

Abstract
Fabry disease is an X-linked lysosomal storage disorder caused by a deficiency of α-galactosidase A (α-gal A). The clinical variability of the phenotypes of Fabry disease in females is still poorly understood. The degree of aberrant methylation of non-mutated alleles is thought to have significant effects on X-chromosome inactivation (XCI). We previously reported that one heterozygous Fabry female showing classical phenotypes had complete methylation of the non-mutated allele of the GLA gene. In this report, we summarized 36 heterozygous females with a clinical severity score based on the FAbry STabilization indEX (FASTEX). We measured their α-gal A activity and plasma/ serum globotriaosylsphingosine (lyso-Gb3) accumulation and performed electron microscopy of skin biopsies. We analyzed the methylation-sensitive restriction enzyme sites throughout the GLA gene, including the 5’UTR, and found a single SacII site and multiple HhaI and HpaII sites aggregated in exon 1 and the 5’UTR. One HpaII sequence in exon 7 was also detected as a methylation-sensitive site. With methylation-sensitive restriction enzymes, methylated and non-methylated alleles could be separated, and the ratio of the methylation was quantified. We found a clear correlation between the severity of the phenotype and lyso-Gb3 accumulation for heterozygous Fabry disease in females. Methylation of the non-mutated allele was also proportionately correlated to the clinical severity score measured by FASTEX.
Keywords: Heterozygous Fabry females, Fabry stabilization indEX, Methylation, X-chromosome inactivation
Abbreviations: α-gal A, α-galactosidase A; Gb3, globotriaosylceramide; lyso-Gb3, globotriaosylsphingosine; XCI, X-chromosomal inactivation
Highlights
-
•
We summarized 36 heterozygous Japanese Fabry females with their clinical severity score.
-
•
We had detected methylation-sensitive restriction enzyme sites in exon 7 along with exon 1 and 5`UTR.
-
•
A clear correlation of patients’ FASTEX scores, sphingolipids accumulations and dysmethylation of the GLA gene was detected.
1. Introduction
Fabry disease (OMIM #301500) is an X-linked disorder characterized by the deficiency of the lysosomal hydrolase α-galactosidase A (α-gal A, E.C. 3.2.1.22) due to mutations in the GLA gene [1,2]. Patients with partial or complete deficiency of α-gal A are unable to degrade glycosphingolipids, such as globotriaosylceramide (Gb3), and related glycosphingolipids, such as globotriaosylsphingosine (Lyso-Gb3), effectively, and these lipids consequently accumulate in body fluids and the lysosomes of a variety of cell types, including the vascular endothelium, kidney, heart, eye, and nerve cells [3,4].
Clinical manifestations in classically affected hemizygotes who have no detectable α-gal A activity include pain and paresthesia in the extremities, angiokeratoma, and hypo- or anhidrosis. Corneal and lenticular opacities are also early findings. With increasing age, proteinuria, hyposthenuria, and lymphedema appear [2]. Severe renal impairment leads to hypertension and uremia. Death usually occurs from renal failure or cardiac or cerebrovascular disease [1].
The clinical course and prognosis of heterozygous females differ from those of hemizygotes [5]. Approximately 70% of heterozygous Caucasians have whorl-like corneal dystrophy as an early manifestation of the disease [6,7]. However, Japanese female Fabry patients have a heterogeneous presentation. The age at onset of disease varies from 4 to 68 years. >50% of the heterozygotes present with acroparesthesia and corneal opacities. Approximately 40% of the patients have proteinuria and left ventricular hypertrophy (LVH). The incidence of LVH in women over 68 years of age is approximately 100% [8]. The diagnoses of heterozygous females are also complicated because of residual α-Gal A activity [9]. Even genetic testing of at-risk patients or patients suspicious for Fabry disease has been compromised by the recently recognized occurrence of various benign missense mutations [[10], [11], [12]].
Various Fabry disease severity scoring systems, such as the Mainz Severity Score Index (MSSI) [13], the Fabry Disease Severity Scoring System (DS3) [14] provide an index of disease severity at a single time point. In our daily practice in Japan, the FAbry STabilization indEX (FASTEX) [15] is commonly used as a mathematical model in order to objectively verify the clinical severity and stability of patients with Fabry disease.
The pathophysiology for the severity of phenotypes in heterozygous Fabry females is still poorly understood. XCI occurs at random within females resulting in an average inactivation ratio of 50:50 with normal distribution [16]. Skewed XCI is described for some other diseases [17]. The existence of skewed XCI in females with Fabry disease was found in a recent study by analyzing the human androgen receptor (HUMARA) gene [18] which demonstrated the existence of skewed inactivation (29%) of heterozygotes females with Fabry disease correlating to Fabry disease severity scores (MSSI and DS3).
In higher order eukaryotes, DNA is methylated only at 5’-CpG-3′ dinucleotides (CpGs), and this DNA modification has important regulatory effects on gene expression, especially in CpG-rich areas (known as CpG islands) located in the promoter regions of many genes [[19], [20], [21]]. As extensive methylation of CpG islands is associated with transcriptional inactivation of several genes on one of the two X-chromosomes in females [22,23], such X chromosomes result in an inactive condition. XCI is accompanied by the changes in DNA methylation at CpG islands, and the methylation of HpaII and HhaI sites correlates with XCI [24,25]. The 5’UTR of the GLA gene contains a CpG island composed of 474 base pairs (chrX:101407840–101,408,313) [26]. Our previous study showed that the methylation of HpaII and HhaI sites in exon 1 of GLA correlates with XCI and the severity of the disease [27].
In this study, we summarized 36 heterozygous Fabry females with their clinical severity score and detected their lyso-Gb3 accumulation in plasma and sphingolipid accumulation with skin biopsies. We obtained the methylation-sensitive restriction enzyme sites (RE) in exon 1, 5’UTR and exon 7 of the GLA gene. We found a clear correlation between the severity of the phenotype, lyso-Gb3 accumulation and methylation of the normal allele detected by nondigestion with methylation-sensitive RE SacII, HhaI and HpaII.
