

Abstract
A predictive understanding of the mechanisms of RNA cleavage is important for the design of emerging technology built from biological and synthetic molecules that have promise for new biochemical and medicinal applications. Over the past 15 years, RNA cleavage reactions involving 2’-O-transphosphorylation have been discussed using a simplified framework introduced by Breaker that consists of four fundamental catalytic strategies (designated α, β, γ, and δ) that contribute to rate enhancement. As more detailed mechanistic data emerge, there is need for the framework to evolve and keep pace. We develop an ontology for discussion of strategies of enzymes that catalyze RNA cleavage via 2’-O-transphosphorylation that stratifies Breaker’s framework into primary (1°), secondary (2°) and tertiary (3°) contributions to enable more precise interpretation of mechanism in the context of structure and bonding. Further, we point out instances where atomic-level changes give rise to changes in more than one catalytic contribution, a phenomenon we refer to as ‘functional blurring’. We hope that this ontology will help clarify our conversations and pave the path forward toward a consensus view of these fundamental and fascinating mechanisms. The insight gained will deepen our understanding of RNA cleavage reactions catalyzed by natural protein and RNA enzymes, as well as aid in the design of new engineered DNA and synthetic enzymes.
Graphical Abstract
1. RNA Cleavage Reactions
The subject of the present Perspective is RNA cleavage reactions1–4 catalyzed by nucleolytic RNA enzymes (ribozymes)5–6, as well as protein enzymes (ribonucleases)7 and several artificially engineered DNA enzymes (DNAzymes)8–9. While illustrative examples and discussion focus on ribozyme mechanisms, the concepts and terminology we develop are equally applicable to protein and DNA enzymes.
RNA strand cleavage by 2′-O-transphosphorylation is universal in biology and has far-reaching implications for medicine10–11. The goal of gaining a predictive understanding of the catalytic mechanisms of RNA cleavage reactions in the context of well-studied biological systems is important from a fundamental scientific perspective, as well as from a technology engineering viewpoint12–13. Elucidating the diverse array of mechanistic strategies exhibited by well-studied biological systems will enable general principles to emerge. These general principles may be transferable outside the biological context and applied to guide the design of synthetic systems, such as xeno nucleic acids14 and Hachimoji DNA/RNA15, which have great promise for new biotechnological applications12–13.
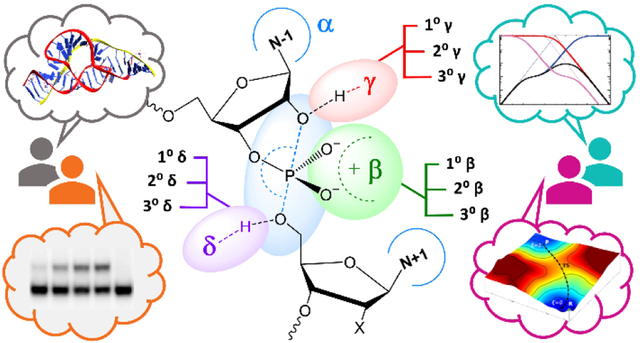
The mechanism and kinetics of RNA cleavage by Brønsted acids/bases and metal ions has been extensively studied2–3, providing a foundation for understanding biological catalysis. In this reaction, the RNA O2′ is activated by deprotonation and makes an in-line nucleophilic attack to the phosphorus atom of the adjacent scissile phosphate to form a pentavalent dianionic transition state (or metastable intermediate), followed by departure of the O5′ leaving group as an oxyanion that ultimately becomes protonated, to form 2′,3′-cyclic phosphate and 5′OH cleavage products (Figure 1, left). Although concerted and stepwise mechanisms are both possible, an idealized transition state (or metastable intermediate) is a dianionic pentavalent phosphorane in a trigonal bipyramidal geometry with nucleophile O2′ and leaving group O5′ ligands occupying apical positions (O2′-P-O5′ angle near17 to 180°) and the formal (2−) charge delocalized between the O2′, O5′ and non- bridge phosphoryl oxygens (NPOs) (Figure 1, right). Naturally occurring protein and RNA enzymes, as well as engineered enzymes such as DNAzymes, use multiple catalytic strategies in concert to enhance the rate of RNA cleavage by factors of typically 105 to 1011-fold1–4, 16. Breaker has suggested a simplified framework for discussion of four basic catalytic strategies for RNA cleavage16, designated α, β, γ, and δ, as illustrated in Figure 1. Concepts and terminology discussed in this Perspective apply to all enzymes catalyzing RNA cleavage via 2’-O-transphosphorylation, however we have focused our discussion and examples on small nucleolytic ribozymes.
Figure 1.

2′-O-transphosphorylation leading to cleavage of the RNA backbone (left of central arrow) and idealized transition state highlighting the general catalytic strategies16 (right of central arrow).  - Arrangement of the O2′ nucleophile, P (scissile phosphorus), and O5′ leaving group in an in-line attack geometry (facilitated by contacts, indicated by blue arcs, that splay the N−1 and N+1 bases).
- Arrangement of the O2′ nucleophile, P (scissile phosphorus), and O5′ leaving group in an in-line attack geometry (facilitated by contacts, indicated by blue arcs, that splay the N−1 and N+1 bases).  - Stabilization (neutralization/protonation) of the negative charge accumulation on the non-bridging phosphoryl oxygens (NPOs).
- Stabilization (neutralization/protonation) of the negative charge accumulation on the non-bridging phosphoryl oxygens (NPOs).  - Activation (deprotonation) of the O2′ nucleophile.
- Activation (deprotonation) of the O2′ nucleophile.  - Stabilization (neutralization/protonation) of the accumulating negative charge on the O5′ leaving group. Although this schematic uses a transition state model to illustrate the fundamental catalytic strategies, these strategies can impact any state along the reaction coordinate. Colored ovals highlight each strategy and encompass the primary atomic positions (defined in section 4 below) associated with the chemical space of bonds for each strategy.
