

Abstract
Artificial metalloenzymes (ArMs) result from the incorporation of an abiotic metal cofactor within a protein scaffold. From the earliest techniques of transition metals adsorbed on silk fibers, the field of ArMs has expanded dramatically over the past 60 years to encompass a range of reaction classes and inspired approaches: Assembly of the ArMs has taken multiple forms with both covalent and supramolecular anchoring strategies, while the scaffolds have been intuitively selected and evolved, repurposed, or designed in silico. Herein, we discuss some of the most prominent recent examples of ArMs to highlight the challenges and opportunities presented by the field.
Short abstract
Artificial metalloenzymes result from the incorporation of an abiotic metal cofactor within a protein scaffold. We summarize recent examples highlighting the challenges and opportunities.
Introduction
In 1956, Fujii and co-workers reported on the use of reduced palladium chloride adsorbed on silk fibers for the asymmetric reduction of dehydro-amino acid derivatives.1 Although these findings have proven challenging to reproduce, this contribution is generally considered to be the first report on a metal-catalyzed asymmetric reaction.2 Interestingly, this protein-modified precious metal catalyst also satisfies the generic definition of an artificial metalloenzyme (ArM hereafter): a hybrid catalyst that results from combining an abiotic metal cofactor with a protein.
Fifty years ago, however, synthesizing and testing enantiopure ligands was more straightforward than isolating or producing proteins and testing the catalytic properties of these in the presence of metal ions. Accordingly, the field of homogeneous catalysis, relying on enantiopure ligands derived from the chiral pool, enjoyed significant growth following Noyori’s seminal 1966 paper entitled “Asymmetric induction in carbenoid reactions by means of a dissymmetric copper chelate”.3
In the 1970s, two groups explored the use of proteins as scaffolds to accommodate non-native metal ions for catalytic purposes. In 1976, Kaiser and Yamamura reported that the hydrolytic enzyme carboxypeptidase A (CPA) could be repurposed into an oxidase upon substitution of the native Zn(II) by Cu(II) for the oxidation of ascorbic acid, Scheme 1a.4 In 1978, Whitesides and Wilson exploited the high affinity of biotinylated probes for avidin to anchor a Rh(diphosphine) within avidin, Scheme 1b.5 Despite the undeniable elegance of both of these pioneering studies, the true potential of ArMs had to await the advent of recombinant protein expression to reveal its full potential. In 1997, Distefano and Davies reported on a covalent scaffold modification of a recombinant adipocyte lipid binding protein (ALBP) with iodoacetamido-1,10-phenanthroline. Upon complexation with Cu(II), the resulting ArM catalyzed the stereoselective hydrolysis of racemic esters, Scheme 1c.6,7 These papers set the stage for the resurgence of interest in artificial metalloenzymes at the turn of the millennium.8
Scheme 1. Pioneering Studies in Artificial Metalloenzymes.

(a) Dative anchoring resulting from metal substitution in carboxypeptidase A (CPA). (b) Supramolecular anchoring based on the biotin-avidin technology. (c) Covalent anchoring to an engineered cysteine within adipocyte lipid binding protein (ALBP).
Four strategies have been used to assemble ArMs.9 (i) Lewis-basic amino acids positioned within a cavity may interact via dative bonds with a coordinatively unsaturated metal (cofactor), Figure 1a. (ii) The native metal of a metalloenzyme may be substituted for another metal, thus conferring novel catalytic activity to the protein. The metal may be part of a prosthetic group (e.g., heme) or bound solely to amino acids, as in the case of carboxypeptidase A, Figure 1b. (iii) A high-affinity inhibitor (or substrate) may be used to anchor a metal cofactor within a host protein via supramolecular interactions, Figure 1c. (iv) Covalent immobilization may be achieved by an irreversible reaction between complementary functional groups on a ligand and on the host protein, respectively, Figure 1d. Relying on these four anchoring strategies, significant progress has been achieved in the assembly and optimization of artificial metalloenzymes.
Figure 1.
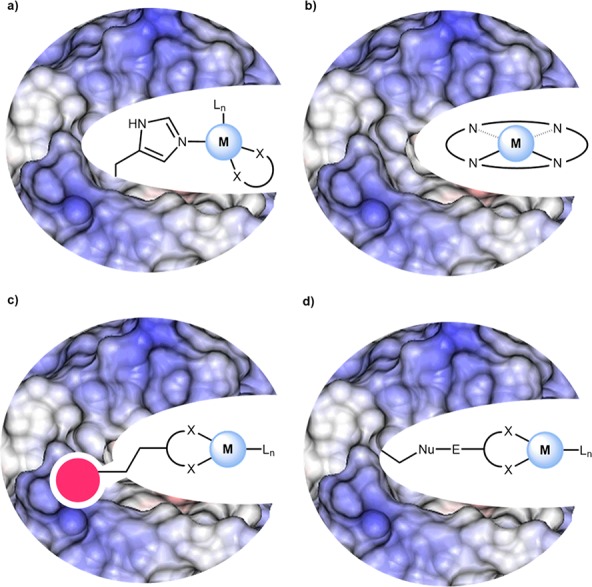
Four general approaches to ArM assembly (a) Dative coordination with an unsaturated metal complex. (b) Metal substitution. (c) Supramolecular coordination using a high-affinity anchor. (d) Covalent immobilization.
Transfer Hydrogenation
Building on the report of Whitesides, the Ward group has extensively exploited streptavidin (Sav) as a host protein to develop artificial metalloenzymes. This remarkably stable homotetrameric protein can be readily expressed in large quantities in Escherichia coli (up to 8 g/L).10 To date, we have reported on 13 different ArMs based on the biotin–streptavidin technology.11−13 Initial optimization efforts often rely on combining a small library of biotinylated cofactors (typically <10) with a focused library of Sav mutants. The chemogenetic optimization strategy allows the rapid identification of promising biotinylated cofactors. Since 2005, we have used Noyori-type piano stool cofactors to develop artificial asymmetric transfer hydrogenases (ATHases), first for ketone, enone, and more recently for imine reduction.14−18 Having singled out the deleterious effect of glutathione on various precious-metal-containing ArMs, we screened several Michael acceptors and oxidizing agents to minimize the glutathione nuisance on Cp*Ir-cofactors. Both diamide and [Cu(gly)2] proved beneficial, allowing us to perform ATHase screening in the presence of cell-free extracts or in the periplasm of E. coli.19−21 This simple strategy permitted us to improve the ATHase activity by directed evolution using cell-free extracts containing Sav isoforms.18 Respectively, four and five rounds of directed evolution led to the identification of both an (R)-selective and an (S)-selective ATHase for the reduction of prochiral imine 1: [Cp*Ir(Biot-L)Cl]·Sav S112A-N118P-K121A-S122M and [Cp*Ir(Biot-L)Cl]·Sav S112R-N118P-K121A-S122M-L124Y (Scheme 2). We were pleased to observe that the evolved ATHases displayed significant organic-solvent tolerance. The reactions could be carried out under biphasic conditions to afford even improved ee’s: from 92% to 95% for (S)-2 and 78% to 85% for (R)-2 (Scheme 2). This remarkably increased organic-solvent stability gives potential for the implementation of ArMs in reactions that are less tolerant to aqueous conditions. Indeed, aqueous compatibility is arguably the most stringent constraint in ArMs development. Engineering or evolving protein scaffolds to operate in (nearly) pure organic solvents would allow this limitation to be overcome.
Scheme 2. Directed Evolution of Sav Applied to the Asymmetric Transfer Hydrogenation of Cyclic Imine 1 To Afford Both (R)- and (S)-Selective ATHases.

