

Abstract
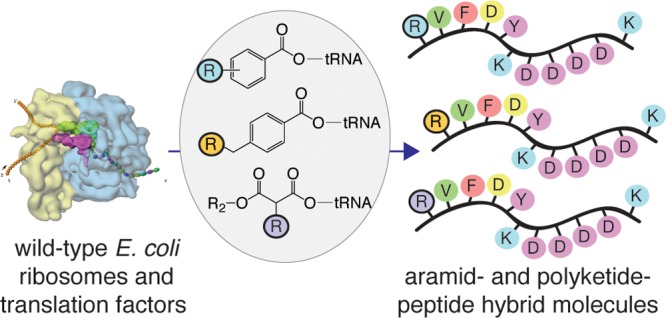
Here, we report that wild type Escherichia coli ribosomes accept and elongate precharged initiator tRNAs acylated with multiple benzoic acids, including aramid precursors, as well as malonyl (1,3-dicarbonyl) substrates to generate a diverse set of aramid-peptide and polyketide-peptide hybrid molecules. This work expands the scope of ribozyme- and ribosome-catalyzed chemical transformations, provides a starting point for in vivo translation engineering efforts, and offers an alternative strategy for the biosynthesis of polyketide-peptide natural products.
Short abstract
Wild type E. coli ribosomes elongate initiator tRNAs acylated with multiple benzoic acids and 1,3-dicarbonyl substrates to generate diverse aramid-peptide and polyketide-peptide hybrid molecules.
Introduction
As far as we know, ribosomes have evolved for billions of years to perform a single reaction—formation of an amide bond between two α-amino acid substrates brought into proximity by tRNAs within the ribosome active site, the peptidyl transferase center (PTC). In cells and extracts, the chemistry possible within a wild type ribosome PTC has expanded to include reactions of more than 200 different nonproteinogenic α-amino and hydroxy acids;1−4 ribosomes containing remodeled PTCs support amide bond formation to and from a small number of β-amino acids5−7 and dipeptides8,9 with limited efficiency. The combination of cell-free in vitro translation systems and ribozyme-catalyzed tRNA acylation reactions offers the opportunity for even greater reaction diversity, including the introduction of multiple N-alkyl,10d-α-,11,12 α-hydroxy,13 and β-amino acids,14,15 the precursors of β-peptide foldamers.16−19
A second family of foldamer-like molecules are aramids, oligomers of substituted aminobenzoic acids.20 Aramids possess remarkably varied properties. Kevlar, a polymer of 1,4-phenylenediamine and terephthaloyl chloride, is a strong and heat-resistant fiber,21 whereas cystobactamids are DNA gyrase inhibitors active against Gram-negative bacteria.22 Many other aramid foldamers with diverse and significant properties have been reported.23−25 Recently, wild type E. coli ribosomes were shown to accept and elongate initiator tRNAs precharged with aromatic foldamer-dipeptide appendages.26 Notably, in this case the foldamer monomers did not themselves react within the PTC, being displaced from the reaction center by a Gly–Phe dipeptide spacer.26,27 Here, we report that wild type E. coli ribosomes accept and elongate precharged initiator tRNAs acylated directly with multiple benzoic acids, including aramid precursors, as well as malonyl (1,3-dicarbonyl) substrates. The result is a diverse set of aramid-peptide and polyketide-peptide hybrid molecules. This work provides new knowledge about the generality of flexizyme-promoted tRNA acylation reactions, expands the scope of ribosome-catalyzed chemical transformations, provides a starting point for in vivo translation engineering efforts, and offers an alternative strategy for biosynthesis of polyketide-peptide natural products.
Results and Discussion
As the first step toward the ribosomal synthesis of aramid-like peptides, we made use of an established microhelix (MH) gel-shift assay28 and high-resolution mass spectrometry (Figure 1A) to evaluate whether the cyanomethyl esters of unsubstituted aminobenzoic acids were substrates for the flexizyme ribozyme eFx.29 Incubation of cyanomethyl esters 1–3 (5 mM) with 25 μM microhelix MH and 25 μM eFx (Table S1) in bicine buffer at pH 9 for 48 h showed little or no evidence of MH acylation when the reaction products were evaluated on an acid-urea PAGE gel (Figure 1B). A low level of MH acylation by the o- and om-analogues 1 and 2 (and a trace with 3) could be observed using a highly sensitive RNase A/LC-HRMS assay30 that detects the acylated adenine nucleoside (Figure 1C). We also investigated the extent of tRNA acylation using the alternative ribozyme dFx31 and the 1,3-dinitrobenzyl esters of p- and o-aminobenzoic acid (4 and 5, respectively, as shown in Scheme S1). These substrates also failed to yield the expected MH products when incubated with dFx under standard conditions14 and analyzed using acid-urea gels or RNase A/LC-HRMS (Figure S1), perhaps due to insolubility. Even the more soluble cyanomethyl ester of ortho-aminonicotinic acid analogue 6 reacted poorly in the presence of eFx (Figure S1).
