

SUMMARY
The development of autoimmune disease type 1 diabetes (T1D) is determined by both genetic background and environmental factors. Environmental triggers include RNA viruses, particularly coxsackie-virus (CV), but how they induceT1D is not understood. Here, we demonstrate that deletion of the transcription factor hypoxia-inducible factor-1α (HIF-1α) from β cells increases the susceptibility of non-obese diabetic (NOD) mice to environmentally triggered T1D from coxsackieviruses and the β cell toxin streptozotocin. Similarly, knockdown of HIF-1α in human islets leads to a poorer response to coxsackievirus infection. Studies in coxsackievirus-infected islets demonstrate that lack of HIF-1α leads to impaired viral clearance, increased viral load, inflammation, pancreatitis, and loss of β cell mass. These findings show an important role for β cells and, specifically, lack of β cell HIF-1α in the development of T1D. These data suggest new strategies for the prevention of T1D.
Graphical Abstract
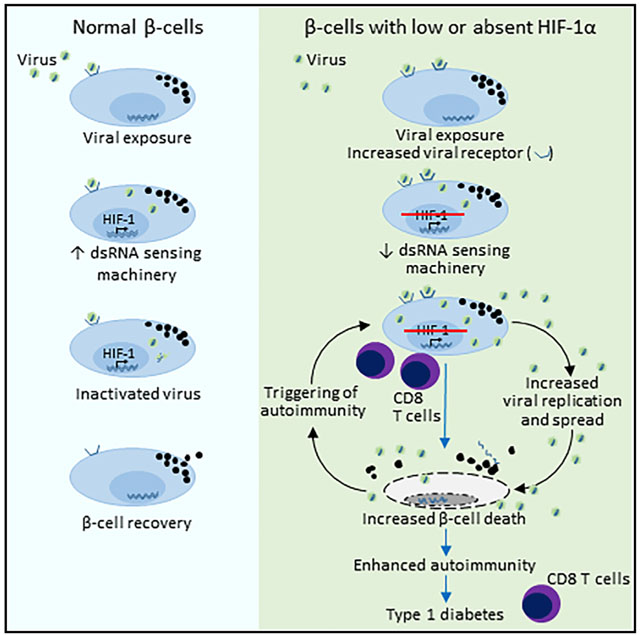
In Brief
Lalwani et al. describe a role for β cell hypoxia-inducible factor-1α (HIF1a) in determining whether β cell injury is followed by resolution and normal function or ongoing injury, autoimmunity, and type 1 diabetes.
INTRODUCTION
Type 1 diabetes (T1D) is characterized by immune-mediated destruction of insulin-producing pancreatic β cells, leading to an inability to maintain normal blood glucose levels (van Belle et al., 2011). The incidence of T1D has been increasing steadily across the world. Although there is a strong genetic component in T1D susceptibility, with identical twins having a 30%−50% concordance (Japan Diabetes Society, Committee on Diabetic Twins, 1988; Metcalfe et al., 2001; Nisticò et al., 2012), genetics alone cannot explain the rapid increases in prevalence. Environmental factors are important for T1D development. It is generally accepted that environmental triggers can initiate pancreatic islet inflammation (insulitis) in susceptible individuals. However, the triggers for β cell autoimmunity and destruction are not clearly understood and almost certainly vary between people and geographic locations (Knip and Simell, 2012; Knip et al., 2005; Piłaciński and Zozulińska-Ziółkiewicz, 2014).
Exposure to RNA viruses is a common environmental event and a proposed trigger of T1D (Rewers and Ludvigsson, 2016). For example, the incidence of T1D in babies with congenital rubella exposure is markedly increased, with 12% already having T1D by 17 years of age (Ginsberg-Fellner et al., 1984). Enteroviruses, particularly coxsackievirus (CV), including strain B4 (CVB4), are strongly associated with T1D (Banatvala et al., 1985; Coppieters et al., 2012; Frisk et al., 1992; Yoon et al., 1978). Coxsackieviruses are positive-sense single-stranded RNA viruses that are β cell tropic and enter β cells via the coxsackievirus and adenovirus receptor (CAR). Once inside the β cell, they replicate and may induce β cell death (Chehadeh et al., 2000; Nair et al., 2010; Foulis et al., 1987; Bottazzo et al., 1985). Coxsackieviruses may stimulate β cell autoimmunity by directly causing β cell death (releasing potential autoantigens), molecular mimicry (peptides that mimic autoantigens), bystander activation (activation and diversification of autoreactive T cells), and viral persistence (persistent viral infection with associated cell damage) (Coppieters et al., 2012; van Belle et al., 2011).
It is becoming increasingly clear that β cells play a role in the initiation and progression of T1D. When β cells die, some of their contents are released, including potential autoantigens that can initiate priming of the immune system in susceptible individuals. This has been elegantly shown in animals with the “wave” of postnatal β cell death and remodeling that occurs at ~2 weeks of age (Turley et al., 2003). Accordingly, greater frequency of β cell death would be associated with a greater opportunity for immune priming and ongoing inflammation. Once self-tolerance to β cell antigens is lost, there is the increased potential for antigen spreading and β cell loss, perpetuating this cycle (von Herrath et al., 2007). Hypoxia-inducible factor-1α (HIF-1α) is an oxygen-sensing transcription factor that coordinates cellular responses to hypoxia (Ratcliffe et al., 1998). We have previously shown that HIF-1α improves β cell function in C57BL/6 mice (Gunton et al., 2005). HIF-1α protein levels are increased not only by hypoxia but also by cytokines, inflammation, reactive oxygen species, and low iron levels (Bilton and Booker, 2003; Hwang and Lee, 2011; Peyssonnaux et al., 2007), all of which would be potential stimuli in the setting of infection.
The role of the β cell in T1D development is an area of increasing interest (Eizirik and Op de Beeck, 2018), and we hypothesized that β cell HIF-1α (βHIF-1α) would assist in halting the cycle of β cell death, release of autoantigens, immune priming, and further β cell death. This was tested using non-obese diabetic (NOD) mice with β cell-specific deletion of HIF-1α (referred to hereafter as bHIF-1α). We found that after exposure to either coxsackieviruses or a β cell toxin, βHIF-1α mice developed T1D much more frequently than controls. This β cell-specific mouse model shows susceptibility to T1D after a range of environmental insults, revealing a critical role for β cells themselves in T1D development. Our results not only demonstrate an important role for β cell HIF-1α in T1D susceptibility but also suggest that coxsackievirus vaccines may prevent T1D in susceptible individuals.
RESULTS
Mice Lacking βHIF-1α Develop T1D after Exposure to CVB4
To examine the role of β cell HIF-1α in virally induced T1D, we infected 8-week-old male control (NOD, FC, or RIP-Cre) and βHIF-1α mice intraperitoneally with CVB4 at a dose of 105 plaque-forming units (pfus) and monitored them for diabetes onset until 105 days post-infection (dpi). Baseline glucose tolerance testing pre-infection showed no differences between 8-week-old male control (NOD, FC, and RIP-Cre) and βHIF-1α mice (Figure 1A). Similarly, baseline islet architecture showed a normal distribution of insulin, glucagon, and somatostatin cells in βHIF-1α mice (Figure S1A). The experimental timeline is shown in Figure S2.
Figure 1. βHIF-1α Mice Develop Diabetes and Pancreatic Fibrosis after CVB4.

(A) Baseline glucose tolerance in NOD controls, floxed controls, RIP-Cre controls, and βHIF-1α mice.
(B) Kaplan-Meier diabetes-free survival in CVB4-infected mice. ***p < 0.001 for βHIF-1α versus controls.
(C) Body weights after CVB4 infection. ***p < 0.001.
(D) Diabetes-free survival after splenocyte adoptive transfer. ***p < 0.001.
(E) Representative H&E photomicrographs of the pancreas from CVB4-infected FC and βHIF-1α mice, at the end of the study. Scale bar, 200 μm.
(F) Random-fed serum insulin concentrations (n = 6 per group). *p < 0.05 versus CVB4-infected FC; ###p < 0.001 versus βHIF-1α non-diabetic mice.
(G) β cell mass in uninfected FC mice and βHIF-1α mice, and in CVB4-infected FC and diabetic and non-diabetic βHIF-1α mice (n = 6 per group). ****p < 0.0001 versus CVB4-infected FC; ##p < 0.01 versus non-diabetic βHIF-1α infected mice.
(H) Sirius Red staining of pancreatic sections from CVB4-infected FC and βHIF-1α mice, showing pancreatic fibrosis (black arrows). Scale bar, 500 μm.
(I) Pancreas fat area (% of total area) in uninfected FC and βHIF-1α mice, and in FC and βHIF-1α mice after CVB4 infection (n = 6 per group). ****p < 0.0001 versus FC infected, ###p < 0.01 versus non-diabetic βHIF-1α infected mice.
(J) Quantification of collagen area (% of pancreas area). **p < 0.01 versus FC infected, ###p < 0.001 versus non-diabetic βHIF-1α infected mice.
Data are means ± SEMs.
However, by 70 dpi, 13 of 22 (59%) βHIF-1α mice had developed florid diabetes (blood glucose level [BGL] ≥20 mmol/L or 360 mg/dL) compared to no NOD, FC, or RIP-Cre controls (p < 0.0001 overall; Figure 1B). No uninfected male βHIF-1α mice developed diabetes. In the diabetic βHIF-1α mice, the increased BGLs were accompanied by a significant reduction in body weight (p = 0.0002, ANOVA) (Figure 1C). Of the 22 infected βHIF-1α mice, 9 required pancreatic enzyme replacement (Creon Forte) because they displayed >10% body weight loss, compared to none of the controls (p < 0.01). Those 9 mice regained normal body weight, and Creon was ceased by 50 days. Of the 9 βHIF-1α mice that received Creon, 8 later developed diabetes.
To confirm that the diabetes in βHIF-1α mice was T1D, we adoptively transferred splenocytes from either diabetic βHIF-1α or duration-matched (non-diabetic) FC mice into diabetes-resistant immunodeficient (NOD-severe combined immunodeficiency [SCID]) recipients (Christianson et al., 1993). As shown in Figure 1D, all of the recipients that were given splenocytes from diabetic βHIF-1α mice developed diabetes within 49 days (100%; 20 of 20). None of the recipients that were given splenocytes from duration-matched FC mice developed diabetes (0 of 10, p = 0.0005 overall).
