but these are typically cleared rapidly in the body. To highlight a clinical example, protamine, a cationic macromolecule that complexes with heparin in the blood, is cleared within minutes in humans (7.4 min half-life)[52]. While protamine may be an extreme, TAT and related peptides tend to exhibit increased clearance and liver/kidney uptake[53, 54]. Plasma protein binding (PPB) can also decrease the cellular uptake of these agents, and care must be used when taking quantitative pharmacology measurements (especially in vitro) to discern what conditions were used. However, plasma proteins tend to have a larger effect on cationic delivery (both peptides and larger cargoes such as PEI-mediated gene delivery) than lipophilic delivery[38, 55]. Even if PPB can be avoided, this could potentially result in rapid kidney clearance as described above. It is important to note that this discussion assumes the molecules are stable compared to the time scale of renal clearance. Otherwise, systemic proteolysis becomes the rate-limiting step for plasma clearance, and efforts should be focused on improving stability to increase systemic exposure.
High cationic charge can also disrupt membranes at higher concentrations, causing toxicity as previously mentioned. This could cause potential issues with parenteral routes of administration, such as subcutaneous injection or even intravenous delivery (e.g. infusion reactions) due to high local concentrations. Therefore, while exceptions may exist[56], cationic ‘cell penetrating peptides’ generally have poor in vivo pharmacokinetics[53]. Lipophilic agents, though less susceptible to causing membrane toxicity, can suffer from issues of solubility. Here, stabilized peptides may be able to take advantage of the ‘chameleon’ like behavior of cyclic peptides, where intramolecular hydrogen bonds can form in membranes, but intermolecular bonds can form with water molecules in an aqueous environment[57]. Extreme lipophilicity may also suffer from slow distribution into different organ systems due to high PPB. Although PPB is important to avoid rapid renal filtration, high PPB can slow the extravasation and diffusion rates of the compounds as seen with smaller molecules (e.g. ~1 kDa fluorescent PARP inhibitors)[26].
Immunogenicity is also a systemic consideration, but with limited clinical experience for this class of agents, it is hard to predict the impact this will have on development. In theory, improved stability should lower the proteolytic processing and MHC display of peptide fragments, while higher doses and slower clearance could increase immunogenicity. The diverse locations where peptides can be degraded (Box 2) makes predicting MHC display challenging but important. Improved mass spectrometry methods may better identify sites of degradation within the body. If an immune response is elicited, the impact on efficacy will also need to be evaluated. The experience with human anti-mouse antibody (HAMA) response necessitated humanization of monoclonal antibodies while other biologics, like the GLP-1 analogue exendin, cause minimal immune response that has little impact on efficacy[58].
Tissue heterogeneity is another consideration for stabilized peptides, since the size of these agents may result in transport limitations over the tens to hundreds of microns length scale. Both small molecules and biologics can exhibit heterogeneity in tissue under certain conditions. Small molecules often exhibit heterogeneous distribution if they have rapid cell uptake and immobilization relative to clearance (e.g. Hoechst 33342)[59] or fast metabolism relative to diffusion[60]. Biologics like antibodies exhibit heterogeneity when their binding rates exceed their diffusion into the tissue[61]. The large gradients in the drug concentration between the region just outside of the blood vessels and regions more distal to the vasculature arise through a competition between immobilization of the agent (receptor binding, cellular internalization, etc.) versus interstitial transport (typically diffusion dominates over convection in this size range). This ratio of an immobilization reaction to diffusion can be captured by a dimensionless number known as the Damköhler number. Because these agents lack an extracellular target, the relevant “immobilization” reaction rate is uptake into the cell. At first glance, it appears that there will be little heterogeneity in the tissue, since cellular uptake rates relative to diffusion are very slow for this class of agents (e.g. even with macromolecular imaging agents)[62]. However, caution must be exercised, since the strategies for increasing cell permeation (high lipophilicity and/or charge) can also dramatically slow the effective diffusion rate. Continuous exposure from the blood can eventually overcome any transient gradients, but degradation of the probe could result in a scenario where the drug is destroyed before it ever reaches distant cells, similar to antibodies[63]. Many tissues have relatively short distances between blood vessels, but tumors are particularly prone to this issue due to their long diffusion distances for efficient delivery[64]. While the stability imparted by cross-linked scaffolds for stabilized peptides and slow cellular uptake of these agents (relative to ~seconds for small molecules) help mitigate the risk of tissue heterogeneity, this is an important consideration when manipulating physicochemical properties to optimize cellular uptake.
Ideally, for any given stabilized peptide scaffold with a designated target, there exists a window of physicochemical properties that meets all the requirements for providing efficient absorption, distribution, and target engagement to elicit the desired therapeutic effect. In fact, there are trade-offs that could generate several ‘windows’ depending on the drug characteristics, such as sustained plasma concentrations and high stability overcoming slow membrane permeability, or prolonged target retention compensating for rapid plasma clearance. Determining where these ‘windows’ exist from animal experiments alone is laborious and time-consuming. The use of a computational model provides a practical approach to explore a more complete landscape of drug delivery. In Box 4, we describe the framework of such a model. The implementation of a computational model combined with experimentally measured rates could be the holistic systems approach needed to aid in higher throughput development of stabilized peptides.
Box 4. Technology Corner: Multiscale Computational Modeling in Stabilized Peptide Development.
Peptide therapeutics are uniquely situated between small molecule drugs and macromolecular biologics based on their molecular weight. Therefore, they share some properties with each, and tools from both fields can be applied. Based on their size and structure, stabilized alpha helix peptides likely share more in common with biologics than small molecules in terms of pharmacokinetic modeling. Unlike small molecule drugs that fit within Lipinski’s Rule of 5, biologics and peptides cannot be assumed to be in equilibrium with the plasma concentration due to transport limitations in the body. For example, antibodies often require high (micromolar) concentrations to drive tumor uptake even though they can saturate their target in vitro at nanomolar concentrations. Likewise, stabilized peptides require high concentrations relative to their target binding affinity to drive delivery to the cytosol. High concentrations can overcome local degradation in the target tissue, such as receptor-mediated uptake and degradation of antibodies or non-specific uptake and degradation of stabilized peptides. These rate-limiting transport steps result in a disconnect between plasma concentrations and concentrations at the site of action (both from delayed uptake and extended drug-target residence time). Computational models developed for biologics are poised to capture these transport limitations and irreversible degradation from a systemic to a molecular level in a quantitative manner to guide stabilized peptide development and experimental design.
