

Abstract
Diabetes is an emerging metabolic disorder. α-Glucosidase inhibitors, such as acarbose, delay the hydrolysis of carbohydrates by interfering with the digestive enzymes. This action decreases the glucose absorption and the postprandial glucose level. We have synthesized 25 tricyclic 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-ones hybrids and evaluated their α-glucosidase inhibitory activity. Compounds 6h and 6d have shown stronger activity than that of acarbose. Compound 6h exhibited the highest inhibition with an IC50 of 104.07 µM. Molecular modelling studies revealed that compound 6h inhibits α-glucosidase due to the formation of a stable ligand-α-glucosidase complex and extra hydrogen bond interactions, and directed in the binding site by Trp329.

25 tricyclic 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-ones hybrids have been synthesized and evaluated their α-glucosidase inhibitory activity. Compounds 6h have shown stronger activity than that of acarbose
Keywords: Pyrido-triazolopyrimidine, α-Glucosidase, Antidiabetic, Acarbose, Molecular docking
Introduction
The number of people with diabetes has increased worldwide. According to the global report in diabetes published by WHO in 2014, the number of the diabetic people has increased from 108 million in 1980 to 422 million. Risk factors for diabetes usually include older age, family history, race or ethnicity, obesity and physical inactivity. The prevalence of diabetes among people over 18 years old has increased from 4.7% in 1980 to 8.5% in 2014 [1]. Diabetes mellitus is a metabolic disease characterized by hyperglycaemia accompanied by carbohydrate, fat and protein metabolism disturbance. The above-mentioned characteristics of diabetes result from insufficient insulin production in Type I diabetes or ineffective action of insulin in Type II diabetes. The increase in blood sugar levels can lead to serious damage to nerves and blood vessels, which causes a major risk on the individual’s health and quality of life.
Several strategies are used for diabetes management and one of them is controlling postprandial hyperglycaemia. Among postprandial hypoglycaemic agents are α-glucosidase inhibitors. α-Glucosidase inhibitors delay the carbohydrate hydrolysis by inhibiting digestive enzymes, and thus reducing postprandial glucose levels. Moreover, α-glucosidase inhibitors have been found to have a range of biological activities as opposed to other antidiabetic drugs (Acarbose, Miglitol and Voglibose). For example, Celgosivir displays antiviral activity against hepatitis C & B viruses and 1-deoxynojirimycin exhibits anticancer activity (Fig. 1). This variety of biological activity makes α-glucosidase inhibitors a class of compounds with multiple therapeutic applications.
Fig. 1.

Important clinically used α-glucosidase inhibitors. The boxed structure is the newly synthesized and tested scaffold
Despite considerable advances in the development of new α-glucosidase inhibitors to treat diabetes, most of the produced compounds were sugar mimetic that requires tedious synthesis. Therefore, a number of compounds mimicking the monosaccharides or oligosaccharides were discovered. Among them, is the N-containing carbasugars such as validamine and valienamine, which have proven to be lead compounds for the development of clinically important agents such as acarbose (Fig. 1) [2]. It has been shown that the valienamine moiety in acarbose mimics the oxocarbenium ion-like transition state that binds to and inhibits α-glucosidase [3]. Therefore, acarbose is an effective inhibitor against glucoamylase, sucrase, dextranase and maltase that prevents the digestion of complex carbohydrates, which are subsequently delivered to the colon. However, bacteria in the colon will digest the complex carbohydrates causing gastrointestinal side effects such as flatulence and diarrhoea. Clinical studies have shown that gastrointestinal side effects are the major cause for reduced patient compliance to the diabetic treatment.
The control of postprandial hyperglycaemia by α-glucosidase inhibitors is clinically effective. Several studies have shown that the addition of acarbose to other oral antidiabetic agents is associated with significant improvements in life expectancy and quality of life, and provides excellent money value over the patient lifetime. However, this advantage of the clinically available α-glucosidase inhibitors is losing its value because of the developed gastrointestinal side effects. Therefore, there is a high demand for effective antidiabetic agents.
We are interested in exploring and developing α-glucosidase inhibitors in an attempt to overcome the side effects associated with sugar mimetic agents. According to our previous results, we decided to modify scaffolds well accepted in medicinal chemistry, such as pyridine, triazole, and pyrimidine. These scaffolds have been incorporated in a variety of compounds that displayed interesting pharmacological activities including antimicrobial [4–6], antiviral [7], anticancer [8], anti-inflammatory [9, 10], antioxidant [11] and anticonvulsant [12] activities. Moreover, pyrido[3,2-e][1,2,4]triazolo[1,5,-a]pyrimidin-5(4H)-ones have been reported as effective fungicidal and herbicidal agents [13, 14].
Herein, we designed two series of phenoxypyrido-triazolopyrimidinones and evaluated their α-glucosidase inhibitory activity. A significant inhibitory activity was observed for the newly synthesized compounds and IC50 values lower than that of acarbose were obtained for two compounds. Moreover, molecular docking studies against α-glucosidase were conducted. We could provide insights of a new lead scaffold for the development of new clinically effective antidiabetic drugs.
Experimental
Chemistry
Melting points were measured in open-glass capillaries using a STUART SMP 10 melting point apparatus and are uncorrected. NMR spectra were recorder in DMSO-d6 either on a Bruker AMX 500 or 700 spectrometer. Chemical shifts are reported relative to TMS as δ ppm values. Spectrometers operated at either 500 or 700 MHz for 1H NMR and 175 MHz for 13C NMR. Coupling constants (J values) are given in Hz. High resolution mass spectra (HREI-MS) were measured on a JEOL MStation JMS-700 system. A Perkin Elmer FT-IR Spectrum BX spectrometer was used to record the IR spectra (KBr, ѵ, cm−1). TLC on DC Mikrokarten polygram SIL G/UV254 (thickness: 0.25 mm from the Macherey–Nagel Firm Duren) was used to monitor the reactions and check the purity of compounds.
Procedure for the synthesis of 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (1)
2-Hydrazino-3-carboxylic pyridine (5.5 mmol) was added portion wise to a solution of diphenyl-N-cyanoimidocarbonate (5 mmol) in ethanol (20 mL) at room temperature. Afterwards, triethylamine (20 mmol) was added dropwise over a 10 min period. After the addition was complete, the reaction mixture was left to stir at room temperature overnight. The mixture was acidified with conc. HCl under ice cooling and then heated for 1–2 h at 80 °C. After cooling, the mixture was poured into ice/water, the resulting solid was collected by filtration, washed with water and dried. The product was obtained as a white amorphous powder (78%); mp: 252–254 °C; IR: 1703 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 13.27 (brs, 1H, NH), 8.82 (dd, J = 4.8, 1.7 Hz, 1H, H-8), 8.55 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.59 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.47 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.34 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.28 (t, J = 7.8 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 166.9 (C-2), 160.1 (C-5), 154.8 (C-3a), 154.5 (C-1′), 149.7 (C-9a), 146.7 (C-8), 138.4 (C-6), 130.3 (C-3′/5′), 125.6 (C-4′), 122.4 (C-5a), 120.3 (C-2′/6′), 113.7 (C-7); HRMS m/z (EI): [M]·+ calcd. for C14H9N5O2, 279.0756; found 279.0797.
General procedure for the synthesis of compounds 2a–l
At room temperature, potassium carbonate (0.6 mmol) was added portion wise over a period of 5 min to a stirred solution of 1 (0.5 mmol) in DMF (5 mL). After 10 min, the appropriate alkyl or heteroalkyl halide (1 mmol) was added dropwise and then the resulting mixture was left stirring at room temperature for 18 h. Afterwards, the mixture was poured into ice/water, the resulting solid was filtered, then washed with water and dried.
4-Ethyl-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2a)
White amorphous powder (73%); mp: 173–175 °C; IR: 1681 (C=O) cm−1; 1H NMR (500 MHz, DMSO-d6): δ 8.84 (dd, J = 4.8, 1.7 Hz, 1H, H-8), 8.61 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.61 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.49 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.37 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.30 (t, J = 7.8 Hz, 1H, H-4′), 4.16 (q, J = 7.2 Hz, 2H, H-1″), 1.31 (t, J = 7.2 Hz, 3H, H-2″); 13C NMR (175 MHz, DMSO-d6): δ 166.6 (C-2), 158.9 (C-5), 154.8 (C-3a), 154.4 (C-1′), 150.4 (C-9a), 145.9 (C-8), 138.7 (C-6), 130.4 (C-3′/5′), 125.7 (C-4′), 122.5 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 39.2 (C-1″), 12.8 (C-2″); HRMS m/z (EI): [M]·+ calcd. for C16H13N5O2, 307.1069; found 307.1098.