2. Materials and methods
2.1. Patients' clinical summary
The information of the 36 heterozygous Japanese Fabry females was summarized based on their previous family history, careful physical examinations, and biochemical and molecular findings. The findings were then categorized to fit FASTEX scoring including the nervous, cardiac and renal systems.
2.2. Calculation of the clinical severity score by FASTEX
The FASTEX score was calculated as described before [15] with some modification. In short, nervous events included pain and other neural events; cardiac events included LVH, ECG/arrhythmia and cardiac functioning categorized by the New York Heart Association (NYHA), and renal events included the albumin-creatinine ratio (ACR)/protein-creatinine ratio (PCR) and eGFR. One of seven events ranged from 0–4. The total raw score was then summed.
2.3. Measurement of α-gal a activity using 4MUGal substrate
The level of α-gal A activity was measured as previously reported [28]. In short, lymphocytes and skin fibroblasts (SFs) were sonicated, and the whole cell lysates were incubated with a mixture containing 700 mM 4-methylumbelliferyl-α-d-galactopyranoside (4 MUGal; Sigma), 0.5 M N-acetyl-d-galactosamine (GalNAc; Sigma), and 50 mM citrate-phosphate (pH 4.5) buffer in a water bath at 37 °C for 3 h. The reactions were stopped with 150 mM ethylenediaminetetraacetic acid (pH 11.5) buffer. The fluorescence (excitation at 355 nm/emission at 460 nm) was measured with a microplate reader. The enzyme activity was calculated as nmol·h−1·mg protein−1.
2.4. Quantification of lyso-Gb3 by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Quantification of lyso-Gb3 was performed as described before [28]. In short, 60 μL fresh plasma was separated from whole blood and placed into a 1.5 mL tube containing 540 μL 2-propanol (HPLC-grade, Kanto, Japan) and 20 μL internal standard. After mixing for 2 min with a microtube mixer (Tommy, Japan), the extraction was centrifuged for 20 min at 12,000g. The supernatant from the extraction was dried under an air stream and reconstituted with 20 μL methanol. The final solution was transferred to autosampler micro vials for measurement by the LCMS-8040 system (Shimadzu, Japan).
The calibration set consisted of LysoGb3 (Matreya LLC, USA) standards with the same amount of N-glycinated LysoGb3 (internal standard, Matreya LLC, USA) in the methanol solvent. The calibrated amount of LysoGb3 was 0.0–0.8 ng (R2 = 0.996), and a calibration set was performed for every 50 samples.
Five microliters of the solution were measured by high-performance liquid chromatography (HPLC) connected to a tandem mass spectrometer (LCMS-8040, Shimadzu, Japan). The mixture was eluted with mobile phase A (10 mM NH4COOH, Wako, Japan) and phase B (10% 10 mM NH4COOH/MeOH) through a C8 column (Inertsil C8–3, 3 μm, 2.1 × 50 mm, GL Sciences Inc., Japan) connected to an in-line filter unit (ACQUITY, Waters, USA). The multiple reaction monitoring (MRM) transitions monitored were 786.5 > 264.3 m/z for LysoGb3 and 843.5 > 264.3 m/z for N-glycinated LysoGb3. The quantification of LysoGb3 was performed by comparing the peak molecular target area with the internal standard area.
2.5. Electron microscopy of skin biopsies
Electron microscopy for skin biopsies for cases 1–5, 7, 12–16, 18, 27–28 was done as described before [29]. In short, a 2 mm skin punch was taken from the left elbow, and small blocks of tissue were fixed in 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.2). Some blocks were directly embedded in Epon or glycol methacrylate. Ultrathin sections were examined either unstained or after various treatments: uranyl acetate and lead citrate, periodic acid‑silver methenamine, periodic acid-thiocarbohydrazide‑silver proteinate (PA-TCH-SP), and hydrogen peroxide-phosphotungistic acid (H202-PTA).
2.6. Detection of methylation sensitivity for SacII, HhaI and HpaII sites in the GLA gene
Methylation-sensitive sites were detected as described before [27]. In short, DNA from peripheral blood and SFs was collected using the Qiagen blood and tissue kit (Cat. No. 69504) and an amount of 400 to 1200 ng of DNA was used for bisulfite modification following the MethylEasy Xceed (Human Genetic Signatures Pty Ltd) protocol. Bisulfite-modified DNA, 40 to 50 ng, was amplified for each exon and the 5’UTR of the GLA gene with respective primer sets. The amplified DNA was then purified and sequenced.
2.7. Quantification of methylation of each allele in the GLA gene
One microgram of DNA from peripheral blood and SFs containing SacII was incubated for 20 h at 37 °C. The digested DNA was then purified by a high pure PCR product purification kit (ref. 11,732,676,001). Purified DNA fragments were used for long PCR amplification with PrimeSTAR GXL (Takara cat# R050A). A pair of primers (forward 5’-AGCGAGACGGTAGACGACGACCAGAACTACTTC-3′, reverse 5’-GGGGTGGGTATCTGGATGAGTAAATATGGGTT-3′) was used to amplify the whole GLA gene 11 kb including the 5’UTRA for all the cases. For HhaI and HpaII sites, 200 ng of DNA were used for the digestion reaction. Purified DNA fragments were used for amplification of exon 1 of the GLA gene for cases 1–9, 32 and 35–36 who had exon 1 mutations by the primers described before [27]. For other cases that had exon 5–7 mutations, exons 5 to 7 were amplified. All amplified PCR fragments were purified by Wizard SV gel and PCR (Promega), and direct sequencing was performed.
Quantification of methylation was performed based on digestion of mutated or non-mutated alleles by methylation-sensitive restriction enzymes. The quantification of methylation was measured by the Mutation Surveyor Softgenetics software (version 5.1.0). In short, the calculation of methylation of the non-mutated allele was performed in digested fragments as the length of non-mutated allele/ (length of mutated allele+ length of non-mutated allele).
