

Abstract
In order to overcome the challenges of microbial resistance as well as to improve the effectiveness and selectivity of chemotherapeutic agents against cancer, a novel series of 4-(4-bromophenyl)-thiazol-2-amine derivatives was synthesized and its molecular structures were confirmed by physicochemical and spectral characteristics. The synthesized compounds were further evaluated for their in vitro antimicrobial activity using turbidimetric method and anticancer activity against oestrogen receptor positive human breast adenocarcinoma cancer cell line (MCF7) by Sulforhodamine B (SRB) assay. The antimicrobial activity results revealed that compound p2, p3, p4 and p6 exhibited promising antimicrobial activity that are comparable to standard norfloxacin (antibacterial) and fluconazole (antifungal). Anticancer screening results demonstrated that compound p2 was found to be the most active one against cancer cell line when compared to the rest of the compounds and comparable to the standard drug (5-fluorouracil). The molecular docking study demonstrated that compounds, p2, p3, p4 and p6 displayed good docking score within binding pocket of the selected PDB ID (1JIJ, 4WMZ and 3ERT) and showed promising ADME properties.
Electronic supplementary material
The online version of this article (10.1186/s13065-019-0575-x) contains supplementary material, which is available to authorized users.
Keywords: Synthesis, Molecular docking, Thiazole derivatives, Antimicrobial, Anticancer
Background
In recent years, epidemiological studies confirmed the significant negative impact of infections caused by pathogenic bacteria and fungi against human health. Large-scale surveillance revealed increasing incidence of drug-resistance that had compromised the efficacy of antimicrobial therapy. The increased emergence of multidrug-resistant pathogenic bacteria has called for exploration of alternative drug therapies [1]. As such, research is now focused towards new antimicrobial agents with expansion of bioactivity of existing drugs and also with novel target so as to address the problem of resistance [2].
In this era, cancer remains as one of the most serious clinical problems and the second primary cause of deaths worldwide. Cancer, which is characterized by uncontrollable division of abnormal cells, could be fatal if proliferation were to occur continuously [3]. Although many effective chemotherapeutic agents are available, they generally exhibit serious side-effects such as toxicity and resistance. With the increasing understanding of drugs’ cytotoxic mechanism of action and the discovery of specific target, novel chemical therapeutic drugs could be designed for treatment of cancer [4].
It has been long since researchers show special interest in heterocyclic compounds that possess sulphur and nitrogen atom [5, 6]. Thiazole, for instance, exhibit widespread biological activities like antibacterial [7, 8], antimycobacterial [9], antileishmanial [10], anticancer [11] and antifungal [12]. The various marketed preparations that contain thiazole nucleus include tiazofurin (antineoplastic agent), ritonavir (antiviral agent), imidacloprid (insecticide), penicillin (antibiotic), nizatidin (antiulcer) and meloxicam (anti-inflammatory) (Fig. 1). The presence of electron withdrawing group at the p-position of phenyl nucleus directly attached to thiazole ring showed good antibacterial activity [8]. Schiff bases exerted various pharmacological activities such as anticancer, antimicrobial and antileishmanial amongst others [13, 14]. Aromatic substitution at para position of thiazole enhanced the anticancer activity. It can thus act as template for further investigation and synthesis of new derivatives [15].
Fig. 1.

Marketed preparation of thiazole nucleus
Structure based drug designing (SBDD) and ligand based drug designing (LBDD) techniques are employed as important drug discovery tools in rational drug designing process [16]. Molecular docking is the advanced computational used techniques in SBDD to obtain optimized conformation of ligand–receptor interaction and to study their relative orientation through the minimized energy free system [17]. Computer aided drug designing (CADD) is fast, economical modernized technique that gives valuable, accurate and deep understandings of experimental findings and new suggestions for molecular structures to be synthesized [18].
Drug molecules might fail during development because of several reasons but as found by the researchers one of the major reasons of failures is related with poor pharmacokinetic and absorption, distribution, metabolism and excretion (ADME) properties [19]. Unexpected drug toxicity is the one of the major factors to withdraw drug from the market. Therefore, ADME properties are the crucial determinants for the clinical success of the drug [20].
Lipinski’s rule of five is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has chemical properties and physical properties that would make it a likely orally active drug in humans. The rule describes molecular properties important for a drug’s pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion (ADME). The rule is important to keep in mind during drug discovery when a pharmacologically active lead structure is optimized step-wise to increase the activity and selectivity of the compound as well as to ensure drug-like physicochemical properties are maintained as described by Lipinski’s rule which states that (i) no more than 5 hydrogen bond donors (ii) no more than 10 hydrogen bond acceptors (iii) a molecular mass less than 500 Da (iv) an octanol–water partition coefficient log P not greater than 5 (https://en.wikipedia.org/wiki/Lipinski%27s_rule_of_five).
Now these days, computational approaches are employed to determine the ADME of the drug molecules. ADME modeling has attracted the considerable attention of the pharmaceutical researchers for the drug discovery as they are high-throughput in nature and cost effective [21]. As a part of our continuous efforts in finding new antimicrobial and anticancer [22, 23] mentioned above, in the present study, 4-(4-bromophenyl)-thiazol-2-amine derivatives were designed for assessment of their antimicrobial and antiproliferative potentials (Fig. 2).
Fig. 2.