A variety of published pharmacokinetic models are available for biologics. One recent model combined the systemic plasma concentrations and organ-level uptake of a physiologically based pharmacokinetic (PBPK) model with the tissue heterogeneity and cellular distribution of a Krogh cylinder model[63] (Fig. I). An advantage of this model is that it requires few simplifying assumptions on rate-limiting steps (e.g. blood flow, permeability, tissue penetration, or cellular processing). Using a ‘systems approach,’ the simulations can initially predict properties for sustained target engagement, and the computational model can be continually refined as experimental data are gathered (e.g. updating cellular uptake/degradation rates, plasma protein binding, and 3D tissue culture penetration early in development to preclinical animal biodistribution and scaling to the clinic). One challenge that arises from complex pharmacokinetics is that there may not be a single well-defined ‘sweet spot’ for stabilized peptide properties. While Lipinski’s Ro5 highlighted properties necessary to avoid major transport limitations, these will likely always be present and need to be overcome with stabilized peptides. The complex PK of biologics can result in properties that are context dependent; for example, the needed membrane permeability depends on the exposure time of the peptide. As an illustration with other biologics, a rapidly internalized and degraded antibody may be better suited for delivering a cytotoxic payload, while a more slowly internalized/degraded antibody would be better for Fc-effector functions and receptor blockade (i.e. the desired internalization rate depends on several factors). Similarly, a more stable peptide compound with slow clearance may be effective even with lower membrane permeability than a readily degraded or fast clearing agent with high permeability. Fortunately, multiscale models like the one described above can help elucidate the sensitivity of pharmacokinetic parameters to focus studies on compounds within an effective parameter/PK space.
Based on current knowledge, if membrane permeability (rather than target organ uptake and/or tissue penetration) is the rate-limiting step for uptake, then close parallels to other biologics occur. Analogous to the clearance modulus and Thiele modulus for antibodies[61], the concentration at the site of action is a ratio between target tissue uptake (membrane permeability for stabilized peptides) and both systemic clearance (from plasma) and local clearance (degradation). The ratios between i) plasma clearance and membrane permeability and ii) degradation/local clearance and membrane permeability determine the intracellular concentration. From there, the binding affinity and intracellular concentration determine the fraction of target engagement. Low membrane permeability can also retain stabilized peptides in cells for long periods of time for extended target engagement, provided they are stable inside the cell. As reports continue to be published on stabilized peptides, our ability to more accurately predict these rates and guide design of new agents with a quantitative systems pharmacology approach will increase. What is needed now is a focused effort on quantifying these mechanistic rates to better guide the field.
Clinical Precedent
In the absence of an FDA-approved drug, limited clinical data is available for intracellular targeted macromolecules. The two examples of drugs mentioned earlier that have been administered to patients – the clinical version of ATSP-7041 (a 1.4 kDa all-hydrocarbon stapled p53 mimetic) and imetelstat (a 4.6 kDa telomerase inhibitor which uses a fatty acid to increase lipophilicity) – illustrate the beneficial impacts of lipophilicity on the quantitative pharmacology and development of these agents. ATSP-7041 (Fig. 3D) was derived from a sequence (pDI) enriched by phage display[65] and outfitted with an i,i+7 hydrocarbon staple51. This agent had high binding affinity to its target (Kd = 900 pM for MDM2), which is likely required due to low intracellular concentrations. The Verdine group had previously developed a similar p53-based stapled peptide inhibitor of MDM2 called SAH-p53–8[16]; however, this peptide proved not potent enough for therapeutic application beyond very controlled in vitro assays[66]. It is therefore useful to look at the differences between ATSP-7041 and SAH-p53–8 that may provide insight into properties needed for success.
Although many physicochemical properties beyond molecular weight are not reported for these peptides, important structural differences and their presumed effect on efficacy are outlined. Firstly, SAH-p53–8 and ATSP-7041 both contain three key hydrophobic residues (Phe19, Trp23, Leu26) required for binding MDM2/MDMX. However, several phage display studies have revealed the importance of another key residue, Tyr22 [67]. This replaced the residue Leu22 in SAH-p53–8, which was preserved from native p53[66]. Furthermore, SAH-p53–8 has additional residues 14–16, all of which are polar, and residues Gln17 and Arg24 are replaced with Leu and Ala, respectively, in ATSP-7041. Chang and colleagues explain that this enlarges the already existing “hydrophobic patch”, leaving only a total of 4 polar residues[66]. This essentially reduces the polar surface area of the peptide while also reducing its total size. These modifications resulted in a peptide with an aqueous solubility too low for peptide characterization. This sequence was therefore further modified to improve solubility without sacrificing amphiphilicity by replacing His21 with the charged Glu residue and adding two c-terminal Ala residues to extend the helix. The resulting sequence is ATSP-7041. Cell viability assays in 10% serum show a more than 50-fold increase in potency between SAH-p53–8 (IC50>30uM) and ATSP-7041 (IC50=600nM).