3. Results
3.1. Clinical characteristics of heterozygous Fabry females
The clinical presentations were widely varied. Some cases were found to have no phenotype, e.g., cases 22, 24–25, 31, 33–36, and some cases were showing a severe phenotype as Hemizygous Fabry males, e.g., cases 1, 4–10, 12–13, 16, 23, 26–28, 30.
3.2. Calculation of the FASTEX score
Based on the clinical characteristics, the FASTEX raw score was summed. Most of the heterozygous female scores (21/36) ranged from 5 to 21. Some of them scored <5, e.g., case 3, 4, 11, 17–20, 22, 24–25, 31, 33–36.
3.3. α-gal A activity
Whole cell lysates from blood lymphocytes and SFs were used to detect α-gal A activity. The enzyme activity was widely varied and irrelevant to clinical severity, and it is listed in Table 1.
Table 1.
Summary of patients' clinical, biochemical and genetic information
| Patient No. | Age | Major clinical characteristics | α-gal A activitya |
Plasma lyso-Gb3b | FASTEX Raw Score | Methylation of normal (non-mutated) allele (%) |
GLA genotypes | Reference (GLA mutation) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lym | SFs | Blood DNA |
SFs DNA |
|||||||||
| SacII site | HhaI (exon 1) site/ HpaII (exon 7) sitec | SacII site | HhaI (exon 1) site/ HpaII (exon 7) sitec | |||||||||
| 1 | 37y | Acroparesthesia, dysmorphism, intellectual disabilities, LVH | 0 | 0 | 61 | 11 | 97.9 | 84.6 | 92.3 | 83.6 | c.36C > A(p.C12X) | [27] |
| 2 | 44y | Mild cardiomegaly | 9.9 | 2.5 | 5.6 | 5 | 46.7 | 29.7 | 35.1 | 21.6 | c.36C > A(p.C12X) | [27] |
| 3 | 42y | Mild pain | 9.1 | 12.9 | 8 | 2 | 10.8 | 7.4 | 1.5 | 44.3 | c.36C > A(p.C12X) | [27] |
| 4 | 11y | Mild acroparesthesia | 2.8 | 0.8 | 28.7 | 1 | 70.6 | 52.5 | 56.8 | 40 | c.36C > A(p.C12X) | [27] |
| 5 | 51y | Chest pain, acroparesthesia, LVH | 8.2 | 4.7 | 21.3 | 11 | 46.7 | 69.2 | 60.1 | 70.7 | c.97G > T(p.D33Y) | This study |
| 6 | 42y | Chest pain, LVH | 4.4 | – | 10.6 | 11 | 52.3 | 69.2 | – | – | c.97G > T(p.D33Y) | This study |
| 7 | 39y | Chest pain, CCF, acroparesthesia | 5.6 | 1.9 | 25 | 12 | 72.2 | 89.5 | 35.5 | 59.2 | c.97G > T(p.D33Y) | This study |
| 8 | 66y | Cerebral infraction, CCF, renal failure | 5.3 | – | 35.5 | 18 | 53 | 56.9 | – | – | c.97G > T(p.D33Y) | This study |
| 9 | 53y | Chest pain, CCF, acroparesthesia | 8.5 | – | 7.1 | 12 | 41.3 | 81.7 | – | – | c.97G > T(p.D33Y) | This study |
| 10 | 55y | Chest pain, cardiomegaly, proteinuria (+) | 7 | – | 7.7 | 10 | 28.3 | 99.9 | – | – | c.668G > C(p.C223S) | [28] |
| 11 | 22y | Pain in extremities | 8.8 | – | 13.1 (S) | 1 | 31.8 | – | – | – | c.668G > C(p.C223S) | [28] |
| 12 | 45y | Pain, mild cardiomegaly, proteinuria (++) | 4.1 | 3.7 | 17.1 | 13 | 74.1 | 45.2 | 4.8 | – | c.950 T > C (p.I317T) | [44] |
| 13 | 53y | Mild pain, mild cardiomegaly, proteinuria (+) | 4.5 | 3.6 | 5.8 | 12 | 49.7 | 74.1 | – | 99.9 | c.950 T > C (p.I317T) | [44] |
| 14 | 35y | Pain, cardiomegaly | 8.3 | 0.1 | 40 | 9 | 82.2 | 38.2 | 93.8 | – | c.901C > T (p.R301X) | [45] |
| 15 | 28y | Acroparesthesia | 3.1 | 6.8 | 26.1 | 5 | 77.9 | 68.3 | 48.7 | – | c.901C > T (p.R301X) | [45] |
| 16 | 45y | Headache, acroparesthesia, chest pain, LVH | 4.2 | 6.0 | 18.2 | 14 | 66.7 | – | – | – | c.334C > T (p.R112C) | [46] |
| 17 | 37y | Mild pain | 5.5 | 5.2 | 1 | 81 | 45.5 | – | – | c.1124G > A(p.G375E) | [47] | |
| 18 | 40y | Mild pain | 8.9 | 1.6 | 25.2 | 2 | 26.7 | 60.6 | – | – | c.749A > C(p.Q250P) | [48] |
| 19 | 37y | Mild pain | 4 | – | 14.2 | 2 | 57.4 | – | – | – | c.749A > C(p.Q250P) | [48] |
| 20 | 50y | Mild proteinuria, mild cardiomegaly | 9.5 | – | 21.5 | 4 | 13.9 | 52.5 | – | – | c.1208 T > C(p.L403S) | [49] |
| 21 | 29y | Mild cardiomegaly | 8.8 | – | 10.5 | 6 | 6.9 | 78.4 | – | – | c.658C > T(p.R220X) | [50] |
| 22 | 30y | Asymptomatic | 3.8 | – | 9.7 | 1 | 39.7 | – | – | – | c.658C > T(p.R220X) | [50] |
| 23 | 62y | Cardiomyopathy, pain, proteinuria (+) | 8.9 | – | 15.1 | 16 | 52 | – | – | – | c.658C > T(p.R220X) | [50] |
| 24 | 5y | Asymptomatic | 4.5 | – | 6.2 (S) | 0 | Non-meth | – | – | – | c.761delTTG | This study |
| 25 | 7y | Asymptomatic | 5.5 | – | 8.9 | 0 | Non-meth | – | – | – | c.761delTTG | This study |
| 26 | 53y | Acroparesthesia, headache, cardiac hypertrophy | 1.8 | – | 24.5 | 14 | Meth | – | – | – | c.802delTTAG | This study |
| 27 | 72y | Proteinuria (+++), creatinine clearance (↓), chest pain, cardiomegaly | 3.4 | 1.7 | 14 | 15 | 92.1 | – | 79.2 | – | c.902G > A(p.R301Q) | [51] |