Design of proposed thiazole molecules for antimicrobial and anticancer potential based on literature
Results and discussion
Chemistry
Synthesis of intermediate and target derivatives (p1–p10) were carried out as per the reactions outlined in Scheme 1. Initially, p-bromoacetophenone and thiourea were reacted in the presence of catalyst iodine to yield the 4-(4-bromophenyl) thiazol-2-amine (Intermediate). The intermediate with corresponding aromatic aldehyde yielded the target compounds (p1–p10). The molecular structures of the synthesized compounds were confirmed by physicochemical properties (Table 1) and spectral characteristics (Table 2). IR spectrum of intermediate showed the characteristic IR band at 817 cm−1 and 666 cm−1 which indicated the presence of N–H str. of NH2 and C–Br str. of C6H5Br, respectively. The presence of stretch at 1265 cm−1 displayed the C–N connectivity and showed the presence of Ar–NH2 linkage. The IR stretch present at 725 cm−1 and 1632 cm−1 showed the C–S and C=N linkage, respectively, therefore these all linkages indicates the existence of thiazole nucleus within the structure of the (Intermediate). The occurrence of band at 3113 cm−1 and 1586 cm−1 indicated the presence of C–H skeletal and C=C skeletal structure, respectively within the phenyl nucleus. The molecular structures of synthesized compounds were further confirmed by 1H NMR spectral data. The 1H-NMR spectrum of intermediate showed singlet at 6.9 δ ppm showed the presence of NH2 group. The 1HNMR spectra of synthesized derivatives displayed multiplet at 6.939–7.52 δ ppm due to presence of aromatic C–H linkage. The presence of singlet at 7.57–9.7 δ ppm displayed the N=CH connectivity, therefore the confirmation of the presence of benzylidene linkage within the synthesized derivatives. The presence of O–CH3 of Ar–OCH3 was confirmed by the appearance of singlet at 3.76–3.9 δ ppm. All compounds showed singlet at 6.9–7.80 δ ppm due to the existence of C–H in thiazole ring. Compound p2 showed singlet at 5.39 δ ppm due to presence of –OH at the para position. Compound p3 showed singlet at 2.91 δ ppm due to presence of –N(CH3)2 at the para position. 13C-NMR spectra of the thiazole derivatives was displayed the fine conformity of their proposed molecular structure i.e. the carbon atoms of phenyl nucleus found around 120.4, 109.4, 128.7, 122.2, 110.3, 150.8 δ ppm, carbon atoms of thiazole around 150.3, 172.4, 109.6 and carbon of N=CH group at 159.4 δ ppm. Elemental analysis results of the thiazole derivatives were lie within the limits of ± 0.5% of the theoretical results.
Scheme 1.

For the synthesis of 4-(4-bromophenyl)thiazol-2-amine derivatives (p1–p10)
Table 1.
Physicochemical properties of the synthesized compounds (p1–p10)
| S. no | Compound | Molecular formula | Color | m.pt. °C | Rf value | % Yield | |
|---|---|---|---|---|---|---|---|
| 1 | (p1) |
4-(4-Bromophenyl)-N-(3,4-dimethoxybenzylidene)thiazol-2-amine |
C18H15N2SBrO2 | Light yellow | 134–136 | 0.45 | 85 |
| 2 | (p2) |
4-((4-(4-Bromophenyl)thiazol-2-ylimino)methyl)-2-methoxyphenol |
C17H13N2SBrO2 | Creamy yellow | 122–125 | 0.38 | 70 |
| 3 | (p3) |
4-(4-Bromophenyl)-N-(4-(dimethylamino)benzylidene)thiazol-2-amine |
C18H16N3SBr | Yellowish white | 117–120 | 0.25 | 65 |
| 4 | (p4) |
N-(3,4,5-Trimethoxybenzylidene)-4-(4-bromophenyl)thiazol-2-amine |
C19H17N2SBrO3 | Greenish yellow | 105–107 | 0.30 | 75 |
| 5 | (p5) |
N-(4-Bromobenzylidene)-4-(4-bromophenyl)thiazol-2-amine |
C16H10N2SBr2 | Dark yellow | 62–64 | 0.38 | 69 |
| 6 | (p6) |
1-((4-(4-Bromophenyl)thiazol-2-ylimino)methyl)naphthalen-2-ol |
C20H13BrN2OS | Light yellow | 105–107 | 0.30 | 72 |
| 7 | (p7) |
N-(4-Chlorobenzylidene)-4-(4-bromophenyl)thiazol-2-amine |
C16H10BrClN2S | Light yellow | 65–70 | 0.23 | 83 |
| 8 | (p8) |
N-(2,4-Dimethoxybenzylidene)-4-(4-bromophenyl)thiazol-2-amine |
C18H15BrN2O2S | Lemon yellow | 71–73 | 0.52 | 72 |
| 9 | (p9) |
N-(3-Methoxybenzylidene)-4-(4-bromophenyl)thiazol-2-amine |
C17H13BrN2OS | Yellowish white | 188–190 | 0.79 | 65 |
| 10 | (p10) |
3-(((4-(4-Bromophenyl)thiazol-2-yl)imino)methyl)phenol |
C16H11BrN2OS | Dark yellow | 106–108 | 0.21 | 71 |
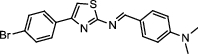
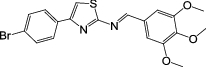
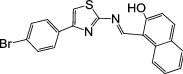
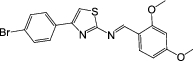
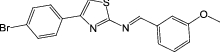
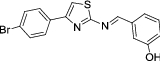
Table 2.
Spectral data of the synthesized compounds and intermediate (Int.)