In vivo studies of ATSP-7041 showed that it is primarily excreted intact by the liver, resulting in 79% collected in the feces and < 3% in the urine. The reported half-life in humans is 5.5 hrs[66]. Imetelstat also has a plasma clearance half-life of around 5 hrs in humans[68] with primarily liver uptake but some kidney/bladder signal as measured by radiolabeling[69]. The relatively long half-lives compared to cationic agents provides some evidence that lipophilicity is a feasible mechanism to improve intracellular delivery for this class of drugs. Clinical plasma clearance is not the only shared trait between these intracellular targeted macromolecules. While the targets and structures are very different, they both contain modifications to achieve high stability, lipophilicity, and binding affinity. Imetelstat has a thio-phosphoramidite backbone to improve stability of the nucleic acid backbone[70] (similar to the function of side-chain crosslinking for ATSP-7041), a conjugated 16 carbon fatty acid to increase lipophilicity and cell penetration[71] (comparable to the all-hydrocarbon staple for ATSP-7041), and high affinity and specificity for its target (45 pM[70] for imetelstat and 900 pM for ATSP-7041) to bind at low intracellular concentrations.
Significant work remains to determine if stabilized peptide scaffolds can generally be used to target intracellular proteins in the clinic. Major outstanding questions remain around the specific mechanisms of cellular uptake and other aspects of development (see Outstanding Questions Box). For example, how important is the distribution of lipophilic groups on the molecular surface versus net-lipophilicity? What is the role of molecular shape? Many stabilized peptides are often seen in endosomes (which are inaccessible to the cytosol). Is this a necessary step for delivery of lipophilic stabilized peptides or simply a function of faster uptake into endosomes than permeability across these membranes?
Outstanding Questions Box.
The largest outstanding question concerns how the physicochemical properties of a stabilized peptide affect the rate of membrane permeability. This question includes how properties such as lipophilicity/amphiphilicity, polar surface area, rotatable bonds/flexibility, helicity/shape, solubility, number of hydrogen bond donors/acceptors, and intramolecular hydrogen bonding all affect membrane permeability. The questions are compounded by the incomplete knowledge surrounding the mechanism(s) of cellular entry. What combination of properties is needed for direct membrane permeability, for escape from endosomes/ lysosomes, or even for retrograde transport through other organelles?
How does membrane permeability interplay with stability and affinity to determine target saturation and efficacy? Ideally, membrane permeability will be expressed in terms of a rate rather than a binary property. Can increased stability and/or higher affinity compensate for slower membrane permeability? Will this increase the concentration by slowing losses from inside the cell and enable higher target binding at lower concentrations, respectively?
What is the most efficient screening strategy for novel binders: a biologically based system with large diversity (e.g., mRNA display with 1015 variants), a chemistry-based system with more accessible synthetic chemical diversity (e.g., peptide arrays and one-bead-one-compound), a ‘parallel’ hybrid approach (e.g., alkylation chemistry on phage), or a ‘series’ hybrid approach (e.g., biological screening to leverage library size followed by chemistry-based screening for local refinement of structure)?
How do the combination of properties enabling cytosolic access impact on the multiscale distribution necessary for in vivo and clinical efficacy? Can the multiscale delivery challenges of cationic agents be overcome in the clinic? Are the current examples of lipophilic agents outliers or generalizable examples of a broader, so far undefined, class of intracellular drugs?
It is clear that both cationic charge and lipophilicity approaches for improving cellular permeability can be effective in cell culture. High throughput studies from the nanoparticle field show a generalizable utility of cationic charge for improving cytosolic delivery with a range of cargos[72–74] that has been extended to smaller (stabilized) peptides[75]. In contrast, lipophilic-mediated delivery is dependent on charge, helicity, and other factors[76] sometimes showing lab and assay variability[11, 77], requiring more stringent design. However, cationic charge on nanoparticles results in rapid clearance and toxicity, requiring shielding or pH sensitive approaches with nanoparticles/polymers[78]—something more challenging to design in small peptides (with Bird et al. 2016 explicitly stating combined cationic charge and lipophilicity should generally be avoided[44]). Therefore, while lipophilicity may be more challenging for cytosolic delivery, given the multiple benefits from a quantitative pharmacology standpoint for clinical translation, this may be the tougher road worth choosing.
Concluding Remarks and Future Directions
Few stabilized peptides thus far have been reported to have in vivo efficacy. However much can be learned from the combined knowledge in the field of small molecule drug development, biologics, other constrained peptide classes, and the few examples of stabilized peptide agents that have exhibited in vivo efficacy. It is clear that in the case of stabilized peptides, sequence optimization must take into account effects on binding affinity, solubility, membrane partitioning, clearance rate, and other pharmacokinetic processes. Hydrophobicity (lipophilicity) and the existence and location of polar residues seem to be important in the context of not only membrane permeability but also for protease degradation, plasma protein binding, and subsequent uptake and clearance rates as in the case of ATSP-7041. Furthermore, even large peptides (> 1 kDa) are not precluded from having in vivo efficacy (and non-intravenous routes of administration), but the barriers are high. Current knowledge on the precise relationship between physicochemical properties and mechanistic rates (e.g. membrane permeability, protease stability, and plasma clearance) is incomplete. However, based on other non-Lipinski molecules, these agents are likely to be on the more lipophilic range of the 0–8 logD obtained for cyclic peptides with protease stability and plasma clearance on the order of hours. This should enable sufficient exposure and membrane permeability based on recent evidence that several of these compounds are taken up within cells on the time scale of hours[38, 79]. The two examples discussed, ATSP-7041 and imetelstat, fit these criteria.
Stabilized peptides stand at the interface between traditional small molecule drugs and larger biologics. Future directions should continue to take advantage of the unique contributions from each of these fields while also acknowledging the possibility of pharmacokinetic and pharmacodynamic outcomes that are specific to stabilized peptides. Directed evolution methods developed for biologics can help in the selection of binders in the much larger ‘chemical space’ of these macromolecules. Similar to biologics, the plasma concentration of these agents is rarely the concentration at the site of action, and the concepts of multi-scale drug delivery can be employed to understand the various transport hurdles in vivo, going beyond the cellular delivery issue. A computational model like the one described in Box 4 would be especially useful here, factoring in important aspects like the relationship between plasma concentration and both systemic exposure and tissue/target engagement. Borrowing from the field of small molecules, leveraging data on lipophilicity, polar-surface area, charge, etc. to understand membrane permeability and partitioning will aid in our understanding of if and how these molecules access the cytosol and their targets. Quantitative measurements of these properties (i.e. rates of membrane permeability rather than binary ‘yes/no’ classification) can also be factored into a model, allowing for more robust analysis and prediction of in vivo outcomes. Ultimately, the hurdles for development are high, but the potential payoff – to be able to specifically hit targets beyond the reach of biologics and small molecules – would open up a vast array of new therapeutics.