| 28 | 74y | CCF, cardiac arrhythmia, pacemaker in situ, proteinuria (++) | 5.1 | 3.6 | 48.7 | 21 | 82.8 | – | – | – | c.679C > T(p.R227X) | [52] |
| 29 | 45y | Mild pain | 6.0 | – | 25.8 | 7 | 94.3 | 99.9 | – | – | c.679C > T(p.R227X) | [52] |
| 30 | 62y | Mild pain, LVH | 8.2 | – | 25 | 13 | 22.5 | – | – | – | IVS3 + 1G > A | [53] |
| 31 | 44y | Asymptomatic | – | – | 23.1 | 0 | 49.4 | – | – | – | c.658C > T(p.R220X) | [50] |
| 32 | 45y | Mild cardiomegaly | 4.5 | – | 4.1 | 5 | 40 | – | – | – | c.118C > T(p.P40S) | [54] |
| 33 | 6y | Asymptomatic | 5.4 | – | 18.5 | 0 | 47.7 | – | – | – | c.1019G > A(p.W340X) | [55] |
| 34 | 6y | Asymptomatic | 7.5 | – | 13 | 0 | 37.5 | – | – | – | c.1019G > A(p.W340X) | [55] |
| 35 | 6y | Asymptomatic | 2.2 | – | 27 | 2 | 83.8 | 81.4 | – | – | c.3G > A(p.M1I) | [56] |
| 36 | 8y | Asymptomatic | 4.5 | – | 7.4 | 1 | 63.9 | 74.9 | – | – | c.3G > A(p.M1I) | [56] |
LVH, left ventricular hypertrophy; α-gal A, α-galactosidase A; LYM, lymphocyte; SFs, skin fibroblasts; y, year; −, not measured; Met, methylated; Non-met, non-methylated.
α-gal A activity was measured with 4-MU substrate and the normal range of α-gal A activity was 10–12 nmol/h/mg protein in lymphocytes and skin fibroblasts. Blood lymphocytes were collected to measure the enzyme activity before two weekly enzyme infusions for patients who were in ERT.
Lyso-Gb3 was measured in the plasma of all the patients except in cases 11 and 24. Their Lyso-Gb3 was measured in blood serum. The normal range of Lyso-Gb3 in plasma/ serum was considered <2.1 ± 2 nmol/L.
Methylation of the HhaI site was measured in exon 1 for the patients who have exon 1 mutations including cases 1–9, and 35–36, and methylation of the HpaII site was measured in exon 7 for the patients who have exon 5–7 mutations including cases 10, 12–15, 17–18, 20–21, and 29.
3.4. Quantification of lyso-Gb3
Fresh plasma/serum were collected from all subjects, and lyso-Gb3 was extracted in methanol. Three consecutive measurements were performed and averaged. The list is summarized in Table 1.
3.5. Electron microscopy
Skin biopsies were taken from the left elbow and prepared for visualization by electron microscopy. Sphingolipid accumulation also varied case by case. Diffuse accumulation was detected in the lysosomes of almost all kind of cells including neuronal Schwann cells, vascular endothelium and fibroblasts (Fig. 1) in case 1. Mainly vascular endothelial and fibroblastic accumulation were found in cases 5, 7 (Fig. 2). Sphingolipid accumulation was detected predominantly in exocrine and fibroblastic cells in case 7 and myelin-like deposition was detected primarily in fibroblasts in case 27 (Fig. 2). Other cases were found to have heterogeneous and irregular or no accumulation (results not shown).
Fig. 1.

Electron micrograph of a section of skin tissue from case 1 showing massive lysosomal accumulation in peripheral neuronal Schwann cells (A), vascular endothelium (B) and fibroblasts (C).
Fig. 2.

In case 7, deposition was detected in the exocrine gland and fibroblastic cells (A, B). Massive and myelin-like depositions were observed in case 27 (C, D).
3.6. Detection of methylation sensitivity for restriction enzyme sites in the GLA gene
To assess differential methylation, genomic DNA was first treated with a methylation-sensitive endonuclease and then PCR-amplified. After PCR of bisulfite-reacted DNA, almost all the unmethylated cytosines (C) were converted to thymine (T), while methylcytosines were not. When we searched a single methylation-sensitive restriction enzyme site, one SacII site was detected in the 5’UTR, and a cluster of HhaI and HpaII sites were detected in exon 1 and 5’UTR of the GLA gene. Other exons also contained HhaI and HpaII sites; however, only one methylation-sensitive HpaI RE site was detected in exon 7 (Fig. 3).
Fig. 3.

Genomic DNA was treated with methylation-sensitive endonuclease and PCR-based amplification was done. Methylation-sensitive SacII, HpaII and HhaI were detected in exon 1, the 5’UTR (before and after treatment, A1 and A2, respectively) and exon 7 (before and after treatment, B1 and B2, respectively).
3.7. Quantification of methylation of individual alleles
The quantification of methylation was performed based on digestion of mutated or non-mutated alleles by methylation-sensitive RE. The digestion of an allele indicated non-methylation. The quantification of methylation of the normal (non-mutated) allele for heterozygous Fabry females was calculated and plotted as a percentage. The quantification is listed in Table 1.