| Sr. no. | IR KBr (cm−1) | 1H-NMR (DMSO-d6, ppm) | 13C-NMR (DMSO-d6, ppm) | MS: m/z (M++1) | Elemental analysis (CHN) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C=C str. | C–Br str. | C–O–C str. OCH3 | C–S str. | C=N str. | Other (str./bending) | |||||
| Int. | 1586 | 666 | – | 725 | 1632 | 817 (N–H str., NH2), 1265 (C–N str., Ar–NH2) | 7.19–7.56 (m, 4H, ArH), 6.90 (s, 1H, CH of thiazole), 4.12 (s, 2H, –NH2 of thiazole) | 127.4, 131.1, 122.6, 130.2, 127.3, 130.8 (6C, phenyl nucleus), 167.7, 100.4, 151.5 (3C, thiazole) | 255 | Theoretical calc: C, 42.37; H, 2.77; N, 10.98; Found: C, 42.31; H, 2.74; N, 10.89 |
| p1 | 1587 | 642 | 1266 | 725 | 1674 | – | 6.93 (m, 7H, aromatic H), 7.42 (s, 1H, CH of thiazole) | 120.4, 109.4, 128.7, 122.2, 110.3, 150.87, 123.2, 110.0, 150.2, 110.6, 148.5, 128.6 (12C, phenyl nucleus), 150.3, 172.41, 109.6 (3C, thiazole), 159.4 (N=CH), 55.2, 54.6 (2C, OCH3) | 404 | Theoretical calc: C, 53.61; H, 3.75; N, 6.95; Found: C, 53.48; H, 3.69; N, 6.81 |
| p2 | 1590 | 601 | 1208 | 756 | 1673 | – | 6.76–7.45 (m, 7H, ArH), 7.43 (s, 1H, C–H of thiazole), 8.85 (s, 1H, N=CH), 3.71 (s, 3H, OCH3), 5.39 (s, 1H, OH) | 130.4, 127.4, 124.7, 132.1, 130.3, 131.8, 119.8, 115.4, 150.2, 147.6, 113.4, 127.9, (12C, phenyl nucleus), 151.3, 173.1, 108.6 (3C, thiazole), 158.2 (N=CH), 55.7 (O–CH3), 53.4 (1C, –OCH3) | 390 | Theoretical calc: C, 52.45; H, 3.37; N, 7.20; Found: C, 52.33; H, 3.41; N, 7.18 |
| p3 | 1588 | 726 | 1229 | 812 | 1658 | 830 [(C–N str., N(CH3)2] | 6.80–7.41 (m, 8H, ArH), 7.80 (s, 1H, –N=CH), 3.43 (s, 3H, OCH3), 7.45 (s, 1H, CH of thiazole) | 130.4, 127.4, 124.7, 132.1, 130.3, 131.8, 124.6, 129.1, 110.8, 152.2, 110.8, 127.3 (12C, phenyl nucleus), 151.3, 173.41, 108.6 (3C, thiazole), 158.2 (N=CH), 40.8, 39.9 (2C, N(CH3)2) | 375 | Theoretical calc: C, 57.00; H, 4.53; N, 10.50; Found: C, 57.04; H, 4.42; N, 10.47 |
| p4 | 1588 | 575 | 1232 | 705 | 1682 | 1325 (C–O str. and O–H in plane bending, phenol) | 6.9–7.39 (m, 6H, ArH), 7.73 (s, 1H, –N=CH), 3.73 (s, 3H, OCH3), 7.47 (s, 1H, CH of thiazole), 2.91 (s, 6H, N(CH3)2) | 121.5, 107.2, 128.7, 123.1, 111.1, 151.2, 151.1, 102.3, 130.1, 103.1, 152.1, 140.5 (12C, phenyl nucleus), 149.3, 171.2, 109.8 (3C, thiazole), 160.1 (N=CH), 59.6, 55.8 (2C, OCH3, 54.3, 58.5 (2C, OCH3) | 422 | Theoretical calc: C, 52.66; H, 3.95; N, 6.46; Found: C, 52.54; H, 3.87; N, 6.44 |
| p5 | 1587 | 589 | – | 829 | 1677 | – | 7.28–7.60 (m, 8H, ArH), 7.90 (s, 1H, N=CH), 7.49 (s, 1H, CH of thiazole) | 120.1, 126.7, 123.1, 111.1, 151.2, 109.2, 130.1, 126.2, 131.2, 122.8, 129.7, 128.6, 131.2 (12C, phenyl nucleus), 149.3, 171.2, 109.8 (3C, thiazole), 160.1 (N=CH), 59.6 (C, –OCH3) | 423 | Theoretical calc: C, 45.52; H, 2.39; N, 6.64; Found: C, 45.48; H, 2.27; N, 6.51 |
| p6 | 1590 | 592 | – | 742 | 1618 | 1395 (C–O str. and O–H in plane bending, phenol), | 7.36–8.01 (m, 10H, ArH), 8.14 (s, 1H, N=CH), 7.43 (s, 1H, CH of thiazole) | 122.6, 126.7, 132.1, 118.1, 151.2, 128.21, 131.4, 119.5, 170.5, 107.9 (12C, naphthalene nucleus), 148.3, 169.1, 109.1 (3C, thiazole), 159.10 (N=CH) | 410 | Theoretical calc: C, 58.69; H, 3.20; N, 6.84; Found: C, 58.55; H, 3.18; N, 6.83 |
| p7 | 1590 | 607 | – | 829 | 1521 | 725 (C–Cl str., ArCl) | 7.24–7.58 (m, 8H, ArH), 7.90 (s, 1H, N=CH), 7.43 (s, 1H, CH of thiazole) | 127.2, 129.4, 134.5, 129.8, 135.6, 126.3, 129.8, 133.2, 131.4, 124.8, 135.5 (12C, phenyl nucleus), 149.5, 173.2, 108.8 (3C, thiazole), 159.1 (N=CH) | 380 | Theoretical calc: C, 50.88; H, 2.67; N, 7.4; Found: C, 50.76; H, 2.51; N, 7.2 |
| p8 | 1583 | 572 | 1263 | 720 | 1675 | – | 6.80–7.35 (m, 7H, ArH), 7.90 (s, 1H, N=CH), 7.36 (s, 1H, CH of thiazole), 3.88 (s, 1H, OCH3) | 124.2, 134.5, 130.2, 133.5, 128.8, 104.5, 132.1, 115.1, 158.3, 100.2, 161.8 (12 C, phenyl nucleus), 149.5, 173.2, 108.8 (3C, thiazole), 161.2 (N=CH), 54.6, 53.5 (2C, OCH3) | 404 | Theoretical calc: C, 53.61; H, 3.75; N, 6.95; Found: C, 53.59; H, 3.64; N, 6.81 |
| P9 | 1532 | 659 | 1265 | 805 | 1583 | – | 6.90–7.27 (m, 8H, ArH), 7.40 (s, 1H, N=CH), 7.38 (s, 1H, CH of thiazole), 3.90 (s, 1H, OCH3) | 125.2, 133.5, 131.2, 127.5, 126.8, 127.6, 120.1, 136.5, 110.1, 159.7 (12C, phenyl nucleus), 151.5, 172.3, 107.6 (3C, thiazole), 160.2 (N = CH), 54.3 (O-CH3) | 374 | Theoretical calc: C, 54.70, H, 3.51; N, 7.50; Found: C, 54.56; H, 3.28; N, 7.38 |
