

Abstract
The Zika virus (ZIKV) has gained attention for the last few years due to the congenital microcephaly and Guillain–Barre Syndrome that resulted in humans. The non-structural protein-3 (NS3) helicase of ZIKV play an important role in viral RNA replication. In this article, we performed hundred nanosecond molecular dynamics simulation and molecular docking of the NS3 helicase of ZIKV with 1,4-benzothiazine derivatives. The root mean square deviation (RMSD) analyses showed the stability of the NS3 helicase. The simulation showed that the flexible and rigid domains of the protein play a crucial role during the RNA replication process. All such domains with ligand binding pockets can be targeted for drug design. The molecular docking showed that the strong hydrogen bonding and arene-cation interactions are responsible for the binding between NS3 and 1,4-benzothiazine derivatives, which provides a new dimension for potent drug design for ZIKV.
Electronic supplementary material
The online version of this article (10.1186/s13065-019-0582-y) contains supplementary material, which is available to authorized users.
Keywords: ZIKV, Microcephaly, Nonstructural protein-3 Helicase, Molecular dynamics simulation, Molecular docking
Introduction
Zika virus (ZIKV) is a mosquito-borne flavivirus like yellow fever virus, dengue virus (DENV), West Nile virus (WNV) and chikungunya [1]. It contains a single-stranded positive RNA with a total of 10,794-nucleotides in its genome [2]. The ZIKV transmission occurs through numerous Aedes spp. mosquitoes, including Aedes africanus, Aedes luteocephalus, Aedes hensilli, and Aedes aegypti [3–6]. Initially the ZIKV was observed in rhesus monkey amid observation of yellow fever in the zika woodland in Uganda in the year 1947 and it was initially reported in people in 1952 [7, 8]. Later on in 1954, it was observed in humans with febrile diseases in West Africa [7, 8]. In 2014 several cases of ZIKV were recorded in Indonesia, Micronesia, Thailand, Philippines, French Polynesia and Easter Island in South Pacific [9, 10]. From May, 2015 onward there was a boom in the spread of ZIKV in Brazil and from there it spread to other countries [11, 12].
In early 2016, the World Health Organization declared the ZIKV spread as a international public health emergency due to the presence of congenital neurological disorders like microcephaly, Guillain–Barre syndrome and cranial nerve dysfunction [13]. The exact mechanism of action of ZIKV that how it causes microcephaly is not known but research efforts are in progress to establish the link between ZIKV and microcephaly [10, 14, 15]. Generally, the mosquito borne flavivirus pathogenesis begins with its replication in dendritic cells close to the site of inoculation from where it move to the lymph centers and blood circulation system [16]. After the initial viral attack, the virions can be observed in the blood, while the viral RNA can been seen after eleven days of ailment [17]. The helicases are an omnipresent exceedingly diverse group of proteins that carry out an astonishing variety of functions in cells [18]. These are ATP-ases which depend on nucleic acid and have the potential to unwind DNA or RNA duplex substrates [18]. Because of this unwinding properties, they are significantly involved in every process of cells linked with DNA like replication, repair, transcription, translation, synthesis of ribosomes, RNA maturation and splicing and the procedures related with the nuclear export [18]. Recently a number of structural as well as non-structural proteins from ZIKV that have enzymatic functions are resolved at high resolution through various techniques and they are useful target for drug design [19]. The NS3 helicase is one of the most thoroughly studied antiviral drug target [20, 21]. In NS3 of flaviviruses, the enzymatic activity is coupled with the C-terminal region, namely an RNA helicase (NS3-Hel) concerned with genome replication and RNA synthesis [22, 23]. The NS3-Hel is part of the superfamily helicases, and its inhibition in DENV makes the virus unable to replicate [24, 25]. The two motor domains formed in the helicase part of the NS3 proteins are identical with all other helicases of superfamily 1 and 2 but the third domain is very different from other helicases. The binding site for ATP is located between the two domains while the RNA binds between the third and motor domain [26]. The inchworm like movement of NS3 helicase in a 3′ to 5′ directions along one strand of RNA was observed in detailed mechanistic studies which involved the displacing of the complementary strand in an ATP utilized reaction. The same type of structural and enzymatic studies of DENV helicase suggest that there is no difference between the mechanism of hepacivirus and flavivirus [27, 28]. The non-nucleoside based viral inhibitors are an active area of research and benzothiazine based compounds have been studied against several flaviviruses [21, 29, 30]. They contains a nitrogen and a sulfur atom in their basic nucleus that makes them a good interacting agent inside the active site of an enzyme [30]. Different research groups have shown that benzothiazine are suitable inhibitors of helicases [31, 32]. Currently there are two MD simulation studies available that explore the dynamics of ZIKV NS3 helicase but to get further insight docking novel inhibitors and further simulation studies are required [33]. In this study we targeted the ZIKV NS3 helicase with 1, 4-benzothiazine analogues (Additional file 1: Figure S1) using molecular dynamics simulation and molecular docking methods. The reason for choosing 1,4-benzothiazine based derivatives was that a number of research groups have used 1,4-benzothiazine analogues both computationally and experimentally against different viral helicase enzymes [31, 34]. Therefore, we design these derivatives and used it for molecular docking and simulation studies. We hope that this study will also be vital in the effort to find suitable inhibitors against the spread of this devastating viral disease.