- Stabilization (neutralization/protonation) of the accumulating negative charge on the O5′ leaving group. Although this schematic uses a transition state model to illustrate the fundamental catalytic strategies, these strategies can impact any state along the reaction coordinate. Colored ovals highlight each strategy and encompass the primary atomic positions (defined in section 4 below) associated with the chemical space of bonds for each strategy.
2. Nucleolytic Ribozymes (small self-cleaving RNAs)
Of key interest to the present discussion is how RNA cleavage is catalyzed by small catalytic RNA molecules known as nucleolytic ribozymes5–6. Nucleolytic ribozymes serve as platforms for the design of new biomedical technology13 and therapeutics10–11, and as models for our understanding of RNA catalysis and its implications for theories of evolution18. The central challenge is to gain a predictive understanding of precisely how these small RNA molecules, with their limited repertoire of chemical functional groups, are able to function as “high-speed” ribozymes16. Such an understanding would enable molecular engineering and guide the design of new catalytic RNA-based technology13 and medicine10–11.
The past 5 years have witnessed the discovery of new classes of nucleolytic ribozymes through comparative genomics19, and a doubling of the number of structurally characterized ribozymes since the first crystal structure almost 30 years prior20. These breakthroughs have fueled intense experimental and theoretical efforts directed toward elucidation of the detailed catalytic mechanisms of these ribozymes. The resulting whirlwind of mechanistic research has sparked much debate and caused some confusion in the literature, creating barriers to progress. Recently, Breaker pointed out contributing factors to these barriers, and made a call to clean up the resulting “mechanistic debris” in order to help the community navigate to calmer conditions.21 The first factor identified by Breaker was that “researchers frequently use different terms to discuss the same catalytic effects”, and suggested that the community adopt the framework illustrated in Figure 1 for discussion of catalytic strategies for RNA cleavage.16
This framework has been useful in facilitating discussion of RNA cleavage reactions16 for over 15 years, but as more detailed mechanistic data emerge, there is need to accommodate increased specificity and resolution of our description of catalysis. Toward that end, we propose to develop an ontology for discussion of catalytic strategies of RNA cleaving enzymes that stratifies Breaker’s original framework16 into different levels of contribution for each strategy in order to enable more precise interpretation of mechanism in the context of structure and bonding. Ontologies are used frequently in science to establish common taxonomy and structured vocabulary, as well as define conceptual entities and their inter-relationships within an application domain22. For example, an RNA ontology consortium has been proposed to describe and characterize RNA sequences, secondary structure, 3D structure, and dynamics pertaining to RNA function23.
3. Clarification of terminology: “Role” vs. “Effect”; and normal, inverse, and rescue effects
In the following section, we define three “levels of contribution” (e.g., primary, secondary and tertiary) for each catalytic strategy. These terms are applied to describe both “catalytic roles” and “catalytic effects”. Before proceeding to the different contribution levels, we clarify terminology that will be used to distinguish between “roles” and “effects”. These definitions pertain to catalytic strategies employed by a native enzyme in comparison to a mutant enzyme used to probe such catalytic strategies. Here a “role in catalysis” or “catalytic role” refers to the chemical mechanism by which an atom or functional group contributes to a specific strategy used by the native enzyme (e.g., the guanine N1 plays a primary role in γ catalysis by acting as a general base to deprotonate the nucleophile). Alternatively, an “effect on catalysis” or “catalytic effect” is used to indicate the outcome of a measurement that results from probing a modification of the native enzyme (e.g., the guanine N1C chemical modification has a primary effect on γ catalysis by knocking out the general base heteroatom). In the presentation below, we frame our discussion in the context of catalytic “effects” that arise due to perturbations to the native system under a set of standard conditions, with the assumption that the translation of their meaning to “roles” in the native system is readily inferred, although exceptions can occur.
Underlying this discussion is the idea that catalytic effects can be probed experimentally by measuring the ratio (k/k′), where k and k′ are the pseudo first-order rate constants for the native and modified substrate/enzyme reactions with no change in rate limiting step, respectively. Implicit in this analysis, and hence on the application of the proposed ontology that follows, is that chemistry is the rate-limiting step in the catalytic reaction, the considerations for which have been described in detail elsewhere.24
Borrowing terminology from the kinetic isotope effect literature25, a normal effect is one that has a ratio greater than unity (k/k′ > 1) and leads to a rate reduction, whereas an inverse effect is one that has a ratio less than unity (k/k′ < 1) and leads to a rate enhancement. For example, removal of a key conserved functional group would impede catalysis and have a normal effect, whereas introduction of an enhanced leaving group or alleviation of an inhibitory interaction would promote catalysis and have an inverse effect. Finally, a rescue effect is one where the deleterious (normal) effect on rate (i.e. k/k′ > 1) due to a single change to the native system (e.g., a mutation, chemical modification or change in reaction conditions) is partially, fully, or over-fully recovered by a second aggregate change (not necessarily of the same type) to the system.
4. Definitions of 1°, 2°, and 3° contribution to β, γ and δ catalytic strategies
Our proposed ontology uses ideas and conventions from other areas of mechanistic enzymology and structural biology to extend the framework for discussion of catalytic strategies illustrated in Figure 1. In-line fitness26 (α catalysis) imposes geometrical requirements needed to satisfy so- called Westheimer’s rules for transition states in phosphate transesterification and hydrolysis.27 Our ontology does not introduce additional stratification of α catalysis from Breaker’s original framework16. On the other hand, β, γ and δ catalysis strategies can be inherently associated with bonding and/or non-bonding interactions involving specific atoms. Using this association, we propose a decomposition of β, γ and δ catalysis into primary (1°), secondary (2°) and tertiary (3°) contributions. As mentioned above, when interpreting the outcomes of measurements (or calculations) used to probe mechanism, we will refer these contributions to a particular (β, γ or δ) strategy as primary, secondary and tertiary catalytic effects.