Thanks to its large cone-shaped hydrophobic funnel leading to the Zn(His)3 active site,22 human carbonic anhydrase II offers an attractive scaffold for the assembly of ArMs. Using arylsulfonamide (ArSulf) as a high-affinity anchor, the Ward group reported on Cp*Ir-based artificial asymmetric transfer hydrogenases (ATHases) and Ru-based artificial metathases.17,23,24 To improve the modest ATHase activity of [(Cp*)Ir(pico-ArSulf)Cl]·hCA II (4·hCA II), we teamed up with the Baker group to computationally redesign the ArM by identifying and mutating residues in hCA II to increase the affinity between the cofactor and the host protein.24 Indeed, an X-ray crystal structure of 4·hCA II revealed a poorly localized cofactor, with only 30% occupation of the Ir moiety.17 We hypothesized that, upon firmly localizing the cofactor in a single and favorable locus, the ArM may display increased activity and selectivity. Aided by Rosetta, four hCA II mutants were identified containing up to eight mutations that were predicted to improve affinity of the protein for the cofactor. Gratifyingly, these designs indeed led to a 50-fold increase in cofactor affinity versus WT hCA II and afforded a 10-fold improvement in TON, with up to 92% ee in favor of (S)-salsolidine 3. To further increase hydrophobic contacts between the cofactor and hCA II, a methyl substituent on the Cp* (4) was replaced by a propyl group in cofactor (5), which further improved the ATHase selectivity: 96% ee (S)-3 and 59 TON at 4 °C, Scheme 3.
Scheme 3. Directed Evolution of an Iridium-Dependent ATHase Using an hCA II Scaffold.
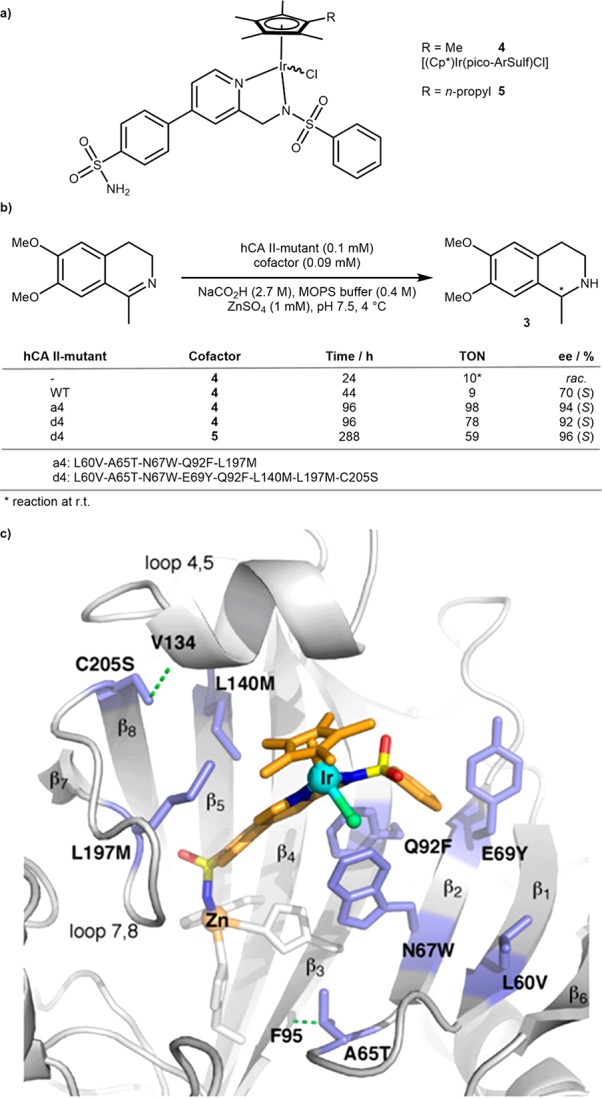
(a) Structure of the piano stool complex cofactor. (b) Reaction conditions and results of imine reduction to 3 with computationally designed mutants. (c) X-ray structure of WT ATHase (PDB 3ZP9) used as a template for computational optimization; residues that stabilize the complex are shown in purple. Reproduced with permission from ref (24). Copyright 2015 American Chemical Society.
In addition to evolving ATHases based either on the Sav or the hCA II scaffold, the Ward group has capitalized on the remarkable stability of three-legged piano stool complexes to evaluate the possibility of integrating ArMs in enzyme networks for metabolic engineering purposes. The following features were demonstrated: enzyme cascades,25 including the use of NADH as a reductant for ATHase,26 zymogens,27 cross-regulation,28 and gene networks.29 These features have recently been reviewed.13
Inspired by the iron uptake properties of siderophores and their associated periplasmic binding proteins (PBPs), the Duhme-Klair group has recently repurposed these scaffolds as redox-responsive ArMs capable of controlled and reversible anchoring of the cofactor.30 Iron-siderophore PBPs, such as CeuE produced by Campylobacter jejuni, are capable of scavenging Fe(III) and releasing the metal following reduction to Fe(II) inside the cell, Scheme 4a. To validate this concept, transfer hydrogenation was selected as a benchmark reaction to assess the reversible binding, relying on enantiomeric excess as a readout of anchoring of the cofactor within CeuE.
Scheme 4. Redox-Triggered Binding of a Siderophore-Inspired Artificial Metalloenzyme for Transfer Hydrogenation.
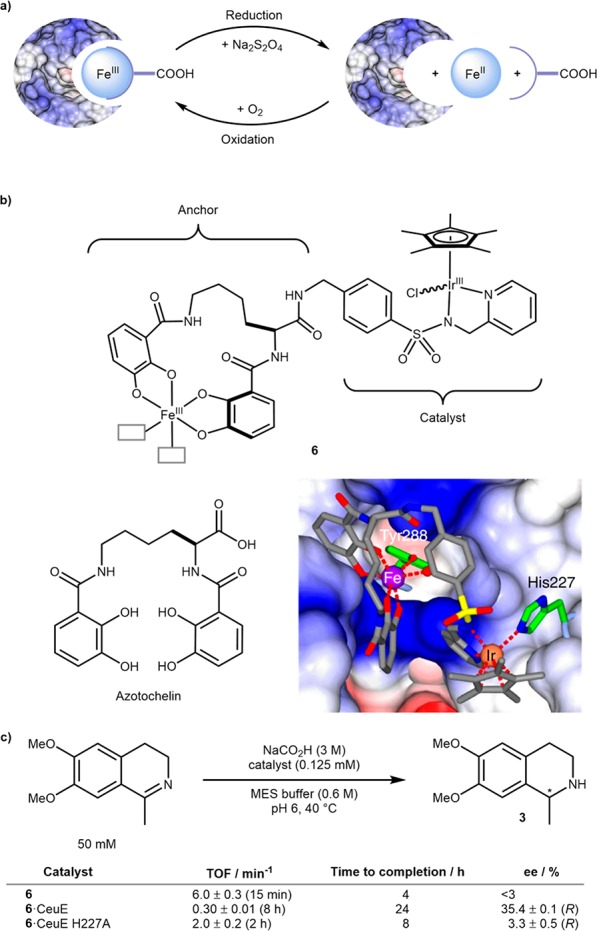
(a) Concept of redox reversible cofactor binding. (b) Structure of cofactor 6 inspired from “azotochelin” the siderophore; crystal structure of the 6·CeuE highlighting the binding of H227 to Ir(III). (c) Selected results for the benchmark reduction to salsolidine 3. Reproduced with permission from ref (30). Copyright 2018 Nature Publishing Group.
Cofactor 6 was synthesized based on the azotochelin siderophore, to act as an iron binding anchor, fused to a catalytic portion based on a highly active Ir complex inspired by previous examples of transfer hydrogenases,12Scheme 4b. The remaining two coordination sites around iron were occupied with solvent in the free cofactor but were hypothesized to bind Y288 and H227 of CeuE, as observed in the crystal structure of Fe(III)azotochelin·CeuE. However, on assembly of 6·CeuE, both mass spectrometry and crystallographic data supported Fe(III) binding solely to Y288. Instead, H227 was found to bind to iridium, displacing the chloride. X-ray crystallography also suggested that the CeuE binding pocket had a preference for a single enantiomer of 6. For the reduction of the salsolidine precursor, 6·CeuE performed with a 20-fold lower TOF than the free cofactor. The authors hypothesized that this may be due to hindrance caused by binding of H227 to the iridium. The formation of (R)-salsolidine 3 with an ee of 35.4% supported the hypothesis that the cofactor 6 was indeed bound to the protein. To further probe the effect of His-Ir coordination, the group expressed mutant H227A which, as expected, increased TOF but at the cost of a substantially lowered ee. To demonstrate the reversibility of binding, the Fe(III) was reduced using sodium dithionite. The free CeuE scaffold could be isolated by gel electrophoresis and was shown to be properly folded. At the same time, the free cofactor could be isolated by extraction, but both recycling methods proved almost mutually exclusive, meaning either cofactor or protein could be recovered at one time. The reactivity of the recycled CeuE was investigated with fresh cofactor 6 and, while conversion dropped to ∼80% of the fresh ArM, the ee remained intact.
Multiple Electron Reductions
Lu and co-workers have recently disclosed their efforts to engineer a highly efficient sulfite reductase dependent on a heme-[4Fe-4S] cofactor introduced by in vitro reconstitution.31 The group began by searching the PDB to identify hemoproteins capable of accommodating an [4Fe-4S] cluster. They eventually selected cytochrome c peroxidase (CcP) as a suitable scaffold. Rosetta matcher was subsequently used to identify strategic sites to introduce cysteine residues to anchor the [4Fe-4S] cluster. Additional mutations allowed relief of steric clashes and interference from a nearby aspartate residue. The modeled sextuple mutant SiRCcP.1 bore a high degree of structural similarity to a native SiR, Scheme 5. The reconstituted heme-[4Fe-4S]·SiRCcP.1 was observed to oxidize methyl viologen (MV+) at a rate of 0.348 (±0.15) min–1, in the presence of sodium sulfite as a measure of sulfite reductase activity. To further improve catalytic performance, inspection of SiR active sites identified key features and conserved residues. This revealed a bias for Lys and Arg residues, which generate an overall positive charge in the active site, thought to be important for substrate coordination. Mutagenesis of three strategic active site residues afforded the next-generation heme-[4Fe-4S]·SiRCcP.1 W51K-H52R-P145K mutant that displayed >5-fold increase in reductase activity compared to SiRCcP.1. Further mutations were carried out in the secondary coordination sphere to further stabilize the [4Fe-4S] cluster by H-bonds to the inorganic sulfur atoms, with D235 found to be oriented toward the [4Fe-4S] cluster. The D235C mutant (SiRCcP.3) displayed a sulfite reduction activity of >20 min–1, a 63-fold increase over that of SiRCcP.1 and a remarkable 18% of the native activity of an SiR from Mycobacterium tuberculosis.
Scheme 5. Engineering of a Heme-[4Fe-4S]-Dependent Sulfite Reductase.

(a) Overall reduction equation of sulfite to sulfide and (b) binding of the siroheme-[4Fe-4S] in native E. coli SiR (PDB ID 2GEP) used to search the PDB for suitable hemoprotein scaffolds (left); the resulting model of the rationally designed heme-[4Fe-4S] binding site with coordinating residues in yeast CcP, identified as a suitable scaffold (right). Reproduced with permission from ref (31). Copyright 2018 The American Association for the Advancement of Science.
Orchestrating the timely delivery of six electrons and seven protons highlights the exquisite level of control that can be achieved with ArMs and makes this example particularly noteworthy, Scheme 5.
Oxidation
The Kamer group has capitalized on the apolar channel of steroid carrier protein (SCP) 2L to assemble an ArM for the oxidation of lignin toward the catalytic depolymerization of this abundant polymer.32 A series of nitrogen-rich ligands bearing a maleimide 7−9 were synthesized for conjugation to an engineered cysteine residue at one of two ends of the hydrophobic tunnel, Scheme 6. Upon addition of Fe(OTf)2, the oxidizing ability of the ArM were evaluated using hydrogen peroxide for the oxidation of the monomeric lignin mimic 10. The combination Fe 7·SPC-2L A100C was found to be the most active cofactor and position for cysteine bioconjugation. Upon lowering the catalyst loading from 5% to 2.5%, the conversion dropped substantially (from 100% to 35%). To improve this, the introduction of coordinating residues, which are known to stabilize metal-oxide intermediates, was investigated. For this purpose, His or Asp at site F94 were selected as this residue was computed to lie in the vicinity of the complex but not integral to maintaining the structure of the channel. These mutants were combined with ligands 7 and 8 and screened against the same substrate 10, Scheme 6b. Interestingly, both histidine mutants showed no improvement over the original ArMs; however, there was noticeable improvement observed with the aspartate mutants. This effect was rationalized with molecular dynamic simulations. These suggested that, while the aspartate would not displace a water molecule on the iron, the carboxylate was significantly closer to the metal than the wild type phenyl and within hydrogen-bonding distance to one of the bound water molecules.
Scheme 6. Iron-Catalyzed Oxidation of Lignin by SCP-2L-Based ArMs.