Figure I. Multiscale PBPK Model for Stabilized Peptides.
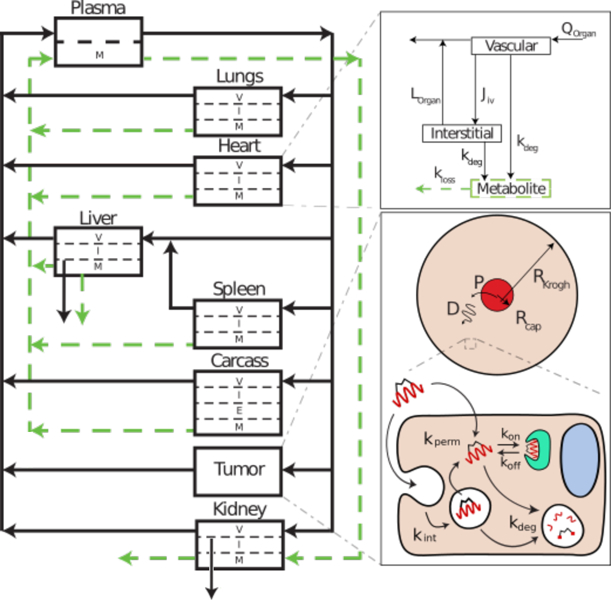
This diagram is a representative illustration of a multi-scale computational physiologically based pharmacokinetic model for a stabilized peptide to help predict the necessary rates and peptide properties for optimal organ distribution, tissue uptake, and target engagement. Each arrow represents a kinetic rate constant that can be measured in vitro or estimated from the literature and updated for a particular drug during the development pipeline (e.g. updating organ transport rates with preclinical biodistribution data). The cellular rate constants can be used to determine if these values are feasible for delivery within the broader organ and systemic distribution. Each organ is shown as a compartment with the relevant sub-compartmental concentrations: vascular (V), interstitial (I), endothelial (E), and metabolite (M). Transport of the peptide drug between these sub-compartments is shown using the heart as an example. The peptide is transported into the vascular sub-compartment via the blood (Q = volumetric blood flow rate). The drug can then be transported convectively (Jiv = transport to interstitium from vascular compartment) into the interstitial portion of the tissue and subsequently exit through lymphatic fluid (Lorgan) or undergo metabolism (kdeg = degradation rate constant) either directly from the vascular compartment (e.g. plasma proteases or uptake into endothelial cells and degradation) or from the interstitium (e.g. extracellular proteases or cellular uptake/degradation). It is often important to track the metabolites, particularly if these include a radiolabel or fluorophore, to properly interpret experimental data. The metabolites can re-enter the blood (kloss) and eventually get eliminated via the kidney or liver. Distribution of the drug in the tissue of interest (e.g. the tumor), is modeled using a Krogh cylinder to analyze potential tissue-level gradients in drug concentration. The Krogh cylinder model is defined using a blood vessel radius (Rcap), cylinder radius (RKrogh), blood vessel permeability (P), and the diffusion coefficient in tissue (D). The tissue of interest is further broken down to the cellular level. The peptide drug enters the cell either by diffusion through the membrane (kperm) or endocytosis (kint) followed by endosomal escape (e.g. kperm). The cytosolic peptide can then bind the target of interest (kon, koff). The peptide will also be degraded, either in the cytosol or in endosomes (kdeg).
References
- [1].Lau JL and Dunn MK, “Therapeutic peptides: Historical perspectives, current development trends, and future directions,” Bioorganic Med. Chem, 2017. [DOI] [PubMed]
- [2].Hopkins AL and Groom CR, “The druggable genome,” Nat. Rev. Drug Discov, vol. 1, no. 9, pp. 727–730, Sep. 2002. [DOI] [PubMed] [Google Scholar]
- [3].Bullock BN, Jochim AL, and Arora PS, “Assessing helical protein interfaces for inhibitor design,” J. Am. Chem. Soc, vol. 133, no. 36, pp. 14220–14223, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lipinski CA, “Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions,” Adv. Drug Deliv. Rev, vol. 101, pp. 34–41, 2016. [DOI] [PubMed] [Google Scholar]
- [5].Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, and Korsmeyer SJ, “Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix.,” Science, vol. 305, no. 5689, pp. 1466–70, Sep. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lau YH, de Andrade P, Quah S-T, Rossmann M, Laraia L, Sköld N, Sum TJ, Rowling PJE, Joseph TL, Verma C, Hyvönen M, Itzhaki LS, Venkitaraman AR, Brown CJ, Lane DP, and Spring DR, “Functionalised staple linkages for modulating the cellular activity of stapled peptides,” Chem. Sci, vol. 5, no. 5, p. 1804, 2014. [Google Scholar]
- [7].Smith GP and Petrenko VA, “Phage Display,” Chem. Rev, vol. 97, no. 2, pp. 391–410, Apr. 1997. [DOI] [PubMed] [Google Scholar]
- [8].Liu R, Barrick JE, Szostak JW, and Roberts RW, “[19] Optimized synthesis of RNA-protein fusions for in vitro protein selection,” 2000, pp. 268–293. [DOI] [PubMed]
- [9].Carvajal LA, Ben-Neriah D, Senecal A, Bernard L, Narayanagari S-R, Kenworthy C, Thiruthuvanathan V, Guerlavais V, Annis DA, Bartholdy B, Will B, Anampa J, Mantzaris I, Aivado MA, Singer RH, Coleman R, Verma A, and Steidl UG, “Dual Inhibition of Mdmx and Mdm2 Using an Alpha-Helical P53 Stapled Peptide (ALRN-6924) As a Novel Therapeutic Strategy in Acute Myeloid Leukemia,” Sci. Transl. Med, vol. 10, no. 437, pp. 1–12, 2018. [Google Scholar]
- [10].Sawyer TK, Partridge AW, Kaan HYK, Juang Y-C, Lim S, Johannes C, Yuen TY, Verma C, Kannan S, Aronica P, Tan YS, Sherborne B, Ha S, Hochman J, Chen S, Surdi L, Peier A, Sauvagnat B, Dandliker PJ, Brown CJ, Ng S, Ferrer F, and Lane DP, “Macrocyclic α helical peptide therapeutic modality: A perspective of learnings and challenges,” Bioorg. Med. Chem, vol. 26, no. 10, pp. 2807–2815, Jun. 2018. [DOI] [PubMed] [Google Scholar]