Methodology
The crystal structure of the NS3 helicase of ZIKV was obtained from Protein Data Bank with the PDB ID 5JRZ [35]. PyMOL software version 1.7 was used for visualization and checking the protein [36]. The ligands were searched from the literature and newly reported compounds were selected for molecular docking [37]. For molecular docking, we used the Molecular Operation Environment (MOE) 2014 docking software program [38]. The prediction of the active site in the protein was carried out with MOE 2014 software, site finder tool which identified the various sites in which one was selected for docking procedure. The removal of water molecules was done from the crystal structure. The addition of missing hydrogen atoms was done, correction of charges and assigning of the correct hybridization state of each residue was done through the preparation program of MOE. The corrected protonation was done using 3D protonate module present in MOE with Generalized Born/Volume Integral (GB/VI) electrostatic function. To explore the potential binding site, the complete structure of the enzyme was used as a receptor. For each ligand, generation of multiple conformations were done by applying a selected torsion angles to all the rotatable bonds in each ligand. For each drug candidate about thirty conformations were generated. For each ligand and the receptor, the accepted conformations were scored by using the London Dock scoring function which calculates the free energy for the binding ligand in a given conformation [39].
The binding affinities were calculated using generalized-Born volume integral/weighted surface area (GBVI/WSA) method present in MOE 2014. Generalized Born interaction energy is the non-bonded interaction energy between the receptor molecule and the ligand. It is composed of Coulomb electrostatic interaction, Van der Waals, and implicit solvent interaction energies. The binding affinity was calculated for each hit after energy minimization, and reported in unit of kcal/mol [40]. The PCA was performed as reported previously [41].
Molecular dynamics simulations
The crystal structure of NS3 helicase of ZIKV, PDB ID 5JRZ having 1.62 Å resolution was used for atomic coordinates for both Apo and in docked complex with derivative 7 saved as PDB file, which was constructed through SYBYLs-X2.1.1 [42]. For molecular dynamics simulation the AMBER12 software package [43, 44] was utilized adjusting all the parameters. The addition of hydrogen atoms was done through LEaP module of AMBER-12. For the maintenance of neutral condition of the system the counter-ions were utilized. A truncated octahedral box of TIP3P molecules of water was used for the solvation of both the systems. The cutoff distance is kept 8 Å for the determination of pairwise interactions (van der Waals and direct coulombic interactions). In order to compute the long range electrostatic interactions the particle mesh Ewald (PME) method [45] of AMBER12 was used while the ff12SB force field was selected for the finding of the intramolecular interactions. For preparations runs a Langevin thermostat was utilized with friction constant of 1 ps−1 while for the production runs a Berendsen thermostat was used [46]. All MD simulations were accelerated using the CUDA version of PMEMD in GPU cores of NVIDIAs Tesla K20. The 500-step steepest descent minimization and 2000-step conjugated gradient minimization were performed for initial minimization with macromolecules frozen in order to avoid further structural clash in the solvated system. The whole system was again passed through the minimization process at the end, keeping 1000-step steepest descent minimization and 19,000-step conjugate gradient minimization. The adjustment of parameters after energy minimization at 400 ps heating up and 200 ps equilibration in the NVT ensemble at 310 K were done before the start of MD simulations in the NPT ensemble at 310 K. In order to carry out a comparison of Apo and complex various simulated trajectories at different nanoseconds were collected at 310 K during the 100 ns simulation timescale.