For each catalytic strategy, we first identify the chemical space of bonds that are either broken or formed along the reaction coordinate, and the primary atomic positions associated with these bonds. Due to the strong influence of divalent metal ions in RNA cleavage28–29, we include ionic bonding (direct inner-sphere coordination) as part of this chemical bonding space. Positions not associated with these bonds are non-primary atomic positions. The designations of the following “primary” and “non-primary” atomic positions are used to facilitate definitions of the primary, secondary and tertiary catalytic effects.
The primary atomic positions are defined for each catalytic strategy as follows:
Primary β atomic positions: the NPO positions themselves, any atoms directly involved in protonation of the NPOs (e.g., the proton itself and the heteroatom of the acid from which the proton was transferred), and any metal ion directly coordinated to the NPOs (atoms under green oval in Figure 1). It does not include atoms on the acid not directly involved in a bond with the proton, nor does it include atoms that hydrogen bond to the NPOs.
Primary γ atomic positions: the O2′ position itself, any atoms directly involved in nucleophile activation (e.g., the O2’ proton itself, and the heteroatom of the base to which the proton is transferred), and any metal ion directly coordinated to the O2′ position (atoms under red oval in Figure 1). It does not include atoms on the base not directly involved in a bond with the proton, nor does it include nearby metal ions (not directly coordinated to the O2’) that electrostatically influence the pKa of the base.
Primary δ atomic positions: the O5′ position itself, any atoms directly involved in leaving group stabilization (e.g., the acid proton itself, and the heteroatom of the acid from which the proton is transferred), and any metal ion directly coordinated to the O5′ position (atoms under purple oval in Figure 1). It does not include atoms on the acid not directly involved in a bond with the proton, nor does it include nearby metal ions (not directly coordinated to the O5’) that electrostatically influence the pKa of the acid.
With these definitions, we now adopt conventions used in the discussion of isotope effects to categorize catalytic effects as primary or secondary,25 and concepts from structural biology to introduce tertiary catalytic effects. Illustrative examples for each effect are given in a general context below, and then are expanded in the next section to include specific examples of perturbations (with measurements and/or predicted values) that in some cases involve coupling of catalytic effects.
1° Catalytic Effects.
A primary catalytic effect is one that results from changes in the identity of the primary atomic positions, as defined above.
Illustrative examples include: 1) disruption of the direct coordination of a catalytic divalent metal ion at the NPO position (e.g. thio substitution disrupting Mg2+ coordination) would give rise to a primary β effect, 2) a chemical modification that removes the general base atom involved in activation of the nucleophile (e.g. guanine general base N1C knockout) would give rise to a primary γ effect, and 3) a chemical modification that removes the general acid atom that donates a proton to the leaving group (e.g. adenine (N1) general acid N1C knockout) would give rise to a primary δ effect.
2° Catalytic Effects.
A secondary catalytic effect, on the other hand, is the change in the electronic environment of the primary atom resulting from changes in the identity of non-primary atomic positions (and thus is exclusively different from a primary catalytic effect). Modifications leading to a secondary catalytic effect have an indirect influence on the bonding environment of the primary atoms without involving any change to their identity. This can occur through electrostatic, inductive or stereoelectronic effects that perturb the underlying electronic structure of the bonds between primary atoms (e.g., through either remote chemical modification or short-ranged non-bonded interactions).
Illustrative examples include: 1) elimination of a stabilizing hydrogen bond to the NPO (while otherwise not changing the structure of the active site), such as deletion of a nucleobase exocyclic amine that hydrogen bonds to the NPO30 would give rise to a secondary β effect, 2) chemical modification at non-primary atomic positions of the general base that changes the pKa of the primary position (e.g. guanine general base N7C modification up-shifting the pKa at the N1 position) would give rise to a secondary γ effect, and 3) replacement of a divalent metal ion acting as a general acid through a coordinated water molecule with a different metal ion that has a shifted pKa would give rise to a secondary δ effect.
3° Catalytic Effects.
A tertiary catalytic effect reflects alteration of the position of the primary atoms resulting from modification of the structural scaffold or hydrogen bond network that organizes the enzyme active site. This alteration can lead to changes in: 1) positions of key residues, functional groups, or protons in the active site, 2) interactions that support active conformations of the substrate itself, 3) binding modes and/or occupations of metal ions or other small molecules required for activity, or 4) orientation of solvent molecules that form hydrogen bond networks important for catalysis.
5. Application of the ontological framework in the context of perturbation studies
Experimental studies where perturbations (mutations, site-specific chemical modifications, or changes in environmental conditions) probe specific catalytic strategies can provide deep insight into mechanism24, 31–33. As the nature of these perturbations and their coupling reach higher precision and greater complexity, there is need for the framework for mechanistic discussions to evolve and keep pace. The present ontology strives to achieve this.
We recognize that in some cases, hydrogen bonds and metal ion binding modes play both a structural and chemical role in catalysis, and it can be challenging to design experiments that allow their effects to be isolated. Nonetheless, theoretical methods can impose constraints that experimental methods cannot, and enable quantitative deconstruction of catalytic effects according to elements of our proposed ontology, with additive or non-additive contributions to catalysis.