| p10 | 1505 | 720 | 1265 | 829 | 1583 | 1397 (C–O str. and O–H in plane bending, phenol) | 6.70–7.38 (m, 8H, ArH), 9.70 (s, 1H, N=CH), 7.52 (s, 1H, CH of thiazole) | 127.2, 132.5, 130.2, 126.5, 125.8, 115.2, 157.3, 112.2, 136.5, 120.0, 131.1 (12C, phenyl nucleus), 150.5, 171.3, 108.5 (3C, thiazole), 160.1 (N=CH) | 360 | Theoretical calc: C, 53.49; H, 3.09; N, 7.80; Found: C, 53.41; H, 3.07; N, 7.72 |
In vitro antimicrobial activity
The antimicrobial potential of synthesized molecules was determined using turbidimetric (tube dilution method). The antibacterial activity was determined against Gram-negative bacterium: Escherichia coli (MTCC443) and Gram-positive bacteria: Staphylococcus aureus (MTCC3160), Bacillus subtilis (MTCC441) and compared to positive control norfloxacin. The antifungal study of compounds was carried out against fungal strains: Candida albicans (MTCC227) and Aspergillus niger (MTCC281) and compared to positive control (fluconazole). The results of antibacterial and antifungal evaluation were recorded in terms of minimum inhibitory concentration (MIC) (Table 3, Figs. 3 and 4).
Table 3.
Antimicrobial and anticancer screening results of synthesized thiazole molecules (p1–p10)
| Compound | (Antimicrobial screening) MIC = µM | aIC50 = µM | ||||
|---|---|---|---|---|---|---|
| Microbial species | ||||||
| Bacterial | Fungal | Cancer cell line | ||||
| S.A. | B.S. | E.C. | C.A. | A.N. | MCF7 | |
| p1 | 31 | 62.0 | 31 | 31 | 31 | 17.4 |
| p2 | 16.1 | 32.1 | 16.1 | 32.1 | 32.1 | 10.5 |
| p3 | 32.4 | 32.4 | 32.4 | 32.4 | 16.2 | 37.4 |
| p4 | 28.8 | 28.8 | 28.8 | 28.8 | 28.8 | 38.0 |
| p5 | 29.6 | 29.6 | 29.6 | 29.6 | 29.6 | 40.3 |
| p6 | 30.5 | 30.5 | 30.5 | 15.3 | 30.5 | 73.3 |
| p7 | 33.1 | 33.1 | 33.1 | 16.5 | 33.1 | 47.6 |
| p8 | 31 | 31.0 | 62.0 | 15.5 | 62.0 | 52.1 |
| p9 | 33.5 | 67.0 | 67.0 | 33.5 | 33.5 | 17.2 |
| p10 | 17.4 | 34.8 | 34.8 | 34.8 | 34.8 | 21.4 |
| Norfloxacin | 4.7 | 4.7 | 4.7 | – | – | – |
| Fluconazole | – | – | – | 5.0 | 5.0 | – |
| 5-Fluorouracil | – | – | – | – | – | 5.2 |
S.A.: Staphylococcus aureus (MTCC3160); B.S.: Bacillus subtilis (MTCC441); E.C.: Escherichia coli (MTCC443); C.A.: Candida albicans (MTCC227) and A.N.: Aspergillus niger (MTCC281)
aIC50 is the concentration required to inhibit 50% of cell growth
Fig. 3.

Graphical representation of antibacterial activity of synthesized compounds
Fig. 4.

Graphical representation of antifungal activity of synthesized compounds
In vitro antimicrobial results of developed compounds p2 (MICsa and MICec = 16.1 µM) showed promising potential against S. aureus and E. coli, respectively. Compound p4 (MICbs = 28.8 µM) displayed potent antibacterial activity against B. subtilis. Antifungal activity results demonstrated that compound p6 (MICca = 15.3 µM) displayed significant antifungal activity against C. albicans and compound p3 (MICan = 16.2 µM) was found to be most potent against A. niger.
In vitro anticancer activity
Anticancer activity of the synthesized thiazole compounds was tested against an oestrogen receptor positive human breast adenocarcinoma cell line (MCF7) using the SRB colorimetric assay in comparison to a standard drug (5-fluorouracil). Anticancer activity results (Table 3 and Fig. 5) revealed that thiazole exhibited good anticancer potential against cancer cell line MCF7. Compound p2 (IC50 = 10.5 μM), in particular, exhibited anticancer activity and almost comparable to the reference drug, 5-fluorouracil (IC50 = 5.2 μM).
Fig. 5.

Graphical representation of anticancer activity of synthesized compounds
Molecular docking
Molecular docking is done to study the binding mode of the synthesized 4-(4-bromophenyl)-thiazol-2-amine derivatives with their respective receptors. The PDB files required were identified through literature survey. Docking studies of the most active compounds were carried out using GLIDE module of docking software Schrodinger v11.5. Docking score values were used to rank the conformations of these ligand–receptor complexes. Molecular docking study of the most active antibacterial compounds (p2 and p4) and standard drug (norfloxacin) was done in the active sites of topoisomerase II (PDB ID: 1JIJ) obtained from the protein data bank. The ligand interaction diagram (2D) and pictorial presentation (3D) of docked compound and standard drug are shown in Fig. 6. The 2D ligand interaction diagrammatic view depicted that these compounds share same homology with standard norfloxacin (Table 4) by interacting with similar amino acid residues. The compound p2 form H-bond with amino acids Tyr36 and Asp177 that is responsible for good antibacterial activity.