Figure 1.

Simple aminobenzoic acid cyanomethyl esters are poor substrates for the eFx ribozyme. (A) Protocol used to detect acylation of microhelix (MH) or tRNA by cyanomethyl esters 1–3. (B) Acid-urea gel-shift analysis of MH acylation by cyanomethyl esters 1–3 in the presence of ribozyme eFx. Yield was estimated by UV densitometry. (C) LC-HRMS analysis of MH acylation reactions after RNase A digestion. Adenine nucleosides acylated on the 2′ or 3′ hydroxyl of the 3′ terminal ribose of MH could be detected in eFx-promoted reactions of the cyanomethyl ester of l-phenylalanine (Phe) and aminobenzoic acid esters 1 and 2; trace levels were detected in reactions containing 3. These products were not observed in analogous reactions containing m-aminobenzoic acid (compound C).
The inability to efficiently acylate MH or tRNA with simple aminobenzoic acids in high yields using eFx or dFx led us to consider chemical acylation methods for the preparation of these materials. Isatoic anhydride can acylate the terminal 2′- or 3′-OH group of an unprotected tRNA, and the resulting anthraniloyl-tRNA (o-AN-tRNA) retains the ability to associate productively with EF-Tu-GTP.32 Inspired by this reactivity, we incubated E. coli tRNAVal (ValT) or initiator tRNA (fMetT) with 8–80 mM isatoic anhydride in 90% CH3CN containing 2–5 mM NaOH for 3 h at 37 °C, digested the products with RNase A, and used LC-HRMS to detect the formation of nucleoside 7 (m/z = 387.1411, Scheme S1); this product will be observed only if reaction occurs at the tRNA 3′-end (Figure S2A). A peak corresponding to this mass was observed only in reactions containing tRNA, isatoic anhydride, and base; in the absence of base, the acylation efficiency dropped by 1–2 orders of magnitude (Figure S2B). Mindful of the fact that isatoic anhydride reagents can also modify RNA on the 2′-OH group of internal ribose residues in SHAPE reactions,33 we also evaluated the reaction using ultra-performance liquid chromatography (UPLC), which (as expected) showed evidence of multiple reaction products, whereas eFx-promoted reactions did not (Figure S2C).
We next made use of a commercial in vitro translation kit (PURExpress Δ; aa, tRNA) to evaluate if an initiator tRNA (fMetT) acylated with o- (prepared using isatoic anhydride) or m-aminobenzoic acid (prepared using eFx) would be accommodated by the P-site of wild type E. coli ribosomes and initiate translation. We supplemented the kit with the requisite amino acids and tRNAs, precharged initiator tRNA (o- or m-AN-tRNA) (50–100 μM), and a duplex DNA template (0.5–1 μg) encoding the FLAG-containing polypeptide MVFDYKDDDDK (MVF-FLAG). After a 6 h incubation, the reaction mixture was treated with Ni-NTA resin to remove all PURExpress Δ components (which are His6-tagged), and the remaining material was analyzed by LC-HRMS (Figure 2A). If the o- or m-AN-tRNA initiates translation in place of an initiator tRNA charged with formyl methionine (fMet), then a polypeptide product containing the sequence AN-VFDYKDDDDK (AN-VF-FLAG) should be observed. Parallel experiments were performed using the elongator tRNA ValT acylated (using eFx) with β-Phe.14 Clear evidence for the formation of a peptide carrying an aminobenzoic acid monomer was observed only in the presence of both DNA template and o-AN-tRNA (Figure 2B). The identity of this product was further confirmed by isotope labeling experiments (Figure S3) that showed the expected mass shift when the reaction was supplemented with 13C-labeled Phe. No AN-VF-FLAG polypeptide was detected in the presence of DNA template and m-AN-tRNA.
Figure 2.