We now illustrate the application of the proposed ontology using several examples that involve experimental measurements. We consider a perturbation that is introduced by a modification to the native system under a standard set of conditions. The measured outcome of a perturbation produces an overall “catalytic effect”, as discussed above, that can have many additive or non-additive contributions. In the present ontology, these contributions can be deconstructed into primary, secondary or tertiary β, γ or δ effects. We thus use the terminology “a primary/secondary/tertiary β, γ or δ (catalytic) effect due to a perturbation” to describe one of possibly many catalytic effects that result from a particular perturbation, rather than using the descriptors to characterize the perturbation itself (e.g., WRONG: “a primary/secondary/tertiary β, γ or δ perturbation”). The specific examples below for β, γ or δ catalytic effects are based on plausible catalytic strategies of the Varkud satellite (VSr), hammerhead (HHr) and twister (Twr) ribozymes, respectively, and are summarized in Table 1. In some cases, a perturbation may produce multiple catalytic effects (referred to as functional blurring). For these, the catalytic effects highlighted in the discussion are shown in bold (and left-aligned) under “Effects” in Table 1, whereas the other catalytic effects, enumerated in the table but not discussed in the text, are shown in regular font and right-aligned. Finally, the last subsection illustrates a more complex example involving the twister ribozyme (Figure 2 and Table 2) that has been a focal point of “mechanistic debris” in the recent literature.21
Table 1.
Summary of exemplary primary, secondary and tertiary catalytic effects.
| Sysa | Variantb | Effectsc | k/k′d | Changee |
|---|---|---|---|---|
| VSr | A621 :S(SP) | 1° β | NPO thio substitution disrupts Mg2+ binding | |
| 2° γ | >103 | pKa up-shift of G638:N1 (due to loss of Mg2+) | ||
| 3° γ | Disrupt anchoring of G638 (due to loss of Mg2+) | |||
| G638I/A756(3cP) | 2° β | NPO charge destabilization (loss of H-bonds) | ||
| 3° γ | 140 | Disruption of G638:N1 (general base) positioning | ||
| 3° δ | Disruption of A756:N1 (general acid) positioning | |||
| G623(7cG) | 3° β | Disruption of Mg2+ binding pocket (G623:N7) | ||
| 2° γ | >103 | pKa up-shift of G638:N1 (due to loss of Mg2+) | ||
| 3° γ | Disrupt anchoring of G638 (due to loss of Mg2+) | |||
| C17(dC) | 1° γ | >103 | Nucleophile knockout (C17:O2’ removal) | |
| HHr | G12(3cG) | 2° γ | >200 | pKa up-shift of G12:N1 (general base) |
| A9G | 3° γ | >300 | Disrupt anchoring of G12 (due to loss of H-bonds) | |
| A1 (3cA) | 1° δ | >>105 | General acid (A1 :N3) knockout | |
| Twr | A1 (1 cA) | 2° γ | 0.05f | pKa up-shift of A1:N3 (general acid) |
| A1 (2AP) | 3° δ | >105 | Disruption of A1 position (due to loss of H-bonds) | |
| 2° δ | pKa down-shift of A1:N3 (due to loss of NPO interactions) |
Ribozyme systems: Varkud satellite (VSr), hammerhead (HHr) and twister (Twr) ribozymes.
Identity of the all chemical modifications to the wild type (WT) ribozyme/substrate.
Exemplary effects (discussed as “specific examples” in the text) are shown in bold and left-aligned and additional effects (indicated by “Note” in text) are right-aligned.
Relative rates of the mutations to the native system for VSr34, HHr35–37 , and Twr38–39. The k/k′ value for A621:S(SP) thio substitution for VSr is equivalent to kO/kS.
Summary of the change in the system caused by the mutation.
Based on A19U variant.
Figure 2.

Application of ontology in describing the effects of G33I mutation and NPO thio substitutions of the substrate in the twister ribozyme (Twr). Summarized in the center are key interactions (1–4) that are highlighted in each variant/substrate by ovals in colors corresponding to their associated catalytic strategies, γ (red) and β (green). The wild-type (WT) Twr is shown in the top left, G33I mutant (top right), S(RP) substrate (bottom left), and G33I/S(RP) (bottom right). Arrows indicate transformations between the different variant/substrates. Relative kinetic rates and catalytic effects resulting from each transformation are indicated with their associated arrows. Lack of color highlighting indicates an absence (or significant weakening) of an interaction present in the wild-type and has a normal catalytic effect, whereas emphasis of color/size of highlighting indicates an inverse effect (e.g., a larger red sphere at G33:N1 (1) indicates a favorable down-shifted pKa).
Table 2.
Observed and relative rates for G33I mutation and substrate thio substitutions, S(RP/SP), in twister ribozyme compared to wild type (WT).
| Sysa | Variantb | Ionsc | kobs (min−1)d | k/k′e | ko/ksf | Rescueg |
|---|---|---|---|---|---|---|
| Twr | WT | 2.45 ± 0.04 | ||||
| S(RP) | Mg2+ | 0.026 ± 0.002 | 94 | 94 | ||
| S(SP) | 3.3 ± 0.2 | 0.7 | 0.7 | |||
| WT | Mg2+ | 0.8 ± 0.1 | 3.1 | (metal) | ||
| S(RP) | + | 0.007 ± 0.002 | 350 | 114 | 0.8 | |
| S(SP) | Mn2+ | 3.7 ± 0.4 | 0.7 | 0.2 | 3.2 | |
| G33I | 0.0057 ± 0.0005 | 430 | (mutational) | |||
| G33I/S(RP) | Mg2+ | 0.0060 ± 0.0003 | 410 | 0.95 | 99 | |
| G33I/S(SP) | 0.004 ± 0.001 | 610 | 1.4 | 0.5 |
Ribozyme system, Twr: twister ribozyme.
Identity of the all chemical modifications to the wild type (WT) ribozyme/substrate.
Molarity of each indicated divalent ion present is 10mM.