Fig. 6.

Pictorial presentation (3D) and Ligand interaction diagram (2D) of most active antibacterial compounds (p2, p3 and p4) and standard norfloxacin
Table 4.
Docking results of most active (antibacterial and antifungal) compounds and standard drugs
| Compound | Docking score | Glide energy (kcal/mol) | Interacting amino acid residues |
|---|---|---|---|
| p2 | − 5.547 | − 49.479 | Asp177, Gln174, Tyr36, Cys37, Ala39, Asp40, Thr42, Lys84, Gly83, Ser82, Asp80 |
| p3 | − 6.513 | − 48.914 | His468, Cys470, Phe463, Leu380, Pro379, Hie378, Thr322, Ser319, Tyr126 |
| p4 | − 4.845 | − 54.654 | His47, Gly49, Hie50, Pro53, Gln174, Asp177, Ile200, Asn199, Gln196, Asp195, Gly193, Gly192 |
| p6 | − 8.342 | − 45.842 | Tyr126, Leu129, Thr130, Tyr140, Ile139, Cys470, Thr322, Ser319, Thr318 |
| Norfloxacin | − 6.18 | − 53.349 | Asp177, Glu174, Leu70, Tyr36, Cys37, Gly38, Ala39, Asp40, Gln190, Val191, Gly192 |
| Fluconazole | − 5.847 | − 40.932 | His468, Arg469, Cys470, Ile471, Tyr126, Hie378, Leu380, Leu383, Arg385 |
Molecular docking study of the most active antifungal compound p3, p6 and standard drug (fluconazole) was done against active sites of lanosterol alpha demethylase (PDB: 4WMZ) obtained from the protein data bank. The ligand interaction diagram (2D) and pictorial presentation (3D) of docked compound and standard drug are as shown in Table 4 and Fig. 7. The diagrammatic view depicted that this compound share similar homology with that of standard fluconazole. The nitrogen atom of thiazole nucleus of compound p3 and p6 form H-bond with Cys470 amino acid residue. The compound p3 also show pi–pi interaction with Tyr126 amino acid residue. The most active anticancer compound p2 was docked in the binding pocket of ER-alpha of MCF7 (PDB ID-3ERT) co-crystallized with tamoxifen ligand. The results were examined based on docking score obtained by molecular docking software (Fig. 8, Table 5). The docking score was illustrated in the negative terms. More negative the docking score better would be the binding affinity of ligand with the receptor.
Fig. 7.

Pictorial presentation (3D) and ligand interaction diagram (2D) of most active antifungal compounds (p3 and p6) and standard fluconazole
Fig. 8.

Pictorial presentation (3D) and ligand interaction diagram (2D) of most active compound (p2) and standard 5-fluorouracil
Table 5.
Docking results of most active anticancer compound p2 with standard drug
| Compound | Docking score | Glide energy (kcal/mol) | Interactive residues |
|---|---|---|---|
| p2 | − 6.732 | − 42.44 | Trp383, Leu384, Leu387, Met388, Leu391, Phe404, Met343, Leu346, Met347 |
| 5-Fluorouracil | − 3.414 | − 24.58 | Glu353, Ala350, Leu349, Leu346, Leu348, Leu387, Met388, Phe404, Leu391, Arg394 |
ADME results
Determination of ADME parameters of the synthesized 4-(4-bromophenyl)-thiazol-2-amine derivatives were done using QikProp module of Schrodinger v11.5. Around five physically relevant and pharmacologically significant parameters of the most active compounds, p2, p3, p4 and p6 were determined and summarized in Table 6. The compound p2 lie within the range of all the parameters of Lipinski rule of five while rest of the potent compounds followed the Lipinski rule of five except the lipophilicity parameter and these compounds can be further optimized to improve their lipophilicity. The results displayed that compounds, p2, p3, p4 and p6 lie within the close agreement with the Lipinski’s rule thus making these derivatives as useful lead molecules for further study.
Table 6.
ADME parameters of most active antimicrobial and anticancer compounds
| Comp. | Molecular structure | ADME parameters | |||||
|---|---|---|---|---|---|---|---|
| Mol MW | QPlogPo/w | DonorHB | AccptHB | Percent human oral absorption | Rule of five | ||
| p2 |
|
389.266 | 4.607 | 1.0 | 4.0 | 100.0 | 0 |
| p3 |
|
400.335 | 5.71 | 0.0 | 3.5 | 100.0 | 1 |
| p4 |
|
433.319 | 5.447 | 0.0 | 4.75 | 100.0 | 1 |
| p6 |
|
409.299 | 5.52 | 1.0 | 3.25 | 100.0 | 1 |
Structure activity relationship (SAR) studies
The in vitro antimicrobial and cytotoxicity outcomes demonstrated the following structure activity relationship for 4-(4-bromophenyl)-thiazol-2-amine derivatives (Fig. 9):
Electron withdrawing group (Br) present at para-position of phenyl nucleus directly attached to thiazole ring improved antimicrobial and anticancer activities of 4-(4-bromophenyl)-thiazol-2-amines.
The presence of electron withdrawing group [–N(CH3)2] on phenyl nucleus of synthesized compound p3 and the pi–pi interaction with the amino acid residues in the binding pocket in receptor, enhanced antifungal activity. Compound p6 (having 2-OH naphthaldehyde), improved antifungal activity against C. albicans.
The presence of electron releasing group [–OCH3] on benzylidene portion of synthesized molecules (compound p4) and the pi–pi interaction with the amino acid residue within the binding pocket produce moderate antimicrobial activity.
The presence of electron releasing groups [OH, –OCH3] on benzylidene portion of synthesized molecules (compound p2) and the pi–pi and hydrogen bond interaction with the amino acid residues enhanced antimicrobial and anticancer activities.