- [11].Li Y, Rodewald LW, Wertman KF, Wahl GM, Li Y, Rodewald LW, Hoppmann C, Wong ET, Lebreton S, and Safar P, “A Versatile Platform to Analyze Low-Affinity and Transient Protein-Protein Interactions in Living Cells in Real Time,” CellReports, vol. 9, no. 5, pp. 1946–1958, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thean D, Ebo JS, Luxton T, Lee XC, Yuen TY, Ferrer FJ, Johannes CW, Lane DP, and Brown CJ, “Enhancing specific disruption of intracellular protein complexes by hydrocarbon stapled peptides using lipid based delivery,” Sci. Rep, vol. 7, no. 1, pp. 1–11, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schafmeister CE, Po J, and Verdine GL, “An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides,” J. Am. Chem. Soc, vol. 122, no. 6, pp. 12364–12365, 2000. [Google Scholar]
- [14].Blackwell HE and Grubbs RH, “Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis,” Angew. Chemie - Int. Ed, vol. 37, no. 23, pp. 3281–3284, 1998. [DOI] [PubMed] [Google Scholar]
- [15].Moll UM, Petrenko O, Moll UM, and Petrenko O, “The MDM2-p53 Interaction The MDM2-p53 Interaction,” pp. 1001–1008.
- [16].Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL, Hughes H, and Har V, “Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide,” J. Am. Chem. Soc, no. 129, pp. 2456–2457, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ng S, Jafari MR, Matochko WL, and Derda R, “Quantitative synthesis of genetically encoded glycopeptide libraries displayed on M13 phage,” ACS Chem. Biol, vol. 7, no. 9, pp. 1482–1487, 2012. [DOI] [PubMed] [Google Scholar]
- [18].Heinis C, Rutherford T, Freund S, and Winter G, “Phage-encoded combinatorial chemical libraries based on bicyclic peptides,” Nat. Chem. Biol, vol. 5, no. 7, pp. 502–507, Jul. 2009. [DOI] [PubMed] [Google Scholar]
- [19].Peraro L, Zou Z, Makwana KM, Cummings AE, Ball HL, Yu H, Lin YS, Levine B, and Kritzer JA, “Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy,” J. Am. Chem. Soc, vol. 139, no. 23, pp. 7792–7802, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Torres O, D. Y??ksel, Bernardina M, Kumar K, and Bong D, “Peptide tertiary structure nucleation by side-chain crosslinking with metal complexation and double ‘Click’ cycloaddition,” ChemBioChem, vol. 9, no. 11, pp. 1701–1705, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tornøe CW, Christensen C, and Meldal M, “Peptidotriazoles on Solid Phase : [ 1 , 2 , 3 ] -Triazoles by Regiospecific Copper ( I ) -Catalyzed 1 , 3-Dipolar Cycloadditions of Terminal Alkynes to Azides,” J. Org. Chem, vol. 67, no. 9, pp. 3057–3064, 2002. [DOI] [PubMed] [Google Scholar]
- [22].Lau YH, de Andrade P, Sköld N, McKenzie GJ, Venkitaraman AR, Verma C, Lane DP, and Spring DR, “Investigating peptide sequence variations for ‘double-click’ stapled p53 peptides.,” Org. Biomol. Chem, vol. 12, no. 24, pp. 4074–7, Jun. 2014. [DOI] [PubMed] [Google Scholar]
- [23].Zhang L, Navaratna T, and Thurber GM, “A Helix-Stabilizing Linker Improves Subcutaneous Bioavailability of a Helical Peptide Independent of Linker Lipophilicity,” Bioconjug. Chem, vol. 27, no. 7, pp. 1663–1672, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ, “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings,” Adv. Drug Deliv. Rev, vol. 64, no. SUPPL., pp. 4–17, 1997. [DOI] [PubMed] [Google Scholar]
- [25].Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, and Weissleder R, “Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo,” Nat. Commun, vol. 4, pp. 1504–1510, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thurber GM, Reiner T, Yang KS, Kohler RH, and Weissleder R, “Effect of Small-Molecule Modification on Single-Cell Pharmacokinetics of PARP Inhibitors,” Mol. Cancer Ther, vol. 13, no. 4, pp. 986–995, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Craik DJ, Fairlie DP, Liras S, and Price D, “The Future of Peptide-based Drugs,” Chem. Biol. Drug Des, vol. 81, no. 1, pp. 136–147, 2013. [DOI] [PubMed] [Google Scholar]
- [28].Nielsen DS, Shepherd NE, Xu W, Lucke AJ, Stoermer MJ, and Fairlie DP, “Orally Absorbed Cyclic Peptides,” Chem. Rev, vol. 117, no. 12, pp. 8094–8128, 2017. [DOI] [PubMed] [Google Scholar]
- [29].Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, and Kopple KD, “Molecular properties that influence the oral bioavailability of drug candidates,” J. Med. Chem, vol. 45, no. 12, pp. 2615–2623, 2002. [DOI] [PubMed] [Google Scholar]
- [30].Tian S, Li Y, Wang J, Zhang J, and Hou T, “ADME evaluation in drug discovery. 9. Prediction of oral bioavailability in humans based on molecular properties and structural fingerprints,” Mol. Pharm, vol. 8, no. 3, pp. 841–851, 2011. [DOI] [PubMed] [Google Scholar]