4-Allyl-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2b)
White amorphous powder (78%); mp: 170–172 °C; IR: 1686 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.85 (dd, J = 4.8, 1.7 Hz, 1H, H-8), 8.62 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.63 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.49 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.37 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.31 (t, J = 7.8 Hz, 1H, H-4′), 5.97 (m, 1H, H-2″), 5.30 (d, J = 17.2 Hz, 1H, H-3a’’), 5.20 (d, J = 10.4 Hz, 1H, H-3b’’), 4.73 (d, J = 4.9 Hz, 1H, H-1″); 13C NMR (175 MHz, DMSO-d6): δ 166.6 (C-2), 158.9 (C-5), 154.9 (C-3a), 154.4 (C-1′), 150.5 (C-9a), 146.0 (C-8), 138.8 (C-6), 131.5 (C-1″), 130.4 (C-3′/5′), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 118.0 (C-3″), 113.1 (C-7), 46.0 (C-1″); HRMS m/z (EI): [M]·+ calcd. for C17H13N5O2, 319.1069; found, 319.1099.
4-Benzyl-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2c)
White amorphous powder (70%); mp: 195–197 °C; IR: 1673 (C=O) cm−1; 1H NMR (500 MHz, DMSO-d6): δ 8.85 (dd, J = 4.8, 1.7 Hz, 1H, H-8), 8.62 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.62 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.49 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.46 (d, J = 7.8 Hz, 2H, H-2″/6″), 7.36 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.34 (t, J = 7.8 Hz, 2H, H-3″/5″), 7.29 (m, H-4′, 2H, 4″), 5.32 (s, 2H, H-7″); 13C NMR (175 MHz, DMSO-d6): δ 166.5 (C-2), 159.3 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.7 (C-9a), 146.1 (C-8), 138.8 (C-6), 135.9 (C-1″), 130.3 (C-3′/5′), 128.9 (C-3″/5″), 128.3 (C-2″/6″), 128.1 (C-4″), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 47.2 (C-7″); HRMS m/z (EI): [M]·+ calcd. for C21H15N5O2, 369.1226; found 369.1276.
4-(2-Methylbenzyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2d)
White amorphous powder (75%); mp: 160–162 °C; IR: 1670 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.87 (dd, J = 4.8, 1.7 Hz, 1H, H-8), 8.63 (dd, J = 7.8, 1.7 Hz, 1H, H-6), 7.64 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.48 (t, J = 7.9 Hz, 2H, H-3′/5′), 7.34 (d, J = 7.9 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.9 Hz, 1H, H-4′), 7.22 (d, J = 7.4 Hz, 1H, H-6″), 7.18 (m, 2H, H-3″/5″), 7.08 (t, J = 7.4 Hz, 1H, H-4″), 5.28 (s, 2H, H-7″), 2.42 (s, 3H, Ar–Me); 13C NMR (175 MHz, DMSO-d6): δ 166.5 (C-2), 159.4 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.8 (C-9a), 146.2 (C-8), 138.9 (C-6), 135.8 (C-1″), 133.8 (C-2″), 130.5 (C-3″), 130.3 (C-3′/5′), 127.7 (C-4″), 126.5 (C-5″), 126.4 (C-6″), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 45.1 (C-7″), 19.4 (Ar–Me); HRMS m/z (EI): [M]·+ calcd. for C22H17N5O2, 383.1382; found 383.1412.
4-(3-Methylbenzyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2e)
White amorphous powder (79%); mp 181–183 °C; IR: 1678 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.87 (d, J = 4.0 Hz, 1H, H-8), 8.64 (d, J = 7.6 Hz, 1H, H-6), 7.65 (dd, J = 7.8, 4.8 Hz, 1H, H-7), 7.51 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.38 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.32 (t, J = 7.8 Hz, 1H, H-4′), 7.28 (br s, 1H, H-2″), 7.25 (d, J = 7.4 Hz, 1H, H-6″), 7.23 (t, J = 7.4 Hz, 1H, H-5″), 7.11 (d, J = 7.4 Hz, 1H, H-4″), 5.29 (s, 2H, H-7″), 2.29 (s, 3H, Ar–Me); 13C NMR (175 MHz, DMSO-d6): δ 166.5 (C-2), 159.3 (C-5), 155.0 (C-3a), 154.4 (C-1′), 150.7 (C-9a), 146.1 (C-8), 138.9 (C-6), 138.1 (C-1″), 135.9 (C-3″), 130.3 (C-3′/5′), 128.8 (C-2″), 128.7 (C-5″), 128.0 (C-4″), 125.7 (C-4′), 125.4 (C-6″), 122.6 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 47.2 (C-7″), 21.5 (Ar–Me); HRMS m/z (EI): [M]·+ calcd. for C22H17N5O2, 383.1382; found 383.1412.
4-(3-Methoxybenzyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2f)
White amorphous powder (83%); mp: 189–191 °C; IR: 1682 (C=O) cm−1; 1H NMR (500 MHz, DMSO-d6): δ 8.85 (dd, J = 4.7, 1.5 Hz, 1H, H-8), 8.63 (dd, J = 7.8, 1.5 Hz, 1H, H-6), 7.62 (dd, J = 7.8, 4.7 Hz, 1H, H-7), 7.48 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.36 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.4 Hz, 1H, H-4′), 7.25 (t, J = 8 Hz, 1H, H-5″), 7.01 (m, 2H, H-2″/6″), 6.86 (dd, J = 8, 1.6 Hz, 1H, H-4″), 5.28 (s, 2H, H-7″), 3.72 (s, 3H, O–Me); 13C NMR (175 MHz, DMSO-d6): δ 166.5 (C-2), 159.8 (C-3″), 159.3 (C-5), 154.9 (C-3a), 154.3 (C-1′), 150.7 (C-9a), 146.1 (C-8), 138.8 (C-6), 137.5 (C-1″), 130.3 (C-3′/5′), 130.0 (C-5″), 125.7 (C-4′), 122.6 (C-5a), 120.4 (C-6″), 120.1 (C-2′/6′), 114.1 (C-4″), 113.3 (C-7), 113.1 (C-2″), 47.1 (C-7″), 55.5 (O–Me); HRMS m/z (EI): [M]·+ calcd. for C22H17N5O3, 399.1331; found 399.1374.
4-((5-Oxo-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-4(5H)-yl)methyl)benzo-nitrile (2g)
White amorphous powder (73%); mp: 149–151 °C; IR: 1688 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.86 (dd, J = 4.9, 1 Hz, 1H, H-8), 8.62 (dd, J = 7.8, 1.1 Hz, 1H, H-6), 7.82 (d, J = 8.1 Hz, 2H, H-3″/5″), 7.66 (d, J = 8.1 Hz, 2H, H-2″/6″), 7.63 (dd, J = 7.8, 4.9 Hz, 1H, H-7), 7.48 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.34 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.8 Hz, 1H, H-4′), 5.39 (s, 2H, H-7″); 13C NMR (175 MHz, DMSO-d6): δ 166.4 (C-2), 159.4 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.6 (C-9a), 146.2 (C-8), 141.6 (C-1″), 138.8 (C-6), 133.2 (C-3″/5″), 130.3 (C-3′/5′), 128.9 (C-2″/6″), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 119.2 (C ≡ N), 113.2 (C-7), 110.8 (C-4″), 46.9 (C-7″); HRMS m/z (EI): [M]·+ calcd. for C22H14N6O2, 394.1178; found 394.1211.
4-(4-Nitrobenzyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2h)
White amorphous powder (77%); mp: 150–152 °C; IR: 1672 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.87 (d, J = 4.9 Hz, 1H, H-8), 8.63 (d, J = 7.8, 1 Hz, 1H, H-6), 8.19 (d, J = 8.5 Hz, 2H, H-3″/5″), 7.74 (d, J = 8.5 Hz, 2H, H-2″/6″), 7.64 (dd, J = 7.8, 4.9 Hz, 1H, H-7), 7.48 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.34 (d, J = 8.2 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.3 Hz, 1H, H-4′), 5.45 (s, 2H, H-7″); 13C NMR (175 MHz, DMSO-d6): δ 166.4 (C-2), 159.4 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.6 (C-9a), 147.4 (C-4″), 146.2 (C-8), 143.7 (C-1″), 138.8 (C-6), 129.3 (C-2″/6″), 130.3 (C-3′/5′), 125.8 (C-4′), 123.9 (C-3″/5″), 122.6 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 46.8 (C-7″); HRMS m/z (EI): [M]·+ calcd. for C21H14N6O4, 414.1077; found 414.1110.
4-(4-Chlorobenzyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2i)
White amorphous powder (70%); mp: 144–146 °C; IR: 1677 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.85 (dd, J = 4.5, 1.1 Hz, 1H, H-8), 8.62 (d, J = 7.8 Hz, 1H, H-6), 7.62 (dd, J = 7.6, 4.8 Hz, 1H, H-7), 7.49 (m, H-3′/5′, 4H, 3″/5″), 7.39 (d, J = 8.3 Hz, 2H, H-2″/6″), 7.35 (d, J = 8.1 Hz, 2H, H-2′/6′), 7.30 (t, J = 7.4 Hz, 1H, H-4′), 5.30 (s, 2H, H-7″); 13C NMR (175 MHz, DMSO-d6): δ 166.5 (C-2), 159.3 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.6 (C-9a), 146.1 (C-8), 138.8 (C-6), 135.0 (C-1″), 132.7 (C-4″), 130.3 (C-3′/5′, 2″/6″), 128.8 (C-3″/5″), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 113.1 (C-7), 46.6 (C-7″); HRMS m/z (EI): [M]·+ calcd. for C21H14ClN5O2, 403.0836; found 403.0872.