Fig. 9.

Structural activity relationship of synthesized thiazole derivatives
From result of structure activity relationship study, we may conclude that different structural requirements are required for a molecule to be effective against different goal. The aforementioned facts are supported by the earlier research findings [8, 24, 25].
Experimental section
Materials and methods
Preparatory material required for carrying out the research work was obtained from the commercial sources [Loba Chemie, Pvt Ltd. Mumbai, India Central Drug House (CDH) Pvt. Ltd., New Delhi, India] and used without further purification. The purity of the synthesized compounds was observed by thin layer chromatography (commercial silica gel plates (Merck), Silica gel F254 on aluminium sheets) using chloroform:toluene (7:3, v/v) as mobile phase. Sonar melting point apparatus was used to determine the melting point of synthesized compounds in open capillary tubes. 1H-NMR (DMSO-d6) and 13C-NMR (DMSO-d6) were recorded on Bruker Avance III 600 NMR spectrometer, 1H at 600 MHz and 13C at150 MHz using appropriate deuterated solvents. The results are conveyed in parts per million (δ, ppm) downfield from tetramethyl silane (internal standard). Proton NMR data are given as multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet) and number of protons. Infrared (IR) spectra were recorded on a Bruker FTIR spectrometer. The mass spectral data was recorded on Waters Q-TOF micromass (ESI–MS). Elemental analysis for synthesized derivatives was performed on CHN analyzer (Additional files 1, 2 and 3).
General procedure for the synthetic scheme 1
Step A: Synthesis of 4-(4-bromophenyl)-thiazol-2-amine (intermediate)
A mixture of p-bromo acetophenone (0.1 mol), thiourea (0.2 mol) and iodine (0.1 mol) was refluxed for 11–12 h. The reaction mixture was cooled and washed with diethyl ether to remove unreacted acetophenone and iodine. The completion of reaction was confirmed by thin layer chromatography. After this reaction mixture was allowed to cool and poured into the solution of ammonium hydroxide, precipitated and then filtered [26].
Step B: Synthesis of final derivatives (p1–p10)
A mixture of 4-(4-bromophenyl)-thiazol-2-amine (0.02 mol) and substituted aldehydes (0.02 mol) was refluxed in minimum amount of ethanol in presence of small amount of glacial acetic acid for 6–7 h. The completion of reaction was monitored by TLC. The mixture was cooled and poured in ice cold water. The solid thus obtained was filtered and dried [27].
Antimicrobial evaluation (in vitro)
The antimicrobial activity of the synthesized molecules 4-(4-bromophenyl) thiazol-2-amine was evaluated against Gram positive bacteria [Staphylococcus aureus (MTCC3160) and Bacillus subtilis (MTCC441)], Gram negative bacterium Escherichia coli (MTCC443), and fungal strains—Aspergillus niger (MTCC281); Candida albicans (MTCC227) by tube dilution method. The stock solution was prepared for the test compounds (p1–p10) and for the standard drugs (norfloxacin and fluconazole) in acetone to get a concentration of 100 μg/mL and this stock solution was further serially tube diluted [28]. Dilution of test and standard compounds were prepared with double strength nutrient broth-I.P (antibacterial) and sabouraud dextrose broth-I.P (antifungal) [29]. The samples were incubated at 37 ± 1 °C for 24 h (bacteria), 25 ± 1 °C for 7 days (A. niger and C. albicans), respectively and results were recorded in terms of MIC.
Anticancer evaluation (in vitro)
The antiproliferative screening of synthesized 4-(4-bromophenyl)thiazol-2-amine molecules was conducted against the oestrogen receptor positive human breast adenocarcinoma, MCF7, in comparison to a standard drug (5-fluorouracil) using the SRB assay. Briefly, MCF7 cells were exposed to the compounds for 72 h. Treated cells were being fixed with trichloroacetic acid and then stained with 0.4% (w/v) SRB in 1% acetic acid. Unbound dye was removed by five washes with 1% acetic acid solution. Protein-bound dye was solubilized with 10 mM Tris base prior to reading of optical density using a computer-interfaced, 96-well microtiter plate reader. The anticancer activity result was expressed as mean IC50 value of at least triplicates [30].
Molecular docking and ADME studies
Molecular docking
The selected target proteins (PDB ID-3ERT, 4WMZ and 1JIJ) required for molecular docking studies were obtained from the RCSB Protein data bank (http://www.rcsb.org/pdb/home/home.do) (Additional file 3). The selected PDB file was prepared for the molecular docking study using Protein Preparation Wizard (preprocessed, optimized and minimized). A grid is generated around the co crystallized ligand so that it can be excluded and new compounds can be attached to the same active site to study their interactions with receptor. The molecular structures of compounds that are to be docked must be in good representations of as they would appear in a protein–ligand complex. LigPrep module of Schrodinger v11.5 was used to prepare the ligand (compound) for docking in Maestro format. The prepared ligand and receptor are docked using extra precision (XP). XP module docked the compounds with better precision and accuracy. The XP parameters like docking score glide energy and glide model value were calculated within the Schrodinger v11.5 (Additional file 1) [17, 31–33].
ADME study
Most of the drug molecules fail during clinical trials, to streamline our study ADME properties determination is the crucial step. Drug likeliness and ADME properties of the most active compounds were determined using QikProp, GLIDE and Schrodinger v11.5. LigPrep module of Schrodinger v11.5 was employed to prepare the ligand (compound) in Maestro format (.maez) for ADME study. Then we went on task, browsed the Qikpro dialogue box and ligand prepared file (.maez) of the synthesized derivatives was inserted to obtain the ADME parameters (Additional file 2) [34, 35].