4. Discussion
After the approval of enzyme replacement therapy (ERT), it become the most popular treatment option for Fabry disease. Orally administered pharmacological chaperone migalastat hydrochloride also provide a beneficial effect on GLA amenable mutations [30]. Recent study showed that >28% Japanese families had amenable mutations to migalastat [31]. However, timing the initiation of therapy is most crucial to get maximum beneficial effects [30,32,33]. The international guidelines for ERT are also being set up for presymptomatic patients [34]. The heterozygous female patients manifest wide variety of clinical features, some have mild and some manifest severe phenotypes in early childhood [7,8,27]. Our current study also showed a heterogeneous clinical presentation; one was asymptomatic, and others showed classical phenotypes as Hemizygous males (Table 1).
By measuring α-gal A enzyme activity, the clinical phenotype of heterozygous females cannot be predicted, and lyso-Gb3 was thought to be a useful biomarker for the diagnosis of Fabry disease heterozygotes [35]. In this study, we also found similar pictures showed in some severe cases of high enzyme activity (Table 1). Because we used whole cell lysates, which contained both the precursor and mature forms, we could not separate their activities [36,37]. However, the clinical severity of heterozygous females correlated to the accumulation of lyso-Gb3 (Table 1) and massive sphingolipid accumulation detected by electron microscopy in different cells of skin biopsies (Fig. 1, Fig. 2).
XCI and skewed XCI favoring the expression of the mutant allele has been proposed as a mechanism to explain the development of severe clinical symptoms in heterozygous Fabry disease and some other X-linked diseases [18,38,39]. The skewed XCI patterns were determined by studying the methylation status of the polymorphic (CAG)n repeat region located within exon 1 of the HUMARA gene [18] for 29% cases. Our study demonstrated direct evidence of methylation in the GLA gene. In most of the cases 27/36 (75%), the methylation of the normal allele (non-mutated allele) directly correlated to the FASTEX severity score (Fig. 4), probably due to DNA methylation in the GLA gene suppressing transcription [40]. One allele was mutated, and another allele was methylated in affected females, which caused complete suppression of GLA gene transcription and mature α-gal A synthesis. We recently reported that the aberrant DNA methylation of the GLA gene is directly associated with autophagy dysfunction [41], which caused massive accumulation of sphingolipids and their derivatives. In this study, we found a clear correlation between methylation of the normal allele and accumulation of lyso-Gb3 (Fig. 1, Fig. 2, Fig. 5). Except in cases 4, 24, 25, 28, 29, all the cases were receiving ERT for 3–10 years, and have a significant correlation between the clinical severity FASTEX score, methylation of the normal allele and deposition of lyso-Gb3.
Fig. 4.
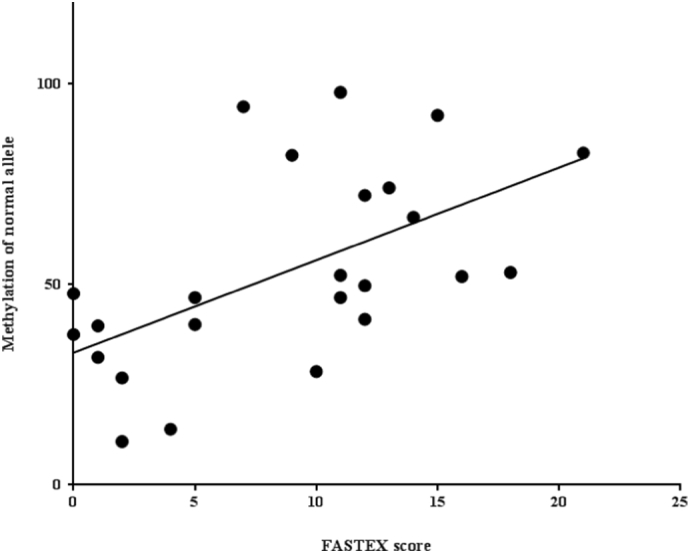
Correlation between the patient severity (FASTEX) score and methylation of the normal allele measured by the digestion ratio of mutated and non-mutated alleles by methylation-sensitive RE SacII. The DNA was extracted from blood lymphocytes. Statistical significance was valued as Spearman (ρ) = 0.6333, p = .0004.
Fig. 5.
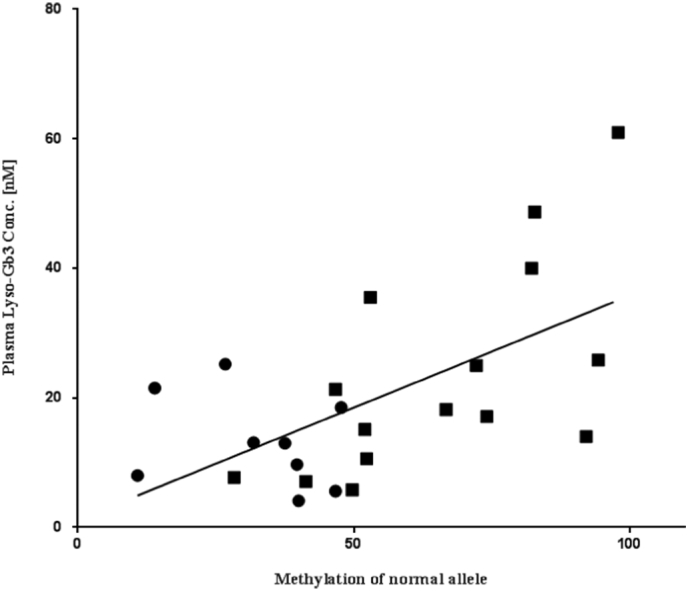
Correlation between the patient lyso-Gb3 accumulation and methylation of the normal allele measured by the digestion ratio of mutated and non-mutated alleles by methylation-sensitive RE SacII. The DNA was extracted from blood lymphocytes. The circle represents mild cases and the square represents severe cases (FASTEX raw score > 10). Statistical significance was valued as Spearman (ρ) = 0.5271, p = .004.