All experimental values from reference38.
k/k′ values are obtained using the WT in 10 mM Mg2+ conditions (first row).
kO/kS values are for oxo and thio substituted substrates for a given variant under a single set of ionic conditions.
Rescue effects are shown for metal ion rescue, (kO/kS)Mg2+/(kO/kS)Mn2+, and mutational rescue, ((kO/kS)WT/(kO/kS)Mut).
5.1. Perturbations Affecting α Catalysis:
Any mutation or chemical modification that impacts the in-line fitness of the reactive atoms (e.g., mutation that disrupts scaffold hydrogen bonds imposing the splay of N−1/N+1 nucleobases flanking the scissile phosphate required for in-line fitness). The present ontology does not distinguish tiers of α catalysis.
5.2. Perturbations Affecting β Catalysis:
-
Primary beta (1° β): substitution of the NPO by another atom (e.g., NPO thio substitution), alteration of conditions that disrupt or re-establish direct (inner-sphere) Mg2+ ion coordination to NPO (e.g., titration with Co(NH3)63+ or thiophilic Cd2+ ions, respectively40), or chemical modification of an acid donor heteroatom that prevents protonation of the NPO.
Specific example (VSr): A621:S(SP) thio substitution34 (kO/kS >103) that leads to disruption of the direct coordination of a Mg2+ ion at the pro-SP NPO of the scissile phosphate (A621) and gives rise to a (normal) primary β effect, and recovery of the rate by titration with a thiophilic (Cd2+) metal ion gives rise to a primary β rescue effect. Note (example of functional blurring): this Mg2+ ion also plays secondary and tertiary roles in γ catalysis, tuning the pKa of G638 (the implicated general base) and orienting it for nucleophile activation.
-
Secondary beta (2° β): Disruption of H-bond donation to the NPO from Mg2+ bound water or nucleobase functional group (e.g., removal of exocyclic amine of guanine, G:N2, or adenine, A:N6).
Specific example (VSr): G638I/A756(3cP) double mutation34 (k/k’ ~140), where 3cP indicates 3-deazapurine, that leads to elimination of hydrogen bond donation from the exocyclic amines of G638 and A756 to the pro-RP NPO of the scissile phosphate and gives rise to a (normal) secondary β effect. Note (example of functional blurring): these functional groups also play a role in tertiary γ and δ catalysis, positioning the general base (G638) and acid (A756), respectively.
-
Tertiary beta (3° β): Change in structural scaffold or hydrogen bond network needed to support primary and/or secondary β catalysis (e.g., mutation of residue that anchors a nucleobase or disrupts a metal ion binding site in position to H-bond or electrostatically stabilize a scissile phosphate NPO).
Specific example (VSr): G623(7cG) mutation34 (k/k’ >103) that leads to disruption of a Mg2+ ion binding site and gives rise to a (normal) tertiary β effect. Note (example of functional blurring): this Mg2+ ion also plays secondary and tertiary roles in γ catalysis, tuning the pKa of G638 (the implicated general base) and orienting it for nucleophile activation.
5.3. Perturbations Affecting γ Catalysis:
-
Primary gamma (1° γ): Modification of the nucleophile itself (e.g., 2′-deoxy, 2′-amino, 2′-O-methyl) or of the general base heteroatom that accepts the proton from the 2′OH (e.g., 1cG), or direct (inner-sphere) coordination of a metal ion to the nucleophile to facilitate its activation (deprotonation).
Specific example (HHr): C17(dC) mutation35 (k/k’ >103) that leads to removal of the nucleophile and gives rise to a (normal) primary γ effect.
-
Secondary gamma (2° γ): Remote (non-primary) chemical modification or metal ion binding that affects nucleophile activation by proton transfer to a general or specific base. This may include a pKa shift of the general base itself (e.g., G→A, 7cG, 6sG, or G:O6 • Mg2+) or change in the acidity of the nucleophile through hydrogen bonding or outer-sphere metal ion coordination.
Specific example (HHr): G12(3cG) mutation36 (k/k’ >200) that leads to up-shifting of the pKa at the N141, and gives rise to a (normal) secondary γ effect.
-
Tertiary gamma (3° γ): Change in structural scaffold or hydrogen bond network needed to support primary and/or secondary contributions to γ catalysis (e.g., G→A mutation that disrupts the sugar/Hoogsteen edge base pair that positions G to act as the general base, or competitive “over-determined” H-bonding that releases the 2′OH from inhibitory interactions with the NPOs upon introduction of a pro-RP thio group, thus giving rise to an inverse thio effect30, 42–43).
Specific example (HHr): A9G mutation37 (k/k’ >300) that leads to disruption of the hydrogen bonding interactions between the sugar edge of G12 with the Hoogsteen edge of A9 that anchors the general base (G12) and gives rise to a (normal) tertiary γ effect.
5.4. Perturbations Affecting δ Catalysis:
-
Primary delta (1° δ): Modification of the leaving group itself (e.g., O5′ thio substitution) or the general acid atom that donates the proton to the O5′ (e.g., 1cA or 3cA chemical modifications in the case these positions act as general acid), or replacement or removal of a divalent metal ion that acts as a Lewis acid by making direct inner-sphere contact with the O5′ leaving group, or elimination of a divalent metal ion that acts as a Brønsted acid by donating a proton from a metal-bound water molecule to the O5′ (e.g., Mg2+→ Na+ or Co(NH3)63+.)
Specific example (Twr): A1(3cA) mutation38 (k/k’ >>105) that leads to knockout of the adenine general acid and gives rise to a (normal) primary δ effect, and (predicted) recovery of the rate by 5′ thio substitution44 would give rise to a primary δ rescue effect.