4-(2-Morpholinoethyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2j)
White amorphous powder (50%); mp: 169–171 °C; IR 1680 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.85 (d, J = 4.6 Hz, 1H, H-8), 8.63 (d, J = 7.8 Hz, 1H, H-6), 7.63 (dd, J = 7.6, 4.8 Hz, 1H, H-7), 7.49 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.33 (d, J = 7.9 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.4 Hz, 1H, H-4′), 4.24 (t, J = 7 Hz, 2H, CH2-8″), 3.51 (br s, 4H, H-3″/5″), 2.69 (t, J = 7 Hz, 2H, CH2-7″), 2.47 (br s, 4H, H-2″/6″); 13C NMR (175 MHz, DMSO-d6): δ 166.9 (C-2), 159.1 (C-5), 155.0 (C-3a), 154.3 (C-1′), 150.6 (C-9a), 146.7 (C-8), 138.8 (C-6), 130.3 (C-3′/5′), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 113.7 (C-7), 66.6 (C-3″/5″), 55.1 (CH2-7″), 53.7 (C-2″/6″), 41.1 (CH2-8″); HRMS m/z (EI): [M]·+ calcd. for C20H20N6O3, 392.1597; found 392.1629.
2-Phenoxy-4-(2-(piperidin-1-yl)ethyl)pyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (2k)
White amorphous powder (45%); mp: 200–202 °C; IR: 1679 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.82 (d, J = 4.3 Hz, 1H, H-8), 8.60 (d, J = 7.8 Hz, 1H, H-6), 7.60 (dd, J = 7.8, 4.3 Hz, 1H, H-7), 7.37 (t, J = 8.3 Hz, 2H, H-3′/5′), 7.33 (d, J = 7.7 Hz, 2H, H-2′/6′), 7.27 (t, J = 7.4 Hz, 1H, H-4′), 4.27 (t, J = 7 Hz, 2H, CH2-8″), 2.79 (t, J = 7 Hz, 2H, CH2-7″), 2.57 (m, 4H, H-2″/6″), 1.48 (m, 4H, H-3″/5″), 1.36 (m, 2H, H-4″); 13C NMR (175 MHz, DMSO-d6): δ 166.6 (C-2), 159.1 (C-5), 154.9 (C-3a), 154.6 (C-1′), 150.5 (C-9a), 146.7 (C-8), 138.7 (C-6), 130.3 (C-3′/5′), 125.7 (C-4′), 122.6 (C-5a), 120.1 (C-2′/6′), 113.7 (C-7), 54.9 (CH2-7″), 54.3 (C-2″/6″), 40.3 (CH2-8″), 25.5 (C3″/5″), 23.9 (C-4″); HRMS m/z (EI): [M]·+ calcd. for C21H22N6O2, 390.1804; found 390.1838.
2-(3-(5-Oxo-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-4(5H)-yl)propyl)isoindo-line-1,3-dione (2l)
White amorphous powder (71%); mp: 155–157 °C; IR: 1670 (C=O) cm−1; 1H NMR (700 MHz, DMSO-d6): δ 8.82 (dd, J = 4.2, 1.1 Hz, 1H, H-8), 8.55 (dd, J = 7.8, 1.1 Hz, 1H, H-6), 7.82 (m, H-5″/6″, 4H, H4″/7″), 7.59 (dd, J = 7.8, 4.2 Hz, 1H, H-7), 7.47 (t, J = 7.8 Hz, 2H, H-3′/5′), 7.33 (d, J = 8.1 Hz, 2H, H-2′/6′), 7.28 (t, J = 7.5 Hz, 1H, H-4′), 4.17 (t, J = 7.1 Hz, 2H, CH2-8″), 3.71 (t, J = 7.1 Hz, 2H, CH2-10″), 2.16 (quint, J = 7.1 Hz, 2H, CH2-9″); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-1″/3″), 166.6 (C-2), 159.2 (C-5), 154.8 (C-3a), 154.3 (C-1′), 150.5 (C-9a), 145.9 (C-8), 138.6 (C-6), 134.9 (C-3a”/7a”), 132.0 (C-5″/6″), 130.3 (C-3′/5′), 125.7 (C-4′), 123.5 (C-4″/7″), 122.6 (C-5a), 120.0 (C-2′/6′), 113.0 (C-7), 41.1 (CH2-8″), 35.7 (CH2-10″), 26.1 (CH2-9″); HRMS m/z (EI): [M]·+ calcd. for C25H18N6O4, 466.1390; found 466.1421.
Procedure for the synthesis of 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine-5(4H)-thione (3)
A mixture of compound 1 (1 mmol) and phosphorus pentasulfide (1 mmol) was refluxed in absolute pyridine (10 mL) for 3 h. After cooling, the mixture was poured into ice/water, the resulting yellow solid was filtered, washed thoroughly with water and dried. Yield (90%); mp: 270–272 °C; IR: 1272 (C=S) cm−1; 1H NMR (500 MHz, DMSO-d6): δ 14.02 (br s, 1H, –NH), 8.92 (dd, J = 8.1, 1.7 Hz, 1H, H-6), 8.88 (dd, J = 4.6, 1.7 Hz, 1H, H-8), 7.63 (dd, J = 8.1, 4.6 Hz, 1H, H-7), 7.48 (td, J = 7.6, 2.3 Hz, 2H, H-3′/5′), 7.35 (d, J = 7.7 Hz, 2H, H-2′/6′), 7.29 (t, J = 7.4 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 186.0 (C-5), 167.3 (C-2), 155.6 (C-3a), 154.5 (C-1′), 148.6 (C-9a), 143.2 (C-8), 141.6 (C-6), 130.3 (C-3′/5′), 125.7 (C-4′), 124.7 (C-5a), 123.1 (C-7), 120.2 (C-2′/6′); HRMS m/z (EI): [M]·+ calcd. for C14H9N5OS, 295.0528; found 295.0563.
General procedure for the synthesis of 5-((3-methoxybenzyl)thio)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine (4)
At room temperature, potassium carbonate (1 mmol) was added portion wise over a period of 10 min to a stirred solution of 1 (0.5 mmol) in DMF (5 mL). After stirring for 10 min, 3-methoxybenzyl bromide (1.5 mmol) was added dropwise and the reaction mixture left to stir at room temperature for 20 h. The mixture was poured into ice/water, the resulting solid was filtered, washed with water and dried. Yield (68%); mp 149–151 °C; 1H NMR (500 MHz, DMSO-d6): δ 9.04 (d, J = 4.5 Hz, 1H, H-8), 8.68 (d, J = 7.9 Hz, 1H, H-6), 7.73 (dd, J = 7.9, 4.5 Hz, 1H, H-7), 7.49 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.38 (d, J = 7.7 Hz, 2H, H-2′/6′), 7.30 (t, J = 7.4 Hz, 1H, H-4′), 7.25 (t, J = 8 Hz, 1H, H-5″), 7.13 (br s, 1H, H-2″), 7.08 (d, J = 7.4 Hz, 1H, H-6″), 6.86 (d, J = 7.4 Hz, 1H, H-4″), 4.66 (s, 2H, H-7″), 3.74 (s, 3H, O–Me); 13C NMR (175 MHz, DMSO-d6): δ 168.5 (C-5), 167.8 (C-2), 159.8 (C-3″), 156.4 (C-9a), 154.5 (C-3a), 153.4 (C-1′), 143.8 (C-8), 138.2 (C-6), 135.8 (C-1″), 130.3 (C-3′/5′), 130.2 (C-5″), 125.7 (C-4′), 122.7 (C-5a), 121.9 (C-7), 120.4 (C-2′/6′), 120.1 (C-6″), 115.4 (C-4″), 113.6 (C-2″), 55.5 (O–Me), 34.1 (C-7″); HRMS m/z (EI): [M]·+ calcd. for C22H17N5O2S, 415.1103; found 415.1136.