Results and discussion
Molecular docking of 1,4-benzothiazine derivatives
On docking several derivatives of 1,4-benzothiazine with ZIKV NS3 helicase, they bind inside the ATP binding pocket of the enzyme. Most of these inhibitor derivatives interact with the enzyme through hydrogen bonding and cation-arene interaction. In the majority of the derivatives docking with the enzyme, Lys-200, Arg-459 and 462, and Thr-201 were involved in interactions (Fig. 1 and Additional file 1: Figure S2) and Additional file 1: Table S1. The involvements of these residues were also previously recorded by other docking and simulation studies [47, 48].
Fig. 1.

2D and 3D structures of 1,4-benzothiazine analogue 7 with ZIKV NS3 helicase. The inhibitor occupies the ATP binding site and interacts mostly with lysine-200 and arginine-459 and 462 of the active binding pocket
Stability of the simulated systems
The molecular dynamics simulation provided us the changes inside the protein under observation during the virtual time and thus provides useful information regarding the protein drug interaction. The longer the time, the better the observations of the different types of interaction and movements of the protein domains. The stability of the system is shown by the changes in the root mean square deviation (RMSD) during the course of simulation time. As shown in Fig. 2, left panel shows the RMSD graphs of the backbone Cα atoms of both Apo and protein in complex with the ligand (1,4-benzothiazine analogue 7). In the start of the simulation there is a 0.5 Å rise in RMSD value for the first few nanosecond, but then it converged for both systems and it is around 1.9 Å. Although there is minor fluctuation in the RMSD value for both the systems, but as a whole both the systems are highly stable during the 100 ns time course (Fig. 2).
Fig. 2.

The C-α backbone RMSD of NS3 helicase APO enzyme and in complex with 1,4-benzothiazine derivative 7. The average C-α RMSD was calculated to be 1.8 Å to 1.9 Å, respectively. The right side panel showed the root mean square fluctuations in Apo and in complex with the ligand, while the lower right side panel showed the superposition of the two upper RMSF graphs for better assesment of the residues involved in fluctuations
Structural fluctuation of the NS3 protein
The root mean square fluctuation (RMSF) analysis of the 449 amino acid of the helicase enzyme was calculated to get an insight into the residues that are involved in the interactions with the inhibitor, secondly the most mobile residues and the non-flexible residues are important as they may be further targeted for drug design. It can be observed from Figs. 2 and 3 that the helicase in complex with the inhibitors (1,4-benzothiazine derivative 7) has less fluctuations as compared to the apo form of the protein. The residues number 25-30 are highly flexible with RMSF of 3.7 Å in the apo form, but this value falls to 2 Å in the complex state, showing the interactions between the helicase and the 1,4-benzothiazine derivative 7 in the docked form. In a similar study, Ramharack et al. showed that the NS3 helicase containing more than six hundred residues in complex with its inhibitor NIT008D has more fluctuation then its apo form [47]. While in our simulation of the ZIKV NS3 helicase has on the average same fluctuation in apo and in complex form but variation are there with increase or decrease in RMSF values.
Fig. 3.
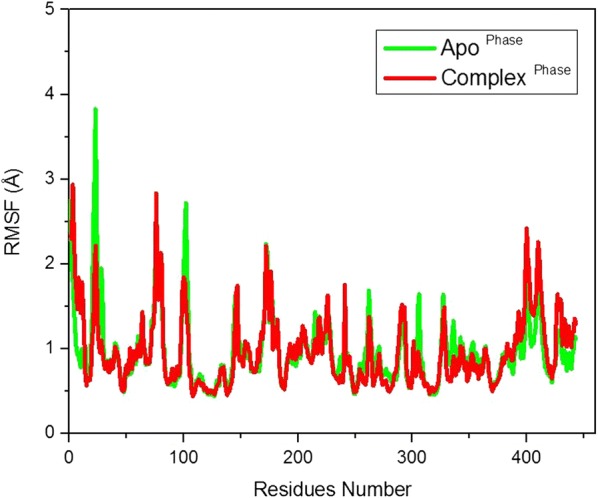
The RMSF of APO protein and its complex with the inhibitor (1,4-benzothiazine derivative 7)
Drug designing involved two opposite views about the drug targets in literature; one is that inhibitors inhibit the most rigid residues and the second view is that inhibitors target highly flexible residues [48]. The rigid residues and flexible regions in enzymes have specified role in various biological reactions [48]. The molecular recognition and catalytic properties are linked with the mobility of specific residues while production of various structures in beta-folds is due to the rigid residues, that are essential for interaction with the substrate and its catalysis.