Consistent with the development of T1D, diabetic βHIF-1α mice had an 86% reduction in β cell mass compared to infected FC mice (Figures 1E and 1F), and random-fed serum insulin concentrations were reduced by 56% in diabetic βHIF-1α mice compared to infected FC mice (Figure 1G).
Previous reports have suggested that the chronic stage of CVB4-induced diabetes is characterized by manifestations such as insulitis, sustained presence of inflammatory infiltrates, fat replacement, and fibrosis (Ramsingh, 1997; Coppieters et al., 2012). Histological examination showed that diabetic βHIF-1α mice had increased fat area per total pancreas area (Figures 1H and 1I). To examine the severity of fibrosis, pancreatic sections were stained with Sirius Red to visualize collagen in red (Figure 1H). Diabetic βHIF-1α mice had an increased collagen area per total pancreas area (Figure 1J). Islets in FC and βHIF-1α mice did not show differences in glucagon-positive or somatostatin-positive cells 21 dpi (Figure S3), indicating no obvious effect of β cell HIF-1α deletion on other islet cells.
T1D in humans has a similar incidence in males and females. As previously mentioned, our studies were performed in male NOD mice unless stated otherwise, as female NOD mice display increased insulitis from an early age (Figure S1B) and a spontaneous T1D incidence of ~70% by 40 weeks. Increased insulitis increases the risk of diabetes (Horwitz et al., 1998, 1999). For completeness, we tested females for diabetes susceptibility after CVB4. Female mice infected at 8 weeks of age with 105 particle-forming unit CVB4 intraperitoneally revealed an increased rate of diabetes in βHIF-1α mice (Figures S4A and S4B). By 41 dpi, 50% of βHIF-1α mice (5 of 10) had developed diabetes compared to 20% of NOD (1 of 5) and 0% of FC (0 of 5) mice (p = 0.049). As in the males, mice that developed diabetes lost weight (Figure S4B). These data confirm that in both males and females, β cell HIF-1α is needed to avoid diabetes after CVB4 infection.
Heterozygous βHIF-1α Mice Also Develop T1D after Coxsackievirus Infection
In some circumstances, genetic heterozygosity may provide host resistance to infectious diseases and viral clearance (Penn et al., 2002). To investigate whether β cell HIF-1α heterozygosity was sufficient to provide resistance against CVB4-induced diabetes, we infected male FC and βHIF-1α heterozygous mice (RIP-Cre+, HIF-1α fl/wild type [WT]) with CVB4 intraperitoneally. Within 4 weeks, 33% (4 of 12) βHIF-1α heterozygotes developed diabetes compared to 0 controls by 15 weeks (0 of 12; p = 0.032, Figure S4C, and weight curves in Figure S4D). This demonstrates that diabetes risk is also increased by heterozygous deletion. It was interesting to observe that diabetes incidence was numerically lower than in homozygous mice, suggesting the possibility of an HIF-1α gene-dose-response effect in β cells.
Oral Exposure to CVB4 Induces T1D in βHIF-1α Mice
In humans, coxsackievirus transmission occurs primarily by the fecal-oral route. To mimic this situation, we tested the effect of oral CVB4 infection (105 particle-forming unit/mouse) in a new cohort of NOD, FC, and bHIF-1α mice. By 95 days, 6 of 13 (46%) of CVB4-infected βHIF-1α mice had developed diabetes compared to 0 of 16 controls (8 NOD and 8 FC; p = 0.0096, Figure S5A, and weight curve in Figure S5B). We note that diabetes onset was somewhat delayed after oral rather than intraperitoneal inoculation of coxsackievirus. Consistent with the previous results, diabetic βHIF-1α mice exhibited reduced β cell mass and serum insulin concentrations compared to infected FC mice and non-diabetic βHIF-1α mice (Figures S5C–S5E). These findings demonstrate that Hif1a is important for β cell resistance to CVB4 infection itself or for β cells to survive the effects of CVB4 infection.
βHIF-1α Mice Also Develop T1D after Exposure to CVB1
To determine whether the effects of βHIF-1α deletion were CVB4 specific or more generalized, we investigated whether a different strain of coxsackievirus would also induce diabetes. A new cohort of control and βHIF-1α mice was infected with CVB1 and monitored for diabetes onset until 105 dpi. Of 9 βHIF-1α mice, 4 (44%) developed diabetes, compared to 0 of 6 NOD and 0 of 8 FC controls (p = 0.02; Figures 2A and 2B). Similar to CVB4 infection, 4 of 9 βHIF-1α mice required pancreatic enzyme replacement (Creon Forte) after CVB1 versus 0 controls (p = 0.014). All of the mice requiring Creon Forte in this experiment went on to develop diabetes.
Figure 2. βHIF-1α Mice Develop T1D When Exposed to CVB1.

(A) Kaplan-Meier plot of diabetes-free survival in mice infected with CVB1. *p < 0.05 overall.
(B) Weight of mice post-CVB1 infection. ****p < 0.0001, ANOVA.
(C) Insulin and H&E pancreatic sections at sacrifice after CVB1 infection. Scale bar, 200 μm.
(D) β cell mass in FC (black bars) and in non-diabetic (gray bars) and diabetic (white bars) βHIF-1α mice (n = 8, 5, and 4 per group, respectively) at the end of the study post-CVB1 infection. **p < 0.01 versus FC mice; ###p < 0.001 versus non-diabetic βHIF-1α mice.
(E) Serum insulin in FC (black bars) and in non-diabetic (gray bars) and diabetic (white bars) βHIF-1α mice (n = 8, 5, and 4 per group, respectively) at the end of the study. *p < 0.05 versus FC mice.
(F) Kaplan-Meier plots for diabetes-free survival in NOD-SCID recipients that were given splenocytes from CVB1-infected FC (black squares, dashed line) or βHIF-1α mice (white circles, solid line). n = 10–15 recipients per group. ****p < 0.0001.
Data are means ± SEMs.
These diabetic βHIF-1α mice also exhibited reduced β cell mass and serum insulin concentrations compared to both infected FC and non-diabetic βHIF-1α mice (Figures 2C–2E). Absolute confirmation that the diabetes was autoimmune was provided by adoptive transfer studies. All of the recipients of splenocytes from diabetic βHIF-1α mice developed diabetes within 42 days of adoptive transfer (10 of 10) compared to 0 FC recipients (0 of 10, p < 0.0001 overall; Figure 2F).
Overall, these results demonstrate that β cell HIF-1α plays an important role in determining the fate of β cells after viral infection. Absence of β cell HIF-1α predisposes to development of T1D after coxsackievirus infection, irrespective of viral strain (B4 or B1), route of infection (intraperitoneal or oral), gender, or homo- or heterozygosity for a null HIF-1α allele in β cells.
Absence of β Cell HIF-1α Modulates Incidence and Course of Disease in MLDS-Induced Diabetes
Next, we hypothesized that β cells lacking HIF-1α would also exhibit susceptibility to other mechanisms of β cell death. The multiple low-dose stretozotocin (MLDS) model gives 5 sub-diabetogenic doses of the β cell toxin streptozotocin and causes only minor β cell death, but MLDS can cause T1D in susceptible mice (Rossini et al., 1977). To examine the role of HIF-1α in this process, we performed MLDS treatment in control and βHIF-1α mice (timeline in Figure S2B). At day 10 post-MLDS, all of the groups of NOD, FC, and βHIF-1α mice had equally mildly impaired glucose tolerance, suggesting that MLDS had an equivalent effect on all of the strains. However, by the end of the study, all 11 βHIF-1α mice (100%) had developed diabetes, compared to 5 of 18 controls (1 of 6 NOD and 4 of 12 FC, p = 0.0010). Proof that MLDS diabetes was immune was provided by adoptive transfer of splenocytes from diabetic FC and βHIF-1α mice into NOD-SCID recipients. By day 21 after adoptive transfer, 100% of the recipients (10 of 10) that were given sple-nocytes from diabetic βHIF-1α mice had developed autoimmune diabetes. At day 35 after adoptive transfer, 100% of the recipients (10 of 10) that received splenocytes from diabetic FC mice also developed autoimmune diabetes. These results demonstrate that increased susceptibility to T1D in βHIF-1α mice was not limited to only viruses.
CVB4-Induced T1D Is Accompanied by Increased Pancreatic Viral Load and Classic Manifestations of Pancreatitis
To investigate how β cell HIF-1α deficiency is involved in the initial steps of viral persistence and clearance, we infected a new cohort of FC and βHIF-1α mice with CVB4 and sacrificed them at 4, 7, and 21 dpi (timeline in Figure S2C). As shown in Figure 3A, immunohistochemistry for the CVB4 capsid protein VP1 showed no differences at 4 or 7 dpi. However, at 21 dpi and at the end of the long-term study, there was more VP1 staining in βHIF-1α mice than in controls. This suggests that β cells in the βHIF-1α mice may have an impaired ability to clear CVB4 or the VP1 protein.
Figure 3. CVB4-Induced Diabetes Is Accompanied by Increased Viral Load and Classic Manifestations of Pancreatitis.

(A) Representative photomicrographs of capsid protein VP1 (brown 3,3′-diaminobenzidine [DAB] chromogen) with H&E counterstain of pancreatic sections from uninfected and CVB4-infected FC and βHIF-1α mice at 4, 7, and 21 dpi and the end of the study. Scale bar, 200 μm.
(B) Pancreatic viral load in CVB4-infected FC (black bars) and βHIF-1α (white bars) at 4, 7, and 21 dpi. Virus was undetectable at 21 dpi. ***p < 0.001 versus FC mice.
(C) β cell mass in uninfected and 21 dpi FC (black bars) and βHIF-1α (white bars) mice (n = 6 per group). ****p < 0.0001 versus FC mice, ####p < 0.0001 versus 0 dpi FC mice.
(D) Pancreas weight normalized to body weight in uninfected and 4,7, and 21 dpi FC (black bars) and βHIF-1α (white bars) mice (n = 6 per group) *p < 0.05 versus FC mice.