Conclusion
A novel series of thiazole derivatives was synthesized, docking study and evaluated in vitro antimicrobial and anticancer activities. Presence of electron releasing (OCH3, OH) and electron withdrawing (p-Br) groups at benzylidene ring of the compound p2 made it as most potent antibacterial and anticancer agent. The presence of electron releasing group (OCH3) in compounds p3 and p4 also made them potent antifungal (A. niger) and antibacterial (B. subtilis) agents, respectively. The presence of fused aromatic nucleus in p6 made it the most active antifungal agent against C. albicans. Molecular docking study of the selected most active compounds exhibited the best docked score and ADME properties with their better potency of antimicrobial and anticancer activities. The active compounds, p2, p3, p4 and p6 may act as useful leads for further development of antimicrobial and anticancer agents.
Additional files
Additional file 1. Molecular Docking study of the synthesized compounds (p1–p10) and standard drugs.
Additional file 2. ADME properties of the most active synthesized compounds (p2–p4 and p6).
Additional file 3. Proteins structures and PDB id link.
Authors’ contributions
BN, DS—performed synthesis, antimicrobial activity and docking study of most active anticancer compounds; SK—performed docking study of most active antimicrobial compounds and KR, SML, SAAS and VM—performed spectral characterization and antiproliferative study of synthesized thiazole compounds. All authors read and approved the final manuscript.
Acknowledgements
The authors are thankful to Head, Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, for providing necessary facilities to carry out this research work.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- LBDD
ligand based drug designing
- SBDD
structure based drug designing
- ADME
absorption, distribution, metabolism and excretion
- CADD
computer aided drug designing
- ER-alpha
estrogen receptor alpha
- NMR
nuclear magnetic resonance
- IR
infrared
- MCF7
Michigan Cancer Foundation7
- SRB
Sulforhodamine B
- MTCC
Microbial Type Culture Collection
- μM
micro mole
- PDB
protein data bank
- XP
extra precision
- 5-Fu
5-fluorouracil
- H-bond
hydrogen-bond
- 2D
2 dimensional
- 3D
3 dimensional
- SAR
structure activity relationship
- MIC
minimum inhibitory concentration
- RCSB
Research Collaboratory for Structural Bioinformatics
Contributor Information
Deepika Sharma, Email: deepikashrma07@gmail.com.
Sanjiv Kumar, Email: sanjiv.pharmchem@gmail.com.
Balasubramanian Narasimhan, Email: naru2000us@yahoo.com.
Kalavathy Ramasamy, Email: kalav922@gmail.com.
Siong Meng Lim, Email: stvlsm@gmail.com.
Syed Adnan Ali Shah, Email: benzene301@yahoo.com.
Vasudevan Mani, Email: vasumpharmacol@gmail.com.
References
- 1.Zhang HJ, Qin X, Liu K, Zhu DD, Wang XM, Zhu DD. Synthesis, antibacterial activities and molecular docking studies of Schiff bases derived from N-(2/4-benzaldehyde-amino) phenyl-N′-phenyl-thiourea. Bioorg Med Chem. 2011;19:5708–5715. doi: 10.1016/j.bmc.2011.06.077. [DOI] [PubMed] [Google Scholar]
- 2.Muhammad YA, Narang R, Nayak SK, Singh SK. Synthesis, antibacterial activity and molecular docking studies of N′-benzylidene/N′-(1-phenylethylidene)-hexa-2,4-dienehydrazide derivatives. J Chem Pharm Res. 2016;8(3):930–937. [Google Scholar]
- 3.Hejmadi M. Introduction to cancer biology, 2nd edn. 2. Frederiksberg: Bookboon.com; 2010. p. 7. [Google Scholar]
- 4.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancers. 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta V, Kant V. A review on biological activity of imidazole and thiazole moeities and their derivatives. Sci Int. 2013;1(7):253–260. doi: 10.17311/sciintl.2013.253.260. [DOI] [Google Scholar]
- 6.Kashyap SJ, Garg VK, Sharma PK, Kumar N, Dudhe R, Gupta JK. Thiazoles: having diverse biological activities. Med Chem Res. 2012;21:2123–2132. doi: 10.1007/s00044-011-9685-2. [DOI] [Google Scholar]
- 7.Mohammad H, Reddy PVN, Monteleone D, Mayhoub AS, Cushman M, Hammac GK, Seleem MN. Antibacterial characterization of novel synthetic thiazole compounds against methicillin-resistant Staphylococcus pseudintermedius. PLoS ONE. 2015;10(6):1–19. doi: 10.1371/journal.pone.0130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng K, Xue JY, Zhu HL. Design, synthesis and antibacterial activity studies of thiazole derivatives as potent ecKAS III inhibitors. Bioorg Med Chem Lett. 2013;23:4235–4238. doi: 10.1016/j.bmcl.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Makam P, Kannan T. 2-Aminothiazole derivatives as antimycobacterial agents: synthesis, characterization, in vitro and in silico studies. Eur J Med Chem. 2014;87:643–656. doi: 10.1016/j.ejmech.2014.09.086. [DOI] [PubMed] [Google Scholar]
- 10.Rodrigues CA, Santos PFD, Costa MOLD, Pavani TFA, Xander P, Geraldo MM, Mengarda A, Moraes JD, Rando DGG. 4-Phenyl-1,3-thiazole-2-amines as scaffolds for new antileishmanial agents. J Venom Anim Toxins Incl Trop Dis. 2018;24(26):1–10. doi: 10.1186/s40409-018-0163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sapkale PV, Patil AV. Thiazoles as an anticancer agent: an overview. Indo Am J Pharm Res. 2016;6(10):6648–6661. [Google Scholar]