- [31].Ahlbach CL, Lexa KW, Bockus AT, Chen V, Crews P, Jacobson MP, and Lokey RS, “Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products,” Future Med. Chem, vol. 7, no. 16, pp. 2121–2130, Oct. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Qian Z, Dougherty PG, and Pei D, “Targeting intracellular protein – protein interactions with cell-permeable cyclic peptides,” Curr. Opin. Chem. Biol, vol. 38, pp. 80–86, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Matsson P, Doak BC, Over B, and Kihlberg J, “Cell permeability beyond the rule of 5,” Adv. Drug Deliv. Rev, vol. 101, pp. 42–61, 2016. [DOI] [PubMed] [Google Scholar]
- [34].Over B, Matsson P, Tyrchan C, Artursson P, Doak BC, Foley MA, Hilgendorf C, Johnston SE, Lee MD, Lewis RJ, McCarren P, Muncipinto G, Norinder U, Perry MWD, Duvall JR, and Kihlberg J, “Structural and conformational determinants of macrocycle cell permeability,” Nat. Chem. Biol, vol. 12, no. 12, pp. 1065–1074, 2016. [DOI] [PubMed] [Google Scholar]
- [35].Villar EA, Beglov D, Chennamadhavuni S, Porco JA, Kozakov D, Vajda S, and Whitty A, “How proteins bind macrocycles,” Nat. Chem. Biol, vol. 10, no. 9, pp. 723–731, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, and Bradner JE, “Direct inhibition of the NOTCH transcription factor complex,” Nature, vol. 462, no. 7270, pp. 182–188, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, and Korsmeyer SJ, “A Stapled BID BH3 Helix Directly Binds and Activates BAX,” Mol. Cell, vol. 24, no. 2, pp. 199–210, 2006. [DOI] [PubMed] [Google Scholar]
- [38].Li YC, Rodewald LW, Hoppmann C, Wong ET, Lebreton S, Safar P, Patek M, Wang L, Wertman KF, and Wahl GM, “A Versatile Platform to Analyze Low-Affinity and Transient Protein-Protein Interactions in Living Cells in Real Time,” Cell Rep, vol. 9, no. 5, pp. 1946–1959, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chu Q, Moellering RE, Hilinski GJ, Kim Y, Grossmann TN, Yeh JT-H, Verdine GL, Yeh T, and Verdine GL, “Towards understanding cell penetration by stapled peptides,” Medchemcomm, vol. 6, no. c, pp. 111–119, 2014. [Google Scholar]
- [40].Huang Y, Feng Q, Yan Q, Hao X, and Chen Y, “Alpha-helical cationic anticancer peptides: a promising candidate for novel anticancer drugs.,” Mini Rev. Med. Chem, vol. 15, no. 1, pp. 73–81, 2015. [DOI] [PubMed] [Google Scholar]
- [41].Rezaei Araghi R, Bird GH, Ryan JA, Jenson JM, Godes M, Pritz JR, Grant RA, Letai A, Walensky LD, and Keating AE, “Iterative optimization yields Mcl-1–targeting stapled peptides with selective cytotoxicity to Mcl-1–dependent cancer cells,” Proc. Natl. Acad. Sci, p. 201712952, 2018. [DOI] [PMC free article] [PubMed]
- [42].Wachter F, Morgan AM, Godes M, Mourtada R, Bird GH, and Walensky LD, “Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting,” Oncogene, vol. 36, no. 15, pp. 2184–2190, Apr. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ward BP, Ottaway NL, Perez-Tilve D, Ma D, Gelfanov VM, Tschöp MH, and DiMarchi RD, “Peptide lipidation stabilizes structure to enhance biological function,” Mol. Metab, vol. 2, no. 4, pp. 468–479, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, and Walensky LD, “Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices,” Nat. Chem. Biol, vol. 12, no. 10, pp. 845–852, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, Cao Y, Lynaugh H, Brown M, Baruah H, Gray LT, Krauland EM, Xu Y, Vásquez M, and Wittrup KD, “Biophysical properties of the clinical-stage antibody landscape,” Proc. Natl. Acad. Sci, vol. 114, no. 5, pp. 944–949, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Giordanetto F and Kihlberg J, “Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties?,” J. Med. Chem, vol. 57, no. 2, pp. 278–295, 2014. [DOI] [PubMed] [Google Scholar]
- [47].Santos GB, Ganesan A, and Emery FS, “Oral Administration of Peptide-Based Drugs: Beyond Lipinski’s Rule,” ChemMedChem, vol. 11, no. 20, pp. 2245–2251, 2016. [DOI] [PubMed] [Google Scholar]
- [48].Chen B, Colgrave ML, Daly NL, Rosengren KJ, Gustafson KR, and Craik DJ, “Isolation and characterization of novel cyclotides from Viola hederaceae: Solution structure and anti-HIV activity of vhl-1, a leaf-specific expressed cyclotide,” J. Biol. Chem, vol. 280, no. 23, pp. 22395–22405, 2005. [DOI] [PubMed] [Google Scholar]
- [49].Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, Neamati N, Shekhtman A, and Camarero JA, “In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide,” J. Am. Chem. Soc, vol. 135, no. 31, pp. 11623–11633, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Christensen EI, Birn H, Storm T, Weyer K, and Nielsen R, “Endocytic Receptors in the Renal Proximal Tubule,” Physiology, vol. 27, no. 4, pp. 223–236, Aug. 2012. [DOI] [PubMed] [Google Scholar]
- [51].Lau J, Bloch P, Schäffer L, Pettersson I, Spetzler J, Kofoed J, Madsen K, Knudsen LB, McGuire J, Steensgaard DB, Strauss HM, Gram DX, Knudsen SM, Nielsen FS, Thygesen P, Reedtz-Runge S, and Kruse T, “Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide,” J. Med. Chem, vol. 58, no. 18, pp. 7370–7380, 2015. [DOI] [PubMed] [Google Scholar]