(E) Serum amylase in CVB4-infected FC and βHIF-1α mice at 4, 7, and 21 dpi (n = 6 per group). *p < 0.05 versus FC mice (n = 6 per group).
(F) Spleen viral load after CVB4 infection at 4, 7, and 21 dpi.
Data are means ± SEMs.
To investigate this, we performed plaque assays, which measure replicating virus. Plaque assays showed that βHIF-1α mice had a significantly higher viral load than FC mice at 7 dpi (0.96 ± 0.12 versus 0.34 ± 0.06 plaque-forming units/μL; p = 0.0001). By 21 dpi, infectious virus was cleared from the pancreas in both groups (Figures 3B and S1B). Plaque assays showed very low levels of virus in the spleen by 21 days, with no difference between genotypes (Figure 3C).
Although it is possible to preserve islet mass during pancreatic infection, this was not the case in βHIF-1α mice; at 21 dpi βHIF-1α mice already had a 77% reduction in β cell mass compared to baseline (p < 0.0001; Figure 3D). CVB4-infected βHIF-1α mice also had extensive loss of exocrine pancreatic tissue compared to FC mice. Total pancreas weight normalized to body weight was reduced by 48% in βHIF-1α versus FC mice (Figure 3E). Elevated serum amylase is a classic marker of pancreatitis. Serum amylase activity was significantly elevated 7 days after CVB4 infection in βHIF-1α mice (50% higher than FC mice, p < 0.026; Figure 3F).
In contrast to the effects on β cell mass, glucagon and somatostatin expression was not differentially affected. Glucagon and somatostatin stains did not reveal significant glucagon or somatostatin depletion in the pancreatic sections scanned from CVB4-infected βHIF-1α mice (Figure S3).
These results indicated that CVB4-induced T1D in βHIF-1α mice included classic manifestations of pancreatitis, with reduced pancreas weight, hyperamylasemia, higher pancreatic viral load, extensive destruction of acinar cells, and replacement by fat. The observation of replacement of tissue by fat is consistent with previous reports in NOD, and studies in other mice have indicated that following coxsackievirus infection, there is a loss of acinar cells and replacement with cells of the fat lineage (e.g., Horwitz et al., 1998).
CVB4 Infection Leads to Increased Insulitis and Pancreatic Fibrosis in βHIF-1α Mice
Previous reports in NOD mice have suggested that autoimmune diabetes is accelerated in the presence of critical threshold of insulitis (Serreze et al., 2005; Horwitz et al., 1998, 1999). Examination of pancreatic sections from uninfected and CVB4-infected FC and βHIF-1α mice revealed an increase in insulitis at the islet periphery of βHIF-1α mice from as early as 4 dpi (Figure 4A; insulitis indicated by yellow arrows). Similarly, more immune cells were present in the insulitis area and in the infiltrate around the is-lets of βHIF-1α mice at 4,7, and 21 dpi, compared to FC mice (Figures 4B and 4C). There were no differences in the number of islets isolated from βHIF-1α and FC mice until 7 dpi, when βHIF-1α mice had significantly fewer islets isolated per pancreas (data not shown). Furthermore, due to significantly reduced pancreatic size and weight, β cell mass, and presence of pancreatic tissue fibrosis in βHIF-1α mice at 21 dpi, islet isolation was challenging for comparison purposes with FC mice (data not shown).
Figure 4. CVB4 Infection Leads to Increased Insulitis and Pancreatic Fibrosis in βHIF-1α Mice.

(A) Insulin and H&E staining of pancreatic sections of uninfected and CVB4-infected FC and βHIF-1α mice at 4, 7, and 21 dpi, showing insulitis (yellow arrows) and fat accumulation. Scale bar, 200 μm.
(B) Number of immune cells in insulitic area in FC (black bars) and βHIF-1α (white bars) mice at 4, 7, and 21 dpi (n = 6 per group). *p < 0.05 and **p < 0.01 versus FC mice.
(C) Immune cells in islet infiltrates in FC (black bars) and βHIF-1α (white bars) mice at 4, 7, and 21 dpi. n = 6 per group. *p < 0.05, **p < 0.01 versus FC mice.
(D) Pancreatic fat area (% of total area) in FC (black bars) and βHIF-1α (white bars) mice at 21 dpi (n = 6 per group). **p < 0.01 versus FC mice.
(E) Collagen area per pancreas (%) in FC (black bars) and βHIF-1α (white bars) mice at 21 dpi (n = 6 per group). ***p < 0.001 versus FC mice.
(F) Representative photomicrographs of Sirius Red staining from FC and βHIF-1α mice at 21 dpi, showing the degree of pancreatic fibrosis. Scale bar, 500 μm.
(G) FACS analysis: proportions of total lymphocytes, CD3+ T cells, CD4+ T helper6 cells, CD8+ cytotoxic T cells, granzyme B, CD19+ B cells, and macrophages at 7 dpi. Data are representative of 3 independent experiments, with n = 6 mice per group in each experiment. #Comparisons with uninfected and infected FC and βHIF-1α mice; *comparisons between infected FC and βHIF-1α mice.
Data are means ± SEMs.
At 21 dpi, pancreata from βHIF-1α mice had more pancreatic fat (72% of histological area versus 48% in controls, p = 0.0025) and were also more fibrotic compared to FC mice, with increased collagen area per pancreas area (9% versus 4% in controls, p = 0.0006; Figures 4D–4F). The immune infiltrates from the islets of FC and βHIF-1α mice were further characterized by flow cytometry. Total hematopoietic cells (CD45+) from FC and βHIF-1α mice at 7 dpi were sorted to yield T cells (CD3+ and CD4+ or CD8+ and CD25+ for activation), B cells (CD19+), and macrophages (F4/80+, CD11c+, and CD11b−). Intracellular staining for granzyme B, an enzyme secreted by CD8+ T cells involved in β cell death (van Belle et al., 2011), was also performed.
CVB4 infection led to a significant increase in total lymphocytes in both βHIF-1α and FC mice at 7 days (Figure 4G), but without significant differences between the 2 groups. However, total T cells (CD3+) were significantly elevated in the infected βHIF-1α mice. CD4+ T cells did not differ between infected βHIF-1α mice compared to FC mice, but infiltrates from infected βHIF-1α mice did contain increased numbers of CD8+ T cells. CD8+ cells play a critical role in immune-mediated cell death (Tsai et al., 2008; van Belle et al., 2011). βHIF-1α mice also had elevated granzyme B staining within their CD8+ T cells (p = 0.0001 versus infected FC mice). This indicates that CD8+ effector T cells from βHIF-1α mice are likely to be more damaging as well as more numerous. B cells (CD19+) were significantly elevated in infiltrates from βHIF-1α mice after infection (p < 0.0001), while this did not occur in infected FC mice. Lastly, CVB4 infection led to increased numbers of macrophages in both βHIF-1α and FC mice; however, this induction was significantly more pronounced in infected βHIF-1α mice.
These results indicate that β cell HIF-1α has a role in the induction of the immune response in response to β cell and islet CVB4 infection. It follows that the elevated inflammatory milieu in βHIF-1α mice would contribute to greater fibrosis and autoimmune diabetes in βHIF-1α mice.
Altered Gene Expression in Islets from CVB4-Infected βHIF-1α Mice In Vivo and In Vitro
As HIF-1α is part of the HIF1 transcription factor, we subsequently investigated changes in gene expression in isolated islets from CVB4-infected bHIF-1α and FC mice, at 4, 7, and 14 dpi. Enterovirus (EV) RNA expression was measured, as well as that of its receptor (Cxadr) and genes involved in endoplasmic reticulum (ER) stress and RNA sensing.
Seven days after CVB4 infection, islets from βHIF-1α mice had increased enterovirus RNA expression compared to islets from FC mice (15-fold higher, p < 0.0001). Consistent with plaque assays, no significant differences were observed on other days (Figure 5A). The expression of Cxadr, which permits viral entry β cells, was markedly increased in islets from βHIF-1α mice at 7 dpi (13.9-fold higher, p < 0.0001) and 14 dpi (31-fold higher, p < 0.0001; Figure 5B).
Figure 5. Gene Expression Changes in FC and βHIF-1α Mouse Islets following CVB4 Infection.

(A) Expression of enterovirus (EV) RNA in uninfected and CVB4-infected FC (black bars) and βHIF-1α (white bars) mice at 4,7, and 14 dpi (n = 5 per group per time). Data are medians and 95% confidence intervals. ***p < 0.0001 versus matching time.
(B) Expression of coxsackievirus and adenovirus receptor (Cxadr) in uninfected and CVB4-infected FC (black bars) and βHIF-1α (white bars) mice at 4, 7, and 14 dpi (n = 5 mice per group per time). Data are medians and 95% confidence intervals. ***p < 0.0001 versus matching time.
(C) Heatmap of real-time PCR of ER stress, oxidative stress, and RNA sensor gene expression in islets from uninfected and CVB4-infected mice at 4,7, and 14 dpi (n = 5 per group). **p < 0.01 compared to control uninfected (day 0). †† p < 0.01 for decreased expression versus time-matched controls.
(D) Average CT (cross-threshold) values for TATA-Box binding protein (Tbp) and 18 s housekeeping genes in islets from FC (black bars) and βHIF-1α (white bars) after 24-h CVB4 infection followed by actinomycin D treatment for the times indicated.
(E) Expression of enterovirus RNA in islets from FC (black bars) and βHIF-1α (white bars) mice after 24-h CVB4 infection and actinomycin D treatment.
(F) Heatmap plot of real-time PCR data of inflammatory and/or apoptotic and RNA sensor gene expression changes in islets from FC and βHIF-1α mice collected after 24-h CVB4 infection followed by treatment with actinomycin D for the time shown. **p < 0.01 compared to control 0 h. ††p < 0.01 for decreased expression versus time-matched FC. ΔΔDecreased expression versus βHIF-1α baseline.
Data are means ± SEMs.