Principal component analysis
The principal component analysis (PCA) was performed for both Apo and complex with the inhibitor (1,4-benzothiazine derivative 7). The simulation results showed that specific movements are present in the domains of the helicase with and without the inhibitor (Fig. 4). In case of Apo it is very clear from the Fig. 4, which in alpha helices and loops of domain II; and loops of domain I there is an anti-clock wise motion which is represented by the blue arrows. It means that there are conformational changes in the enzyme after 100 ns simulations. The motion showed the catalytic importance of that particular region in the enzyme function. In case of the ZIKV helicase in complex with the inhibitor (1,4-benzothiazine derivative 7), a clockwise motion is present in alpha helices and β-sheets of domain II. A similar kind of domain motions were also observed by Ramharack et al. in their simulation studies of ZIKV NS3 helicase [47]. From the PCA analysis it can be concluded that conformational changes are present in both Apo and complex system but their direction and magnitudes are totally different, showing the effects of the inhibitor.
Fig. 4.

PCA analyses of both Apo and protein in complex with the 1,4-benzothiazine derivative 7 after 100 ns simulations time
Interaction analysis
The MD simulation of the complex of the NS3 helicase with inhibitor showed that the 1,4-benzothiazine analogue 7 interacted with Lysine200, Arginine462 and Arginine459 in the active site as shown in Fig. 5. In Fig. 5 the colored ball model is derivative 7 while others are important interacting residues of the protein. The docking score of the derivative is − 13.09 kcal/mol with binding free energy of − 12.34 kcal/mol. The good docking score elucidate that the inhibitor is strongly bound in the pocket with a favorable binding energy value. This binding pocket is the same ATP binding site where the inhibitor NITD008, a Flavivirus adenosine analogue bind in the simulation and docking studies performed by Ramharack et al. [47]. The ATP binding site is a hydrophobic pocket and is present between domain I and II [35, 49]. Lysine and arginine residues are playing important role in the interaction and catalysis at this site [35, 49]. Similarly this ATP binding site is the same as in the DENV helicase enzyme [26].
Fig. 5.
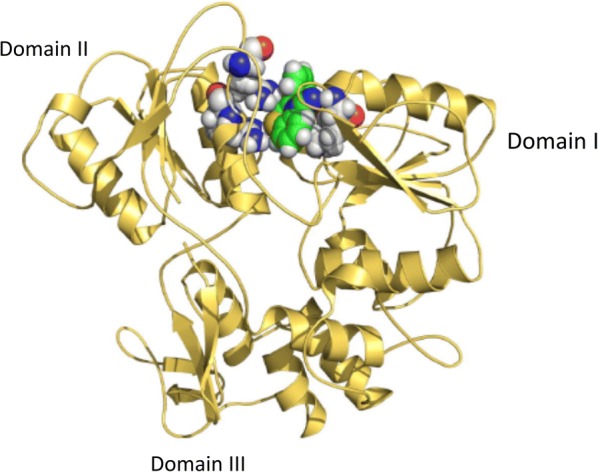
Interaction of 1,4-benzothiazine derivative 7 in the predicted active site of NS3 helicase
Superposition of Apo and complex before and after simulation
The superposition of the Apo and complex form were performed before and after simulation of 100 ns as shown in Fig. 6a. The sphere in the model indicated the positions where variations occur with respect to the simulation time. Each sphere showed different degree of motion and also changes in their orientations. The change in position of the spheres showed a displacement toward the center of the axis of the protein from left and right side. This type of motion may be essential for processing the RNA molecule when it enters the helicase and probably a common feature of the flaviviridae NS3 helicases.
Fig. 6.

a Superposition of Apo NS3 helicase before and after 100 ns simulation. b Superposition of complex before and after 100 ns simulation. c Superposition of complex before and after 100 ns simulation in a closer view
In case of the complex form, before the simulation time, the inhibitors was represented by green color while the portions of three domains were represented by dark brown color and after the simulation the ligand was shown by blue color and the protein mobile spheres were represented by cyan color (Fig. 6b, c). When we performed the superposition of the NS3 helicase with the inhibitor, there is a difference in the position of the mobile residues which showed that the inhibitor produced conformational changes when it binds inside the active site of the NS3 helicase which was previously confirmed from RMSF analysis. Secondly, again the amount of displacement and orientation in the spheres is different from one another in the same protein. While an internal motion towards the center of axis from left and right side is also observed in the complex form after the simulation time.