(a) Nitrogen-bearing ligands for anchoring to an engineered cysteine residue and structure of lignin and (b) selected results for the lignin oxidation with different SCP-2L mutants and cofactors.
Hydrolysis
In stark contrast to natural metallohydrolases, few homogeneous transition metal complexes efficiently catalyze hydrolytic reactions. Thanks to their well-understood mechanisms and ease of implementing a high-throughput screen or selection platform for such reactions, the zinc-catalyzed amide and (phospho)ester hydrolysis has proven a fertile playground for the design and evolution of artificial metallohydrolases.
With the aim of repurposing metalloproteins for catalytic purposes, the Baker group has relied on the Rosetta suite of algorithms. In the quest for a de novo hydrolase, the PDB was first screened for enzymes containing open zinc coordination sites, favoring catalytic over structural zinc atoms, to act as Lewis acids for hydrolysis. Transition states were computed for the hydrolysis of two representative organic phosphates, and these were used to dock into promising candidates. A search for scaffolds capable of accommodating at least two beneficial hydrogen bonds to either the nucleophilic hydroxyl-, phosphoryl-, or leaving-group-oxygen of the computed transition state was carried out. Any matches satisfying these criteria were then assessed for their transition state shape-complementarity. This yielded a library of twelve designer enzymes with one, an octuple mutant of a native adenosine deaminase (Zn·PT3), displaying moderate catalytic activity toward the hydrolysis of phosphate 11 (kcat/KM = 4 M–1 s –1). This hit was further improved by saturation mutagenesis of twelve strategic sites around the active site, including five that were predicted by computation, to increase the transition state complementarity. Beneficial mutations were identified at 3 positions (I62L, V218F, and V299E) which, when combined, gave Zn·PT3.1 with a ∼40-fold increase in activity. Following error prone PCR and additional mutagenesis of another computationally highlighted problematic residue, this was improved to a final efficiency of (kcat/KM = 9750 ± 1534 M–1 s–1) in the final Zn·PT3.3 (overall PT3 S57D-Q58V-P59K-I62L-E186D-V218F-V299E) enzyme, Scheme 7.33 It should be noted that the elegant strategy delineated above does not fulfill the requirements of an artificial metalloenzyme. Indeed, it is rather a repurposing strategy whereby a Zn-containing deaminase is converted to a phosphatase.
Scheme 7. An in Silico Repurposed Deaminase Was Further Optimized by Directed Evolution To Improve Its Catalytic Performance.
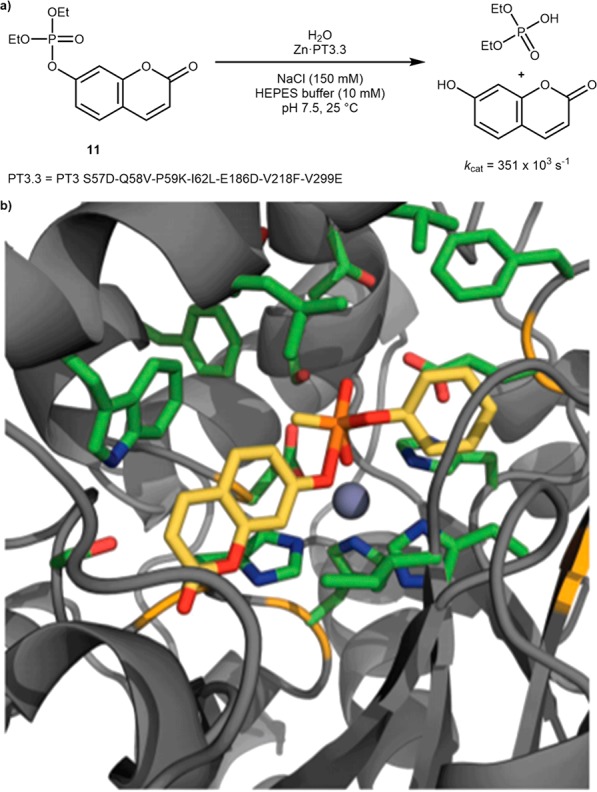
(a) Hydrolysis of a coumarin-derived phosphate ester 11 by Zn·PT3.3 and (b) model of the transition state of 11-hydrolysis docked into the Zn·PT3 crystal structure. Reproduced with permission from ref (33). Copyright 2012 Nature Publishing Group.
Capitalizing on their expertise in metal-mediated protein assembly,34 Tezcan and co-workers have exploited zinc ions as both templating and catalytic ions upon dative anchoring within a protein scaffold. They developed a structurally robust and in vivo active β-lactamase by assembling four cytochrome cb562 (cyt cb562) units through incorporation of Zn(II) coordination sites comprising histidines, aspartates, and glutamates. The supramolecular assembly, Zn8·AB34 K104A-E57G, comprised eight zinc ions, four for structural integrity and four as catalytic sites, Scheme 8a. Following directed evolution, performed in the periplasm of E. coli, the evolved ArM included sixteen mutations and catalyzed the hydrolysis of ampicillin with a remarkable kcat/KM of 350 min–1 M–1, Scheme 8b.35
Scheme 8. Assembly of de Novo Designed Zinc-Containing Enzymes as Artificial Metallohydrolases.
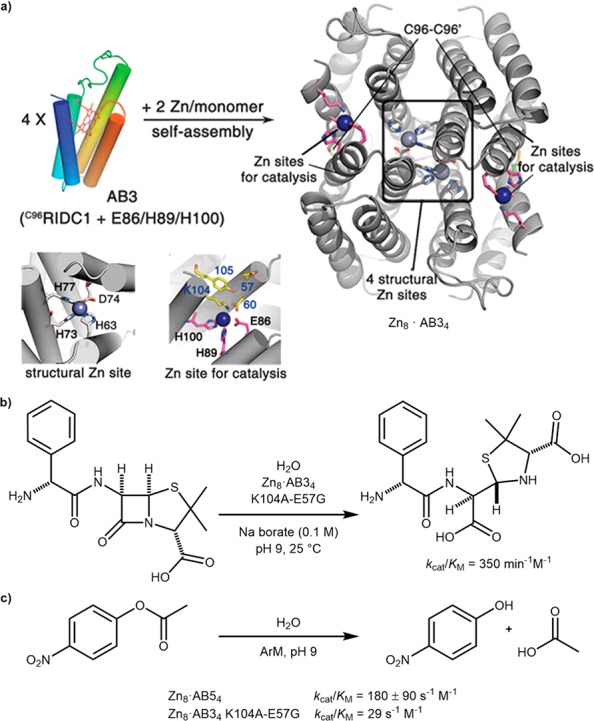
(a) Zn8·AB34 with structural and catalytic zinc sites highlighted. (b) Hydrolysis of ampicillin by Zn8·AB34 K104A-E57G mutant. (c) Comparison of hydrolysis rates of p-nitrophenol by Zn8·AB34 K104A-E57G and Zn8·AB54. Adapted with permission from ref (35). Copyright 2014 The American Association for the Advancement of Science.
More recently, the group has also assembled another cyt cb562-derived tetramer Zn8·AB54 which shows structural similarity to Zn8·AB34 K104A-E57G but with different catalytic zinc microenvironments.36 While sharing two of the three Zn-binding ligands with Zn8·AB34 K104A-E57G, the coordinated catalytic zinc atoms were instead oriented toward the exterior of the tetramer. This change in secondary coordination sphere was made evident by the several-fold higher rate of p-nitrophenyl acetate hydrolysis by Zn8·AB54 (kcat/KM = 180 ± 90 s–1 M–1 at pH 9 versus kcat/KM = 29 s–1 M–1) (Scheme 8c). The unoptimized Zn8·AB54 was also demonstrated to be a β-lactamase for ampicillin (kcat = 1.03 ± 0.06 min–1 and kcat/KM = 120 ± 10 min–1 M–1) and displayed Michaelis–Menten behavior, which was not observed with Zn8·AB34 until further optimization was carried out. For the directed evolution of Zn8·AB54, four residues (K85, E92, Q103, and K104) lining the active site were selected for saturation mutagenesis. However, no single or double mutant revealed any significant improvement of β-lactamase efficiency. It was hypothesized that, with the new zinc binding sites present, the structural C96 disulfide bridges may be unnecessary; hence, the Zn8·AB54 C96T mutant was prepared. This was observed to have tighter Zn binding sites than the more open Zn8·AB34 assembly and improved hydrolysis activity toward ampicillin (kcat/KM = 210 min–1 M–1 for Zn8·AB54 C96T versus kcat/KM = 130 min–1 M–1 for Zn8·AB54). Combined, these studies highlight the effect of active site positioning in two homologous assemblies and the importance of the secondary coordination sphere in enzyme efficiency and evolvability.
In a related approach, the Hilvert group has recently demonstrated the versatility of combining various evolutionary techniques to optimize a highly active and specific esterase.37 The starting peptide MID1 dimer, initially reported by Kuhlman,38,39 was assembled from two computationally designed helix-turn-helix fragments containing two interfacial Zn(II)His3 sites. These zinc sites acted both to template peptide assembly as well as to facilitate ester-bond hydrolysis in the neighboring hydrophobic pockets. Following fusion of the dimer subunits, the zinc site farthest from the linker was deleted through computationally guided mutations to afford MID1sc, which contained a single zinc ion that catalyzed the hydrolysis of p-nitrophenyl acetate at rates similar to that of the MID1 dimer (kcat/KM = ∼25 M–1 s–1 versus 40 M–1 s–1 for MID1). MID1sc was subsequently subjected to multiple rounds of computational redesign, DNA shuffling, and random and cassette mutagenesis to yield MID1sc10. This resulted in a significantly deepened hydrophobic pocket, Scheme 9b, and spectacular activity for the hydrolysis of (S)-12, exhibiting a kcat of 1.64 ± 0.04 s–1 and a kcat/KM of 980 000 ± 110 000 M–1 s –1, thus rivalling the best natural hydrolases, Scheme 9a. The substrate specificity was demonstrated by the selective binding of (S)-nitrophenyl-phosphonate 13 in the active site. The X-ray structure revealed the remarkable complementary fit for this transition state mimic, Scheme 9c.37
Scheme 9. Design and Directed Evolution of an Artificial Metallohydrolase MID1sc.
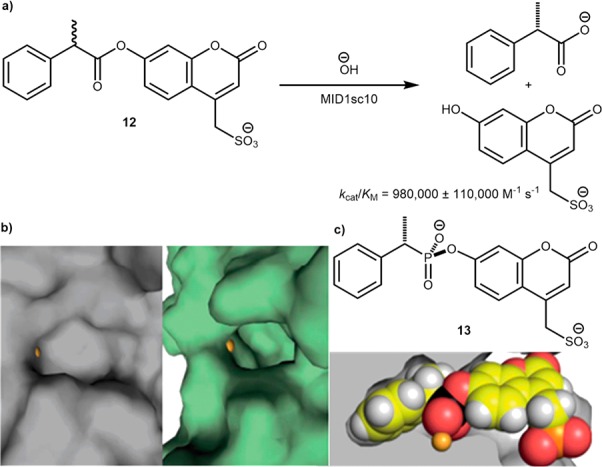
(a) Hydrolysis of profluorescent ester 12 by mutant MID1sc10. (b) Deepening of the hydrophobic binding pocket in MID1sc10 (right) compared to MID1 (left) as a result of directed evolution. (c) Cut-away crystal structure of MID1sc highlighting the tight binding of the enantiopure phosphonate transition state mimic 13. Reproduced with permission from ref (37). Copyright 2018 The American Association for the Advancement of Science.
These studies highlight the great potential of combining computational design with directed evolution tools. As zinc displays very limited background activity and is not inhibited by cellular extracts, these studies could be performed either in vivo or with clarified cell extracts, thus allowing a straightforward high-throughput screening or selection protocol.
Hydration
To expand its scope of ligands for enzymatic purposes, nature may resort to the use of either a prosthetic group (heme, corrin, etc.) or post-translational modification of natural amino acids. As an alternative, chemists may introduce unnatural amino acids (UAAs) into host proteins to fine-tune the catalytic properties of ArMs. Thanks to the development of the stop codon suppression methodology, the incorporation of the UAAs in vivo has become increasingly accessible.40 To ensure tight dative anchoring of Cu(II) to its host protein, Roelfes and co-workers engineered a bipyridine-alanine (BpyA) in the hydrophobic pocket of the lactococcal multidrug resistance regulator (LmrR) homodimeric scaffold.41 Complementation with Cu(II) afforded an ArM (Cu·LmrR M89BpyA) that catalyzed the enantioselective hydration of prochiral enones. Introduction of a glutamate residue (i.e., V15E) in the secondary coordination sphere afforded up to 64% ee for the hydration of enone 14, Scheme 10.42,43 The authors speculate that this residue contributes to steer the enantioselective delivery of the hydroxide to the β-position of the enone 14. The isosteric glutamine mutation (i.e., V15Q) afforded the β-hydroxyketone in 15% ee, Scheme 10.
Scheme 10. Unnatural Amino Acids Bearing a Chelating Group (i.e., Biypridine-alanine BpyA) Allow the Anchoring of Copper into Homodimeric Lactococcal Multidrug Resistance Regulator (LmrR), and Site-Directed Mutagenesis of Second Coordination Sphere Residues Leads to a Significantly Improved Artificial Hydratase.