Next, we assessed the role of βHIF-1α in ER and oxidative stress after CVB4 infection. Following infection, a panel of ER and oxidative stress genes was significantly altered in islets (Figure 5C, top and center panels). These genes included ER stress chaperones binding immunoglobulin protein (Bip) and glucose-regulated protein 94 (Grp94), pro-apoptotic ER stress genes, cyclic AMP-dependent transcription factor (Atf3), and the oxidative stress genes glutathione peroxidase 1 (Gpx), superoxide dismutase 1 (Sod1), and catalase (Cat).
In contrast to control islets, which increased the expression of RNA virus-sensing genes, islets from βHIF-1α mice showed a decreased expression of key intracellular sensors for viral RNA, including retinoic acid-inducible gene-I (Rig-i), Laboratory of Genetics and Physiology 2 (Lgp2), melanoma differentiation-associated protein 5 (Mda5), and nucleotide-binding oligomerization domain-containing protein 2 (Nod2) (Figure 5C, bottom panel) following CVB4 infection.
These results suggest that the absence of β cell HIF-1α diminishes the appropriate response of βcells to RNA virus infection. This would be predicted to impair viral clearance, as we observed with the plaque assay and with the enterovirus 5′ UTR PCR.
To investigate viral clearance in the setting of CVB4-induced islet death, we compared the expression of enterovirus RNA and apoptotic genes in isolated FC and βHIF-1α islets that were infected with CVB4 in vitro. Actinomycin D was used to inhibit transcription. To test its efficacy, expression of the house-keeping genes TATA-Box binding protein (Tbp) and 18 was measured. Tbp and 18 s showed a progressive increase in the cross-threshold (CT) values, meaning a decrease in expression, with increasing actinomycin D duration (Figure 5D). At 24 h, enterovirus RNA expression was significantly increased in βHIF-1α islets (>10-fold higher, p = 0.0021; Figure 5E), again, consistent with impaired viral clearance.
Although islets from βHIF-1α started with higher expression of the protective gene A20 pre-infection, expression became significantly decreased in these islets after CVB4 infection and actinomycin D (Figure 5F). Consistent with the previous in vivo islet gene expression (Figures 5C), βHIF-1α islets also had a significantly decreased expression of viral RNA sensors Rig-I, Lgp2, and Mda5, in contrast to FC mice, in which Rig-I, Lgp2, and NOD2 increased (Figure 5F, bottom panel). This shows that in β cells, expression of the HIF-1α component of the HIF1 transcription factor is necessary for the normal expression of viral RNA sensors.
Knockdown of HIF-1α in Human Islets Increases Their Susceptibility to β cell Death after CVB4 Infection
Infection of human islets with CVBs is known to lead to β cell dysfunction (Chehadeh et al., 2000; Kim et al., 2016; Gallagher et al., 2015). We investigated whether human islets with knockdown of HIF-1α would have a poorer response to CVB4 infection. Human islets were obtained from 4 donors who did not have diabetes. Their islets were subjected to CVB4 with or without preceding HIF-1α knock down by lentiviral small hairpin RNA (shRNA), as shown in Figure S2G.
There were no obvious differences in the number and morphology of human islets after short-term (24 h) treatment, as shown in Figure 6A. The efficacy of HIF-1α knock down by shRNA was measured by real-time PCR and was >80% (Figure 6B, top panel). Islets from the 4 different human donors showed similar patterns of gene expression. All 4 donors had a significant increase in CXADR expression with CVB4 infection, which was further increased by HIF-1α knockdown. Similarly, expression of pro-apoptotic BAX was also increased by concurrent CVB4 and HIF-1α knockdown. In addition, shRNA against HIF-1α in human islets markedly increased viral load (Figure 6C). These findings are consistent with our observations in mouse tissue, demonstrating that HIF-1α is important for the intrinsic antiviral response of β cells following CVB4 infection.
Figure 6. Gene Expression Changes in Human Islets after HIF-1α Knockdown and CVB4 Infection.

(A) Bright-field microscopic images ofisolated human control islets in culture ± HIF-1α knockdown and ± CVB4 infection at 24 h. Magnification x20.
(B) Heatmap of gene expression changes (n = 4 individual human donors, triplicate samples from each donor per time point). *p < 0.05 and **p < 0.01 for increase versus media. ††p < 0.01 for decrease versus media control.
(C) Expression of enterovirus RNA in human control islets from 4 distinct donors, after HIF-1α knockdown and CVB4 infection collected at 24 h (n = 4 individual human donors, triplicate samples from each donor per time point). Enterovirus RNA was not detected in uninfected islets. ****p < 0.0001 versus islets infected with CVB4 alone by 2-way ANOVA with Dunnett’s correction.
Data are means ± SEMs.
DISCUSSION
T1D is an immune-mediated disease, with incidence increasing steadily in recent decades around the world (Maahs et al., 2010). Recently, attention has turned to identifying β cell factors that confer resistance against stress and insults (Colli et al., 2010; Eizirik et al., 2012; Moore et al., 2009).
Here, we demonstrate that deletion of the transcription factor HIF-1α from the pancreatic β cells of NOD mice increases their susceptibility to T1D, after coxsackieviral insult (CVB1 or CVB4). While male NOD mice typically have an ~10% incidence of T1D by 40 weeks of age (Gale and Gillespie, 2001; Atkinson and Leiter, 1999), the male βHIF-1α NOD mice exposed to coxsackievirus had an ~50% rate of T1D by only 23 weeks of age. Notably, the protective effects of having normal β cell HIF-1α on CVB4-induced T1D were observed independently of mouse gender or route of administration (intraperitoneal versus oral) and with 2 different viral types (CVB4 and CVB1). Heterozygosity for a null HIF-1α allele also led to an increased risk of T1D in mice, with a suggestion of a gene-dose-response effect.
Our data indicate that viral clearance was impaired by deleting HIF-1α from β cells. Relative to FC mice, β cell HIF-1α mice had increased enteroviral RNA expression in their islets at 7 dpi. This was supported by plaque assays and by pancreatic VP1 capsid protein staining. In addition, increased expression of the CVB4 receptor Cxadr was detected in β cell HIF-1α mice compared to FC mice at 7 and 14 dpi. These findings are consistent with increased viral attachment (Figure 3A), infection, and/or re-infection (Figure 3B) in βHIF-1α mice. Furthermore, the mice also showed a reduced expression of important viral sensors Rig-I, Lgp2, Mda5, and Nod2. These sensors detect viral products and activate distinct signaling pathways that in turn lead to an antiviral response by the induction of Dicer (McCartney and Colonna, 2009).
Impaired viral clearance in β cell HIF-1α mice is accompanied by increased pancreatitis. This finding suggests that the β cell-specific defect led to greater exocrine pancreatitis. This is likely to be mediated by the greater viral load in βHIF-1α mice. It is a seldom-recognized fact that people with T1D have substantial reductions in pancreatic mass and/or volume. The magnitude of this loss is far greater than would be expected with the loss of β cells alone. This is consistent with prior pancreatitis, presumably at a subclinical level (Williams et al., 2012; Campbell-Thompson et al., 2016).
Increased viral load and pancreatitis both have obvious potential to increase islet and β cell death, thereby further priming autoimmunity. HIF-1α is induced in pancreatitis (Gomez et al., 2004). During acute pancreatitis, HIF-1α facilitates the restoration of oxygen homeostasis and regulates the expression of cell-cycle and anti-apoptotic genes (Gomez et al., 2004; Carmeliet et al., 1998; Semenza, 2001; Ladoux and Frelin, 1993). In βHIF-1α mice, this protective response is lost in the β cells, and our data show that this causes increased β cell death, with the eventual development of T1D.
HIF-1α activity is increased by cytokines and inflammation, as well as by pancreatitis. In other tissues such as kidney (Higgins et al., 2007; Haase, 2012), liver (Moon et al., 2009; Roth and Copple, 2015), and white adipose tissue (Halberg et al., 2009; Sun et al., 2013), HIF-1α decreases fibrotic response. βHIF-1α mice showed a marked increase in fibrosis. People with T1D also have increased pancreatic fibrosis, predominantly in the periductal regions (Meier et al., 2005). The fibrosis is also consistent with increased pancreatitis. One human autopsy study of 47 people with T1D reported pancreatic fibrosis in 66% of cases and fatty infiltration in 32% (Waguri et al., 1997). A much older paper did not report on fatty infiltration but found pancreatitis in 17 of 54 patients (32%) and fibrosis in 39 of the 54 cases (72%) (Gepts, 1965). We believe it is likely that subclinical pancreatitis may be more prevalent in people with T1D than previously realized.
Both viruses and β cell toxins cause a range of deleterious changes in β cells that can promote increased cytokine and chemokine production and ER stress leading to increased β cell apoptosis (Akerfeldt et al., 2008; Fonseca et al., 2011; Gallagher et al., 2015). Lack of HIF-1α and, in human islets, knockdown of HIF-1α caused decreased expression of the RNA virus-sensing machinery, particularly Mda5 and Nod2. These changes are consistent with the increased viral load and impaired viral clearance seen in βHIF-1α mice.
We characterized the immune cell populations in the islet infiltrates of βHIF-1α and control mice at the peak of viral load. More detailed studies revealed that a lack of β cell HIF-1α led to a larger and more aggressive immune infiltrate, accompanying increased viral load, and exocrine pancreatitis. Lack of β cell HIF-1α led to increased proportions of cytotoxic T cells, B cells, and macrophages in the islet infiltrate. Increased viral load, impaired ER stress response, impaired viral processing genes, and pancreatitis together would contribute to a greater inflammatory response and the more aggressive infiltrate seen. The effects of HIF-1α knockdown in human islets exhibited robust and consistent reduced expression of RNA sensor and anti-apoptotic genes and increased expression of enterovirus RNA and CXADR in all of the donors tested. This confirms important effects of HIF-1α in human islets.
Genetic association studies performed in both Japanese (Yamada et al., 2005) and caucasian (Hungarian) populations (Nagy et al., 2009) have shown that polymorphisms in the human HIF- 1α gene is associated with diabetes. In addition, other studies have shown the impact of HIF-1α polymorphisms in diabetes complications such as nephropathy (Gu et al., 2013), cardiomyopathy (Alidoosti et al., 2011), and microvascular and diabetic foot complications (Pichu et al., 2015). Our studies demonstrate an important role for β cells and, particularly, β cell HIF-1α in response to environmental triggers for T1D. To our knowledge, this is the first demonstration of a role for β cell-specific knockout in T1D development after environmental triggers and of a role for HIF-1α in viral clearance.