Conclusion
Helicase enzyme of various viruses is an important drug target and in this study, we reported the utilization of 1-4 benzothiazine derivatives as inhibitor of ZIKV helicase for the docking study. The docking results indicated that these compounds were in interactions mainly with residues Thr201, Lys200, Arg462 and Arg459 of the NS3 which was reported hotspots by making hydrogen bonds and arene-cation interactions. The 1,4-benzothiazine derivative 7 showed maximum interactions with best docking scores and binding energies while its validation was further carried out with molecular dynamics simulation for 100 ns and the results were found excellent including, RMSD, RMSF values, principal component analysis and superposition expressed the stability of the drug inside the binding pocket. From the above study, we can conclude that this class of compounds can be used as possible novel inhibitors of ZIKV helicase (NS3) and it’s a way forward toward virtual screening and pharmacophore mapping tools to explore the treasures of biological important class of compounds to inhibit the genome replication of ZIKV. The molecular dynamic simulation confirmed that the docked conformation is reliable. Binding energy calculations through the MOE docking program showed that van der Waal interaction and hydrogen bonding provided the most substantial force for the binding of the inhibitor.
Additional file
Additional file 1: Figure S1. The nine different 1,4-benzothiazine derivatives from D-1 to D-9 used in molecular docking against ZIKV NS3 helicase. Figure S2. The molecular docking interaction of 1,4-benzothiazine derivatives 1-9 in 2D and 3D. Table S1. Docking score and binding energy of 1,4-benzothiazine derivatives.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for its funding this prolific research Group No. (R.G.P.2/23/40/2019).
Authors’ contributions
SB and NA contributed equally. SB, NA and AR performed the molecular docking and simulation. NA, KH, SB, AU, AM, AA and YM wrote the manuscript. All authors read and approved the final manuscript.
Funding
King Khalid University grant awarded to Dr. Yahia Mabkhot.
Availability of data and materials
Data and material are available on request.
Competing interests
All the authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Syed Lal Badshah, Email: shahbiochemist@gmail.com.
Nasir Ahmad, Email: nasirshah121@yahoo.com.
Ashfaq Ur Rehman, Email: serendifity_rehman@outlook.com.
Khalid Khan, Email: drkhalidchem@yahoo.com.
Asad Ullah, Email: asad_icp@yahoo.com.
Abdulrhman Alsayari, Email: alsayari@kku.edu.sa.
Abdullatif Bin Muhsinah, Email: ajmohsnah@kku.edu.sa.
Yahia N. Mabkhot, Email: ygaber@kku.edu.sa.
References
- 1.Moulin E, Selby K, Cherpillod P, et al. Simultaneous outbreaks of dengue, chikungunya and Zika virus infections: diagnosis challenge in a returning traveller with nonspecific febrile illness. New Microbes New Infect. 2016;11:6–7. doi: 10.1016/j.nmni.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunha MS, Esposito DLA, Rocco IM, et al. First complete genome sequence of Zika virus (Flaviviridae, Flavivirus) from an Autochthonous transmission in Brazil. Genome Announc. 2016;4:2015–2016. doi: 10.1128/genomeA.00032-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner LM, Chen N, Sarkar S. Global risk of Zika virus depends critically on vector status of Aedes albopictus. Lancet Infect Dis. 2016;16:522–523. doi: 10.1016/S1473-3099(16)00176-6. [DOI] [PubMed] [Google Scholar]
- 4.Wong PSJ, Li M, Zhi I, Chong CS, et al. Aedes (Stegomyia) albopictus (Skuse): a potential vector of Zika virus in Singapore. PLoS Negl Trop Dis. 2013;1:1. doi: 10.1371/journal.pntd.0002348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leandro H, Dutra C, Rocha MN, et al. Wolbachia blocks currently circulating Zika virus isolates in Brazilian Aedes aegypti mosquitoes. Cell Host Microbe. 2016;19:1–4. doi: 10.1016/j.chom.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bajpai S, Nadkar MY. Zika virus infection, the recent menace of the Aedes mosquito. J Assoc Physicians India. 2016;64:42–45. [PubMed] [Google Scholar]