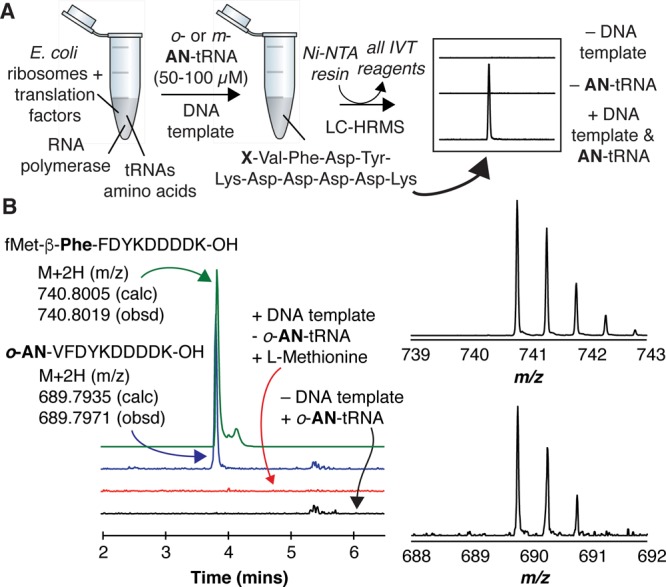
Initiator tRNA (fMetT) acylated with o-aminobenzoic acid can initiate translation within the PTC of wild type E. coli ribosomes. (A) Protocol used to evaluate whether an initiator tRNA (fMetT) acylated with o- (prepared using isatoic anhydride) or m-aminobenzoic acid (prepared using eFx) (AN-tRNA) could support translation in vitro. (B) LC-HRMS analysis of reaction products showing DNA template-dependent translation of a polypeptide whose mass corresponds to that of o-AN-VFDYKDDDDK (o-AN-VF-FLAG). No such polypeptide is observed in the absence of DNA template or in the presence of l-methionine. LC-HRMS analysis of an analogous β-Phe-containing polypeptide is shown for comparison.
Aminobenzoate esters hydrolyze exceptionally slowly,34 suggesting that the electron-rich aromatic ring contributes to the low reactivity of 1–3. In addition, the structure of the ethyl ester of l-phenylalanine bound to Fx (as an Fx-tRNA fusion)35 shows pi-stacking between Fx base guanine 24 and the l-phenylalanine aromatic ring; this stacking would be less favorable with an electron-rich arene.36,37 To investigate whether reactivity in eFx-promoted reactions was correlated with arene electron density, we prepared a diverse set of substituted benzoic acid cyanomethyl esters (Figure 3A) and evaluated the extent to which eFx reactivity correlated with the sign and magnitude of the relevant sigma factor, which measures the inductive effect of the aromatic substituent.38 As expected, benzoic acid cyanomethyl esters possessing strong electron-withdrawing substituents, such as penta-fluoro 8, p-nitro 9 (σ = +0.78), or p-Cl 10 (σ = +0.23), were excellent eFx substrates in model MH reactions, with 78-99% yields (Figure 3B,C). However, other factors are clearly important: a benzoic acid cyanomethyl ester possessing a weak electron-withdrawing substituent, such as p-azido 11 (σ = +0.08), was also an excellent substrate (yield of acylated MH = 74%), as were analogues possessing both strong and weak electron-donating substituents, such as p-methoxy 12 (σ = −0.27; yield of acylated MH = 62%) and p-methyl 13 (σ = −0.17; yield of acylated MH = 54%). Notably, the poorest yields were observed in eFx-promoted reactions of substrates 6 (yield of acylated MH = 25%) and 15 (yield of acylated MH = 23%), all of which contain one or more acidic protons/hydrogen bond donors, just like amino benzoic acids 1, 2, and 3. These results imply that the presence of hydrogen bond donors in certain positions contributes to the poor reactivity of amino benzoic acids 1–3. Consistent with this notion, p-hydroxybenzoic acid 16 [pKa = 8.3 (p-hydroxybenzoic acid methyl ester)] was a poor substrate, whereas alcohol 17 [pKa = 15 (benzyl alcohol)] and aldehyde 18 reacted well (Scheme S1 and Figure S4). It is possible that certain hydrogen bond donors alter the position of the aromatic ring in the eFx active site or coordinate and inactivate functional groups involved in catalysis. Determining the exact nature of these interactions is beyond the scope of this discussion but will be essential to effectively engineer new ribozymes that accept diverse foldamer building blocks in vitro and in vivo.