Methylation of the SacII and HhaI sites for heterozygous females is highly correlated to the FASTEX severity score and lyso-Gb3 accumulation (statistically significant) [Fig. 4, Fig. 5], probably because these sites were close to the promoter region, which contains 474 base pairs of CpG island [26]. In some cases (cases 10, 13, 15, 17, 21, and 29) the methylation of the normal allele measured in the HpaII site in exon 7 was correlated to the FASTEX severity score although the statistical significance could not be detected due to a small number of cases.
Several limitations existed in our study. We could not elucidate the phenotype-specific methylation effects, and the severity scores were not recorded when the cases were first diagnosed. We could also not confirm the progression of cardiomyopathy, renal failure or related effects in relation to the methylation ratio of the heterozygous females. However, our current study, which correlated the clinical severity to methylation of the normal allele and accumulation of lyso-Gb3, would be an effective tool for early prediction of phenotypes and early initiation of therapy in females heterozygous for Fabry disease. In addition, several targeted genetic editing techniques were reported in recent years [42,43]. By detecting the methylation sites in the GLA gene, the methylation site can be edited and might allow gene therapy applications for Fabry disease patients.
5. Conclusion
We summarized 36 heterozygous Fabry females with their clinical severity score, their lyso-Gb3 accumulation in plasma and sphingolipid accumulation with skin biopsy. We had detected methylation sensitive RE site in exon 7 along with exon 1 and 5`UTR and of the GLA gene. We found a clear correlation between the severity of the phenotype, lyso-Gb3 accumulation and methylation of normal allele detected by non-digestion with methylation sensitive RE.
Declaration of Competing Interest
Nothing to be declared for MAH, CW, HY, TM and KA. YE has been awarded grants, research support and honoraria from Actelion, BioMarin, Sanofi, Shire and Dainippon Sumitomo Pharmaceuticals Limited.
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Science, Sports and Technology Type C (H26-28) and Ministry of Health, Labour and Welfare of Japan Intractable disease- Lysosomal disease (H26-H28).
Contributor Information
Mohammad Arif Hossain, Email: arif@jikei.ac.jp.
Yoshikatsu Eto, Email: yosh@sepia.ocn.ne.jp.
References
- 1.Brady R.O., Gal A.E., Bradley R.M., Martensson E., Warshaw A.L., Laster L. Enzymatic defect in Fabry's disease: ceramide trihexosidase deficiency. New Eng. J. Med. 1967;276:1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- 2.Desnick R.J., Ioannou Y.A., Eng C.M. A-Galactosidase a deficiency: Fabry disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. eighth ed. McGraw Hill; New York: 2001. pp. 3733–3774. [Google Scholar]
- 3.Nance C.S., Klein C.J., Banikazemi M., Dikman S.H., Phelps R.G., McArthur J.C., Rodriguez M., Desnick R.J. Later-onset Fabry disease: an adult variant presenting with the cramp-fasciculation syndrome. Arch. Neurol. 2006;63:453–457. doi: 10.1001/archneur.63.3.453. [DOI] [PubMed] [Google Scholar]
- 4.Smid B.E., van der Tol L., Biegstraaten M., Linthorst G.E., Hollak C.E., Poorthuis B.J. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015;52:262–268. doi: 10.1136/jmedgenet-2014-102872. [DOI] [PubMed] [Google Scholar]
- 5.Desnick R.J., Bishop D.F. Fabry disease α-galactosidase a deficiency and Schindler disease. α-N-acetylgalactosaminidase deficiency. In: Stanbury J.B., Wyngaarden J.B., Fredrickson D.S., Goldstein J.L., Brown M.S., editors. The Metabolic Basis of Inherited Disease. 6th ed. McGraw-Hill; New York: 1989. p. 1751. [Google Scholar]
- 6.Franceschetti A.T. Fabry disease ocular manifestations. Birth Defects Orig Artic Ser. 1976;12:195–208. [PubMed] [Google Scholar]
- 7.Guffon N. Clinical presentation in female patients with Fabry disease. J. Med. Genet. 2003;40 doi: 10.1136/jmg.40.4.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi M., Ohashi T., Sakuma M., Ida H., Eto Y. Clinical manifestations and natural history of Japanese heterozygous females with Fabry disease. J. Inherit. Met. Dis. 2008;31(Suppl. 3):S483–S487. doi: 10.1007/s10545-007-0740-6. [DOI] [PubMed] [Google Scholar]
- 9.Linthorst G.E., Poorthuis B.J., Hollak C.E. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J. Am. Coll. Cardiol. 2008;51:2082–2083. doi: 10.1016/j.jacc.2008.02.050. [DOI] [PubMed] [Google Scholar]
- 10.Yasuda M., Shabbeer J., Benson S.D., Maire I., Burnett R.M., Desnick R.J. Fabry disease: characterization of alpha-galactosidase a double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum. Mutat. 2003;22:486–492. doi: 10.1002/humu.10275. [DOI] [PubMed] [Google Scholar]
- 11.Smid B.E., Hollak C.E., Poorthuis B.J., van den Bergh Weerman M.A., Florquin S., Kok W.E., Lekanne Deprez R.H., Timmermans J., Linthorst G.E. Diagnostic dilemmas in Fabry disease: a case series study on GLA mutations of unknown clinical significance. Clin. Genet. 2015;88:161–166. doi: 10.1111/cge.12449. [DOI] [PubMed] [Google Scholar]
- 12.Oder D., Uceyler N., Liu D., Hu K., Petritsch B., Sommer C., Ertl G., Wanner C., Nordbeck P. Organ manifestations and long-term outcome of Fabry disease in patients with the GLA haplotype D313Y. BMJ Open. 2016;6 doi: 10.1136/bmjopen-2015-010422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giannini E.H., Mehta A.B., Hilz M.J. A validated disease severity scoring system for Fabry disease. Mol. Genet. Metab. 2010;99:283–290. doi: 10.1016/j.ymgme.2009.10.178. [DOI] [PubMed] [Google Scholar]