Procedure for the synthesis of 5-chloro-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine (5)
Compound 1 (2 mmol) was refluxed with phosphorus oxychloride (2 mL) in benzene (14 mL) for 3 h. After evaporation of the solvent, the residue was treated with a saturated aqueous solution of potassium carbonate. The resulting solid was filtered, washed thoroughly with water and dried. The product was obtained as a white amorphous powder (70%); mp: 190–192 °C; 1H NMR (500 MHz, DMSO-d6): δ 9.16 (dd, J = 4.6, 1.6 Hz, 1H, H-8), 8.84 (dd, J = 8.3, 1.6 Hz, 1H, H-6), 7.89 (dd, J = 8.3, 4.6 Hz, 1H, H-7), 7.50 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.39 (d, J = 7.7 Hz, 2H, H-2′/6′), 7.33 (t, J = 7.4 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 168.8 (C-2), 157.4 (C-5), 156.7 (C-9a), 154.3 (C-3a), 152.5 (C-1′), 145.4 (C-8), 138.5 (C-6), 130.4 (C-3′/5′), 125.9 (C-4′), 123.8 (C-5a), 120.4 (C-2′/6′), 114.3 (C-7); HRMS m/z (EI): [M]·+ calcd. for C14H8ClN5O, 297.0417; found 297.0451.
Procedure for the synthesis of compounds 6a–d
Compound 5 (1 mmol) was refluxed with the appropriate amine (5 mmol) in pyridine (10 mL) for 5–7 h. After cooling, the mixture was poured into ice/water, the resulting solid was filtered, washed with water and dried.
2-Phenoxy-N-phenylpyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-amine (6a)
Brown amorphous powder (61%); mp: 251–253 °C; 1H NMR (500 MHz, DMSO-d6): δ 10.15 (br s, 1H, –NH), 9.14 (d, J = 7.8 Hz, 1H, H-6), 8.95 (d, J = 4.0 Hz, 1H, H-8), 7.86 (d, J = 7.8 Hz, 2H, H-2″/6″), 7.74 (dd, J = 8.1, 4.6 Hz, 1H, H-7), 7.46 (m, H-3′/5′, 4H, 3″/5″), 7.34 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.27 (t, J = 7.4 Hz, 1H, H-4′), 7.22 (t, J = 7.4 Hz, 1H, H-4″); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-2), 155.2 (C-9a), 154.8 (C-5, 3a), 154.7 (C-1′), 145.6 (C-8), 138.9 (C-6), 135.4 (C-1″), 130.2 (C-3′/5′), 129.1 (C-3″/5″), 125.4 (C-4′), 125.0 (C-4″), 123.15 (C-2″/6″), 121.7 (C-7), 120.4 (C-2′/6′), 108.3 (C-5a); HRMS m/z (EI): [M]·+ calcd. for C20H14N6O, 354.1229; found 354.1264.
2-Phenoxy-N-(p-tolyl)pyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-amine (6b)
Pale green amorphous powder (60%); mp: 162–164 °C; 1H NMR (700 MHz, DMSO-d6): δ 10.10 (br s, 1H, –NH), 9.12 (d, J = 8.2 Hz, 1H, H-6), 8.94 (d, J = 4.1 Hz, 1H, H-8), 7.73 (m, 3H, H-7, H-2″/6″), 7.47 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.34 (d, J = 8.3 Hz, 2H, H-2′/6′), 7.27 (t, J = 7.4 Hz, 1H, H-4′), 7.24 (d, J = 8.1 Hz, 2H, H-3″/5″), 2.33 (s, 3H, Ar–Me); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-2), 155.2 (C-9a), 154.8 (C-5), 154.7 (C-3a), 154.6 (C-1′), 145.6 (C-8), 136.3 (C-6), 135.3 (C-1″), 134.2 (C-4″), 130.2 (C-3′/5′), 129.5 (C-3″/5″), 125.3 (C-4′), 123.2 (C-2″/6″), 121.6 (C-7), 120.4 (C-2′/6′), 108.3 (C-5a), 21.1 (Ar–Me); HRMS m/z (EI): [M]·+ calcd. for C21H16N6O, 368.1386; found 368.1416.
N-(4-Methoxyphenyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-amine (6c)
Green amorphous powder (64%); mp: 174–176 °C; 1H NMR (700 MHz, DMSO-d6): δ 10.10 (br s, 1H, –NH), 9.09 (d, J = 8.2 Hz, 1H, H-6), 8.94 (d, J = 4.6 Hz, 1H, H-8), 7.72 (m, 3H, H-7, H-2″/6″), 7.46 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.33 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.27 (t, J = 7.4 Hz, 1H, H-4′), 7.02 (d, J = 7.8 Hz, 2H, H-3″/5″), 3.79 (s, 3H, O–Me); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-2), 156.8 (C-4″), 155.3 (C-9a), 154.9 (C-5), 154.8 (C-3a), 154.7 (C-1′), 145.6 (C-8), 135.2 (C-6), 131.7 (C-1″), 130.2 (C-3′/5′), 125.3 (C-4′), 125.1 (C-2″/6″), 121.6 (C-7), 120.4 (C-2′/6′), 114.3 (C-3″/5″), 108.3 (C-5a), 55.7 (O–Me); HRMS m/z (EI): [M]·+ calcd. for C21H16N6O2, 384.1335; found 384.1369.
N-(4-Ethoxyphenyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-amine (6d)
Green amorphous powder (68%); mp: 154–156 °C; 1H NMR (700 MHz, DMSO-d6): δ 10.18 (br s, –1H, NH), 9.08 (d, J = 7.4 Hz, 1H, H-6), 8.93 (d, J = 4.1 Hz, 1H, H-8), 7.71 (m, 3H, H-7, H-2″/6″), 7.46 (t, J = 7.7 Hz, 2H, H-3′/5′), 7.33 (d, J = 8.2 Hz, 2H, H-2′/6′), 7.27 (t, J = 7.3 Hz, 1H, H-4′), 6.99 (d, J = 7.8 Hz, 2H, H-3″/5″), 4.04 (q, J = 7 Hz, 2H, –CH2–CH3), 1.35 (t, J = 7 Hz, 3H, –CH2–CH3); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-2), 156.1 (C-4″), 155.2 (C-9a), 154.9 (C-5), 154.7 (C-3a/1′), 145.5 (C-8), 135.2 (C-6), 131.6 (C-1″), 130.2 (C-3′/5′), 125.3 (C-4′), 125.0 (C-2″/6″), 121.6 (C-7), 120.4 (C-2′/6′), 114.7 (C-3″/5″), 108.2 (C-5a), 63.6 (–CH2–CH3), 15.2 (–CH2–CH3); HRMS m/z (EI): [M]·+ calcd. for C22H18N6O2, 398.1491; found 398.1524.
Procedure for the synthesis of N-(2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-yl)hydroxylamine (6e)
Compound 5 (1 mmol) was refluxed with hydroxylamine hydrochloride (3 mmol) in pyridine (10 mL) for 5 h. After cooling, the mixture was poured into ice/water, the resulting solid was collected, washed with water and dried. White amorphous powder (63%); mp: 249–251 °C; 1H NMR (500 MHz, DMSO-d6): δ 11.55 (br s, 1H, –OH), 10.80 (br s, 1H, –NH), 9.08 (dd, J = 4.4, 1.1 Hz, 1H, H-8), 8.27 (dd, J = 7.8, 1.1 Hz, 1H, H-6), 7.45 (t, J = 8.2 Hz, 2H, H-3′/5′), 7.35 (dd, J = 7.8, 4.4 Hz, 1H, H-7), 7.30 (d, J = 7.9 Hz, 2H, H-2′/6′), 7.25 (t, J = 7.4 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 166.7 (C-2), 154.6 (C-9a/5), 151.1 (C-3a), 150.2 (C-1′), 143.9 (C-8), 139.8 (C-6), 130.2 (C-3′/5′), 125.4 (C-4′), 122.1 (C-7), 120.2 (C-2′/6′), 112.6 (C-5a); HRMS m/z (EI): [M]·+ calcd. for C14H10N6O2, 294.0865; found 294.0899.
Procedure for the synthesis of 5-hydrazinyl-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine (6f)
Compound 5 (2 mmol) was refluxed with hydrazine hydrate (20 mmol) in ethanol (15 mL) for 3 h. After cooling, the resulting solid was filtered, washed with water and dried. The product was obtained as a pale brown solid (74%); mp: 241–243 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.82 (dd, J = 4.4, 1.1 Hz, 1H, H-8), 8.74 (d, J = 8.0 Hz, 1H, H-6), 7.58 (dd, J = 8.2, 4.4 Hz, 1H, H-7), 7.45 (t, J = 8.3 Hz, 2H, H-3′/5′), 7.33 (d, J = 7.8 Hz, 2H, H-2′/6′), 7.26 (t, J = 7.40 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 168.2 (C-2), 155.7 (C-9a), 154.7 (C-3a/5), 154.3 (C-1′), 145.1 (C-8), 134.1 (C-6), 130.1 (C-3′/5′), 125.2 (C-4′), 121.4 (C-7), 120.2 (C-2′/6′), 106.7 (C-5a); HRMS m/z (EI): [M]·+ calcd. for C14H11N7O, 293.1025; found 293.1060.
Procedure for the synthesis of compounds 6g and 6h
A mixture of 6f (1 mmol) and 2-hydroxy-5-methoxy benzaldehyde (1 mmol) or 2-methoxy benzaldehyde (1 mmol) was refluxed in ethanol (10 mL) for 3–5 h. Then, the solvent was removed under reduced pressure and the resulting solid was filtered, washed with water and dried.