-
Secondary delta (2° δ): Remote (non-primary) chemical modification or metal ion binding that affects leaving group departure by proton transfer from a general or specific acid. This may include pKa shift of the general acid itself (e.g., A→G, or 7cA) or replacement (not removal) of a divalent metal ion that acts as a Brønsted acid by donating a proton from a metal-bound water molecule to the O5′ leaving group (e.g., Mg2+→ Mn2+ or Cd2+), or action of buffer through a water wire to protonate the leaving group when protonation by the general acid (e.g., A:N3) is not productive (neutral and higher pH)45.
Specific example (Twr): A1(1cA) chemical modification38 (k/k’ ~0.0013) that leads to tuning of the pKa at the N3 position (pKa ~ 2.6) and gives rise to a (inverse) secondary δ effect.
-
Tertiary delta (3° δ): Change in structural scaffold or hydrogen bond network needed to support primary and/or secondary δ contributions to catalysis (e.g., A→P mutation that eliminates key structural hydrogen bonds that position A to act as a general acid to donate a proton to the O5′ leaving group).
Specific example (Twr): A1(2AP) mutation39 (k/k’ >105) that leads to disruption of a critical anchoring interaction between A1:N2 and the NPOs of C16 and C17. Note (example of functional blurring): these interactions also play a role in secondary δ catalysis by shifting the pKa of A1:N3 toward neutrality in the ribozyme environment.
5.5. Specific Illustrative Example: the Twister Ribozyme
In this section we explore in more detail chemical modifications in Twr to demonstrate application of the proposed ontology. Unlike the twister examples given in the previous sections we now turn our attention to a more complex set of examples involving a mutation of the general base to inosine (G33I) together with stereospecific thio substitutions of the NPO positions of the scissile phosphate of the substrate (Figure 2). Kinetic data for these mutations, including under ionic conditions (10 mM Mn2+) that examine metal ion rescue effects, are summarized in Table 2. These effects have been measured experimentally and interpreted computationally,46 and here we apply the ontology to discuss the interpretation of these results using a model whereby G33 is held in position by hydrogen bonds with A2 to act as a general base to activate the nucleophile through the N1 position, while the exocyclic amine (N2) donates a hydrogen bond to the pro-RP NPO of the scissile phosphate.
WT → G33I (top edge): k/k′ ~43038.
This mutation eliminates the exocyclic amine (−NH2) of guanine and down-shifts the pKa at the N1 position. The expected catalytic effects include:
(Position 1) A slightly inverse 2° γ effect due to the shift in pKa towards neutrality from 9.5 (inferred from activity-pH profiles38) to 9.1 (estimated from a ΔpKa ~ −0.4 shift of G to I41), which could be detected from measurement of the mutant activity-pH profile. This scenario assumes the N1 of G33 is the general base. The inverse effect due to pKa shift has been predicted by computation to be attenuated by a very slight normal effect due to an increase in the barrier to proton transfer from the nucleophile to the N1 of G3344. Alternately stated, the thermodynamic gain of pKa down-shifting outweighs the kinetic penalty of decreased basicity, leading to the overall slight inverse effect.
(Position 2) A normal 3° γ effect due to the disruption of the G33/A2 base pair that helps to structurally position (anchor) the general base (G33:N1 in close proximity to O2′) for nucleophile activation.
(Position 3) A normal 2° β / 3° γ effect due to loss of H-bonding of G33:N2 with the pro-RP oxygen that provides both electrostatic stabilization of the NPO (2° β) and structural organization (positioning) of G33 (3° γ).
WT → S(RP) (left edge): kO/kS ~9438.
This chemical modification introduces a thio substitution at the pro-RP position of the substrate scissile phosphate, and exhibits negligible Mn2+ rescue (kO/kS)Mg2+/(kO/kS)Mn2+ ~0.8. The expected catalytic effects include:
(Position 3) A normal 2° β / 3° γ effect due to weakened H-bonding of G33:N2 with the thio-substituted S(RP) non-bridge position of the substrate, similar to the 2° β / 3° γ effect for WT → G33I, as well as steric effects involving the larger sulfur atom that may further disrupt positioning of the general base.
(Position 4) A near-negligible 1° β effect due to thio substitution at the NPO position.
S(RP) → G33I/S(RP) (bottom edge): k/k′ ~4.338.
This mutation is identical to WT → G33I except for the inclusion of the thio-substituted substrate, with expected catalytic effects:
(Position 1) A slightly inverse 2° γ effect due to the shift in pKa towards neutrality from 9.5 to (estimated) 9.1 as in WT → G33I.
-
(Position 2) A normal 3° γ effect due to the disruption of the G33/A2 base pair as in WT → G33I.
Note: the 2° β / 3° γ effects in the WT → G33I mutation at position 3 due to loss of H-bonding of G33:N2 with the pro-RP oxygen are largely absent with the S(RP) substrate since this hydrogen bond is already disrupted due to the presence of the thio-substituted NPO with which it is interacting.
G33I → G33I/S(RP) (right edge): kO/kS ~0.9538.
This perturbation involves a S(RP) thio substitution of the substrate for the G33I mutant, with no significant expected overall catalytic effects.
(Position 4) A near-negligible (inverse) 1° β effect due to thio substitution at the NPO position. Note: With the elimination of the N2 exocyclic amine of inosine, the H-bonding interaction that was disrupted by the S(RP) substrate in the WT → S(RP) is already absent. The subtle differences in 2° β / 3° γ effects in position 3 are expected to be small.
Theoretical methods47 can, in principle, aid in the deconstruction of some mutational effects by isolating different contributions. For example, the normal 2° γ effect at position 1 resulting from G33I mutation could be decomposed into its thermodynamic (pKa down-shifting) component by measuring or calculating the microscopic pKa shift at the N1 position due to G33I mutation, and its kinetic (decreased basicity) component by calculating separately the relative free energy barriers for proton transfer to N1 for each mutation. Finally, the 3° γ (position 2) and 2° β / 3° γ (position 3) effects in the WT → G33I mutation could be decoupled by removal of the exocyclic amine of G33 and calculating the new free energy barrier for the reaction under artificial structural restraints that maintain anchoring of its position to act as general base, followed by repeating the calculation releasing these restraints (requiring additional free energy to properly position the general base) to recover the aggregate effect.