- 14.Whybra C., Kampmann C., Krummenauer F. The mainz severity score index: a new instrument for quantifying the Anderson–Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004;65:299–307. doi: 10.1111/j.1399-0004.2004.00219.x. [DOI] [PubMed] [Google Scholar]
- 15.Mignani R., Pieruzzi F., Berri F., Burlina A., Chinea B., Gallieni M., Pieroni M., Salviati A., Spada M. FAbry STabilization indEX (FASTEX): an innovative tool for the assessment of clinical stabilization in Fabry disease. Clin. Kidney J. 2016;9:739–747. doi: 10.1093/ckj/sfw082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amos-Landgraf J.M., Cottle A., Plenge R.M., Friez M., Schwartz C.E., Longshore J., Willard H.F. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am. J. Hum. Genet. 2006;79:493–499. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minks J., Robinson W.P., Brown C.J. A skewed view of X chromosome inactivation. J. Clin. Invest. 2008;118:20–23. doi: 10.1172/JCI34470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Echevarria L., Benistan K., Toussaint A., Dubourg O., Hagege A.A., Eladari D., Jabbour F., Beldjord C., De Mazancourt P., Germain D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016;89:44–54. doi: 10.1111/cge.12613. [DOI] [PubMed] [Google Scholar]
- 19.Razin A., Riggs A.D. DNA methylation and gene function. Science. 1980;210:604–610. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 20.Bird A. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 21.Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 22.Pfeifer G.P., Steigerwald S.D., Mueller P.R., Wold B., Riggs A.D. Genomic sequencing and methylation analysis by ligation mediated PCR. Science. 1989;246:810–813. doi: 10.1126/science.2814502. [DOI] [PubMed] [Google Scholar]
- 23.Riggs A.D., Pfeifer G.P. X-chromosome inactivation and cell memory. Trends Genet. 1992;8:169–174. doi: 10.1016/0168-9525(92)90219-t. [DOI] [PubMed] [Google Scholar]
- 24.Sharp A.J., Stathaki E., Migliavacca E., Brahmachary M., Montgomery S.B., Dupre Y., Antonarakis S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21:1592–1600. doi: 10.1101/gr.112680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of Hpall and Hhal sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 26.Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 27.Hossain M.A., Yanagisawa H., Miyajima T., Wu C., Takamura A., Akiyama K., Itagaki R., Eto K., Iwamoto T., Yokoi T., Kurosawa K., Numabe H., Eto Y. The severe clinical phenotype for a heterozygous Fabry female patient correlates to the methylation of non-mutated allele associated with chromosome 10q26 deletion syndrome. Mol. Genet. Metab. 2017;120:173–179. doi: 10.1016/j.ymgme.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Okada J., Hossain M.A., Wu C., Miyajima T., Yanagisawa H., Akiyama K., Eto Y. Ten-year-long enzyme replacement therapy shows a poor effect in alleviating giant leg ulcers in a male with Fabry disease. Mol. Genet. Metab. Rep. 2017;14:68–72. doi: 10.1016/j.ymgmr.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faraggiana T., Churg J., Grishman E., Strauss L., Prado A., Bishop D.F., Schuchman E., Desnick R.J. Light- and electron-microscopic histochemistry of Fabry's disease. Am. J. Pathol. 1981;103:247–262. [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes D.A., Nicholls K., Sunder-Plassmann G. Safety of switching to migalastat from enzyme replacement therapy in Fabry disease: experiencefrom the phase 3 ATTRACT study. Am. J. Med. Genet. A. 2019;179:1069–1073. doi: 10.1002/ajmg.a.61105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi M., Ohashi T., Kaneshiro E., Higuchi T., Ida H. Mutation spectrum of α-galactosidase gene in Japanese patients with Fabry disease. J. Hum. Genet. 2019;64:695–699. doi: 10.1038/s10038-019-0599-z. [DOI] [PubMed] [Google Scholar]
- 32.Biegstraaten M., Arngrímsson R., Barbey F. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry working group consensus document. Orphanet. J. Rare Dis. 2015;10:36. doi: 10.1186/s13023-015-0253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopkin R.J., Jefferies J.L., Laney D.A. The management and treatment of children with Fabry disease: a United States-based perspective. Mol. Genet. Metab. 2016;117:104–113. doi: 10.1016/j.ymgme.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Ortiz A., Germain D.P., Desnick R.J., Politei J., Mauer M., Burlina A., Eng C., Hopkin R.J., Laney D., Linhart A., Waldek S., Wallace E., Weidemann F., Wilcox W.R. Fabry disease revisited: management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018;123:416–427. doi: 10.1016/j.ymgme.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Nowak A., Mechtler T.P., Desnick R.J., Kasper D.C. Plasma LysoGb3: a useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol. Genet. Metab. 2017;120:57–61. doi: 10.1016/j.ymgme.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Park J.Y., Kim G.H., Kim S.S., Ko J.M., Lee J.J., Yoo H.W. Effects of chemical chaperone on genetic mutations in alpha-galactosidase a in Korean patients with Fabry disease. Exp. Mol. Med. 2009;41:1–7. doi: 10.3858/emm.2009.41.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hossain M.A., Higaki K., Saito S., Ohno K., Sakuraba H., Nanba E., Suzuki Y., Ozono K., Sakai N. Chaperone therapy for Krabbe disease: potential for late-onset GALC mutations. J. Hum. Genet. 2015;60:539–545. doi: 10.1038/jhg.2015.61. [DOI] [PubMed] [Google Scholar]