- [52].Butterworth J, Lin YA, Prielipp R, Bennett J, and James R, “The pharmacokinetics and cardiovascular effects of a single intravenous dose of protamine in normal volunteers,” Anesth. Analg, vol. 94, no. 3, pp. 514–522, 2002. [DOI] [PubMed] [Google Scholar]
- [53].Sarko D, Beijer B, Boy RG, Nothelfer EM, Leotta K, Eisenhut M, Altmann A, Haberkorn U, and Mier W, “The pharmacokinetics of cell-penetrating peptides,” Mol. Pharm, vol. 7, no. 6, pp. 2224–2231, 2010. [DOI] [PubMed] [Google Scholar]
- [54].Huang C-W, Li Z, and Conti PS, “In Vivo Near-Infrared Fluorescence Imaging of Integrin 2 1 in Prostate Cancer with Cell-Penetrating-Peptide-Conjugated DGEA Probe,” J. Nucl. Med, vol. 52, no. 12, pp. 1979–1986, Dec. 2011. [DOI] [PubMed] [Google Scholar]
- [55].Phan NN, Li C, and Alabi CA, “Intracellular Delivery via Noncharged Sequence-Defined Cell-Penetrating Oligomers,” Bioconjug. Chem, vol. 29, no. 8, pp. 2628–2635, Aug. 2018. [DOI] [PubMed] [Google Scholar]
- [56].Collado Camps E and Brock R, “An opportunistic route to success: Towards a change of paradigm to fully exploit the potential of cell-penetrating peptides,” Bioorg. Med. Chem, vol. 26, no. 10, pp. 2780–2787, Jun. 2018. [DOI] [PubMed] [Google Scholar]
- [57].Naylor MR, Bockus AT, Blanco MJ, and Lokey RS, “Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets,” Curr. Opin. Chem. Biol, vol. 38, pp. 141–147, 2017. [DOI] [PubMed] [Google Scholar]
- [58].Fineman MS, Mace KF, Diamant M, Darsow T, Cirincione BB, Booker Porter TK, Kinninger LA, and Trautmann ME, “Clinical relevance of anti-exenatide antibodies: safety, efficacy and cross-reactivity with long-term treatment,” Diabetes, Obes. Metab, vol. 14, no. 6, pp. 546–554, Jun. 2012. [DOI] [PubMed] [Google Scholar]
- [59].Bhatnagar S, Deschenes E, Liao J, Cilliers C, and Thurber GM, “Multichannel Imaging to Quantify Four Classes of Pharmacokinetic Distribution in Tumors,” J. Pharm. Sci, vol. 103, no. 10, pp. 3276–3286, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hicks KO, Siim BG, Jaiswal JK, Pruijn FB, Fraser AM, Patel R, Hogg A, Liyanage HDS, Dorie MJ, Brown JM, Denny WA, Hay MP, and Wilson WR, “Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors,” Clin. Cancer Res, vol. 16, no. 20, pp. 4946–4957, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thurber GM, Schmidt MM, and Wittrup KD, “Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance,” Adv. Drug Deliv. Rev, vol. 60, no. 12, pp. 1421–1434, Sep. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Thurber GM, Figueiredo JL, and Weissleder R, “Multicolor Fluorescent Intravital Live Microscopy (FILM) for Surgical Tumor Resection in a Mouse Xenograft Model,” PLoS One, vol. 4, no. 11, p. e8053, Nov. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cilliers C, Guo H, Liao J, Christodolu N, and Thurber GM, “Multiscale Modeling of Antibody-Drug Conjugates: Connecting Tissue and Cellular Distribution to Whole Animal Pharmacokinetics and Potential Implications for Efficacy,” AAPS J, vol. 18, no. 5, pp. 1117–1130, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Baish JW, Gazit Y, Berk DA, Nozue M, Baxter LT, and Jain RK, “Role of Tumor Vascular Architecture in Nutrient and Drug Delivery: An Invasino Percolation-Based Network Model,” Microvasc. Res, vol. 51, pp. 327–346, 1996. [DOI] [PubMed] [Google Scholar]
- [65].Phan J, Li Z, Kasprzak A, Li B, Sebti S, Guida W, Schönbrunn E, and Chen J, “Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX,” J. Biol. Chem, vol. 285, no. 3, pp. 2174–2183, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To K-H, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, and Sawyer TK, “Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy.,” Proc. Natl. Acad. Sci. U. S. A, vol. 110, no. 36, pp. E3445–54, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pazgier M, Liu M, Zou G, Yuan W, Li C, Li C, Li J, Monbo J, Zella D, Tarasov SG, and Lu W, “Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX,” Proc. Natl. Acad. Sci, vol. 106, no. 12, pp. 4665–4670, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Thompson PA, Drissi R, Muscal JA, Panditharatna E, Fouladi M, Ingle AM, Ahern CH, Reid JM, Lin T, Weigel BJ, and Blaney SM, “A phase I trial of imetelstat in children with refractory or recurrent solid tumors: a Children’s Oncology Group Phase I Consortium Study (ADVL1112).,” Clin. Cancer Res, vol. 19, no. 23, pp. 6578–84, Dec. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P, Wright WE, and Shay JW, “In vivo inhibition of lung cancer by GRN163L: A novel human telomerase inhibitor,” Cancer Res, vol. 65, no. 17, pp. 7866–7873, 2005. [DOI] [PubMed] [Google Scholar]
- [70].Asai A, Oshima Y, Yamamoto Y, aki Uochi T, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan R, Wunder E, Piatyszek M, Li S, Chin AC, Harley CB, and Gryaznov S, “A novel telomerase template antagonist (GRN163) as a potential anticancer agent,” Cancer Res, vol. 63, no. 14, pp. 3931–3939, 2003. [PubMed] [Google Scholar]