6. Summary
The end goal of experimental/computational work on a catalytic RNA system is to gain insight from the development of an atomically detailed model of mechanism that enables prediction. Thus, it is essential to have an ontology that enables interpretation of experimental and computational data, including from precision chemical modifications and molecular simulations, in terms of specific interactions within an active site and their contributions to catalysis. Toward this end, the proposed ontology has several advantages. First, it adheres to the original framework introduced by Breaker16 that has served the community for the last 15 years. Second, it adopts familiar terminology from mechanistic enzymology (particularly isotope effects) and structural biology. Third, it allows different catalytic strategies to be further categorized into a tiered hierarchy of primary, secondary, and tertiary contributions (even though the coupling of such effects occasionally become blurred experimentally) that enable more precise characterization of mechanistic details. It is the hope that the community will adopt this ontology as a common framework for discussion of catalytic strategies of RNA-cleaving enzymes and will help pave the path forward toward a consensus view of these fundamental and fascinating mechanisms.
Acknowledgements
The authors are grateful for financial support provided by the National Institutes of Health (MIRA R35-GM127064 to P.C.B., GM127100 to M.E.H, GM131568 to J.A.P., and GM62248 to D.M.Y.). The authors further thank Ron Breaker, David Lilley, Andrew Veenis, Kyle Messina, and McCauley Meyer for helpful comments and suggestions.
References
- 1.Perreault DM; Anslyn EV, Unifying the current data on the mechanism of cleavage– transesterification of RNA. Angew. Chem., Int. Ed 1997, 36 (5), 432–450. [Google Scholar]
- 2.Oivanen M; Kuusela S; Lönnberg H, Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by Brønsted acids and bases. Chem. Rev 1998, 98 (3), 961–990. [DOI] [PubMed] [Google Scholar]
- 3.Korhonen H; Williams NH; Mikkola S, βLG values in mechanistic studies on the transesterification of RNA models and their application in a metal ion complex promoted transesterification. J. Phys. Org. Chem 2013, 26 (2), 182–186. [Google Scholar]
- 4.Lassila JK; Zalatan JG; Herschlag D, Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem 2011, 80 (1), 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lilley DM, How RNA acts as a nuclease: some mechanistic comparisons in the nucleolytic ribozymes. Biochem. Soc. Trans 2017, 45 (3), 683–691. [DOI] [PubMed] [Google Scholar]
- 6.Ward WL; Plakos K; DeRose VJ, Nucleic acid catalysis: Metals, nucleobases, and other cofactors. Chem. Rev 2014, 114 (8), 4318–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raines RT, Ribonuclease A Chem. Rev 1998, 98 (3), 1045–1066. [DOI] [PubMed] [Google Scholar]
- 8.Breaker RR; Joyce GF, The expanding view of RNA and DNA function. Chem. Biol 2014, 21 (9), 1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman SK, Deoxyribozymes: Selection design and serendipity in the development of DNA catalysts. Acc. Chem. Res 2009, 42 (10), 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullenger BA; Nair S, From the RNA world to the clinic. Science 2016, 352 (6292), 1417–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim CM; Smolke CD, Biomedical applications of RNA-based devices. Curr. Opin. Chem. Biol 2017, 4, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z; Hutter D; Sheng P; Sismour AM; Benner SA, Artificially expanded genetic information system: a new base pair with an alternative hydrogen bonding pattern. Nucleic Acids Res. 2006, 34 (21), 6095–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J; Cao Z; Lu Y, Functional nucleic acid sensors. Chem. Rev 2009, 109 (5), 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anosova I; Van Horn WD; Kowal EA; Egli M; Dunn MR; Chaput JC, The structural diversity of artificial genetic polymers. Nucleic Acids Res. 2015, 44 (3), 1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshika S; Leal NA; Kim M-J; Kim M-S; Karalkar NB; Kim H-J; Bates AM; Watkins NE; SantaLucia HA; Meyer AJ; DasGupta S; Piccirilli JA; Ellington AD; SantaLucia J; Georgiadis MM; Benner SA, Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 2019, 363 (6429), 884–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emilsson GM; Nakamura S; Roth A; Breaker RR, Ribozyme speed limits. RNA 2003, 9 (8), 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez X; Dejaegere A; Leclerc F; York DM; Karplus M, Nucleophilic attack on phosphate diesters: a density functional study of in-line reactivity in dianionic, monoanionic, and neutral systems. base-catalyzed-hydrolysis; biological phosphoryl transfer; polarizable continuum model; methyl ethylene phosphate; gas-phase basicities; ab-initio; ester hydrolysis; aqueous-solution; ribonuclease-a; transphosphorylation reactions 2006, 110, 11525–11539. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert W, Origin of life: The RNA world. Nature 1986, 319 (6055), 618–618. [Google Scholar]
- 19.Weinberg Z; Kim PB; Chen TH; Li S; Harris KA; Lünse CE; Breaker RR, New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat. Chem. Biol 2015, 11 (8), 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pley HW; Flaherty KM; McKay DB, Three-dimensional structure of a hammerhead ribozyme. Nature 1994, 372 (6501), 68–74. [DOI] [PubMed] [Google Scholar]