Hydroformylation
The Kamer group has recently taken advantage of the apolar channel of steroid carrier protein (SCP) 2L as a cavity for the hydroformylation of long-chain alkenes.44 An initial conjugation of maleimide 15 with cysteine V83C or A100C, located at either end of the hydrophobic tunnel Scheme 11c, afforded 15·SCP-2L V83C or A100C bearing a free hydrazine. Hydrazone formation with one of three aldehyde-bearing phosphine ligands 16–18 and further incubation with Rh(acac)(CO)2 yielded the final ArMs: Rh(acac)(16–18)∼15·SCP-2L V83C/A100C, Scheme 11a. The selected regioisomer of the phosphine was shown to play an essential role in determining the hydroformylase activity of the resulting ArM: the meta17 and the para18 isomers displayed a 30- and 70-fold increased activity compared to the ortho16 isomer, respectively. The best performing mutant Rh(acac)18∼15·SCP-2L A100C catalyzed the hydroformylation of 1-octene with TONs of over 400 and 79% selectivity in favor of linear (n-)nonanal, Scheme 11b. Although the free Rh(acac)(CO)2 yielded higher TONs (>500) for the hydroformylation of 1-octene under anhydrous conditions, the regioselectivity of the free complex was much lower (55% n-nonanal). These findings suggest that the protein cavity favors the formation of the linear aldehyde. Overall, these ArMs proceeded under notably milder conditions than those typically required by industrial processes (35 versus 125 °C).
Scheme 11. Steroid Carrier Protein (SCP) 2L as a Host Protein for the Hydroformylation of Long-Chain Alkenes.
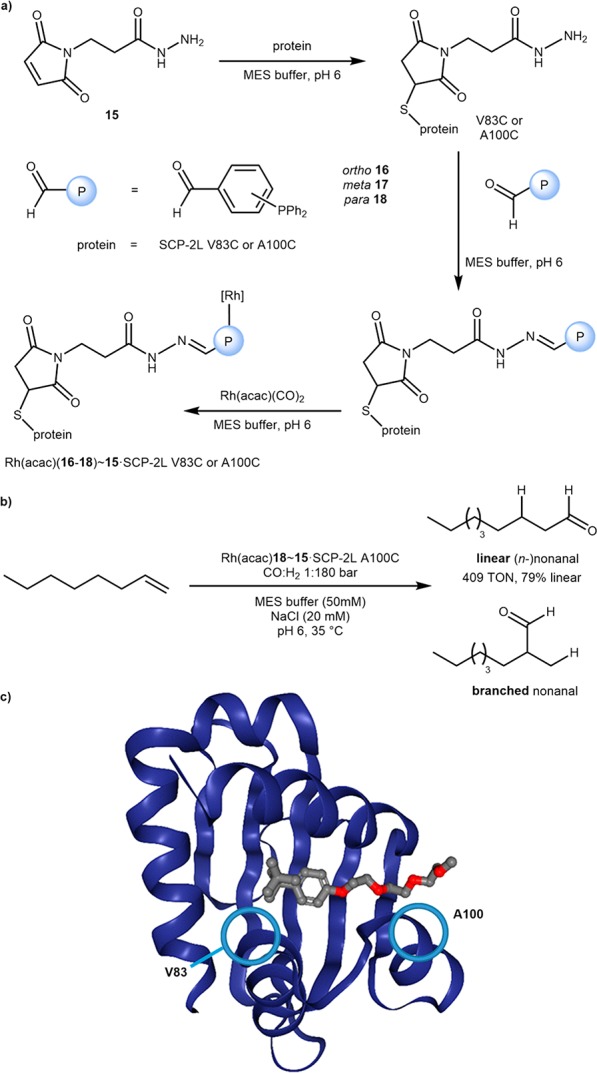
(a) Sequential (bio)conjugation of maleimide derivative 15, phosphines 16–18, and rhodium to assemble the final artificial hydroformylases. (b) The hydroformylation of 1-octene by Rh(acac)-18∼15·SCP-2L A100C yields preferentially linear nonanal. (c) Crystal structure of SCP-2L with Triton X-100 bound in the hydrophobic tunnel (PDB 1IKT) with the positioning of engineered cysteine residues for bioconjugation at either opening shown. The authors hypothesize that the hydrophobic channel forces the insertion of the CO at the terminal position of the alkene, resulting in the linear aldehyde.
Diels–Alder
Mahy and co-workers have reported an interesting approach to assemble a copper-catalyzed Diels–Alderase based on naturally surface-expressed guanine nucleotide receptors, specifically the adenosine-binding subunit A2A.45 Being a therapeutic target, the structure–activity relationship of several adenosine-like antagonists has been well-studied. Hence, a suitable scaffold was selected and repurposed into a copper binding cofactor on the surface of HEK-A2A mammalian cells. Accordingly, the furane-bearing anchor was tethered to phenanthroline via two different linkers to afford ligands 19 and 20. The corresponding Cu(II) complexes Cu·19 and Cu·20 were combined with HEK-A2A cells expressing the A2A receptor on their surface. The reaction of cyclopentadiene with chelating substrate 21 was selected for the evaluation of the Diels–Alderase activity. Control experiments revealed that cofactors alone showed high activity (both ∼20 TON) and endo/exo selectivity (84/16) as a racemate. In the presence of HEK-293 cells not expressing the A2A receptor, low Diels–Alderase activity was observed (both ∼6 TON, endo/exo 84/16, 0% ee). The authors speculate that this may be caused by unspecific binding to the cell surface. In the presence of cells expressing the target receptor, the longer-chain cofactor Cu·20 gave results comparable to the free cofactor, suggesting specific binding but very limited influence of the host protein on the catalytic performance. In the presence of the cofactor bearing the shorter chain 19, a similar activity with a slightly biased endo/exo selectivity (82/18) and a modest ee (14%) was observed, Scheme 12.
Scheme 12. Diels–Alderase Based on Targeting the Surface-Expressed Adenosine Receptor A2A with Selected Results of the Copper-Catalyzed Diels–Alder Reaction of Cyclopentadiene and Chelating Substrate 21 in the Presence of Complexes Cu·19 and Cu·20.

Friedel–Crafts Alkylation
In addition to its hydratase activity (Scheme 10), the Cu·LmrR M89BpyA was found to catalyze the enantioselective Friedel–Crafts alkylation of indoles with a chelating enone 22. Up to 80% ee and 10 TONs were achieved, Scheme 13a.41 In addition, Roelfes showed that the presence of two symmetry-related, native tryptophan 96 residues in the hydrophobic pocket of LmrR were sufficient to anchor via π-stacking interactions, either a [Cu(phen)(NO3)2]2+46 (Scheme 13b) or a heme cofactor (Scheme 14).47 The corresponding [Cu(phen)(NO3)2]2+·LmrR outperformed the UAA-bearing ArM, Cu·LmrR M89BpyA,41 for the Friedel–Crafts alkylation of 2-methylindole giving full conversion (11 TON) and 93% ee, Scheme 13b, versus the 92% conversion (10 TON) and 80% ee, Scheme 13a. The critical involvement of W96 in the tight association between [Cu(phen)(NO3)2]2+ and LmrR was confirmed by mutagenesis: [Cu(phen)(NO3)2]2+·LmrR W96 (Kd = 2.6 μM) versus [Cu(phen)(NO3)2]2+·LmrR W96A (Kd = 45 μM).46
Scheme 13. Lactococcal Multidrug Resistance Regulator (LmrR) as a Scaffold for Two Different Approaches to Copper-Catalyzed Enantioselective Friedel–Crafts Alkylation of Indole Derivatives and Enone 22.

(a) Unnatural amino acid bearing a chelating group BpyA allows the anchoring of labile copper and b) Entirely supramolecular anchoring of a copper-phenanthroline complex.
Scheme 14. Supramolecular Anchoring of Heme into the Lactococcal Multidrug Resistance Regulator (LmrR) Affords a Stereoselective Cyclopropanase.

Cyclopropanation
Following from the above example from Roelfes’ group, further mutagenesis of several hydrophobic residues in the vicinity of the tryptophan pair, the Heme·LmrR M8A was observed to catalyze the cyclopropanation of p-methoxystyrene to 23 with 449 TONs and 51% ee with small amounts of diethyl fumarate as a side product, Scheme 14.
In a contrasting approach, Lewis and co-workers relied on the UAA p-azidophenylalanine (AzF) to covalently anchor a dirhodium-tetracarboxylate bearing a terminal alkyne. The thermostable prolyl oligopeptidase (POP) scaffold was engineered with AzF and conjugated to the dirhodium cofactor 24 via strain-promoted azide–alkyne cycloaddition (24·POP-Z), Scheme 15a. Four bulky residues lining the mouth of the active site were mutated to alanine to aid cofactor association, and a histidine residue (L328H) was introduced to favor two-point binding of the rhodium. Following two additional mutations, the resulting ArM 24·POP-ZA4 L328H-G99F-G549F catalyzed the enantioselective olefin cyclopropanation of terminal olefins with up to 92% ee and 74 TONs.48 Thanks to a streamlined expression and screening protocol, the authors could expand the directed evolution campaign to include distant mutations. A later identified mutant 3-VRVH, containing twelve mutations from 24·POP-Z was shown by individually reverting each mutation to have only three residues contributing directly to selectivity (S301G, G99S, and Y326H). 3-VRVH afforded the cyclopropane 25 in 92% ee and 76 TON versus 91% ee and 37 TON for 24·POP A4 S301G-G99S-Y326H with only the selectivity-essential mutations.49 Interestingly, after solving the crystal structure, only the latter two mutations were identified as residing in the active site, highlighting the difficulty in selecting beneficial modification sites and the importance of secondary-coordination sphere mutations in improving efficiency. A further interesting observation was the effect on ArM reactivity of the L328X mutation to increasingly coordinating residues: Phe, Cys, Met, and His. Counterintuitive to the Lewis-acidic nature of these metal coordinating residues ordinarily lowering metal reactivity, both this and the selectivity of carbene insertion into the olefins over water were improved, Scheme 15b. With the L328H mutant, the ee of cyclopropane 25 was improved from 38% to 85% over the WT, and the chemoselectivity of 25/26 also improved from 0.6 to 1.6. This was alongside evidence of chloride binding in the ArM active site, observed in the crystal structure, being beneficial to ArM selectivity, Scheme 15b. With no L328 mutation, 0.1 M NaBr gives 18% ee (23 TON) versus 1.75 M NaBr giving 38% ee (29 TON) in the formation of 25. MD simulations predicted that both of these interactions work synergistically to cause a conformational change creating a single Rh active site in a more hydrophobic cavity, Scheme 15c.50
Scheme 15. p-Azidophenylalanine (AzF) Can Be Used To Covalently Anchor a Dirhodium Tetracarboxylate Cofactor within Prolyl Oligopeptidase (POP).