- 7.Dick GWA. Zika virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- 8.Wikan N, Smith DR. Zika virus: history of a newly emerging arbovirus. Lancet Infect Dis. 2016;16:e119–e126. doi: 10.1016/S1473-3099(16)30010-X. [DOI] [PubMed] [Google Scholar]
- 9.Musso D. Zika virus transmission from French Polynesia to Brazil. Emerg Infect Dis. 2015;21:1887–1889. doi: 10.3201/eid2110.151125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cauchemez S, Besnard M, Bompard P, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–2015: a retrospective study. Lancet. 2016;387:2125–2132. doi: 10.1016/s0140-6736(16)00651-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonaldo MC, Ribeiro IP, Lima NS, et al. Isolation of infective Zika virus from urine and saliva of patients in Brazil. PLoS Negl Trop Dis. 2016;1:1. doi: 10.1371/journal.pntd.0004816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Syed S, Mabkhot YN, Naeem A, et al (2018) Zika virus, microcephaly and its possible global spread. In: Current topics in Zika
- 13.Gulland A. Zika virus is a global public health emergency, declares WHO. BMJ. 2016;352:i657. doi: 10.1136/bmj.i657. [DOI] [PubMed] [Google Scholar]
- 14.De Góes Cavalcanti LP, Tauil PL, Alencar CH, et al. Zika virus infection, associated microcephaly, and low yellow fever vaccination coverage in Brazil: is there any causal link. J Infect Dev Ctries. 2016;10:563–566. doi: 10.3855/jidc.8575. [DOI] [PubMed] [Google Scholar]
- 15.Chan JFW, Choi GKY, Yip CCY, et al. Zika fever and congenital Zika syndrome: an unexpected emerging arboviral disease. J Infect. 2016;72:507–524. doi: 10.1016/j.jinf.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond MS, Shrestha B, Mehlhop E, et al. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003;16:259–278. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- 17.Filipe AR, Martins CMV, Rocha H. Laboratory infection with Zika virus after vaccination against yellow fever. Archiv für die gesamte Virusforschung. 1973;43:315–319. doi: 10.1007/bf01556147. [DOI] [PubMed] [Google Scholar]
- 18.Gorbalenya AE, Koonin EV. Helicases: amino acid sequence comparisons and structure-function relationships. Curr Opin Struct Biol. 1993;3:419–429. doi: 10.1016/S0959-440X(05)80116-2. [DOI] [Google Scholar]
- 19.Badshah S, Naeem A, Mabkhot Y. The new high resolution crystal structure of NS2B-NS3 protease of Zika virus. Viruses. 2017;9:7. doi: 10.3390/v9010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Clercq E. Current race in the development of DAAs (direct-acting antivirals) against HCV. Biochem Pharmacol. 2014;89:441–452. doi: 10.1016/j.bcp.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Fang J, Li H, Kong D, et al. Structure-based discovery of two antiviral inhibitors targeting the NS3 helicase of Japanese encephalitis virus. Sci Rep. 2016;6:34550. doi: 10.1038/srep34550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frick DN, Lam AMI. Understanding helicases as a means of virus control. Curr Pharm Des. 2006;12:1315–1338. doi: 10.2174/138161206776361147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo D, Vasudevan SG, Lescar J. The flavivirus NS2B-NS3 protease-helicase as a target for antiviral drug development. Antiviral Res. 2015;118:148–158. doi: 10.1016/j.antiviral.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Matusan AE, Pryor MJ, Davidson AD, Wright PJ. Mutagenesis of the Dengue virus type 2 NS3 protein within and outside helicase motifs: effects on enzyme activity and virus replication. J Virol. 2001;75:9633–9643. doi: 10.1128/JVI.75.20.9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 26.Luo D, Xu T, Watson RP, et al. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 2008;27:3209–3219. doi: 10.1038/emboj.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frick DN. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug target. Curr Issues Mol Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 28.Gu M, Rice CM. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc Natl Acad Sci USA. 2010;107:521–528. doi: 10.1073/pnas.0913380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vicente J, Hendricks RT, Smith DB, et al. Non-nucleoside inhibitors of HCV polymerase NS5B. Part 2: synthesis and structure-activity relationships of benzothiazine-substituted quinolinediones. Bioorganic Med Chem Lett. 2009;19:3642–3646. doi: 10.1016/j.bmcl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Badshah Syed L, Naeem A. Bioactive thiazine and benzothiazine derivatives: green synthesis methods and their medicinal importance. Molecules. 