(E)-4-Methoxy-2-((2-(2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-yl)hydrazono)methyl)phenol (6g)
Yellow amorphous powder (77%); mp: 220–222 °C; 1H NMR (700 MHz, DMSO-d6): δ 12.51 (br s, 1H, –NH), 10.61 (br s, 1H, –OH), 8.96 (br s, H-6, 2H, H-7″), 8.74 (d, J = 4.5 Hz, 1H, H-8), 7.75 (dd, J = 7.9, 4.4, 1H, H-7), 7.48 (t, J = 8.3 Hz, 2H, H-3′/5′), 7.36 (d, J = 7.5 Hz, 2H, H-2′/6′), 7.28 (br s, 2H, H-4′, 6″), 7.02 (d, J = 8.3 Hz, 1H, H-4″), 6.93 (d, J = 8.3 Hz, 1H, H-3″), 3.75 (s, 3H, O–Me); 13C NMR (175 MHz, DMSO-d6): δ 168.4 (C-2), 162.5 (C-5), 154.9 (C-9a), 154.8 (C-3a), 154.7 (C-1′), 153.2 (C-5″), 152.7 (C-2″), 152.6 (C-7″), 145.9 (C-8), 135.9 (C-6), 130.2 (C-3′/5′), 125.4 (C-4′), 121.5 (C-7), 120.9 (C-4″), 120.3 (C-2′/6′), 118.8 (C-1″), 117.9 (C-6″), 113.4 (C-3″), 107.0 (C-5a), 55.9 (O–Me); HRMS m/z (EI): [M]·+ calcd. for C22H17N7O3, 427.1393; found 427.1426.
(E)-5-(2-(2-Methoxybenzylidene)hydrazinyl)-2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine (6h)
Yellow amorphous powder (72%); mp: 111–113 °C; 1H NMR (700 MHz, DMSO-d6): δ 12.04 (br s, 1H, –NH), 8.94 (br s, H-6, 2H, H-7″), 8.98 (d, J = 4.5 Hz, 1H, H-8), 7.73 (dd, J = 7.9, 4.4, 1H, H-7), 7.53 (t, J = 7.5 Hz, 1H, H-4″), 7.46 (m, 3H, H-3′/5′, 6″), 7.36 (d, J = 7.9 Hz, 2H, H-2′/6′), 7.28 (t, J = 7.4 Hz, 1H, H-4′), 7.15 (d, J = 8.3 Hz, 1H, H-3″), 7.04 (t, J = 7.5 Hz, 1H, H-5″), 3.89 (s, 3H, O–Me); 13C NMR (175 MHz, DMSO-d6): δ 168.5 (C-2), 159.2 (C-5), 157.0 (C-9a, 2″), 154.8 (C-3a), 154.7 (C-1′), 152.1 (C-7″), 145.6 (C-8), 135.9 (C-6), 133.5 (C-4″), 132.3 (C-6″), 130.2 (C-3′/5′), 126.9 (C-5″), 125.4 (C-4′), 121.6 (C-7), 121.2 (C-1″), 120.3 (C-2′/6′), 112.5 (C-3″), 106.9 (C-5a), 56.3 (O–Me); HRMS m/z (EI): [M]·+ calcd. for C22H17N7O2, 411.1444; found 411.1477.
Procedure for the synthesis of N’-(2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5-yl)isonicotinohydrazide (6i)
A mixture of 5 (0.5 mmol) and isoniazid (1.1 mmol) was refluxed in toluene (10 mL) for 4 h. After cooling, the resulting solid was collected by filtration, washed and dried. The product was obtained as a pale green amorphous powder (74%); mp: 163–165 °C; 1H NMR (500 MHz, DMSO-d6): δ 11.30 (br s, 1H, –NH), 9.00 (d, J = 4.3, 2H, H-3″/5″), 8.83 (d, J = 4.5 Hz, 1H, H-8), 8.55 (d, J = 7.8 Hz, 1H, H-6), 8.23 (d, J = 4.3 Hz, 2H, H-2″/6″), 7.89 (dd, J = 8.0, 4.5 Hz, 1H, H-7), 7.51 (t, J = 8.1 Hz, 2H, H-3′/5′), 7.39 (d, J = 8.1 Hz, 2H, H-2′/6′), 7.25 (t, J = 7.40 Hz, 1H, H-4′); 13C NMR (175 MHz, DMSO-d6): δ 168.7 (C-2), 166.9 (C-7″), 155.5 (C-9a), 154.7 (C-3a) 154.6 (C-5), 154.3 (C-1′), 149.7 (C-3″/5″), 145.4 (C-8), 138.5 (C-6), 137.8 (C-1″), 130.3 (C-3′/5′), 125.4 (C-4′), 122.4 (C-3″/5″), 122.0 (C-7), 120.2 (C-2′/6′), 113.7 (C-5a); HRMS m/z (EI): [M]·+ calcd. for C20H14N8O2, 398.1240; found 398.1273.
α-Glucosidase inhibitory assay
Preparation of reagents
Phosphate-buffered saline pH 6.5 was prepared at 20 °C by dissolving sodium phosphate monobasic (1.4 g), sodium chloride (8 g), potassium chloride (0.2 g) and potassium dihydrogen phosphate (0.2 g) in deionized water (1 L). The solution was adjusted to pH 6.5 at 20 °C with 1 M sodium hydroxide (NaOH) or hydrochloric acid (HCl). A 20-mM solution of para-nitrophenyl-α-D-glucopyranoside (p-NPG substrate) was prepared by dissolving 6 mg of p-NPG in 10 mL of buffer solution. The α-glucosidase enzyme solution was prepared by dissolving α-glucosidase enzyme type 1 from Baker’s yeast (1 mg) in cold phosphate-buffered saline (1000 µL), pH 6.5; 50 µL of this solution was mixed into 12 mL of cold phosphate-buffered saline to give a concentration of 0.125 unit/mL. For the screening of samples, 200 µg/mL concentration was prepared in 100% dimethyl sulfoxide (DMSO). Then, two-fold serial dilutions with 5% DMSO were prepared in a 96-well microplate to give final concentrations of 100, 50, 25, 12.5, 6.25, 3.13 and 1.56 µg/mL. Acarbose was used as positive control and prepared at a concentration of 200 µg/mL in 100% DMSO. Then, two-fold serial dilutions with 5% DMSO were prepared in a 96-well microplate to give final concentrations of 100, 50, 25, 12.5, 6.25, 3.13 and 1.56 µg/mL.
Method
A volume of 10 µL of 5% DMSO was added to the 96 wells of the microplate. Then, 10 µL of the stock sample was added to the first well and mixed. Subsequently, 10 µL of the sample from the first well was taken and added to the second well and this procedure was repeated for the two-fold serial dilutions. Next, 20 µL of the α-glucosidase enzyme, 40 µL of phosphate buffered saline at pH 6.5 and 20 µL of deionized water were added to each well in the 96-well microtiter plate and mixed. The mixture was pre-incubated at 37 °C for 10 min. Then, 10 µL of 20 mM p-NPG solution was added to the mixture and the absorbance at 0 min. was measured at 405 nm. The reaction was incubated for 30 min. at 37 °C and the absorbance was then measured. For the negative control, the sample was replaced with 5% of DMSO and acarbose was used as positive control. Experiments were performed in triplicate. The inhibition activity of α-glucosidase was determined based on the described method by Ahmad et al. [15, 16]. The percent inhibition (%) of α-glucosidase inhibitory activity was calculated using the equation:
Statistical analysis
IC50 values are the mean values of three experiments represented as SEM (n = 3). IC50 values are significantly different (P ≤ 0.05). Statistical analysis using one-way ANOVA was applied to the results. Nonlinear regression analysis using a four-parameter logistic curve was performed to calculate the IC50 values. The statistical software used were Microsoft Excel 2010 and Sigma Plot (version 14.0 Notebook, Systat Software Inc.).
Molecular docking
The Autodock software package was used to investigate the α-glucosidase inhibitory activity of the pyrido-triazolopyrimidines through molecular docking analysis [17]. The original docked protein were downloaded from the RCSB data bank web site (PDB file with code 3W37) [18]. The detailed steps for the molecular docking studies were reported by Abuelizz et al. [12].
Results and discussion
Synthesis of fused pyrimidine derivatives
Figure 2 illustrates the synthetic route towards fused pyrimidine derivatives. The starting material (A) was prepared according to a reported procedure [19]. The incorporation of a hydrazine moiety to N-cyanoimidocarbonates to generate triazoles has been successfully achieved and is well documented [20–22]. Thus, 1,2,4-triazole intermediate B was obtained when an ethanolic solution of diphenyl-N-cyanoimidocarbonate was allowed to react with A in a basic medium (triethylamine) at room temperature. Upon the treatment of B with conc. HCl, initially at room temperature followed by heating, afforded novel 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-one (1) in 78% yield. The chemical structure of compound 1 was established on the basis of NMR, IR and mass spectra. The IR spectrum of 1 was characterized by a band at 1703 cm−1 due to a strong stretching vibration of a C=O group.