(a) Assembly of the ArM and representative activity of either cyclopropanation or X–H insertion reactions. (b) Effect of coordinating residues and salt concentration on the activity and selectivity in the cyclopropanation reaction. (c) Postulated open and closed conformation of POP, with the closed conformation favored by both histidine and halide binding and giving a single cofactor pose in a more hydrophobic pocket.
The natural cofactor of hemoproteins may be altered in three ways to achieve novel reactivity: modification of the scaffold, addition of functional groups, and/or substitution of the metal.51 The first approach has been utilized by Hayashi and co-workers to assemble a myoglobin (Mb)-based ArM that catalyzes cyclopropanation.52 Myoglobin was expressed, the heme removed and replaced with the iron porphycene 27, Scheme 16a. The cyclopropanation of styrene with ethyl diazoacetate 28, Scheme 16b, was accelerated 26-fold by 27·Mb relative to heme·Mb. Importantly, the elusive metallocarbene species 29 could be spectroscopically identified.53,54 In a related approach, Fasan and co-workers have reconstituted and evolved Mb with iron chlorin e6 30 that catalyzed efficient and stereoselective olefin cyclopropanation reactions under aerobic conditions, Scheme 16c.55 Using a previously identified mutant (Mb H64V-V68A) for stereoselective cyclopropanation with heme,56 the group showed that the TONs could be more than doubled using 30·Mb H64V-V68A over heme·Mb H64V-V68A, (>990 versus 434 TON) and ∼17-fold over the WT 30·Mb (>990 versus 57 TON), Scheme 16b. With the best mutant-cofactor combination, 30·Mb H64V-V68A, de’s of up to 99.4% and ee’s of up to 98% were observed for a dozen styrene derivatives and analogues including various heterocycles.
Scheme 16. Reconstitution of Myoglobin (Mb) with Porphyrins and Analogues Affording Increased Activity and Selectivity in Fe-Catalyzed Cyclopropanation.
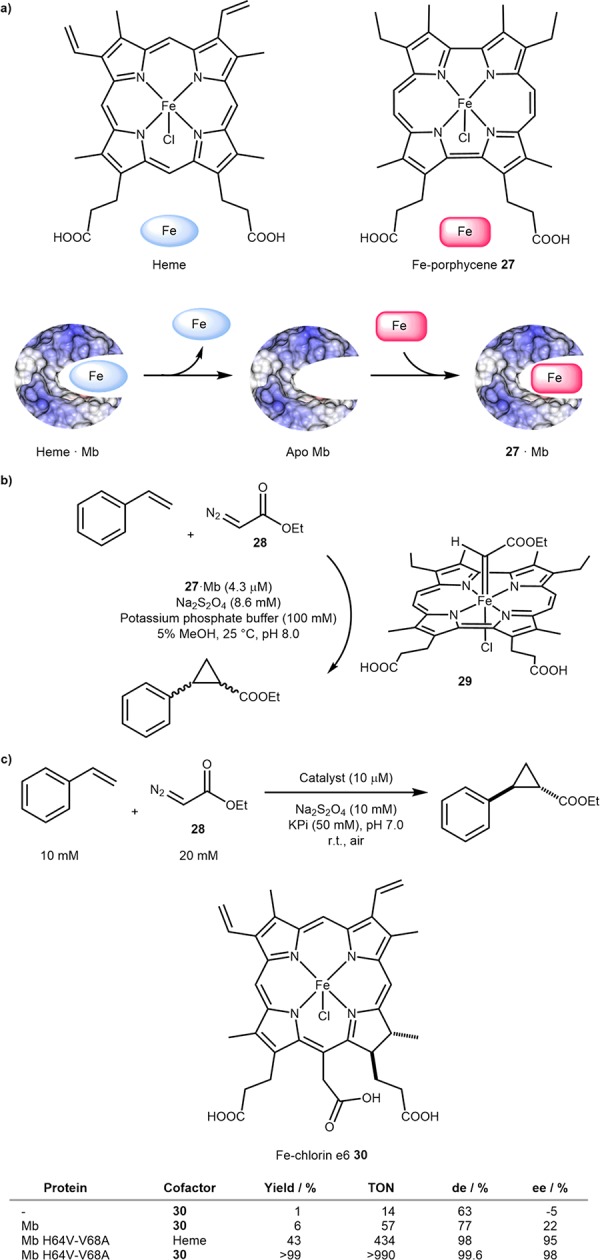
(a) Structure of heme and Fe-porphyrin 27 and reconstitution inside Mb. (b) Cyclopropanation of styrene with diazoacetate 28 and metallocarbene intermediate 29. (c) Selected results of Fasan’s stereoselective cyclopropanation catalyzed by 30·Mb and mutants thereof.
The final method by which a heme may be modified is by the natural iron being substituted by an alternative, potentially noble, metal which may endow the metalloenzyme with novel catalytic properties. Following the pioneering work of Kaiser (Scheme 1a),4 the groups of Soumillon, Kazlauskas, and Hartwig57 substituted the zinc ion present in human carbonic anhydrase by either Mn(II) or Rh(I) to afford ArMs for epoxidation,58,59 hydrogenation,60 and hydroformylation.61 More recently, Hartwig and co-workers substituted the iron in protoporphyrin IX (Fe-PIX, aka Heme) with various metals [M]-PIX and investigated the emerging catalytic activity.62−65 To speed up the screening protocol, the authors developed an E. coli expression system with various apo-hemoprotein hosts (myoglobin Mb, cytochrome P450, etc.) devoid of iron, thus leading to the overexpression of the corresponding apo-hemoproteins to be complemented with [M]-PIX. The Ir(Me)-PIX ArMs displayed remarkable cyclopropanation and C–H activation properties (discussed vide infra). The proximal H93, that usually coordinates the Fe-PIX·Mb, was initially mutated, and the H93A and H93G were identified as the most active mutants. Subsequent iterative rounds of mutagenesis were carried out targeting further residues in the active site (L32, F33, F43, H64, V68, H97, and I99) and focusing on their replacement with hydrophobic and uncharged residues, with almost 500 mutants screened in total. The intermolecular addition of ethyl diazoacetate to alkenes could be carried out to form the enantio- and diastereoenriched cyclopropanes, Scheme 17a.
Scheme 17. Iridium Substitution in Heme Proteins Combined with Directed Evolution Affords Versatile ArMs.

(a) Cyclopropanation with an (IrMe-PIX)-Substituted Myoglobin (Mb). (b) C–H Insertion by an (IrMe-PIX)-Substituted Myoglobin. (c) Dihydrobenzofuran Synthesis with High TON Catalyzed by (IrMe-PIX)·CYP119-Max.
C–H Activation
In recent years, C–H activation has attracted significant attention from the synthetic community: exploiting an inert C–H bond as a functional group amenable to selective derivatization offers fascinating perspectives for late-stage functionalization purposes.66 In addition to cyclopropanation, the library of Ir(Me)-PIX ArMs assembled by Hartwig and co-workers was concomitantly screened as catalysts for intramolecular C–H insertion. Various diazoalkanoates containing an ortho alkylether moiety were shown to undergo intramolecular cyclization to afford the corresponding enantioenriched dihydrobenzofurans, Scheme 17b.
The same group subsequently applied the same strategy to evolve a quadruple mutant of the cytochrome P450 CYP119 C317G-T213G-L69V-V254L (CYP119-Max). This construct displayed remarkable versatility for the insertion into both activated secondary and unactivated primary C–H bonds, sterically hindered bonds, and even intermolecular C–H activation. TONs of >35 000, rivalling natural P450s (36 000 TON),67 and ee’s of up to 94% were obtained for the intramolecular reaction of diazoester 31 (kcat/KM = 269 min–1 mM–1), Scheme 17c. Bespoke mutants for individual substrates were also evolved giving ee’s of up to 98% as well as opposite enantioselectivity in both a mutant- and substrate-dependent fashion.
Suzuki Coupling
To identify a suitable position of the organometallic moiety within its host protein, the Ward group typically relies on a chemogenetic optimization strategy. We illustrate this versatile strategy with an artificial Suzukiase. A small library of biotinylated NHC and monophosphine ligands, combined with [Pd(η3-cinnamyl)Cl]2 (32–34), were screened in the presence of Sav mutants for their cross-coupling activity to produce enantioenriched binaphthyls. Both the activity and the selectivity proved highly mutant-dependent. As for purely synthetic homogeneous catalysts, the bulkiness and electron-donating ability of the phosphine ligands were found to be crucial for activity. Unexpectedly, the best double mutant resulted from combining a highly (R)-selective mutant (i.e., K121E) with a moderately (S)-selective isoform (i.e., S112Y): 32·Sav S112Y-K121E. This mutant gave 90% ee (R)-37 with 50 TON at 4 °C in the reaction between naphthyliodide 35 and boronic acid 36. Introduction of an additional methylene between the biotin anchor and the P(t-Bu)2 afforded preferentially the binaphthyl (S)-37 with 33·Sav K121A or S112M giving 47% and 44% ee, respectively, Scheme 18.68
Scheme 18. Chemogenetic Optimization of an Artificial Suzukiase for the Synthesis of Enantioenriched Biaryls and Selected Results from Directed Evolution.

Benzannulation
In this context, the so-called concerted metalation–deprotonation (CMD) mechanism occupies a place of choice. Following coordination of a metal to a directing group, a C–H group interacts with the metal and is acidified. The presence of an external base leads to deprotonation and metalation. The Ward and Rovis groups joined forces to develop an ArM for enantioselective benzannulation. We hypothesized that introduction of a Lewis-basic residue in the immediate proximity of the precious-metal cofactor, a biotinylated [biot-Cp*RhCl2(H2O)], may allow the performance of the benzannulation at neutral pH, thus alleviating the use of a large excess of base to affect the CMD step. Incorporation of [biot-Cp*RhCl2(H2O)] within streptavidin afforded a benzannulase in the presence of 0.69 M acetate in a 4:1 water–MeOH mixture. Introduction of an aspartate residue (Sav K121D) in the proximity of the computed Rh-position afforded a benzannulase that did not require the addition of an external base. Further fine-tuning was achieved by the introduction of additional mutations within the biotin-binding vestibule: [biot-Cp*RhCl(H2O)]2+·Sav S112Y-K121E afforded the dihydroisoquinolone 38 in 82% ee (R), as a 19:1 regioisomeric mixture and 48 TONs, Scheme 19a–c.69
Scheme 19. Exploiting the CMD Mechanism for C–H Activation To Engineer an Artificial Benzannulase Based on the Biotin–Streptavidin Technology.