Our studies outline a potential pathway for the development of T1D in susceptible individuals. βHIF-1α has an important role in determining the fate of islet β cells in the context of stress and viral infection and provides insights into the unpredicted and unexplored connection between viral infections and the development of autoimmune diabetes. Since HIF-1α is primarily regulated at a protein level, these findings have translational potential and provide insights into potential therapeutic strategies for the prevention and treatment of human T1D. Based on these studies, it would be interesting to speculate that previous coxsackievirus immunization with or without increased HIF-1α protein could provide protection against diabetes in some individuals.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jenny E. Gunton (jenny.gunton@sydney.edu.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics approvals
All mouse studies and procedures complied with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes at Garvan Institute (approval #12.19) and Western Sydney Local Health District (WSLHD, approval #4224) Animal Ethics Committees. Studies involving human islets were approved by WSLHD Ethics Committee. Human pancreatic islets were purified using a modified Ricordi method (Ricordi et al., 1988) as previously reported (O’Connell et al., 2013).
Animals
β-cell-specific Hypoxia Inducible Factor-1α NOD (βHIF-1α) mice are a conditional knockout mouse on the non-obese diabetic (NOD) background that lacks HIF-1α in pancreatic β-cells. While female NOD mice have a 70% spontaneous T1D incidence by ~40 weeks of age, males have only 10% risk of spontaneous diabetes (Leiter et al., 1987; Atkinson and Leiter, 1999). All studies were performed in male mice unless stated otherwise. βHIF-1α mice were created by crossing βHIF-1α C57BL/6 mice (Cheng et al., 2010) with NOD mice for > 12 generations. Baseline histology in male βHIF-1α mice was normal, including for islet glucagon and somatostatin (Figure S1A). Controls were NOD, floxed control (FC) littermates (HIF-1α fl/fl, RIP-Cre negative NOD mice) or RIP-Cre-alone NOD mice (HIF-1α WT/WT, RIP-Cre+ NOD mice) as specified in each experiment. The recipients for adoptive transfer studies were immunodeficient NOD-SCID mice, obtained from the Animal Resource Centre (ARC, Canning Vale, WA, Australia). Generation of NOD-SCID mice is described in (Christianson et al., 1993).
All mice were housed in individually-ventilated cages with sterile bedding and ad libitum access to standard chow and water. Chow provided 60%, 27% and 13% calories from carbohydrate, protein and fat respectively (Agrifood Technology, Werribee, VIC, Australia). The facility was temperature-controlled (22–25°C) with a 12-hour light-dark cycle.
METHOD DETAILS
Viruses and infection of mice
Coxsackievirus B4 (CVB4, Edward’s strain 2 of human origin) was derived from stocks obtained from Professor Malin Flodström-Tullberg, (Karolinska Institute, Sweden). Coxsackievirus B1 (CVB1) was isolated from stool samples obtained from patients without T1D, as previously described (Craig et al., 200; Nair et al., 2010). CVs were propagated in HeLa cell cultures, and culture supernatants containing the viruses were stored at −80°C. At eight weeks of age, male mice (either βHIF-1α, NOD, floxed control (FC) or RIP-Cre-alone) were infected intraperitoneally with 105 plaque-forming units (pfu) of CVB4 or CVB1 in 200 μl saline. Male mice were used for all studies unless specified because female βHIF-1α mice already had significantly higher insulitis scores at baseline (Figure S1B). A separate cohort of mice was inoculated orally (via gavage route) with 105 pfu CVB4 in 200 ml saline. For long-term studies; the mice were followed until diabetes development or 105 days post infection (dpi) (Figure S2A) and for short-term studies; they were sacrificed at 4, 7, 14 or 21 dpi (Figure S2B).
Mouse monitoring
Mice were monitored for signs of distress, weighed and BGLs were checked twice a week after CV infection. If BGLs were > 15mmol/L, mice were commenced on daily insulin (0.5U/kg of Actrapid, administered after BGL testing which was also performed daily). BGLs were measured using the FreeStyle Lite glucometer (Abbott Diabetes Care, Macquarie Park, New South Wales, Australia). Mice that had random-fed BGLs of ≥ 20mmol/l (360mg/dl) on 2 separate occasions were considered diabetic. Intraperitoneal glucose tolerance tests (GTT, 2g/kg) were performed in mice fasted for 4 hours, as previously described (Lalwani et al., 2014; Stokes et al., 2013; Scott et al., 2014).
Pancreatic enzyme replacement and insulin therapy
Creon Forte (Abbott Laboratories, Illinois USA) which is a combination of lipase, protease, and amylase was administered in mice that showed clinical signs of weight loss and food malabsorption after CVB1 or CVB4 infection. All mice that lost > 10% of their pre-virus body weight were treated until baseline weight was regained. Mice were orally administered granules daily in the afternoon (approx. 1 mg each) when required.
CV plaque assay
Pancreatic and spleen viral load were measured by plaque assay (plaque-forming units (pfu) per milliliter). On the day prior to the assay, 500,000 HeLa cells were seeded in 6-well plates containing 2 ml/well of complete medium (RPMI 1640 + 10% BCS + 2mM L-glutamine + 100U/ml Penicillin). Experimental mice were sacrificed and pancreases were collected and homogenized using a Dounce tissue grinder. Serial dilutions were made using serum-free RPM11640 medium. Afterward, 400 μl of diluted virus or tissue homogenate was added to each well of HeLa cells and incubated at 37°C for 1 hour with gentle rocking every 15 minutes. After incubation, the infectious media was removed and 3 mL of agar/2× MEM medium with BCS (1:1 ratio) was added per well. Plates were incubated at 37°C for 3 days and then 3mL Carnoy’s reagent was added per well and plates were incubated at room temperature for an hour. After incubation, the agar/media plug was removed and the cells were stained with 800 uL of 0.1% Crystal Violet for 60 s. Wells were gently rinsed with water and air-dried before counting plaques (examples of plates are shown in Figure S1C). The viral titer was determined as pfu/ml using the following calculation:
Plaque Assay count = number of plaques counted × dilution of the particular well × volume of the diluted virus added to the well
Biochemical measurements
Insulin was determined by ELISA (#90080, Crystal Chem, Illinois, USA). Serum amylase activity was measured using an amylase activity kit from Sigma-Aldrich (catalog# MAK009–1KT, Sydney, New South Wales, Australia),
Isolation of mouse islets and analysis of infiltrates
Mouse islets were isolated and purified from uninfected and CV infected 8-week-old male FC and βHIF-1α mice as described previously (Cheng et al., 2010; Gunton et al., 2005; Lalwani et al., 2014; Stokes et al., 2013). The pancreas was distended with 3mls of liberase containing solution, and incubated at 37°C for 16.5 minutes. After this the samples were physically disrupted, passed through a series of increasingly small sieves and then the islets were separated using a Ficoll density gradient.
Islets were dispersed into single cell suspensions using 5 mL of 0.2 mM EDTA solution for 5 minutes at room temperature For all experiments, n = 5–6 mice per group and 100 islets per mouse were used. After dispersion, the cells were stained for surface molecules/markers with one or more of the fluorochrome-labeled antibodies (details in Table S1). For intracellular cytokine staining, BD Cytofix/Cytoperm Kit (#554714, BD Biosciences, North Ryde, New South Wales, Australia) was used according to manufacturer’s instructions. Total proportions of different immune cells within the islets (islet infiltrate) were quantified by flow cytometry using an LSRFortessa (BD Biosciences, North Ryde, New South Wales, Australia). Data analysis was performed using FACSdiva software (BD Biosciences, North Ryde, New South Wales, Australia).
Adoptive transfer studies
Donors were sacrificed; spleens were excised and dispersed into single cells. Splenocytes were transferred to NOD-SCID recipient mice via tail vein injection, at a dose of 2 × 107 cells/mouse. Recipients were monitored at least twice a week. As above, mice with BGLs ≥ 15mmol/L were commenced on daily insulin and were considered diabetic with BGLs ≥ 20mmol/L on two separate occasions.
Infection of human islets with lentivirus carrying HIF-1α shRNA
For knockdown of HIF-1α, human islets were infected with lentivirus carrying HIF-1α shRNA (#sc-35561-SH, Santa Cruz Biotechnology, Dallas, Texas, USA) at a dose of 20 MOI (multiplicity of infection) and cultured in human islet culture medium as used in (Stokes et al., 2013) and 6μg/ml of polybrene (#sc-134220, Santa Cruz Biotechnology, Dallas, Texas, USA) at 37°C for 48 hours.
In vitro studies with mouse and human islets
Isolated islets from FC and βHIF-1α mice were handpickedand cultured in media (control) or media with CVB4at 104 pfu/islet at 37°C for 24 hours. This was followed by Actinomycin D treatment (10nmol/l) for 2, 4, 8, 16 and/or 24 hours (Figure S2D). In separate experiments, islets were stimulated with or without 200 U/ml (10pg/ml) IL-1β (#401-ML-025, R&D Systems, Minneapolis, Minnesota, USA) at 37°C for 1, 2, or 24 hours (Figure S2E).
Human islets were stimulated with or without 200 U/ml (10pg/ml) IL-1β at 37°C for 1, 2, or 24 hours (Figure S2F). In separate experiments, human islets were treated with lentivirus HIF-1α shRNA (Santa Cruz Biotechnology, Dallas, Texas, USA) for 48 hours for HIF-1α knockdown followed by vehicle or CVB4 infection at a dose of 104 pfu/islet at 37°C in the second 24 hours of lentiviral exposure (Figure S2G). At the end of the study islets were collected, snap-frozen in liquid nitrogen and stored at −80°C for further analysis.