- [71].Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, and Gryaznov SM, “Lipid modification of GRN163, an N3ʹ→P5ʹ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition,” Oncogene, vol. 24, no. 33, pp. 5262–5268, 2005. [DOI] [PubMed] [Google Scholar]
- [72].Siegwart DJ, Whitehead KA, Nuhn L, Sahay G, Cheng H, Jiang S, Ma M, Lytton-Jean A, Vegas A, Fenton P, Levins CG, Love KT, Lee H, Cortez C, Collins SP, Li YF, Jang J, Querbes W, Zurenko C, Novobrantseva T, Langer R, and Anderson DG, “Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery,” Proc. Natl. Acad. Sci, vol. 108, no. 32, pp. 12996–13001, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Akinc A, Lynn DM, Anderson DG, and Langer R, “Parallel synthesis and biophysical characterization of a degradable polymer library for gene delivery,” J. Am. Chem. Soc, vol. 125, no. 18, pp. 5316–5323, 2003. [DOI] [PubMed] [Google Scholar]
- [74].Haining WN, Anderson DG, Little SR, von Berwelt-Baildon MS, Cardoso AA, Alves P, Kosmatopoulos K, Nadler LM, Langer R, and Kohane DS, “pH-Triggered Microparticles for Peptide Vaccination,” J. Immunol, vol. 173, no. 4, pp. 2578–2585, Aug. 2004. [DOI] [PubMed] [Google Scholar]
- [75].Grossmann TN, Yeh JT-H, Bowman BR, Chu Q, Moellering RE, and Verdine GL, “Inhibition of oncogenic Wnt signaling through direct targeting of -catenin,” Proc. Natl. Acad. Sci, vol. 109, no. 44, pp. 17942–17947, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Walensky LD and Bird GH, “Hydrocarbon-stapled peptides: Principles, practice, and progress,” J. Med. Chem, vol. 57, no. 15, pp. 6275–6288, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, Choong ML, Lee MA, Yurlova L, Zolghadr K, Joseph TL, Verma CS, and Lane DP, “Stapled Peptides with Improved Potency and Specificity That Activate p53,” ACS Chem. Biol, vol. 8, no. 3, pp. 506–512, Mar. 2013.23214419 [Google Scholar]
- [78].Shmueli RB, Anderson DG, and Green JJ, “Electrostatic surface modifications to improve gene delivery,” Expert Opin. Drug Deliv, vol. 7, no. 4, pp. 535–550, Apr. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Peraro L, Deprey KL, Moser MK, Zou Z, Ball HL, Levine B, and Kritzer JA, “Cell Penetration Profiling Using the Chloroalkane Penetration Assay,” J. Am. Chem. Soc, p. jacs.8b06144, Aug. 2018. [DOI] [PMC free article] [PubMed]
- [80].O’Neil KT, Hoess RH, Jackson SA, Ramachandran NS, Mousa SA, and DeGrado WF, “Identification of novel peptide anagonists for GPIIb/IIIa from a conformationally constrained phage peptide library,” vol. 14, pp. 509–515, 1992. [DOI] [PubMed] [Google Scholar]
- [81].Kruziki MA, Sarma V, and Hackel BJ, “Constrained combinatorial libraries of Gp2 proteins enhance discovery of PD-L1 binders,” ACS Comb. Sci, 2018. [DOI] [PMC free article] [PubMed]
- [82].Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LGJ, Muyldermans S, Wyns L, and Matagne A, “Single-domain antibody fragments with high conformational stability,” Protein Sci, vol. 11, no. 3, pp. 500–515, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fox JM, Zhao M, Fink MJ, Kang K, and Whitesides GM, “The Molecular Origin of Enthalpy/Entropy Compensation in Biomolecular Recognition,” Annu. Rev. Biophys, vol. 47, no. 1, pp. 223–250, May 2018. [DOI] [PubMed] [Google Scholar]
- [84].Sia SK, a Carr P, Cochran AG, Malashkevich VN, and Kim PS, “Short constrained peptides that inhibit HIV-1 entry.,” Proc. Natl. Acad. Sci. U. S. A, vol. 99, no. 23, pp. 14664–14669, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang J, Yadav V, Smart AL, Tajiri S, and Basit AW, “Toward oral delivery of biopharmaceuticals: An assessment of the gastrointestinal stability of 17 peptide drugs,” Mol. Pharm, vol. 12, no. 3, pp. 966–973, 2015. [DOI] [PubMed] [Google Scholar]
- [86].Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He X, Gavathiotis E, Sodroski JG, and Walensky LD, “Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic.,” Proc. Natl. Acad. Sci. U. S. A, vol. 107, no. 32, pp. 14093–8, Aug. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Novo Nordisk, “Novo Nordisk successfully completes the first phase 3a trial, PIONEER 1, with oral semaglutide,” no. February, pp. 24–26, 2018. [Google Scholar]
- [88].Doak BC, Over B, Giordanetto F, and Kihlberg J, “Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates,” Chem. Biol, vol. 21, no. 9, pp. 1115–1142, 2014. [DOI] [PubMed] [Google Scholar]
- [89].Thompson SJ, Hattotuwagama CK, Holliday JD, and Flower DR, “On the hydrophobicity of peptides: Comparing empirical predictions of peptide log P values.,” Bioinformation, vol. 1, no. 7, pp. 237–41, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Valko KL, Ivanova-Berndt G, Beswick P, Kindey M, and Ko D, “Application of biomimetic HPLC to estimate lipophilicity, protein and phospholipid binding of potential peptide therapeutics,” ADMET DMPK, vol. 6, no. 2, p. 162, Jun. 2018. [Google Scholar]