- 38.Beutler E., Yeh M., Fairbanks V.F. The normal human female as a mosaic of X-chromosome activity: studies using the gene for C-6-PD-deficiency as a marker. Proc. Natl. Acad. Sci. U. S. A. 1962;48:9–16. doi: 10.1073/pnas.48.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobyns W.B., Filauro A., Tomson B.N., Chan A.S., Ho A.W., Ting N.T., Oosterwijk J.C., Ober C. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am. J. Med. Genet. A. 2004;129A:136–143. doi: 10.1002/ajmg.a.30123. [DOI] [PubMed] [Google Scholar]
- 40.Tate P.H., Bird A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 41.Yanagisawa H., Hossain M.A., Miyajima T., Nagao K., Miyashita T., Eto Y. Dysregulated DNA methylation of GLA gene was associated with dysfunction of autophagy. Mol. Genet. Metab. 2019;126:460–465. doi: 10.1016/j.ymgme.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Morita S., Horii T., Hatada I. Editing of DNA methylation using dCas9- peptide repeat and scFv-TET1 catalytic domain fusions. Methods Mol. Biol. 2018;1767:419–428. doi: 10.1007/978-1-4939-7774-1_23. [DOI] [PubMed] [Google Scholar]
- 43.Liu X.S., Wu H., Krzisch M., Wu X., Graef J., Muffat J., Hnisz D., Li C.H., Yuan B., Xu C., Li Y., Vershkov D., Cacace A., Young R.A., Jaenisch R. Rescue of fragile X Syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979–992. doi: 10.1016/j.cell.2018.01.012. (e976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shabbeer J., Yasuda M., Luca E., Desnick R.J. Fabry disease: 45 novel mutations in the alpha-galactosidase a gene causing the classical phenotype. Mol. Genet. Metab. 2002;76:23–30. doi: 10.1016/s1096-7192(02)00012-4. [DOI] [PubMed] [Google Scholar]
- 45.Eng C.M., Niehaus D.J., Enriquez A.L., Burgert T.S., Ludman M.D., Desnick R.J. Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase a gene. Hum. Mol. Genet. 1994;3:1795–1799. doi: 10.1093/hmg/3.10.1795. [DOI] [PubMed] [Google Scholar]
- 46.Ishii S., Sakuraba H., Suzuki Y. Point mutations in the upstream region of the alpha-galactosidase a gene exon 6 in an atypical variant of Fabry disease. Hum. Genet. 1992;89:29–32. doi: 10.1007/BF00207037. [DOI] [PubMed] [Google Scholar]
- 47.Iwafuchi Y., Maruyama H., Morioka T., Noda S., Nagata H., Oyama Y., Narita I. Enzyme replacement therapy in a patient of heterozygous Fabry disease: clinical and pathological evaluations by repeat kidney biopsy and a successful pregnancy. CEN Case Rep. 2017;6:210–214. doi: 10.1007/s13730-017-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagasaki A., Nishie W., Sato K., Oba I., Noguchi E., Akitsu H., Sawamura D., Shimizu H. Clinical and genetic analysis of Fabry disease: report of six cases including three heterozygous females. J. Dermatol. Sci. 2008;52:61–64. doi: 10.1016/j.jdermsci.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Shimotori M., Maruyama H., Nakamura G., Suyama T., Sakamoto F., Itoh M., Miyabayashi S., Ohnishi T., Sakai N., Wataya-Kaneda M., Kubota M., Takahashi T., Mori T., Tamura K., Kageyama S., Shio N., Maeba T., Yahagi H., Tanaka M., Oka M., Sugiyama H., Sugawara T., Mori N., Tsukamoto H., Tamagaki K., Tanda S., Suzuki Y., Shinonaga C., Miyazaki J., Ishii S., Gejyo F. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 2008;29:331. doi: 10.1002/humu.9520. [DOI] [PubMed] [Google Scholar]
- 50.Meaney C., Blanch L.C., Morris C.P. A nonsense mutation (R220X) in the alpha-galactosidase a gene detected in a female carrier of Fabry disease. Hum. Mol. Genet. 1994;3:1019–1020. doi: 10.1093/hmg/3.6.1019. [DOI] [PubMed] [Google Scholar]
- 51.Sakuraba H., Oshima A., Fukuhara Y., Shimmoto M., Nagao Y., Bishop D.F., Desnick R.J., Suzuki Y. Identification of point mutations in the alpha-galactosidase a gene in classical and atypical hemizygotes with Fabry disease. Am. J. Hum. Genet. 1990;47:784–789. [PMC free article] [PubMed] [Google Scholar]
- 52.Davies J.P., Winchester B.G., Malcolm S. Mutation analysis in patients with the typical form of Anderson-Fabry disease. Hum. Mol. Genet. 1993;2:1051–1053. doi: 10.1093/hmg/2.7.1051. [DOI] [PubMed] [Google Scholar]
- 53.Ashton-Prolla P., Tong B., Shabbeer J., Astrin K.H., Eng C.M., Desnick R.J. Fabry disease: twenty-two novel mutations in the alpha-galactosidase a gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J. Investig. Med. 2000;48:227–235. [PubMed] [Google Scholar]
- 54.Koide T., Ishiura M., Iwai K., Inoue M., Kaneda Y., Okada Y., Uchida T. A case of Fabry's disease in a patient with no alpha-galactosidase a activity caused by a single amino acid substitution of Pro-40 by Ser. FEBS Lett. 1990;259:353–356. doi: 10.1016/0014-5793(90)80046-l. [DOI] [PubMed] [Google Scholar]
- 55.Eng C.M., Resnick-Silverman L.A., Niehaus D.J., Astrin K.H., Desnick R.J. Nature and frequency of mutations in the alpha-galactosidase a gene that cause Fabry disease. Am. J. Hum. Genet. 1993;53:1186–1197. [PMC free article] [PubMed] [Google Scholar]
- 56.Blanch L.C., Meaney C., Morris C.P. A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the alpha-galactosidase a gene. Hum. Mutat. 1996;8:38–43. doi: 10.1002/(SICI)1098-1004(1996)8:1<38::AID-HUMU5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]