Gene expression by real-time PCR
Islet RNA was extracted either with the QIAGEN RNeasy Mini kit (#74106, QIAGEN, Valencia, California, USA) or TRI Reagent solution (#T9424, Sigma-Aldrich, Sydney, New South Wales, Australia), according to the manufacturer’s instructions. cDNA was generated from 500 ng of RNA and random hexamer primers using the Maxima First Strand cDNA Synthesis kit for RT-qPCR (#K1641, ThermoFisher Scientific, Scoresby, Victoria, Australia). Real-time PCR was performed in ABI Prism 7900HT Sequence Detection System (Life Technologies Australia Pty Ltd, Mulgrave, Victoria, Australia) using specific primers (mouse sequences in Table S2 and human sequences in Table S3) and Power SYBR Green mastermix (#4367659, Life Technologies Australia Pty Ltd, Mulgrave, Victoria, Australia) as previously described (Lalwani et al., 2014). Differences in gene expression were calculated using the ΔΔCT method. Primers for enterovirus (EV) mRNA targeted the highly conserved 5′ untranslated region (UTR) of the EV genome as previously described (Craig et al., 2003). Therefore, this measures viral load rather than individual translated viral RNAs. For many of the gene expression changes after viral infection, fold changes were very large (> 10-fold) and for those genes, data were log10 transformed for statistical analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Histological methods and insulitis scoring
The collected pancreases were fixed in neutral buffered formalin overnight, processed with standard progressive ethanol rehydration steps, and then embedded in paraffin. Insulin immunohistochemistry with hematoxylin and eosin (H&E) counterstain, and Sirius Red staining were performed as previously described (Scott et al., 2015; Lalwani et al., 2014). Immunostaining for the enterovirus (EV) capsid protein VP1 was performed as previously described with exception of secondary antibody detection with DAB chromogen (Craig et al., 2013; Nair et al., 2013; Yeung et al., 2011). Coxsackievirus and adenovirus receptor (CAR) immunohistochemistry with hematoxylin counterstain was performed as previously described (Gunton et al., 2005; Lalwani et al., 2014) with exception of primary antibody probing with anti-coxsackie and adenovirus receptor (Abcam, ab100811, 1:100). Similarly, glucagon immunohistochemistry with hematoxylin and eosin (H&E) counterstain were performed as previously described (Gunton et al., 2005; Lalwani et al., 2014) with exception of primary antibody probing for glucagon with anti-glucagon (Invitrogen, 18–0064, 1:200). Detection was performed using anti-rabbit secondary conjugated to horseradish peroxidase (Dako, K4003). Somatostatin immunofluorescence used overnight incubation at 4°C with anti-somatostatin (Sigma, SAB4502861) and insulin antibodies (guinea-pig anti-insulin, DAKO, A0564) with appropriate secondary antibodies (Cy2 anti-rabbit and AlexaFluor 568 anti-Guinea-pig) and DAPI (Bio-Rad 1351303). Images were acquired using an Olympus VS120 slide scanner.
Insulitis was evaluated by bright field microscopy on a Leica DM 4000 microscope with a DFC450 camera (Leica Biosystems, Mount Waverly, Victoria, Australia). At least 6 widely-separated pancreatic sections were immunostained for insulin and counter-stained with hematoxylin and eosin (H&E). Insulitis was measured by an observer blinded to experimental group, and a score of 0 to 4 was assigned based on islet infiltration. Scoring was 0; normal islets, 1; mild mononuclear infiltration (≤ 25%) at the islet periphery, 2; 25%−50% of the islet infiltrated, 3; ≥ 50% of the islet infiltrated and lastly 4; ≥ 75% infiltration or islet with no insulin positive cells. At least 120 islets were evaluated per strain/group.
Quantitative histology methods
β-cell mass was calculated as previously described (Gunton et al., 2005). The total number of immune cells in insulitis areas were counted with Image-Based Tool for Counting Nuclei (ITCN), a cell counting Java plug in for ImageJ (NIH freeware) (https://bioimage.ucsb.edu/itcn.html). Total area of fat was quantified with MRI Adipocytes Tools, a fat cell counting Java plug in for ImageJ (NIH freeware) (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Adipocytes_Tool). Necrotic area was measured using ImageJ (NIH Freeware) by freehand drawing around the necrotic area within the islet.
Statistical analysis
Data were evaluated using either Microsoft Excel or GraphPad Prism 7.0 (San Diego, California, USA). Groups were compared using either Student’s unpaired t test or ANOVA, as appropriate. Unless otherwise specified, data are presented as mean ± SEM. Where multiple comparisons were made, post hoc testing used Bonferroni or Turkey’s correction. Logrank (Mantel-Cox) test was used to plot Kaplan-Meier survival curves to compare diabetes free-survival. A p value of ≤ 0.05 was considered significant.
DATA AND SOFTWARE AVAILABILITY
Contact the lead author Professor Gunton for data enquiries.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD111b-AF488 (clone: M1/70) | BD Biosciences | 557672; RRID:AB_396784 |
| CD11c-APC (clone: HC3) | BD Biosciences | 550261; RRID:AB_398460 |
| CD19-PE-CY7 (clone: 1D3) | BD Biosciences | 552854; RRID:AB_394495 |
| CD25-APC (clone: PC61) | BD Biosciences | 557192; RRID:AB_398623 |
| CD27-BV421 (clone: LG.3A10) | BD Biosciences | 740028; RRID:AB_2739800 |
| CD3-BUV737 (clone: 17A2) | BD Biosciences | 564380; RRID:AB_2738781 |
| CD4-FITC (clone:GK1.5) | BD Biosciences | 553729; RRID:AB_395013 |
| CD4-PE (clone: LT3T4) | eBioscience | RRID:AB_12-0041-82 |
| CD45-BUV395 (clone: 30-F11) | BD Biosciences | 564279; RRID:AB_2651134 |
| CD8-V650 (clone: 53–6.7) | BD Biosciences | 563234; RRID:AB_2738084 |
| F4–80-BV421 (clone: T45–2342) | BD Biosciences | 565411; RRID:AB_2734779 |
| Granzyme B-PE (clone: NGZB) | eBioscience | RRID:AB_12-8898-80 |
| anti-mouse enterovirus (clone: 5-D8/1) | Dako | M7064; RRID:AB_2118128 |
| rabbit insulin antibody | Cell Signaling Technology Inc. | 4590; RRID:AB_659820 |
| rabbit anti-cleaved caspase 3 | R&D Systems | RRID:AB_AF835 |
| Bacterial and Virus Strains | ||
| Coxsackie Virus B1 (CVB1) | Professor Maria Craig (Westmead Children’s Hospital, University of Sydney, NSW, Australia) | Obtained from clinical stool samples from patients |
| Coxsackie Virus B1–E2 strain (CVB4) | Associate Professor Malin Flodström-Tullberg, Karolinska Institute, Sweden | N/A |
| Biological Samples | ||
| Human pancreatic primary islets | Wayne Hawthorne of the Westmead National Pancreas and Transplant Unit (NPTU, Australia) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-2-2-Tribromoethanol | Sigma-Aldrich | T48402 |
| 2-mercaptoethanol | Life Technologies | 21985–023 |
| 2-methyl-2-butanol | Lomb Scientific Australia Pty Ltd | 15114 |
| Actinomycin D (Act D) | Sigma-Aldrich | A1410 |
| Accu-Chek Advantage II glucose test strips | Roche Diagnostics, North Ryde, NSW, Australia | N/A |
| Ammonium chloride | Thermo Fisher Australia Pty Ltd | AJA318 |
| Ammonium sulfate | Lomb Scientific Australia Pty Ltd | 56–500G |
| Antigen retrieval solution | Dako, Carpinteria, California, USA. | S1699 |
| Boric acid powder | Thermo Fisher Australia Pty Ltd | B6768 |
| Bovine Calf Serum (BCS) (Australian origin) | Serana, Australia Pty Ltd, Bunbury, Western Australia, Australia. | S-FBS-AU-015 |
| Bovine Serum Albumin (BSA) | Bovogen Biologicals Australia Pty Ltd, Keilor East, Victoria, Australia. | BSAS1.0 |
| Citrate buffer (pH 4.5) | In-house laboratory | N/A |
| Chloroform | Sigma-Aldrich | C2432 |
| Creon Forte (pancrelipase) (prescription drug) | Abbott Laboratories, Lake Bluff, Illinois, USA. | 90010 |
| Crystal violet stain | Sigma-Aldrich | C3886 |
| Direct Red 80 | Sigma-Aldrich | 365548 |
| Disodium hydrogen orthophosphate | Thermo Fisher Australia Pty Ltd | 478 |
| Dimethyl sulfoxide (DMSO) | Lomb Scientific Australia Pty Ltd | 2225 |
| Ethylenediaminetetraacetic acid Disodium (EDTA) | Thermo Fisher Australia Pty Ltd | AJA180 |
| Eosin Y stain solution with Phloxine | Sigma-Aldrich | HT110332 |
| Ethanol (100% molecular grade Ethyl alcohol) | Sigma-Aldrich | E7023 |
| Fast Green FCF | Sigma-Aldrich | F7258 |
| Ficoll-Plaque Plus 1.077 | GE Healthcare, Parramatta, NSW, Australia. | 17–1440 |
| Fluoromount aqueous mounting medium | Sigma-Aldrich | F4680 |
| Formamide(≥99.0%) | Sigma-Aldrich | F7503 |
| Glacial acetic acid | Thermo Fisher Australia Pty Ltd | AJA1 |
| HEPES(1M) | Life Technologies | 15630–080 |
| Hydrochloric acid | Thermo Fisher Australia Pty Ltd | AJA256 |
| Insulin Actrapid 100U/ml (short acting) | Novo Nordisk Pharmaceuticals Pty. Ltd Baulkham Hills, NSW, Australia. | N/A |
| Insulin Levemir 100U/ml (long acting) | Novo Nordisk Pharmaceuticals Pty. Ltd, Baulkham Hills, NSW, Australia. | N/A |
| Isopropanol (2-propanol) | Sigma-Aldrich | 19516 |
| L-glutamine | Life Technologies | 25030–081 |
| Liberase-Enzyme Blend-RI | Roche Diagnostics, Indianapolis, Indiana, USA. | 05989132001 |
| Medium 199 (M199) | Life Technologies | 31100–035 |
| Magnesium sulfate | Sigma-Aldrich | M7506 |
| MEM Non-Essential Amino Acids | Life Technologies | 11140–050 |
| Methanol | Thermo Fisher Australia Pty Ltd | AJA318 |
| Neutral buffered formalin 10% (NBF) | Sigma-Aldrich | HT50-1-1 |
| Normocin | Integrated Sciences Pty Ltd, Chatswood, NSW, Australia. | ANT-NR-1 |
| Optimal Cutting Temperature compound (OCT) | Tissue-Tek, Sakura Finetek, Torrance, California, USA. | 4583 |
| Penicillin/Streptomycin | Life Technologies | 15070–063 |
| Picric acid solution | Sigma-Aldrich | P6744 |
| Proteinase K | Roche Diagnostics, Mannheim, Germany. | 03115828001 |
| Potassium chloride | Thermo Fisher Australia Pty Ltd | AJA383 |