Figure 3.

Probing structure–activity relationships for cyanomethyl esters of substituted benzoic acids in eFx-promoted acylation reactions. (A) Substituted benzoic acid cyanomethyl esters studied herein. (B) Acid-urea gel-shift analysis of MH acylation by cyanomethyl esters 6 and 8–15 in the presence of ribozyme eFx. Yield was estimated by UV densitometry. (C) LC-HRMS analysis of MH acylation reactions containing cyanomethyl esters 6 and 8–15 after RNase A digestion. Exact masses are reported in Table S2.
With a new set of aramid substrates in hand, we used the PURExpress Δ (aa, tRNA) in vitro translation kit to evaluate if initiator tRNAs acylated with diverse benzoic acids could be accommodated in the ribosomal P-site and initiate translation of an AR-VF-FLAG polypeptide carrying an aramid monomer (AR) at the N-terminus (Figure 4). Every benzoic acid cyanomethyl ester that acylated the microhelix MH with a yield >50% in an eFx-promoted reaction (Figure 3) was used to acylate fMetT, and translation reactions were performed and analyzed as described above. With one exception, every single AR-fMetT initiated translation of an AR-VF-FLAG peptide whose mass corresponded to incorporation of the prescribed substituted benzoic acid. The singular exception was p-azidobenzoic acid 11; in this case the mass of the isolated polypeptide was consistent with in situ reduction of the azide to an amine. These results demonstrate that diverse aramid-like monomers can be accommodated directly within the ribosomal P-site and act as acceptors for a natural α-amino acid in the A-site. They show further that use of p-azidobenzoic acid 11 effectively circumvents the poor reactivity of p-aminobenzoic acid 3 to generate a polypeptide with a p-aminobenzoic acid monomer at the N-terminus. The observation that wild type E. coli ribosomes can initiate translation using tRNAs acylated with diverse aramid-like monomers significantly expands the scope of in vitro translation reactions beyond that of Kawakami39 and lays the initial groundwork for the biosynthesis of genetically encoded, sequence-defined polyaramid oligomers.
Figure 4.

Initiator tRNAs acylated with diverse benzoic acids are accommodated in the ribosomal P-site and are elongated into AR-VF-FLAG polypeptides. LC-HRMS analysis of reaction products whose masses correspond to AR-VFDYKDDDDK (AR-VF-FLAG) polypeptides containing diverse substituted benzoic acid monomers.
We next sought to evaluate the relative efficiency of PURExpress reactions initiated with differentially acylated fMetT derivatives. To begin, we monitored the yield of fMet-VF-FLAG (approximated as the extracted ion abundance) as a function of time in PURExpress Δ reactions supplemented with either 50 μM precharged fMetT-fMet (charged using the dFx substrate fMet-DBE) or 50 μM l-methionine. The bulk of both reactions was complete within 100 min, but the yield of fMet-VF-FLAG in reactions supplemented with precharged fMetT-fMet was 1.5% of that obtained in reactions supplemented with l-methionine (Figure S5A). Next, we compared the extracted ion abundance (after 30–90 min) of the AR-VF-FLAG peptide initiated with fMetT precharged with benzoic acid ester 8. The yield of this AR-VF-FLAG polypeptide (C6F5-VF-FLAG) was 25–30% of the yield of fMet-VF-FLAG (generated in reactions supplemented with precharged fMetT-fMet) (Figure S5B) and within the range observed when translation was initiated with fMetT precharged with natural amino acids.40 The relative yields of AR-VF-FLAG peptides initiated with other precharged fMetT derivatives were also comparable (Figure S5D), suggesting similar initiation efficiencies. We note that when ValT was precharged with β-Phe, the yield of fMet-β-Phe-F-FLAG was 5-fold higher than the yield of fMet-VF-FLAG generated with precharged fMetT (Figure S5C). As initiation complex assembly is the rate limiting step during translation,41 the higher yield of fMet-β-Phe-F-FLAG relative to fMet-VF-FLAG is likely due to the difficulty assembling the translation initiation complex using non-natural fMetT derivatives. Benzoic acid monomers that were poor eFx substrates (yields <50%) in model MH reactions, such as 6 and 15 (Figure 3), failed to detectably initiate peptide synthesis from WT ribosomes in vitro. This observation suggests that the ribosome is largely agnostic of aramid structure, and that the concentration of non-natural fMetT derivative, rather than monomer structure, determines the reaction yield in PURExpress reactions.42