- 12.Bharti SK, Nath G, Tilak R, Singh SK. Synthesis, anti-bacterial and anti-fungal activities of some novel Schiff bases containing 2,4-disubstituted thiazole ring. Eur J Med Chem. 2010;45:651–660. doi: 10.1016/j.ejmech.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Yeranale NG, Mathada MBH. Synthesis, characterization, antimicrobial, DNA cleavage and in vitro cytotoxic studies of some metal complexes of Schiff base ligand derived from thiazole and quinoline moiety. Bioinorg Chem Appl. 2014;2014:1–17. doi: 10.1155/2014/314963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vinusha HM, Shiva Prasad K, Chandan S, Begum M. Imino-4-methoxyphenol thiazole derived Schiff base ligands: synthesis, spectral characterization and antimicrobial activity. Chem Sci J. 2015;6(3):1–4. [Google Scholar]
- 15.Hassan GS, El-Messery SM, Al-Omary FAM, El-Subbagh HI. Substituted thiazoles VII. Synthesis and antitumor activity of certain 2-(substituted amino)-4-phenyl-1,3-thiazole analogs. Bioorg Med Chem Lett. 2012;22:6318–6323. doi: 10.1016/j.bmcl.2012.08.095. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira LG, Santos RND, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muchtaridi M, Dermawan D, Yusuf M. Molecular docking, 3d structure-based pharmacophore modeling, and ADME prediction of alpha mangostin and its derivatives against estrogen receptor alpha. J Young Pharm. 2018;10(3):252–259. doi: 10.5530/jyp.2018.10.58. [DOI] [Google Scholar]
- 18.Liao C, Sitzmann M, Pugliese A, Nicklaus MC. Software and resources for computational medicinal chemistry. Future Med Chem. 2011;3(8):1057–1085. doi: 10.4155/fmc.11.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Urban L. The impact of early ADME profiling on drug discovery and development strategy. Drug Discov World Fall. 2004;5:73–86. [Google Scholar]
- 20.Li AP. Screening for human ADME/Tox drug properties in drug discovery. Res Focus. 2001;6(7):357–366. doi: 10.1016/s1359-6446(01)01712-3. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Xing J, Xu Y, Zhou N, Peng J, Xiong Z, Liu X, Luo X, Luo C, Chen K, Zheng M, Jiang H. In silico ADME/T modelling for rational drug design. Q Rev Biophys. 2015 doi: 10.1017/s0033583515000190:1-28. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S, Lim SM, Ramasamy K, Mani V, Shah SAA, Narasimhan B. Design, synthesis, antimicrobial and cytotoxicity study on human colorectal carcinoma cell line of new 4,4′-(1,4-phenylene)bis(pyrimidin-2-amine) derivatives. Chem Cent J. 2018;12(73):1–13. doi: 10.1186/s13065-018-0440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Lim SM, Ramasamy K, Vasudevan M, Shah SAA, Selvaraj M, Narasimhan B. Synthesis, molecular docking and biological evaluation of bis-pyrimidine Schiff base derivatives. Chem Cent J. 2017;11(89):1–16. doi: 10.1186/s13065-017-0322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karki R, Rao GK, Gupta A, Mariappan G, Adhikari S. Synthesis, characterization and antimicrobial activities of Schiff bases of 2-amino-4-(O-chloroanilino)-1, 3-thiazole. J Appl Pharm Sci. 2013;3(07):093–096. [Google Scholar]
- 25.Braga SFP, Fonseca NC, Ramos JP, Souza- Fagundes EM, Oliveira RB. Synthesis and cytotoxicity evaluation of thiosemicarbazones and their thiazole derivatives. Braz J Pharm Sci. 2016;52:299–307. doi: 10.1590/S1984-82502016000200008. [DOI] [Google Scholar]
- 26.Shruthy VS, Yusuf S. In silico, design, docking, synthesis and evaluation of thiazole Schiff bases. Int J Pharm Pharm Sci. 2014;6(3):271–275. [Google Scholar]
- 27.Khan KM, Ambreen N, Karim A, Saied S, Amyn A, Ahmed A, Perveen S. Synthesis of Schiff bases of thiazole as antibacterial and antifungal agents. J Pharm Res. 2012;5:651–656. [Google Scholar]
- 28.Cappuccino JC, Sherman N. Microbiology—a laboratory manual. California: Addison Wesley; 1999. p. 263. [Google Scholar]
- 29.Pharmacopoeia of India, vol Ӏ (2007) Controller of publication, Ministry of Health Department, Govt. of India, New Delhi, pp 37
- 30.Skehan P, Storeng R, Scudiero D, Monks A, Mcmohan J, Vistica D, Wareen JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 31.Gullapelli K, Brahmeshwari G, Ravichander M, Kusuma U. Synthesis, antibacterial and molecular docking studies of new benzimidazole derivatives. Egypt J Basic Appl Sci. 2017;4:303–309. doi: 10.1016/j.ejbas.2017.09.002. [DOI] [Google Scholar]
- 32.Stana A, Vodnar DC, Tamaian R, Au AP, Vlase L, Ionut L, Oniga O, Tiperciuc BS. Design, synthesis and antifungal activity evaluation of new thiazolin-4-ones as potential Lanosterol 14 α-demethylase inhibitors. Int J Mol Sci. 2017;18(177):1–25. doi: 10.3390/ijms18010177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar S, Singh J, Narasimhan B, Shah SAA, Lim SM, Ramasamy K, Mani V. Reverse pharmacophore mapping and molecular docking studies for discovery of GTPase HRas as promising drug target for bis-pyrimidine derivatives. Chem Cent J. 2018;12(106):1–11. doi: 10.1186/s13065-018-0475-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vadivelu A, Gopal V, Reddy CUM. Molecular docking studies of 1,3,4-thiadiazoles as novel peptide deformylase inhibitors as potential antibacterial agents. Int J Pharm Sci Rev Res. 2015;31(1):58–62. [Google Scholar]
- 35.Meraj K, Mahto MK, Christina NB, Desai N, Shahbaz S, Bhaskar M. Molecular modeling, docking and ADMET studies towards development of novel disopyramide analogs for potential inhibition of human voltage gated sodium channel proteins. Bioinformation. 2012;8(23):1139–1146. doi: 10.6026/97320630081139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Molecular Docking study of the synthesized compounds (p1–p10) and standard drugs.
Additional file 2. ADME properties of the most active synthesized compounds (p2–p4 and p6).
Additional file 3. Proteins structures and PDB id link.