(a) Example of benzannulation resulting from iterative saturation mutagenesis. (b) Structure resulting from docking a computed cofactor in a partial X-ray structure. The positions of the rhodium and the nitrogen-amide were superimposed with the electron density from the X-ray structure. The protein is displayed as a solvent-accessible surface with mutated residues S112Y and K121E highlighted as green/red sticks. The cofactor is displayed as a stick and the rhodium as an orange sphere. (c) Cooperation from basic residue binding the active rhodium center. (d) Rovis’s benzannulation of representative hydroxamate ester 39 and styrene 40 by WT mSav:Cp*biotRhCl2 to afford δ-lactam 41.70
Recently, Rovis and co-workers significantly expanded the scope of this reaction. As homotetrameric Sav mutants only catalyzed the C–H activation of acrylate derivatives to yield δ-lactam 41 in low yield, the group relied on an engineered monomeric streptavidin (WT mSav), where the hydrophobic residues at the tetramer interface were mutated to charged ones to deter association. The [biot-Cp*RhCl2(H2O)]·WT mSav, with no additional active site mutations, catalyzed the reaction between acrylamide hydroxamate esters and styrenes to afford δ-lactams with yields of up to 99% and 97% ee. These can be readily reduced to afford pharmaceutically relevant piperidine scaffolds (Scheme 19d). The free cofactor [biot-Cp*RhCl2(H2O)] showed no enantioselectivity (0% ee) and very modest conversion in the reaction between 39 and 40 (15% yield of 41 for [biot-Cp*biotRhCl2(H2O)] versus 99% for [biot-Cp*RhCl2(H2O)]·WT mSav). To gain some insight into the origins of reactivity and stereocontrol derived from π-stacking in the binding pocket, the neighboring aromatic Y112 was removed (Y112A). This mutant was evaluated in the reaction between 39 and 40 and was observed to maintain biotin affinity but give much lower reactivity and selectivity (37% yield and 61% ee of 41 versus 99% yield and 91% ee with WT mSav), consistent with Y112 playing a crucial rigidifying role for the complex in the binding pocket.70
Metathesis
Although there are no naturally occurring metathases, the aqueous tolerance of ruthenium-based metathesis catalysts makes them attractive candidates for the implementation of ArMs. All artificial metathases reported to date rely on Hoveyda–Grubbs-type catalysts (HG-Ru) that are equipped with an anchoring moiety. The first metathases were reported simultaneously by Ward and Hilvert in 2011,71,72 using either the biotin–streptavidin (supramolecular) technology or a covalent modification of the heat-shock protein from Methanocaldococcus jannaschii (MjHSP), respectively. Since these initial reports, olefin metathesis has served as a propitious playground for testing novel concepts in ArMs and was recently reviewed.73,74
The M75L-H76L-Q96C-M148L-H158L mutant of nitrobindin (NB4), a dimeric 10-stranded β-barrel protein, was used by the Hayashi, Okuda, and Schwaneberg groups to create a metathase by covalent anchoring via a maleimide-cysteine coupling. Initially, the conjugation of a cofactor with three different linker lengths 42–44 was attempted with only the longest linker 44 showing a modest coupling yield (25%) to NB4 (44·NB4) via an engineered cysteine Q96C, Scheme 20. To improve the coupling efficiency, a nitrobindin variant was engineered with a larger cavity: NB4 L75A-L158A (NB11). Gratifyingly, it displayed increased conjugation efficiency: cofactors 42–43 with shorter linkers could be covalently coupled in high yield. This mutant conjugated to the longest cofactor 44·NB11 displayed high metathesis activity: >9000 TONs for the ROMP of norbornene derivative 46 and >100 TONs in the RCM of diene 45, Scheme 20.75
Scheme 20. Artificial Metathase Resulting from Covalent Anchoring within Engineered Nitrobindin (NB).
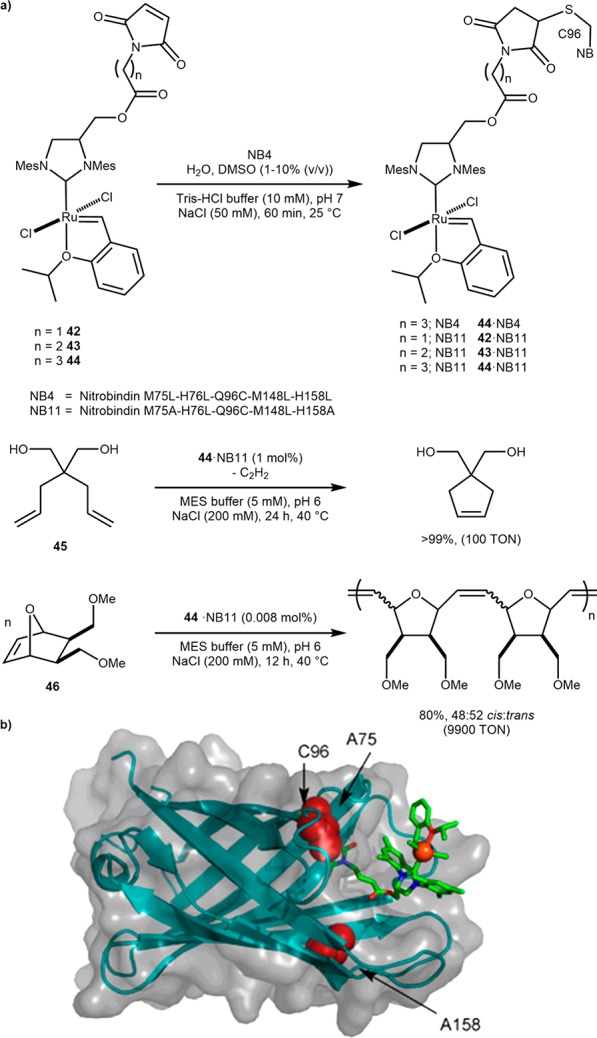
(a) Selected results and b) Transparent solvent-accessible surface representation of 44·NB11 resulting from docking, highlighting selected amino acids. Reproduced with permission from ref (75). Copyright 2015 American Chemical Society.
More recently, efforts have aimed at implementing and evolving an artificial metathase in whole cells. Having demonstrated that Hoveyda–Grubbs-type catalysts are irreversibly deactivated by glutathione, present in mM concentration in aerobic cells, Ward and Panke compartmentalized Sav in the periplasm of E. coli, where glutathione is mostly oxidized to the corresponding disulfide.76 Upon adding an OmpA leader peptide to Sav, the protein is efficiently secreted to the periplasm and shown to assemble into its homotetrameric state. Relying on a profluorescent diolefin 47, we optimized metathase activity in cellulo by directed evolution. Upon ring-closing metathesis, umbelliferone 48 is produced which can readily be detected by fluorescence in a 96-well plate format. The fifth-generation ArM [biot-HG-Ru]·Sav V47A-N49K-T114Q-A119G-K121R ([biot-HG-Ru]·Savmut) displayed 5-fold increased activity compared to [biot-HG-Ru]·WT Sav. Gratifyingly, the evolved metathase also proved more active toward more common diolefinic substrates: up to 680 TONs were observed with Savmut for the RCM of 45 and up to 45 TONs for cationic substrate 49. For the latter, one additional round of directed evolution was required to identify the beneficial mutation R121L in [biot-HG-Ru]·Savmut2 (i.e., V47A-N49K-T114Q-A119G-K121L) which, presumably, reduced charge repulsion in the active site, improving activity ∼2-fold versus [biot-HG-Ru]·Savmut (85 TONs), Scheme 21.
Scheme 21. Whole Cell Directed Evolution of an Artificial Metathase Based on the Biotin–Streptavidin Technology.

(a) Secretion of Sav into the periplasm for in cellulo evolution of the artificial metathase relying on the formation of fluorescent product 48 for expedited screening and b) Selected results for the RCM of dienes 45 and 49.
Alkyne Polymerization
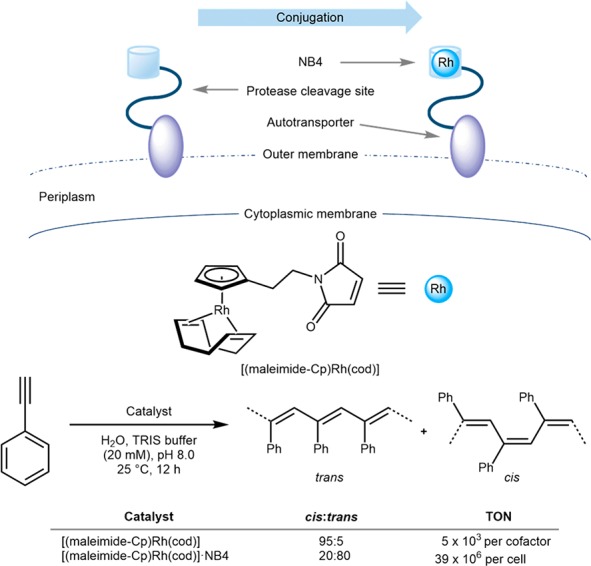
With the aim of further improving cofactor and substrate accessibility in the presence of E. coli whole cells, the groups of Okuda and Schwaneberg displayed nitrobindin on the outer membrane of E. coli. For this purpose, the nitrobindin variant scaffold NB4 (Scheme 20) was fused with an esterase autotransporter to display the host protein on the outer membrane of E. coli, Scheme 22. Following covalent anchoring of [(maleimide-Cp)Rh(cod)] to NB4, they were able to significantly increase the alkyne polymerization efficiency to afford poly(phenylacetylene): from a 5 × 103 TONs (per cofactor) for the free cofactor to 39 × 106 TONs (per cell) with [(maleimide-Cp)Rh(cod)]·NB4, Scheme 22. The cis:trans ratio was also markedly affected from 95% cis (the free cofactor) to 80% trans (for the ArM), Scheme 22.77 This outer-membrane display of ArMs complements the periplasm-compartmentalization strategy for the implementation of thiol-sensitive ArMs in vivo. The lower host-protein copy number that can be displayed on the outer-membrane versus in the periplasm seems to be compensated by the cofactor’s higher accessibility for surface-displayed ArMs.77,78 In a systematic study, Ward and co-workers quantified the cofactor uptake by ICP-MS analysis for both periplasm and surface-displayed ArMs. Both the cofactor concentrations and the TON were very similar in both cases: up to 90 TONs/cofactor could be achieved with both Sav and hCA II-based ArMs.21,79,80
Scheme 22. Surface Display of an Artificial Alkyne Polymerase Resulting from a Maleimide Cysteine Covalent Anchoring to an Engineered Nitrobindin (NB4).
Outlook
The fusion of enzymatic with homogeneous catalysis has resulted in the evolution of a powerful tool—ArMs—to address the drawbacks associated with both fields. The introduction of unnatural metal cofactors into well-defined coordination spheres has allowed the naturally evolved reaction repertoire to be greatly expanded. This simultaneously permits previously synthetically restricted reactions to be carried out under stringent control with equally impressive catalyst lifetimes. Indeed, ArMs hold many potential applications including becoming increasing practical candidates for industrial asymmetric synthesis, adding value in late-stage functionalization, and complementing natural enzymes for chemomimetic biocatalysis. Inspection of some of the most attractive features of both fields of catalysis suggests that ArMs combine some of the distinctive attributes of both worlds, Table 1. These characteristics have been emphasized where relevant vide supra, but all examples displaying the beneficial trait in question (bold) have been highlighted, Table 1.
Table 1. Artificial Metalloenzymes Combine Advantageous Features (in Bold) of Both Homogeneous Catalysts and Enzymes, Where Listed Numbers Refer to the Schemes That Summarize the Corresponding Feature.