Fig. 2.

Synthetic routes towards pyrimidine derivatives (1–6). i: Et3N, EtOH, overnight stirring; ii: conc. HCl, 80 °C; iii: DMF, K2CO3, alkyl or heteroalkyl halides; iv: P2S5, pyridine; v: DMF, K2CO3, 3-methoxybenzylbromide 90 °C; vi: POCl3, benzene; vii: pyridine, aromatic amines; pyridine, hydroxylamine HCl; toluene, isoniazid; EtOH, hydrazine hydrate; EtOH, glacial acetic acid, aldehydes
Under suitable conditions, the reaction of compound 1 with alkyl or heteroalkyl halides gave the expected N-alkylated pyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-ones (2a–l) in 45–83% yields (Fig. 2). The IR and NMR data confirmed the formation of N-alkylated products 2a–l. Compounds 2a–l were obtained as coloured amorphous powders and their IR spectra displayed absorption bands at 1670 − 1688 cm−1 due to the C=O group. Boiling equimolar amounts of target 1 and phosphorous pentasulfide in absolute pyridine furnished the desired 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-thione (3) as a yellow amorphous powder in excellent yield (90%). The IR and 13C NMR spectra of compound 3 display a weak absorption band (1272 cm−1) and a peak resonance (186.0 ppm), respectively, due to the C=S group. The conversion of 3 into 4 was carried out in basic medium using benzyl bromide. The transformation of 1 into 5 was successfully achieved employing phosphorous oxychloride as chlorinating agent in boiling benzene followed by treatment with a saturated basic solution. The formation of 5 was followed by NMR and IR spectroscopy; the latter confirmed the completely disappearance of the C=O band characteristic in compound 1 at 1703 cm−1. The conversion of the lactam group in 1 into an imidoyl chloride moiety (5) was expected to provide a valuable intermediate for further displacement reactions with several nucleophiles. Reaction of 5 with hydroxylamine HCl in a molar ratio of 1:3 in boiling pyridine provided the corresponding product 6e in 63% yield (Fig. 2), while treatment of target 5 with several aromatic amines in pyridine in a molar ratio of 1:5 afforded the corresponding compounds 6a–d in 60–68% yields. The chlorine was replaced in 5 by isoniazid in boiling toluene to give amidrazone 6i in good yield (74%). Hydrazinolysis of 5 in boiling ethanol produced the corresponding compound 6f, which upon treatment with equimolar amounts of aldehydes resulted in the formation of hydrazone derivatives 6g and 6h in good yields (Fig. 2).
The 1H-NMR spectra of targets 1–6 were characterized by two consistent spin coupling systems, each consisting of three types of protons, indicative of the main 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine structure. The first system consists of a triplet (H-3′/5′, 2H), doublet (H-2′/6′, 2H) and triplet (H-4′, 1H) in the range δ 7.50–7.25 ppm, corresponding to the five protons of the 2-phenoxy moiety. The second one corresponds to the three protons of the pyridine-ring (H-6, H-7 and H-8), mostly in the form of two doublets (H-8 & H-6) and a double of doublets (H-7) in the range of δ 8.9–7.5 ppm (see the experimental data). This main structure of phenoxy-pyrido-triazolopyrimidine was also clearly interpretable from its 12 characteristic 13C-resonances, including five resonances of the fused pyrido ring, four of the phenoxy moiety along with the characteristic three signals of C-2, C-3a and C-5. The successful synthesis of the different groups of products (1; 2a–l; 3; 4; 5 and 6a–i) was separately supported by the characteristic splitting patterns, δ and J values of the newly added moieties (alkyl or aryl) in the corresponding 1H and 13C NMR spectra. In addition, the change of structural features among all groups was mainly validated in the form of different δ values for the C-5 due to its structural change from C=O (1 & 2a–l) to C=S (3), C-S-R (4), C–Cl (5) or C–NHR (6a–i) (see the experimental data).
Biological evaluation
α-Glucosidase inhibitory assay
To explore the potential biological activity of the synthesized pyrimidine fused derivatives (1–6), in vitro screening of α-glucosidase inhibition was performed. The enzyme was treated with equal concentrations (100 µg/mL) of each compound. The test was repeated three times for each compound and the average percent inhibition was obtained. The inhibitory activity of all the tested compounds was between 30% and 63%. The eight compounds (out of the 25 tested) that exhibited more than 50% of the average percent inhibition were further evaluated and their IC50 values compared to that of acarbose. Compounds 3, 6c and 6d showed average percent inhibition of more than 60%. On the other hand, compounds 2a, 2d, 6a, 6b and 6h showed average percent inhibition of more than 50% but less than 60%.
In this study, we demonstrated that the introduction of a branch to the pyrimidine scaffold through a nitrogen bridge results in a series of synthetic compounds with significant inhibitory activity against α-glucosidase. The open site available for N-substitution at the fused pyrido-triazolo-pyrimidine rings is on the nitrogen at position 4. However, these derivatives did not exhibit any inhibitory activity except for compounds 2a and 2d. This suggests that the derivatization site has no significant effect on the inhibitory activity. The average percent inhibitions of 2a and 2d were 50.36% and 57.48%, respectively. The replacement of the carbonyl group of the pyrimidinone with amino-bridged substitutes resulted in the production of compounds 6a–6i. Compounds 6a–d and 6h exhibited average percent inhibition of more than 50% (Fig. 3). The comparison between the average percent inhibition of compounds 6a–6i and 2a–2l suggests that the amino-bridge substitution of the pyrimidine ring at position 4 is responsible for the strong inhibitory activity.
Fig. 3.

α-Glucosidase inhibitory assay of the synthesized fused pyrimidine derivatives (1–6). The IC50 was calculated for compounds that exhibited average percent inhibition of more than 50%
IC50 values of the active compounds
The pharmaceutical parameters of promising inhibitors, including the corresponding IC50 values, were calculated. Acarbose, a commonly used antidiabetic drug due to its inhibitory activity against α-glucosidase, was used as positive control. Compound 3 at a concentration of 100 µg/mL exhibited good activity against α-glucosidase enzyme (60.38%). Instead, products derivatized at position 4 has displayed weak inhibitory activity. Only two N-alkylated derivatives (ethyl and 2-methylbenzyl) showed more than 50% average percent inhibition (50.36% and 57.48%, respectively). The IC50 values of these compounds followed a similar trend; the IC50 of compound 3 was 167.82 µM and those of compound 2a and 2d were 304.13 µM and 173.83 µM, respectively (Fig. 4).
Fig. 4.