2016;21:1054. doi: 10.3390/molecules21081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin K. Development of novel antiviral therapies for hepatitis c virus. Virol Sin. 2010;25:246–266. doi: 10.1007/s12250-010-3140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rehman S, Ashfaq UA, Javed T. Antiviral drugs against hepatitis C virus. Genet Vaccines Ther. 2011;9:11. doi: 10.1186/1479-0556-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mottin M, Braga RC, da Silva RA, et al. Molecular dynamics simulations of Zika virus NS3 helicase: insights into RNA binding site activity. Biochem Biophys Res Commun. 2017 doi: 10.1016/j.bbrc.2017.03.070. [DOI] [PubMed] [Google Scholar]
- 34.Hendricks RT, Fell JB, Blake JF, et al. Non-nucleoside inhibitors of HCV NS5B polymerase. Part 1: synthetic and computational exploration of the binding modes of benzothiadiazine and 1,4-benzothiazine HCV NS5b polymerase inhibitors. Bioorg Med Chem Lett. 2009;19:3637–3641. doi: 10.1016/j.bmcl.2009.04.119. [DOI] [PubMed] [Google Scholar]
- 35.Jain R, Coloma J, Garcia-Sastre A, Aggarwal AK. Structure of the NS3 helicase from Zika virus. Nat Struct Mol Biol. 2016;23:752–754. doi: 10.1038/nsmb.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLano WL (2002) The PyMOL molecular graphics system. Schrödinger LLC version 1. http://www.pymol.org
- 37.Chu J-J, Hu B-L, Liao Z-Y, Zhang X-G. Copper-catalyzed three-component tandem cyclization for one-pot synthesis of 1,4-benzothiazines. J Org Chem. 2016;81:8647–8652. doi: 10.1021/acs.joc.6b01668. [DOI] [PubMed] [Google Scholar]
- 38.Molecular Operating Environment (MOE), Version 2009.10, Chemical Computing Group Inc., Montreal, Quebec, Canada
- 39.Kumar YN, Kumar PS, Sowjenya G, et al. Comparison and correlation of binding mode of ATP in the kinase domains of Hexokinase family. Bioinformation. 2012;8:543–547. doi: 10.6026/97320630008543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labute P. The generalized born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem. 2008;29:1693–1698. doi: 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- 41.ur Rahman M, Liu H, Wadood A, Chen H-F. Allosteric mechanism of cyclopropylindolobenzazepine inhibitors for HCV NS5B RdRp via dynamic correlation network analysis. Mol BioSyst. 2016;12:3280–3293. doi: 10.1039/C6MB00521G. [DOI] [PubMed] [Google Scholar]
- 42.Ash S, Cline MA, Homer RW, et al. SYBYL line notation (SLN): a versatile language for chemical structure representation. J Chem Inf Model. 1997;37:71–79. doi: 10.1021/ci960109j. [DOI] [Google Scholar]
- 43.Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wiley Interdiscip Rev Comput Mol Sci. 2013;3:198–210. doi: 10.1002/wcms.1121. [DOI] [Google Scholar]
- 44.Pearlman DA, Case DA, Caldwell JW, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. doi: 10.1016/0010-4655(95)00041-D. [DOI] [Google Scholar]
- 45.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 46.Ruhle V (2007) Berendsen and Nose-Hoover thermostats. Thesis 1–4
- 47.Ramharack P, Ogundate S, Soliman M. Delving into Zika virus structural dynamics—a closer look at NS3 helicase loop flexibility and its role in drug discovery. RSC Adv. 2017;7:22133. doi: 10.1039/C7RA01376K. [DOI] [Google Scholar]
- 48.Badshah SL, Naeem A, Mabkhot Y. Molecular dynamics simulation of cholera toxin A-1 polypeptide. Open Chem. 2016;14:188–196. doi: 10.1515/chem-2016-0021. [DOI] [Google Scholar]
- 49.Tian H, Ji X, Yang X, et al. The crystal structure of Zika virus helicase: basis for antiviral drug design. Protein Cell. 2016;7:450–454. doi: 10.1007/s13238-016-0275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. The nine different 1,4-benzothiazine derivatives from D-1 to D-9 used in molecular docking against ZIKV NS3 helicase. Figure S2. The molecular docking interaction of 1,4-benzothiazine derivatives 1-9 in 2D and 3D. Table S1. Docking score and binding energy of 1,4-benzothiazine derivatives.
Data Availability Statement
Data and material are available on request.