In the authors’ opinion, the most attractive feature offered by ArMs is the possibility of improving the catalytic performance of a new-to-nature organometallic reaction by genetic means. We like to coin this “endowing organometallic catalysis with a genetic memory”.9 The greatest challenge associated with this feature is the pronounced susceptibility of precious-metal cofactors being irreversibly poisoned by cellular components. Accordingly, the directed evolution efforts in the field of ArMs most often rely on screening purified protein samples rather than lysed cells, cell-free extracts, or even performing catalysis in vivo. This imposes a significant bottleneck on the optimization effort. To overcome this limitation, it is highly desirable to use base metal-containing cofactors that are fully compatible with the complex environment present in a cell.
Acknowledgments
T.R.W. acknowledges continued support from the Swiss National Science Foundation (Grant 200020_182046), the NCCR Molecular Systems Engineering (Molecular Systems Engineering), and an ERC advanced grant (the DrEAM, Grant 694424). H.J.D. thanks the Marie Skłodowska-Curie H2020-MSCA-IF-2018 for a postdoctoral fellowship.
The authors declare no competing financial interest.
References
- Akabori S.; Sakurai S.; Izumi Y.; Fujii Y. An Asymmetric Catalyst. Nature 1956, 178, 323–324. 10.1038/178323b0.13358737 [DOI] [Google Scholar]
- Kagan H. B.Historical perspective. In Comprehensive Asymmetric Catalysis; Pfaltz A., Jacobsen E. N., Eds.; Springer-Verlag: Berlin Heidelberg, Germany, 1999; Vol. 1, pp 9–30. [Google Scholar]
- Nozaki H.; Moriuti S.; Takaya H.; Noyori R. Asymmetric Induction in Carbenoid Reaction by Means of a Dissymmetric Copper Chelate. Tetrahedron Lett. 1966, 7, 5239–5244. 10.1016/S0040-4039(01)89263-7. [DOI] [Google Scholar]
- Yamamura K.; Kaiser E. T. Studies on the Oxidase Activity of Copper(II) Carboxypeptidase A. J. Chem. Soc., Chem. Commun. 1976, 830–831. 10.1039/c39760000830. [DOI] [Google Scholar]
- Wilson M. E.; Whitesides G. M. Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc. 1978, 100, 306–307. 10.1021/ja00469a064. [DOI] [Google Scholar]
- Davies R. R.; Distefano M. D. A Semisynthetic Metalloenzyme Based on a Cavity that Catalyzes the Hydrolysis of Ester and Amide Substrates. J. Am. Chem. Soc. 1997, 119, 11643–11652. 10.1021/ja970820k. [DOI] [Google Scholar]
- Ory J. J.; Mazhary A.; Kuang H.; Davies R. R.; Distefano M. D.; Banaszak L. J. Structural Characterization of Two Synthetic Catalysts Based on Adipocyte Lipid-Binding Protein. Protein Eng., Des. Sel. 1998, 11, 253–261. 10.1093/protein/11.4.253. [DOI] [PubMed] [Google Scholar]
- Qi D.; Tann C.-M.; Haring D.; Distefano M. D. Generation of New Enzymes via Covalent Modification of Existing Proteins. Chem. Rev. 2001, 101, 3081–3111. 10.1021/cr000059o. [DOI] [PubMed] [Google Scholar]
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Köhler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Jeschek M.; Bahls M. O.; Schneider V.; Marliere P.; Ward T. R.; Panke S. Biotin-Independent Strains of Escherichia coli for Enhanced Streptavidin Production. Metab. Eng. 2017, 40, 33–40. 10.1016/j.ymben.2016.12.013. [DOI] [PubMed] [Google Scholar]
- Ward T. R. Artificial Metalloenzymes Based on the Biotin-Avidin Technology: Enantioselective Catalysis and Beyond. Acc. Chem. Res. 2011, 44, 47–57. 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- Heinisch T.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Challenges and Opportunities. Acc. Chem. Res. 2016, 49, 1711–1721. 10.1021/acs.accounts.6b00235. [DOI] [PubMed] [Google Scholar]
- Liang A. D.; Serrano-Plana J.; Peterson R. L.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res. 2019, 52, 585–595. 10.1021/acs.accounts.8b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letondor C.; Humbert N.; Ward T. R. Artificial Metalloenzymes Based on Biotin-Avidin Technology for the Enantioselective Reduction of Ketones by Transfer Hydrogenation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4683–4687. 10.1073/pnas.0409684102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creus M.; Pordea A.; Rossel T.; Sardo A.; Letondor C.; Ivanova A.; LeTrong I.; Stenkamp R. E.; Ward T. R. X-ray Structure and Designed Evolution of an Artificial Transfer Hydrogenase. Angew. Chem., Int. Ed. 2008, 47, 1400–1407. 10.1002/anie.200704865. [DOI] [PubMed] [Google Scholar]
- Dürrenberger M.; Heinisch T.; Wilson Y. M.; Rossel T.; Nogueira E.; Knörr L.; Mutschler A.; Kersten K.; Zimbron M. J.; Pierron J.; Schirmer T.; Ward T. R. Artificial Transfer Hydrogenases for the Enantioselective Reduction of Cyclic Imines. Angew. Chem., Int. Ed. 2011, 50, 3026–3029. 10.1002/anie.201007820. [DOI] [PubMed] [Google Scholar]
- Monnard F. W.; Nogueira E. S.; Heinisch T.; Schirmer T.; Ward T. R. Human Carbonic Anhydrase II as Host Protein for the Creation of Artificial Metalloenzymes: The Asymmetric Transfer Hydrogenation of Imines. Chem. Sci. 2013, 4, 3269–3274. 10.1039/c3sc51065d. [DOI] [Google Scholar]
- Hestericova M.; Heinisch T.; Alonso-Cotchico L.; Marechal J. D.; Vidossich P.; Ward T. R. Directed Evolution of an Artificial Imine Reductase. Angew. Chem., Int. Ed. 2018, 57, 1863–1868. 10.1002/anie.201711016. [DOI] [PubMed] [Google Scholar]
- Wilson Y. M.; Dürrenberger M.; Nogueira E. S.; Ward T. R. Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts. J. Am. Chem. Soc. 2014, 136, 8928–8932. 10.1021/ja500613n. [DOI] [PubMed] [Google Scholar]
- Mallin H.; Hestericova M.; Reuter R.; Ward T. R. Library Design and Screening Protocol for Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology. Nat. Protoc. 2016, 11, 835–852. 10.1038/nprot.2016.019. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Rebelein J. G.; Mallin H.; Trindler C.; Pellizzoni M. M.; Ward T. R. Genetic Engineering of an Artificial Metalloenzyme for Transfer Hydrogenation of a Self-Immolative Substrate in Escherichia coli’s Periplasm. J. Am. Chem. Soc. 2018, 140, 13171–13175. 10.1021/jacs.8b07189. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Kaufman G. K.; Urbach A. R.; Gitlin I.; Gudiksen K. L.; Weibel D. B.; Whitesides G. M. Carbonic Anhydrase as a Model for Biophysical and Physical Organic Studies of Protein and Protein-Ligand Binding. Chem. Rev. 2008, 108, 946–1051. 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Kajetanowicz A.; Ward T. R. Carbonic Anhydrase II as Host Protein for the Creation of a Biocompatible Artificial Metathesase. Org. Biomol. Chem. 2015, 13, 5652–5655. 10.1039/C5OB00428D. [DOI] [PubMed] [Google Scholar]
- Heinisch T.; Pellizzoni M.; Dürrenberger M.; Tinberg C. E.; Köhler V.; Klehr J.; Haussinger D.; Baker D.; Ward T. R. Improving the Catalytic Performance of an Artificial Metalloenzyme by Computational Design. J. Am. Chem. Soc. 2015, 137, 10414–10419. 10.1021/jacs.5b06622. [DOI] [PubMed] [Google Scholar]
- Köhler V.; Wilson Y. M.; Dürrenberger M.; Ghislieri D.; Churakova E.; Quinto T.; Knörr L.; Häussinger D.; Hollmann F.; Turner N. J.; Ward T. R. Synthetic Cascades are Enabled by Combining Biocatalysts with Artificial Metalloenzymes. Nat. Chem. 2013, 5, 93–99. 10.1038/nchem.1498. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Köhler V.; Ward T. R. An NAD(P)H-Dependent Artificial Transfer Hydrogenase for Multienzymatic Cascades. J. Am. Chem. Soc. 2016, 138, 5781–5784. 10.1021/jacs.6b02470. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Lebrun V.; Kitanosono T.; Mallin H.; Köhler V.; Haussinger D.; Hilvert D.; Kobayashi S.; Ward T. R. Upregulation of an Artificial Zymogen by Proteolysis. Angew. Chem., Int. Ed. 2016, 55, 11587–11590. 10.1002/anie.201605010. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Ward T. R. Cross-Regulation of an Artificial Metalloenzyme. Angew. Chem., Int. Ed. 2017, 56, 10156–10160. 10.1002/anie.201702181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y.; Kojima R.; Schwizer F.; Bartolami E.; Heinisch T.; Matile S.; Fussenegger M.; Ward T. R. A Cell-Penetrating Artificial Metalloenzyme Regulates a Gene Switch in a Designer Mammalian Cell. Nat. Commun. 2018, 9, 1943. 10.1038/s41467-018-04440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines D. J.; Clarke J. E.; Blagova E. V.; Dodson E. J.; Wilson K. S.; Duhme-Klair A.-K. Redox-Switchable Siderophore Anchor Enables Reversible Artificial Metalloenzyme Assembly. Nat. Catal. 2018, 1, 680–688. 10.1038/s41929-018-0124-3. [DOI] [Google Scholar]
- Mirts E. N.; Petrik I. D.; Hosseinzadeh P.; Nilges M. J.; Lu Y. A Designed Heme-[4Fe-4S] Metalloenzyme Catalyzes Sulfite Reduction like the Native Enzyme. Science 2018, 361, 1098–1101. 10.1126/science.aat8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble M. V.; Jarvis A. G.; Ward A. C. C.; Colburn J. D.; Götze J. P.; Bühl M.; Kamer P. C. J. Artificial Metalloenzymes as Catalysts for Oxidative Lignin Degradation. ACS Sustainable Chem. Eng. 2018, 6, 15100–15107. 10.1021/acssuschemeng.8b03568. [DOI] [Google Scholar]
- Khare S. D.; Kipnis Y.; Greisen P. J.; Takeuchi R.; Ashani Y.; Goldsmith M.; Song Y.; Gallaher J. L.; Silman I.; Leader H.; Sussman J. L.; Stoddard B. L.; Tawfik D. S.; Baker D. Computational Redesign of a Mononuclear Zinc Metalloenzyme for Organophosphate Hydrolysis. Nat. Chem. Biol. 2012, 8, 294–300. 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchfield L. A.; Tezcan F. A. Design and Construction of Functional Supramolecular Metalloprotein Assemblies. Acc. Chem. Res. 2019, 52, 345–355. 10.1021/acs.accounts.8b00617. [DOI] [PubMed] [Google Scholar]
- Song W. J.; Tezcan F. A. A Designed Supramolecular Protein Assembly with In Vivo Enzymatic Activity. Science 2014, 346, 1525–1528. 10.1126/science.1259680. [DOI] [PubMed] [Google Scholar]
- Song W. J.; Yu J.; Tezcan F. A. Importance of Scaffold Flexibility/Rigidity in the Design and Directed Evolution of Artificial Metallo-β-lactamases. J. Am. Chem. Soc. 2017, 139, 16772–16779. 10.1021/jacs.7b08981. [DOI] [PubMed] [Google Scholar]
- Studer S.; Hansen D. A.; Pianowski Z. L.; Mittl P. R. E.; Debon A.; Guffy S. L.; Der B. S.; Kuhlman B.; Hilvert D. Evolution of a Highly Active and Enantiospecific Metalloenzyme from Short Peptides. Science 2018, 362, 1285–1288. 10.1126/science.aau3744. [DOI] [PubMed] [Google Scholar]