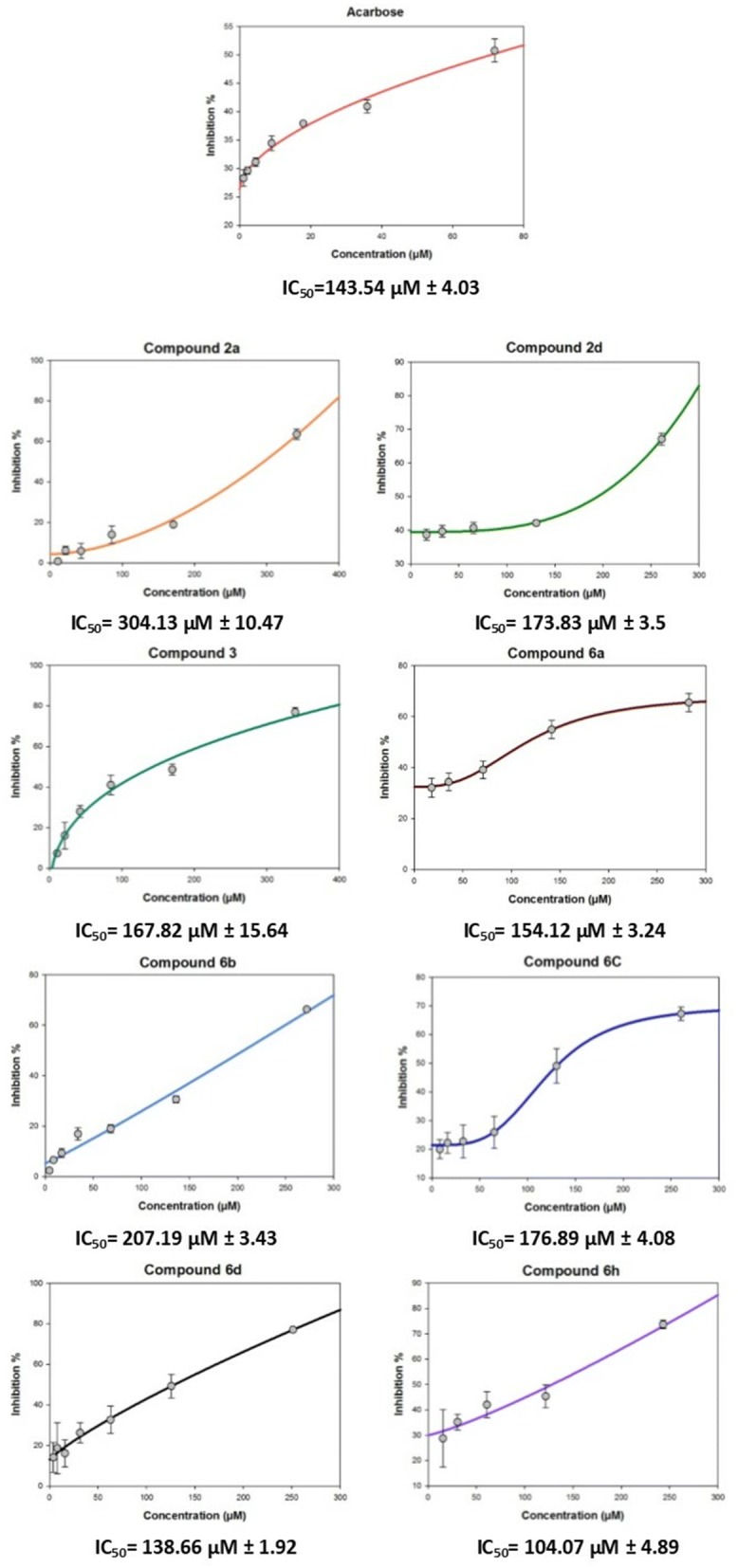
IC50 values of 2a, 2d, 3, 6a, 6b, 6c, 6d and 6h with average percent inhibition of more than 50%. More than five concentrations were evaluated and the inhibitory activity compared to that of acarbose, IC50 values ± SEM (P ≤ 0.05)
On the other hand, the replacement of the chloro group in 5 by different nucleophiles provided derivatives with similar or lower IC50 values than that of acarbose. This is exemplified by compounds 6a, 6b, 6c, 6d and 6h. The rank order of potency according to the IC50 values, including acarbose is 6h ˃ 6d ˃ acarbose ˃ 6a ˃ 6c ˃ 6b. The 2-methoxy-benzyl-methanimine (6h; IC50 = 104.07 µM) and 4-ethoxy-aniline (6d; IC50 = 138.66 µM) substituted derivatives exhibit the best activity when compared to acarbose (IC50 = 143.54 µM). The IC50 values of the amino substituted derivatives ranked as aniline ˃ 4-methoxy-aniline ˃ 4-methyl-aniline. While the IC50 value was slightly reduced by the parent aniline compound 6a (IC50 = 154.12 µM), the 4-methyl-aniline and 4-methoxy-aniline derivatives showed IC50 values of 207.19 and 176.89 µM, respectively.
Molecular docking
To understand the observed α-glucosidase inhibitory activities of the synthesized compounds, molecular docking calculations were performed to study the binding modes of the target enzyme and docked synthesized derivatives. Therefore, the number of established intermolecular hydrogen bonds, binding energies of stable ligand-α-glucosidase complexes and number of closest amino acid residues surrounding the binding site of the α-glucosidase enzyme were determined for the synthesized compounds (Table 1 and Fig. 5).
Table 1.
Inhibitory activities and binding energies of docked 2-phenoxypyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine derivatives and acarbose into the active site of α-glucosidase
| Name of synthesized derivatives | Free binding energy (kcal/mol) | Number of hydrogen bonds | Number of closest residues |
|---|---|---|---|
| 2a | − 8.12 | 1 | 10 |
| 3 | − 9.17 | 3 | 6 |
| 6a | − 9.76 | 1 | 7 |
| 6b | − 9.42 | 3 | 9 |
| 6d | − 9.64 | 3 | 8 |
| 6h | − 10.55 | 3 | 7 |
| Acarbose | − 10.56 | 10 | 10 |
Fig. 5.
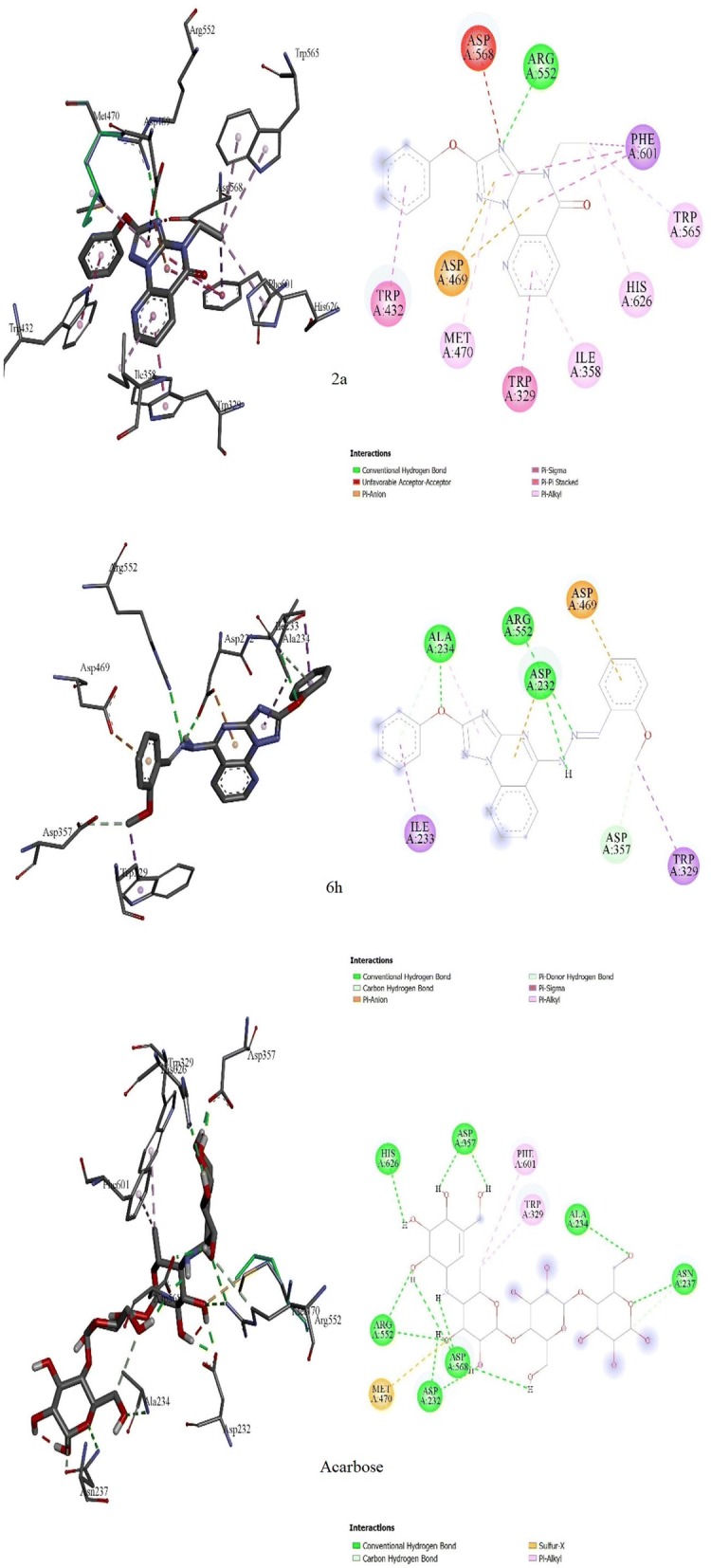
Molecular docking comparison of acarbose with compounds 2a and 6h. 3D binding conformation (right) and 2D binding conformation (left) showing the closest interactions between the active site residues of α-glucosidase and the most active (6h), least active (2a) synthesized derivatives and acarbose
All the synthesized derivatives formed a complex with the target enzyme. The relative standard deviation of the binding energy of the formed complex was less than 2.34 kcal/mol. From Table 1 and Fig. 5, it can be assumed that the maximal inhibition activity of compound 6h is mainly attributable to its binding energy and the stability of the ligand-α-glucosidase complex. Thus, the binding energy of docked 6h was found to be the lowest (− 10.55 kcal/mol) among the energies of the docked derivatives (Table 1 and Fig. 5). Compounds 6a and 6b differ from compound 6h only at the substitution at the amino group (phenyl-, 4-Me-phenyl and N-(2-methoxy benzyl)methanimine, respectively; Fig. 2). The inhibitory activity of compound 6a was higher than that of 6b, and this is may be ascribed to the higher stability of the 6a-α-glucosidase complex (binding energy = − 9.76 kcal/mol) than the 6b-α-glucosidase complex (binding energy = − 9.42 kcal/mol). The number of established hydrogen bonds can be invoked by the reference drug acarbose. The inhibition activity of acarbose correlates with the number hydroxyl groups and the number of the established hydrogen bonds between the ligand and the active site of the α-glucosidase enzyme (Fig. 5). Acarbose can establish 10 hydrogen bonds with Asp232, Ala234, Asn237, Asp357, Arg552, Asp568 and His626. Moreover, two hydrophobic interactions are observed between the methyl group of the deoxyglucosamine moiety and residues Phe601 and Trp329. Although compound 2a can interact with 10 residues at the active site, it can only form one hydrogen bond between the nitrogen group on the triazole ring and Arg552. However, this binding is not stable and provides a free binding energy of − 8.12 kcal/mol. Instead, three residues appear to interact with compound 6h, Ala234, Arg552 and Asp469, to which 6h can establish hydrogen bonds. These three hydrogen bonds enhance the ligand-α-glucosidase complex stability and reduce the free binding energy of compound 6h to − 10.55 kcal/mol. Trp329 can also form hydrophobic bonds with compounds 2a and 6h as in the case of acarbose, which is responsible for the binding conformation of the compounds in the active site.