- 21.Breaker RR, Mechanistic debris generated by twister ribozymes. ACS Chem. Biol 2017, 12 (4), 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith B, Beyond concepts: Ontology as reality representation In Formal Ontology in Information Systems (FOIS), Varzi AC; Vieu L, Eds. 2004; pp 1–12. [Google Scholar]
- 23.Leontis NB; Altman RB; Berman HM; Brenner SE; Brown JW; Engelke DR; Harvey SC; Holbrook SR; Jossinet F; Lewis SE; Major F; Mathews DH; Richardson JS; Williamson JR; Westhof E, The RNA ontology consortium: An open invitation to the RNA community. RNA 2006, 12 (4), 533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fersht A, Enzyme structure and mechanism. 2 ed.; W.H. Freeman: New York, 1985; Vol. 13, p 146–146. [Google Scholar]
- 25.Hengge AC, Isotope effects in the study of phosphoryl and sulfuryl transfer reactions. Acc. Chem. Res 2002, 35 (2), 105–112. [DOI] [PubMed] [Google Scholar]
- 26.Soukup GA; Breaker RR, Relationship between internucleotide linkage geometry and the stability of RNA. RNA 1999, 5 (10), 1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dennis EA; Westheimer F, The geometry of the transition state in the hydrolysis of phosphate esters1. J Am. Chem. Soc 1966, 88 (14), 3432–3433. [DOI] [PubMed] [Google Scholar]
- 28.Schnabl J; Sigel RKO, Controlling ribozyme activity by metal ions. Curr. Opin. Chem. Biol 2010, 14 (2), 269–275. [DOI] [PubMed] [Google Scholar]
- 29.Lönnberg T; Lönnberg H, Chemical models for ribozyme action. Curr. Opin. Chem. Biol 2005, 9 (6), 665–673. [DOI] [PubMed] [Google Scholar]
- 30.Bingaman JL; Zhang S; Stevens DR; Yennawar NH; Hammes-Schiffer S; Bevilacqua PC, The GlcN6P cofactor plays multiple catalytic roles in the glmS ribozyme. Nat. Chem. Biol 2017, 13 (4), 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris ME; Piccirilli JA; York DM, Enzyme transition states from theory and experiment. Biochim. Biophys. Acta 2015, 1854 (11), 1727–1728. [DOI] [PubMed] [Google Scholar]
- 32.Anslyn EV; Dougherty DA, Modern Physical Organic Chemistry. University Science Books: Sausalito, CA, 2006. [Google Scholar]
- 33.Jencks WP, Catalysis in chemistry and enzymology. Courier Corporation: 1987. [Google Scholar]
- 34.Ganguly A; Weissman BP; Giese TJ; Li N-S; Hoshika S; Rao S; Benner SA; Piccirilli JA; York DM, Theory and experiment converge to define the active site configuration and catalytic mechanism of the largest known nucleolytic ribozyme. Nat. Chem 2019, in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blount KF; Uhlenbeck OC, The structure-function dilemma of the hammerhead ribozyme. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 415–440. [DOI] [PubMed] [Google Scholar]
- 36.Nelson JA; Uhlenbeck OC, Hammerhead redux: does the new structure fit the old biochemical data? RNA 2008, 14 (4), 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruffner DE; Stormo GD; Uhlenbeck OC, Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry 1990, 29, 10695–10712. [DOI] [PubMed] [Google Scholar]
- 38.Wilson TJ; Liu Y; Domnick C; Kath-Schorr S; Lilley DMJ, The novel chemical mechanism of the twister ribozyme. J. Am. Chem. Soc 2016, 138 (19), 6151–6162. [DOI] [PubMed] [Google Scholar]
- 39.Košutić M; Neuner S; Ren A; Flür S; Wunderlich C; Mairhofer E; Vu\usurović N; Seikowski J; Breuker K; Höbartner C; Patel DJ; Kreutz C; Micura R, A mini-twister variant and impact of residues/cations on the phosphodiester cleavage of this ribozyme class. Angew. Chem. Int. Ed 2015, 54, 15128–15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frederiksen JK; Li N-S; Das R; Herschlag D; Piccirilli JA, Metal-ion rescue revisited: Biochemical detection of site-bound metal ions important for RNA folding. RNA 2012, 18 (6), 1123–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izatt RM; Christensen JJ; Rytting JH, Sites and thermodynamic quantities associated with proton and metal ion interaction with ribonucleic acid, deoxyribonucleic acid, and their constituent bases, nucleosides, and and nucleotides. Chem. Rev 1971, 71, 439–481. [DOI] [PubMed] [Google Scholar]
- 42.Seith DD; Bingaman JL; Veenis AJ; Button AC; Bevilacqua PC, Elucidation of catalytic strategies of small nucleolytic ribozymes from comparative analysis of active sites. ACS Catal. 2018, 8, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bingaman JL; Gonzalez IY; Wang B; Bevilacqua PC, Activation of the glmS ribozyme nucleophile via overdetermined hydrogen bonding. Biochemistry 2017, 56 (33), 4313–4317. [DOI] [PubMed] [Google Scholar]
- 44.Gaines CS; Giese TJ; York DM, Cleaning up mechanistic debris generated by twister ribozymes using computational RNA enzymology. ACS Catal. 2019, 9, 5803–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Messina KJ; Bevilacqua PC, Cellular small molecules contribute to twister ribozyme catalysis. J. Am. Chem. Soc 2018, 140 (33), 10578–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaines CS; York DM, Ribozyme catalysis with a twist: Active state of the twister ribozyme in solution predicted from molecular simulation. J Am. Chem. Soc 2016, 138 (9), 3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panteva MT; Dissanayake T; Chen H; Radak BK; Kuechler ER; Giambaşu GM; Lee T-S; York DM, Multiscale methods for computational RNA enzymology In Methods in Enzymology, Chen S-J; Burke-Aguero DH, Eds. Academic Press: 2015; Vol. 553, pp 335–374. [DOI] [PMC free article] [PubMed] [Google Scholar]