- Der B. S.; Machius M.; Miley M. J.; Mills J. L.; Szyperski T.; Kuhlman B. Metal-Mediated Affinity and Orientation Specificity in a Computationally Designed Protein Homodimer. J. Am. Chem. Soc. 2012, 134, 375–385. 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der B. S.; Edwards D. R.; Kuhlman B. Catalysis by a De Novo Zinc-Mediated Protein Interface: Implications for Natural Enzyme Evolution and Rational Enzyme Engineering. Biochemistry 2012, 51, 3933–3940. 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764–806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Drienovska I.; Rioz-Martinez A.; Draksharapu A.; Roelfes G. Novel Artificial Metalloenzymes by In Vivo Incorporation of Metal-Binding Unnatural Amino Acids. Chem. Sci. 2015, 6, 770–776. 10.1039/C4SC01525H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drienovska I.; Alonso-Cotchico L.; Vidossich P.; Lledos A.; Marechal J. D.; Roelfes G. Design of an Enantioselective Artificial Metallo-Hydratase Enzyme Containing an Unnatural Metal-Binding Amino Acid. Chem. Sci. 2017, 8, 7228–7235. 10.1039/C7SC03477F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfes G. LmrR: A Privileged Scaffold for Artificial Metalloenzymes. Acc. Chem. Res. 2019, 52, 545–556. 10.1021/acs.accounts.9b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis A. G.; Obrecht L.; Deuss P. J.; Laan W.; Gibson E. K.; Wells P. P.; Kamer P. C. J. Enzyme Activity by Design: An Artificial Rhodium Hydroformylase for Linear Aldehydes. Angew. Chem., Int. Ed. 2017, 56, 13596–13600. 10.1002/anie.201705753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghattas W.; Dubosclard V.; Wick A.; Bendelac A.; Guillot R.; Ricoux R.; Mahy J. P. Receptor-Based Artificial Metalloenzymes on Living Human Cells. J. Am. Chem. Soc. 2018, 140, 8756–8762. 10.1021/jacs.8b04326. [DOI] [PubMed] [Google Scholar]
- Bos J.; Browne W. R.; Driessen A. J.; Roelfes G. Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold. J. Am. Chem. Soc. 2015, 137, 9796–9799. 10.1021/jacs.5b05790. [DOI] [PubMed] [Google Scholar]
- Villarino L.; Splan K. E.; Reddem E.; Alonso-Cotchico L.; Gutierrez de Souza C.; Lledos A.; Marechal J. D.; Thunnissen A. W. H.; Roelfes G. An Artificial Heme Enzyme for Cyclopropanation Reactions. Angew. Chem., Int. Ed. 2018, 57, 7785–7789. 10.1002/anie.201802946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P.; Yang H.; Ellis-Guardiola K.; Lewis J. C. Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation. Nat. Commun. 2015, 6, 7789. 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Swartz A. M.; Park H. J.; Srivastava P.; Ellis-Guardiola K.; Upp D. M.; Lee G.; Belsare K.; Gu Y.; Zhang C.; Moellering R. E.; Lewis J. C. Evolving Artificial Metalloenzymes via Random Mutagenesis. Nat. Chem. 2018, 10, 318–324. 10.1038/nchem.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. C. Beyond the Second Coordination Sphere: Engineering Dirhodium Artificial Metalloenzymes To Enable Protein Control of Transition Metal Catalysis. Acc. Chem. Res. 2019, 52, 576–584. 10.1021/acs.accounts.8b00625. [DOI] [PubMed] [Google Scholar]
- Oohora K.; Onoda A.; Hayashi T. Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts. Acc. Chem. Res. 2019, 52, 945–954. 10.1021/acs.accounts.8b00676. [DOI] [PubMed] [Google Scholar]
- Oohora K.; Meichin H.; Zhao L.; Wolf M. W.; Nakayama A.; Hasegawa J. Y.; Lehnert N.; Hayashi T. Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species. J. Am. Chem. Soc. 2017, 139, 17265–17268. 10.1021/jacs.7b10154. [DOI] [PubMed] [Google Scholar]
- Lewis R. D.; Garcia-Borras M.; Chalkley M. J.; Buller A. R.; Houk K. N.; Kan S. B. J.; Arnold F. H. Catalytic Iron-Carbene Intermediate Revealed in a Cytochrome C Carbene Transferase. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7308–7313. 10.1073/pnas.1807027115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Tinzl M.; Mori T.; Krengel U.; Proppe J.; Soetbeer J.; Klose D.; Jeschke G.; Reiher M.; Hilvert D. Capture and Characterization of a Reactive Haem-Carbenoid Complex in an Artificial Metalloenzyme. Nat. Catal. 2018, 1, 578–584. 10.1038/s41929-018-0105-6. [DOI] [Google Scholar]
- Sreenilayam G.; Moore E. J.; Steck V.; Fasan R. Stereoselective Olefin Cyclopropanation under Aerobic Conditions with an Artificial Enzyme Incorporating an Iron-Chlorin e6 Cofactor. ACS Catal. 2017, 7, 7629–7633. 10.1021/acscatal.7b02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux M.; Tyagi V.; Fasan R. Highly Diastereoselective and Enantioselective Olefin Cyclopropanation using Engineered Myoglobin-Based Catalysts. Angew. Chem., Int. Ed. 2015, 54, 1744–1748. 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key H. M.; Clark D. S.; Hartwig J. F. Generation, Characterization, and Tunable Reactivity of Organometallic Fragments Bound to a Protein Ligand. J. Am. Chem. Soc. 2015, 137, 8261–8268. 10.1021/jacs.5b04431. [DOI] [PubMed] [Google Scholar]
- Okrasa K.; Kazlauskas R. J. Manganese-Substituted Carbonic Anhydrase as a New Peroxidase. Chem. - Eur. J. 2006, 12, 1587–1596. 10.1002/chem.200501413. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gacio A.; Codina A.; Fastrez J.; Riant O.; Soumillion P. Transforming Carbonic Anhydrase into Epoxide Synthase by Metal Exchange. ChemBioChem 2006, 7, 1013–1016. 10.1002/cbic.200600127. [DOI] [PubMed] [Google Scholar]
- Jing Q.; Okrasa K.; Kazlauskas R. J. Stereoselective Hydrogenation of Olefins Using Rhodium-Substituted Carbonic Anhydrase - A New Reductase. Chem. - Eur. J. 2009, 15, 1370–1376. 10.1002/chem.200801673. [DOI] [PubMed] [Google Scholar]
- Jing Q.; Kazlauskas R. J. Regioselective Hydroformylation of Styrene Using Rhodium-Substituted Carbonic Anhydrase. ChemCatChem 2010, 2, 953–957. 10.1002/cctc.201000159. [DOI] [Google Scholar]
- Key H. M.; Dydio P.; Clark D. S.; Hartwig J. F. Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron. Nature 2016, 534, 534–537. 10.1038/nature17968. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Nazarenko A.; Rha J. Y. E.; Seyedkazemi V.; Clark D. S.; Hartwig J. F. An Artificial Metalloenzyme with the Kinetics of Native Enzymes. Science 2016, 354, 102–106. 10.1126/science.aah4427. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Hayashi H.; Clark D. S.; Hartwig J. F. Chemoselective, Enzymatic C-H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc. 2017, 139, 1750–1753. 10.1021/jacs.6b11410. [DOI] [PubMed] [Google Scholar]
- Key H. M.; Dydio P.; Liu Z. N.; Rha J. Y. E.; Nazarenko A.; Seyedkazemi V.; Cark D. S.; Hartwig J. F. Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes. ACS Cent. Sci. 2017, 3, 302–308. 10.1021/acscentsci.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudesly F.; Oble J.; Poli G. Metal-Catalyzed C-H Activation/Functionalization: The Fundamentals. J. Mol. Catal. A: Chem. 2017, 426, 275–296. 10.1016/j.molcata.2016.06.020. [DOI] [Google Scholar]
- Bar-Even A.; Noor E.; Savir Y.; Liebermeister W.; Davidi D.; Tawfik D. S.; Milo R. The Moderately Efficient Enzyme: Evolutionary and Physicochemical Trends Shaping Enzyme Parameters. Biochemistry 2011, 50, 4402–4410. 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- Chatterjee A.; Mallin H.; Klehr J.; Vallapurackal J.; Finke A. D.; Vera L.; Marsh M.; Ward T. R. An Enantioselective Artificial Suzukiase Based on the Biotin-Streptavidin Technology. Chem. Sci. 2016, 7, 673–677. 10.1039/C5SC03116H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K.; Knörr L.; Ward T. R.; Rovis T. Biotinylated Rh(III) Complexes in Engineered Streptavidin for Rate Enhanced Asymmetric C-H Activation. Science 2012, 338, 500–503. 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan I. S.; Ta A. N.; Danneman M. W.; Semakul N.; Burns M.; Basch C. H.; Dippon V. N.; McNaughton B. R.; Rovis T. Asymmetric δ-Lactam Synthesis with a Monomeric Streptavidin Artificial Metalloenzyme. J. Am. Chem. Soc. 2019, 141, 4815–4819. 10.1021/jacs.9b01596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C.; Ringenberg M. R.; Gnandt D.; Wilson Y.; Ward T. R. Artificial Metalloenzymes for Olefin Metathesis Based on the Biotin-(Strept)avidin Technology. Chem. Commun. 2011, 47, 12065–12067. 10.1039/c1cc15004a. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Gillingham D. G.; Ward T. R.; Hilvert D. An Artificial Metalloenzyme for Olefin Metathesis. Chem. Commun. 2011, 47, 12068–12070. 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]
- Sabatino V.; Ward T. R. Aqueous Olefin Metathesis: Recent Developments and Applications. Beilstein J. Org. Chem. 2019, 15, 445–468. 10.3762/bjoc.15.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer D. F.; Schiffels J.; Hayashi T.; Schwaneberg U.; Okuda J. Olefin Metathesis Catalysts Embedded in β-barrel Proteins: Creating Artificial Metalloproteins for Olefin Metathesis. Beilstein J. Org. Chem. 2018, 14, 2861–2871. 10.3762/bjoc.14.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer D. F.; Himiyama T.; Tachikawa K.; Fukumoto K.; Onoda A.; Mizohata E.; Inoue T.; Bocola M.; Schwaneberg U.; Hayashi T.; Okuda J. A Highly Active Biohybrid Catalyst for Olefin Metathesis in Water: Impact of a Hydrophobic Cavity in a β-Barrel Protein. ACS Catal. 2015, 5, 7519–7522. 10.1021/acscatal.5b01792. [DOI] [Google Scholar]
- Jeschek M.; Reuter R.; Heinisch T.; Trindler C.; Klehr J.; Panke S.; Ward T. R. Directed Evolution of Artificial Metalloenzymes for In Vivo Metathesis. Nature 2016, 537, 661–665. 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- Grimm A. R.; Sauer D. F.; Polen T.; Zhu L.; Hayashi T.; Okuda J.; Schwaneberg U. A Whole Cell E. coli Display Platform for Artificial Metalloenzymes: Poly(phenylacetylene) Production with a Rhodium-Nitrobindin Metalloprotein. ACS Catal. 2018, 8, 2611–2614. 10.1021/acscatal.7b04369. [DOI] [Google Scholar]
- Heinisch T.; Schwizer F.; Garabedian B.; Csibra E.; Jeschek M.; Vallapurackal J.; Pinheiro V. B.; Marliere P.; Panke S.; Ward T. R. E. coli Surface Display of Streptavidin for Directed Evolution of an Allylic Deallylase. Chem. Sci. 2018, 9, 5383–5388. 10.1039/C8SC00484F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschek M.; Panke S.; Ward T. R. Periplasmic Screening for Artificial Metalloenzymes. Methods Enzymol. 2016, 580, 539–556. 10.1016/bs.mie.2016.05.037. [DOI] [PubMed] [Google Scholar]
- Rebelein J. G.; Cotelle Y.; Garabedian B.; Ward T. R. Chemical Optimization of Whole-Cell Transfer Hydrogenation Using Carbonic Anhydrase as Host Protein. ACS Catal. 2019, 9, 4173–4178. 10.1021/acscatal.9b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]