Conclusion
We have prepared twenty-five compounds containing fused pyrimidine, pyrido and triazole rings. The synthesized compounds were characterized and subjected to α-glycosidase inhibitory assay. Six promising compounds that showed more than 50% of the average percent inhibition were subjected to further assays to determine their IC50. Acarbose was used as reference drug to compare with the synthesized compounds. Compounds 6d and 6h showed a significant inhibitory activity against α-glycosidase with lower IC50 values than that of acarbose. Molecular docking studies were performed and showed that the number of established intermolecular hydrogen bonds, the binding energies of the stable ligand-α-glucosidase complexes and the number of closest amino acid residues surrounding the binding site are responsible for the inhibitory activity of compound 6h, which is higher than that of acarbose. Moreover, Trp329 plays a significant role in directing the ligand in the binding site of the α-glucosidase enzyme. This study provides a new structural insight for the development of novel α-glucosidase inhibitors, which differ from the commonly employed carbasugars, and which can eventually be transformed into new clinical drugs for the treatment of type II diabetes.
Authors’ contributions
RA and HAA designed the work, the protocol of the study, synthesized-analyzed of compounds and wrote the manuscript. MM interpreted the NMR data and wrote the manuscript. RAh and NNII performed the biological studies and wrote their experimental data. AE performed and the molecular docking study. All the authors revised the whole manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No. RG-1439-011.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group No. RG-1439-011.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hatem A. Abuelizz, Email: Habuelizz@ksu.edu.sa
Nor Azman N. I. Iwana, Email: atywana.ii@gmail.com
Rohaya Ahmad, Email: royhaya2020@yahoo.com.
El-Hassane Anouar, Email: anouarelhassane@yahoo.fr.
Mohamed Marzouk, Email: msmarzouk@yahoo.co.uk.
Rashad Al-Salahi, Email: ralsalahi@ksu.edu.sa.
References
- 1.WHO. Global report on diabetes. Diabetes world health day; 2016
- 2.Asamizu S, Yang J, Almabruk KH, Mahmud T. Pseudoglycosyltransferase catalyzes nonglycosidic C-N coupling in validamycin a biosynthesis. J Am Chem Soc. 2011;133(31):12124–12135. doi: 10.1021/ja203574u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abuelizz HA, Mahmud T. Distinct substrate specificity and catalytic activity of the pseudoglycosyltransferase VldE. Chem Biol. 2015;22(6):724–733. doi: 10.1016/j.chembiol.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hossan AS, Abu-Melha HM, Al-Omar MA, Amr Ael G. Synthesis and antimicrobial activity of some new pyrimidinone and oxazinone derivatives fused with thiophene rings using 2-chloro-6-ethoxy-4-acetylpyridine as starting material. Molecules. 2012;17(11):13642–13655. doi: 10.3390/molecules171113642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Salahi R, Abuelizz HA, El Dib RA, Marzouk M, Alshammari MB. Antimicrobial activity of New 2-Thioxo-benzo[g]quinazolin-4(3H)-one derivatives. Med Chem. 2017;13(1):85–92. doi: 10.2174/1573406412666160610095706. [DOI] [PubMed] [Google Scholar]
- 6.Al-Salahi R, Abuelizz HA, Wadi M, El Dib RA, Alotaibi MA, Marzouk M. Antimicrobial activity of synthesized 2-methylthiobenzo[g][1,2,4]- triazolo[1,5-a]quinazoline derivatives. Med Chem. 2016;12(8):760–766. doi: 10.2174/1573406412666160502155106. [DOI] [PubMed] [Google Scholar]
- 7.Al-Salahi R, Abuelizz HA, Ghabbour HA, El-Dib R, Marzouk M. Molecular docking study and antiviral evaluation of 2-thioxo-benzo[g]quinazolin-4(3H)-one derivatives. Chem Cent J. 2016;10:21. doi: 10.1186/s13065-016-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abuelizz HA, Marzouk M, Ghabbour H, Al-Salahi R. Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm J. 2017;25(7):1047–1054. doi: 10.1016/j.jsps.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abuelizz HA, Al-Salahi R, Al-Asri J, Mortier J, Marzouk M, Ezzeldin E, Ali AA, Khalil MG, Wolber G, Ghabbour HA, Almehizia AA, Abdel Jaleel GA. Synthesis, crystallographic characterization, molecular docking and biological activity of isoquinoline derivatives. Chem Cent J. 2017;11(1):017–0321. doi: 10.1186/s13065-017-0321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abuelizz HA, Anouar EH, Marzouk M, Ezzeldin E, Ali AA, Al-Salahi R. Molecular modeling, enzyme activity, anti-inflammatory and antiarthritic activities of newly synthesized quinazoline derivatives. Future Med Chem. 2017;9(17):1995–2009. doi: 10.4155/fmc-2017-0157. [DOI] [PubMed] [Google Scholar]
- 11.Al-Salahi R, Anouar EH, Marzouk M, Taie HA, Abuelizz HA. Screening and evaluation of antioxidant activity of some 1,2,4-triazolo[1,5-a]quinazoline derivatives. Future Med Chem. 2017;10(4):379–390. doi: 10.4155/fmc-2017-0224. [DOI] [PubMed] [Google Scholar]
- 12.Abuelizz HA, El Dib R, Marzouk M, Anouar EH, Maklad YN, Attia H, Al-Salahi R. Molecular docking and anticonvulsant activity of newly synthesized quinazoline derivatives. Molecules. 2017;22(7):1094. doi: 10.3390/molecules22071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bereznak JF, Chan DM-T, Geffken D, Hanagan MA, Lepone GE, Pasteris RJ, Swann SL (2008) Preparation of fungicidal tricyclic 1,2,4-triazoles. WO 2008103357A120080828
- 14.Wegner P, Krueger M, Krueger G, Head J, Rees R, Johann G (1990) Pyrido[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine-2-sulphonamides, processes for their preparation, and their use as herbicides, plant growth regulators and fungicides. Ger. Offen DE 3825041 A1 19900215
- 15.Ahmad R, Hashim HM, Noor ZM, Ismail NH, Salim Y, Lajis NH, Shaari K. Antioxidant and antidiabetic potential of Malaysian Uncaria. Res J Med Plant. 2011;5(5):587–595. doi: 10.3923/rjmp.2011.587.595. [DOI] [Google Scholar]
- 16.Al-Salahi R, Ahmad R, Anouar E, Iwana Nor Azman NI, Marzouk M, Abuelizz HA. 3-Benzyl(phenethyl)-2-thioxobenzo[g]quinazolines as a new class of potent α-glucosidase inhibitors: synthesis and molecular docking study. Future Med Chem. 2018;10(16):1889–1905. doi: 10.4155/fmc-2018-0141. [DOI] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tagami T, Yamashita K, Okuyama M, Mori H, Yao M, Kimura A. Molecular basis for the recognition of long-chain substrates by plant alpha-glucosidases. J Biol Chem. 2013;288(26):19296–19303. doi: 10.1074/jbc.M113.465211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meszaros LK, Dose A, Biagini SC, Blower PJ. Synthesis and evaluation of analogues of HYNIC as bifunctional chelators for technetium. Dalton Trans. 2011;40(23):6260–6267. doi: 10.1039/c0dt01608j. [DOI] [PubMed] [Google Scholar]
- 20.Al-Salahi R, El-Tahir KE, Alswaidan I, Lolak N, Hamidaddin M, Marzouk M. Biological effects of a new set 1,2,4-triazolo[1,5-a]quinazolines on heart rate and blood pressure. Chem Cent J. 2014;8(1):3. doi: 10.1186/1752-153X-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Salahi R, Geffken D. Synthesis of novel 2-methylsulfanyl-4H-[1,2,4]triazolo[1,5-a]quinazolin-5-one and derivatives. Synth Commun. 2011;41(23):3512–3523. doi: 10.1080/00397911.2010.518780. [DOI] [Google Scholar]
- 22.Al-Salahi RA, Geffken D. Synthesis and reactivity of [1,2,4]triazolo-annelated quinazolines. Molecules. 2010;15(12):8143. doi: 10.3390/molecules15118143. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.