| Potassium dihydrogen phosphate | Thermo Fisher Australia Pty Ltd | AJA391 |
| Recombinant IL-1β/IL-1F2 | R&D Systems, Minneapolis, Minnesota, USA. | 401-ML-025 |
| Red blood cell lysis buffer | Sigma-Aldrich. | R7757 |
| Roswell Park Memorial lnstitute-1640 medium (RPMI 1640) | Life Technologies | 1875–119 |
| SeaKem® LE Agarose | Lonza Australia Pty Ltd, Mount Waverley, Victoria, Australia. | 50004 |
| SeaPlaque® Agarose | Lonza Australia. | 50101 |
| Shandon Instant Hemotoxylin | Thermo Fisher Australia Pty Ltd | 6765015 |
| Sodium bicarbonate | Life Technologies | 25080–094 |
| Sodium chloride | Thermo Fisher Australia Pty Ltd | 465 |
| Sodium hydrogen carbonate | Thermo Fisher Australia Pty Ltd | AJA475 |
| Sodium pyruvate | Life Technologies Australia Pty Ltd, Mulgrave, Victoria, Australia. | 11360–070 |
| Streptozotocin | Sigma-Aldrich | S0130 |
| TRI Reagent | Sigma-Aldrich | T9424 |
| Tris (hydroxymethyl) aminomethane base | Merck Millipore Bayswater, Victoria, Australia. | CM0054 |
| Triton X-100 | Sigma-Aldrich | 234729 |
| Trypan blue | Sigma-Aldrich | T8154 |
| Trypsin-EDTA | Life Technologies | 25300–054 |
| Water for irrigation 1L | Baxter Healthcare Pty Ltd, Old Toongabbie, NSW, Australia. | AHF7114 |
| Critical Commercial Assays | ||
| Amylase activity kit | Sigma-Aldrich | MAK009–1KT |
| Crystal Chem rat insulin ELISA kit | Crystal Chem Inc., Downers Grove, Illinois, USA. | 90010 |
| DakoCytomation EnVision+Dual Link System-HRP (DAB+) kit (DAKO kit) | Dako, Carpinteria, California, USA. | K4011 |
| QIAGEN RNeasy Mini kit | QIAGEN, Valencia, California, USA. | 74106 |
| QIAshredder spin column | QIAGEN, Valencia, California, USA. | 79654 |
| Power SYBR green master mix | Life Technologies. | 4367659 |
| Experimental Models: Cell Lines | ||
| HeLa cells (originally obtained by Assoc. Prof Cecile King) | ATCC | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Animal Resource Centre (ARC, Canning Vale, WA, Australia) | N/A |
| Non-Obese Diabetic (NOD) mice | ARC, Canning Vale, WA, Australia | N/A |
| Severe Combined ImmunoDeficient SCID mice | ARC, Canning Vale, WA, Australia | N/A |
| NOD-SCID mice | ARC, Canning Vale, WA, Australia | N/A |
| βHIF-1α NOD mice | Backcrossed with original βHIF-1α mice on C57BL/6 background | N/A |
| Oligonucleotides | ||
| Primers for mouse PCRs, see Table S2 | This paper | N/A |
| Primers for human PCRs, see Table S3 | This paper | N/A |
| shRNA targeting sequence: HIF-1 alpha | This paper (Santa Cruz Biotechnology) | sc-35561-SH |
| Recombinant DNA | ||
| Not applicable | N/A | N/A |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | |
| FACSdiva software | BD Biosciences | http://www.bdbiosciences.com/ |
Highlights.
Type 1 diabetes is increasing worldwide, which must be due to environmental changes
Lack of β cell HIF1a increases risk of T1D after viral infection
β Cell HIF1a also decreases T1D after low doses of the β cell toxin streptozotocin
β Cell HIF1a is a major factor in determining whether insult leads to T1D or resolution
ACKNOWLEDGMENTS
This work was funded by the National Health and Medical Research Council (NHMRC) of Australia and the Diabetes Australia Research Trust (DART). The Human Islet Transplant Program was funded by the Juvenile Diabetes Research Foundation Ltd (JDRF). The authors would like to thank the histology departments at WIMR and Garvan, the flow cytometry department at WIMR, the animal services facilities at WIMR and Garvan, and the Animal BioResources Centre (ABR) at Moss Vale for breeding and maintaining the mouse lines. The authors would also like to thank Dr. Ki-Wook Kim (UNSW), Kailun Lee (Garvan), and Kuan Min Cha (WIMR) for experimental help; Lindy Williams and Dr. Yi Vee Chew (NPTU) for their assistance with the preparation of human islets; and Dr. Christian Girgis (WIMR) and Arlyn-Pan Lalwani for proofreading the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.086.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, and Laybutt DR (2008). Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 57, 3034–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alidoosti M, Ghaedi M, Soleimani A, Bakhtiyari S, Rezvanfard M, Golkhu S, and Mohammadtaghvaei N (2011). Study on the role of environmental parameters and HIF-1A gene polymorphism in coronary collateral formation among patients with ischemic heart disease. Clin. Biochem 44, 1421–1424. [DOI] [PubMed] [Google Scholar]
- Atkinson MA, and Leiter EH (1999). The NOD mouse model of type 1 diabetes: as good as it gets? Nat. Med 5, 601–604. [DOI] [PubMed] [Google Scholar]
- Banatvala JE, Bryant J, Schernthaner G, Borkenstein M, Schober E, Brown D, De Silva LM, Menser MA, and Silink M (1985). Coxsackie B, mumps, rubella, and cytomegalovirus specific IgM responses in patients with juvenile-onset insulin-dependent diabetes mellitus in Britain, Austria, and Australia. Lancet 1, 1409–1412. [DOI] [PubMed] [Google Scholar]
- Bilton RL, and Booker GW (2003). The subtle side to hypoxia inducible factor (HIFalpha) regulation. Eur. J. Biochem 270, 791–798. [DOI] [PubMed] [Google Scholar]
- Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PGF, and Gamble DR (1985). In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N. Engl. J. Med 313, 353–360. [DOI] [PubMed] [Google Scholar]
- Campbell-Thompson ML, Kaddis JS, Wasserfall C, Haller MJ, Pugliese A, Schatz DA, Shuster JJ, and Atkinson MA (2016). The influence of type 1 diabetes on pancreatic weight. Diabetologia 59, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. (1998). Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394, 485–490. [DOI] [PubMed] [Google Scholar]
- Chehadeh W, Kerr-Conte J, Pattou F, Alm G, Lefebvre J, Wattré P, and Hober D (2000). Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J. Virol 74, 10153–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, O’Connell PJ, Loudovaris T, Kay TW, Kulkarni RN, et al. (2010). Hypoxia-inducible factor-1α regulates β cell function in mouse and human islets. J. Clin. Invest 120, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson SW, Shultz LD, and Leiter EH (1993). Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 42, 44–55. [DOI] [PubMed] [Google Scholar]
- Colli ML, Moore F, Gurzov EN, Ortis F, and Eizirik DL (2010). MDA5and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic β-cell responses to the viral by-product double-stranded RNA. Hum. Mol. Genet 19, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters KT, Boettler T, and von Herrath M (2012). Virus infections in type 1 diabetes. Cold Spring Harb. Perspect. Med 2, a007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig ME, Robertson P, Howard NJ, Silink M, and Rawlinson WD (2003). Diagnosis of enterovirus infection by genus-specific PCR and enzyme-linked immunosorbent assays. J. Clin. Microbiol 41, 841–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig ME, Nair S, Stein H, and Rawlinson WD (2013). Viruses and type 1 diabetes: a new look at an old story. Pediatr. Diabetes 14, 149–158. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, and Op de Beeck A (2018). Coxsackievirus and Type 1 Diabetes Mellitus: The Wolf’s Footprints. Trends Endocrinol. Metab 29, 137–139.29326001 [Google Scholar]
- Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML, Barthson J, et al. (2012). The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 8, e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca SG, Gromada J, and Urano F (2011). Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab 22, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulis AK, Farquharson MA, and Hardman R (1987). Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 30, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk G, Friman G, Tuvemo T, Fohlman J, and Diderholm H (1992). Coxsackie B virus IgM in children at onset of type 1 (insulin-dependent) diabetes mellitus: evidence for IgM induction by a recent or current infection. Diabetologia 35, 249–253. [DOI] [PubMed] [Google Scholar]
- Gale EA, and Gillespie KM (2001). Diabetes and gender. Diabetologia 44, 3–15. [DOI] [PubMed] [Google Scholar]
- Gallagher GR, Brehm MA, Finberg RW, Barton BA, Shultz LD, Greiner DL, Bortell R, and Wang JP (2015). Viral infection of engrafted human islets leads to diabetes. Diabetes 64, 1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepts W (1965). Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14, 619–633. [DOI] [PubMed] [Google Scholar]
- Ginsberg-Fellner F, Witt ME, Yagihashi S, Dobersen MJ, Taub F, Fe-dun B, McEvoy RC, Roman SH, Davies RG, Cooper LZ, et al. (1984). Congenital rubella syndrome as a model for type 1 (insulin-dependent) diabetes mellitus: increased prevalence of islet cell surface antibodies. Diabetologia 27 (Suppl), 87–89. [DOI] [PubMed] [Google Scholar]
- Gomez G, Englander EW, Wang G, and Greeley GH Jr. (2004). Increased expression of hypoxia-inducible factor-1alpha, p48, and the Notch signaling cascade during acute pancreatitis in mice. Pancreas 28, 58–64. [DOI] [PubMed] [Google Scholar]
- Gu HF, Zheng X, Abu Seman N, Gu T, Botusan IR, Sunkari VG, Lokman EF, Brismar K, and Catrina SB (2013). Impact of the hypoxia-inducible factor-1 α (HIF1A) Pro582Ser polymorphism on diabetes nephropathy. Diabetes Care 36, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH, Roberson RS, Ricordi C, O’Connell PJ, Gonzalez FJ, and Kahn CR (2005). Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 122, 337–349. [DOI] [PubMed] [Google Scholar]
- Haase VH (2012). Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair 5 (Suppl 1), S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol 29, 4467–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest 117, 3810–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, and Sarvetnick N (1998). Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat. Med 4, 781–785. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Fine C, Krahl T, Lee J, and Sarvetnick N (1999). Coxsackie virus-mediated diabetes is the direct result of bystander lymphocyte activation and requires both tissue damage and local Th1 immune responses. FASEB J 13, A999. [Google Scholar]
- Hwang AB, and Lee SJ (2011). Regulation of life span by mitochondrial respiration: the HIF-1 and ROS connection. Aging (Albany N.Y.) 3, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japan Diabetes Society, Committee on Diabetic Twins (1988). Diabetes mellitus in twins: a cooperative study in Japan. Diabetes Res. Clin. Pract 5, 271–280. [DOI] [PubMed] [Google Scholar]
- Kim KW, Ho A, Alshabee-Akil A, Hardikar AA, Kay TW, Rawlinson WD, and Craig ME (2016). Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 65, 996–1003. [DOI] [PubMed] [Google Scholar]
- Knip M, and Simell O (2012). Environmental triggers of type 1 diabetes. Cold Spring Harb. Perspect. Med 2, a007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knip M, Veijola R, Virtanen SM, Hyöty H, Vaarala O, and Akerblom HK (2005). Environmental triggers and determinants of type 1 diabetes. Diabetes 54 (Suppl 2), S125–S136. [DOI] [PubMed] [Google Scholar]
- Ladoux A, and Frelin C (1993). Hypoxia is a strong inducer ofvascular endothelial growth factor mRNA expression in the heart. Biochem. Biophys. Res. Commun 195, 1005–1010. [DOI] [PubMed] [Google Scholar]
- Lalwani A, Stokes RA, Lau SM, and Gunton JE (2014). Deletion ofARNT (Aryl hydrocarbon receptor nuclear translocator) in β-cells causes islet transplant failure with impaired β-cell function. PLoS One 9, e98435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH, Prochazka M, and Coleman DL (1987). The non-obese diabetic (NOD) mouse. Am. J. Pathol 128, 380–383. [PMC free article] [PubMed] [Google Scholar]
- Maahs DM, West NA, Lawrence JM, and Mayer-Davis EJ (2010). Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. North Am 39, 481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney SA, and Colonna M (2009). Viral sensors: diversity in pathogen recognition. Immunol. Rev 227, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Bhushan A, Butler AE, Rizza RA, and Butler PC (2005). Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48, 2221–2228. [DOI] [PubMed] [Google Scholar]
- Metcalfe KA, Hitman GA, Rowe RE, Hawa M, Huang X, Stewart T, and Leslie RD (2001). Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care 24, 838–842. [DOI] [PubMed] [Google Scholar]
- Moon JO, Welch TP, Gonzalez FJ, and Copple BL (2009). Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol 296, G582–G592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore F, Colli ML, Cnop M, Esteve MI, Cardozo AK, Cunha DA, Bugliani M, Marchetti P, and Eizirik DL (2009). PTPN2, a candidate gene for type 1 diabetes, modulates interferon-γ-induced pancreatic β-cell apoptosis. Diabetes 58, 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kovacs-Nagy R, Kereszturi E, Somogyi A, Szekely A, Nemeth N, Hosszufalusi N, Panczel P, Ronai Z, and Sasvari-Szekely M (2009). Association of hypoxia inducible factor-1 alpha gene polymorphism with both type 1 and type 2 diabetes in a Caucasian (Hungarian) sample. BMC Med. Genet 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Al-Shabeeb A, and Craig ME (2013). Enterovirus Infection, β-cell apoptosis and Type 1 Diabetes. Microbiol. Aust 34, 153–156. [Google Scholar]
- Nair S, Leung KC, Rawlinson WD, Naing Z, and Craig ME (2010). Enterovirus infection induces cytokine and chemokine expression in insulin-producing cells. J. Med. Virol 82, 1950–1957. [DOI] [PubMed] [Google Scholar]
- Nisticò L, Iafusco D, Galderisi A, Fagnani C, Cotichini R, Toccaceli V, and Stazi MA; Study Group on Diabetes of the Italian Society of Pediatric Endocrinology and Diabetology (2012). Emerging effects of early environmental factors over genetic background for type 1 diabetes susceptibility: evidence from a Nationwide Italian Twin Study. J. Clin. Endocrinol. Metab 97, E1483–E1491. [DOI] [PubMed] [Google Scholar]
- O’Connell PJ, Holmes-Walker DJ, Goodman D, Hawthorne WJ, Loud-ovaris T, Gunton JE, Thomas HE, Grey ST, Drogemuller CJ, Ward GM, et al. ; Australian Islet Transplant Consortium (2013). Multicenter Australian trial of islet transplantation: improving accessibility and outcomes. Am. J. Transplant 13, 1850–1858. [DOI] [PubMed] [Google Scholar]
- Penn DJ, Damjanovich K, and Potts WK (2002). MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc. Natl. Acad. Sci. USA 99, 11260–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, and Johnson RS (2007). Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest 117, 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichu S, Sathiyamoorthy J, Krishnamoorthy E, Umapathy D, and Viswanathan V (2015). Impact of the hypoxia inducible factor-1 α (HIF-1α) pro582ser polymorphism and its gene expression on diabetic foot ulcers. Diabetes Res. Clin. Pract 109, 533–540. [DOI] [PubMed] [Google Scholar]
- Pitaciński S, and Zozulińska-Ziółkiewicz DA (2014). Influence of lifestyle on the course of type 1 diabetes mellitus. Arch. Med. Sci 10, 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsingh AI (1997). Coxsackieviruses and pancreatitis. Front. Biosci 2, e53–e62. [DOI] [PubMed] [Google Scholar]
- Ratcliffe PJ, O’Rourke JF, Maxwell PH, and Pugh CW (1998). Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol 201, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Rewers M, and Ludvigsson J (2016). Environmental risk factors for type 1 diabetes. Lancet 387, 2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricordi C, Lacy PE, Finke EH, Olack BJ, and Scharp DW (1988). Automated method for isolation of human pancreatic islets. Diabetes 37, 413–420. [DOI] [PubMed] [Google Scholar]
- Rossini AA, Like AA, Chick WL, Appel MC, and Cahill GF Jr. (1977). Studies of streptozotocin-induced insulitis and diabetes. Proc. Natl. Acad. Sci. USA 74, 2485–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KJ, and Copple BL (2015). Role of Hypoxia-Inducible Factors in the Development of Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol 1, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C, Bonner J, Min D, Boughton P, Stokes R, Cha KM, Walters SN, Masolowski K, Sierro F, Grey ST, et al. (2014). Reduction of ARNT in myeloid cells causes immune suppression and delayed wound healing. Am. J. Physiol. Cell Physiol 307, C349–C357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott C, Cha K, Rao R, Liddle C, George J, and Gunton JE (2015). Hepatocyte-specific deletion of ARNT (aryl hydrocarbon Receptor Nuclear Translocator) results in altered fibrotic gene expression in the thioacetamide model of liver injury. PLoS One 10, e0121650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2001). Regulation of hypoxia-induced angiogenesis: a chaperone escorts VEGF to the dance. J. Clin. Invest 108, 39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serreze DV, Wasserfall C, Ottendorfer EW, Stalvey M, Pierce MA, Gauntt C, O’Donnell B, Flanagan JB, Campbell-Thompson M, Ellis TM, and Atkinson MA (2005). Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and gamma interferon. J. Virol 79, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes RA, Cheng K, Deters N, Lau SM, Hawthorne WJ, O’Connell PJ, Stolp J, Grey S, Loudovaris T, Kay TW, et al. (2013). Hypoxia-inducible factor-1α (HIF-1α) potentiates β-cell survival after islet transplantation of human and mouse islets. Cell Transplant 22, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clément K, and Scherer PE (2013). Fibrosis and adipose tissue dysfunction. Cell Metab 18, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S, Shameli A, and Santamaria P (2008). CD8+ T cells in type 1 diabetes. Adv. Immunol 100, 79–124. [DOI] [PubMed] [Google Scholar]
- Turley S, Poirot L, Hattori M, Benoist C, and Mathis D (2003). Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med 198, 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belle TL, Coppieters KT, and von Herrath MG (2011). Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol. Rev 91, 79–118. [DOI] [PubMed] [Google Scholar]
- von Herrath M, Sanda S, and Herold K (2007). Type 1 diabetes as a relapsing-remitting disease? Nat. Rev. Immunol 7, 988–994. [DOI] [PubMed] [Google Scholar]
- Waguri M, Hanafusa T, Itoh N, Miyagawa J, Imagawa A, Kuwajima M, Kono N, and Matsuzawa Y (1997). Histopathologic study of the pancreas shows a characteristic lymphocytic infiltration in Japanese patients with IDDM. Endocr. J 44, 23–33. [DOI] [PubMed] [Google Scholar]
- Williams AJK, Thrower SL, Sequeiros IM, Ward A, Bickerton AS, Triay JM, Callaway MP, and Dayan CM (2012). Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J. Clin. Endocrinol. Metab 97, E2109–E2113. [DOI] [PubMed] [Google Scholar]
- Yamada N, Horikawa Y, Oda N, Iizuka K, Shihara N, Kishi S, and Takeda J (2005). Genetic variation in the hypoxia-inducible factor-1alpha gene is associated with type 2 diabetes in Japanese. J. Clin. Endocrinol. Metab 90, 5841–5847. [DOI] [PubMed] [Google Scholar]
- Yeung W-CG, Rawlinson WD, and Craig ME (2011). Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342, d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JW, Onodera T, and Notkins AL (1978). Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J. Exp. Med 148, 1068–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Contact the lead author Professor Gunton for data enquiries.