Like aramid natural products,22 polyketide-peptide hybrid molecules are biosynthesized by mega-assemblies of complex protein enzymes;43−45 the combination of peptide and polyketide-based functionality can translate into highly unique biological functions.46−48 To evaluate whether wild type E. coli ribosomes are capable of biosynthesizing a polyketide-peptide hybrid, we prepared malonate derivatives 19–23 (Figure 5A). Model microhelix MHacylation reactions were analyzed using acid-urea gels (Figure 5B) and RNase A/LC-HRMS (Figure 5C) as described above. Although the malonic acid half esters 19, 20, and 21 were poor substrates for the requisite Fx analogue, cyanomethyl ester 22 was a moderate substrate, acylating the acylated MH in 40% yield. Although no gel-shift was observed in the eFx-promoted MH acylation reaction of cyanomethyl ester 23 (perhaps because of low molecular weight and/or polarity),49 strong evidence for reaction was observed using RNase A/LC-HRMS (Figure 5C). Indeed, the addition of fMetT derivatives acylated with 22 and 23 (50–100 μM) to PURExpress Δ (aa, tRNA) in vitro translation reactions led to the isolation of polypeptides carrying malonates 22 and 23 (22-VF-FLAG and 23-VF-FLAG, respectively), whose masses were confirmed by LC-HRMS (Figure 5D). The yield of 23-VF-FLAG, estimated as described above, was approximately 20% of the yield of fMet-VF-FLAG produced in reactions supplemented with precharged fMetT (Figure S5A). We conclude that extant E. coli ribosomes have the capacity to biosynthesize simple polyketide-peptide hybrid molecules.
Figure 5.

Wild type E. coli ribosomes support the biosynthesis of polyketide-peptide hybrid molecules. (A) Malonic esters 19–23 evaluated as substrates for eFx or dFx. (B) Acid-urea gel-shift analysis of MH acylation by esters 19–23 in the presence of eFx (19, 22) or dFx (20, 21,23). Yield was estimated by UV densitometry. (C) LC-HRMS analysis of MH acylation reactions containing esters 19–23 after RNase A digestion. (D) LC-HRMS analysis of reaction products whose masses corresponds to Mal-VFDYKDDDDK (Mal-VF-FLAG) polypeptides containing methyl and nitrobenzyl malonates 23 and 22. ND = not determined due to lack of separation from unacylated microhelix. Exact masses are reported in Table S2.
In summary, here we report that wild type E. coli ribosomes accept precharged initiator tRNAs acylated with multiple substituted benzoic acids, including the monomeric unit of Kevlar, as well as malonyl (1,3-dicarbonyl) substrates. The ribosome then elongates these substrates to generate a diverse set of aramid-peptide and polyketide-peptide hybrid molecules. This work expands the scope of reactions catalyzed by both flexizyme and wild type ribosomes, provides a starting point for in vivo translation engineering efforts, and offers an alternative strategy for biosynthesis of polyketide-peptide natural products.
Safety Statement
No unexpected or unusually high safety hazards were encountered during the execution of these experiments.
Acknowledgments
This work was supported by the Center for Genetically Encoded Materials, an NSF Center for Chemical Innovation (NSF CHE-1740549). O.A. was supported in part by Agilent Technologies as an Agilent Fellow.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00460.
Synthesis and characterization of flexizyme substrates; and procedures for formation, characterization, purification, and analysis of tRNA acylation and IVT reactions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Guo J.; Wang J.; Anderson J. C.; Schultz P. G. Addition of an α-hydroxy acid to the genetic code of bacteria. Angew. Chem., Int. Ed. 2008, 47, 722–725. 10.1002/anie.200704074. [DOI] [PubMed] [Google Scholar]
- Chin J. W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- Vargas-Rodriguez O.; Sevostyanova A.; Söll D.; Crnković A. Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 115–122. 10.1016/j.cbpa.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. D.; Schultz P. G. Playing with the molecules of life. ACS Chem. Biol. 2018, 13, 854–870. 10.1021/acschembio.7b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini R.; Nguyen D. T.; Chen S.; Dedkova L. M.; Chowdhury S. R.; Alcala-Torano R.; Hecht S. M. Incorporation of β-amino acids into dihydrofolate reductase by ribosomes having modifications in the peptidyltransferase center. Bioorg. Med. Chem. 2013, 21, 1088–1096. 10.1016/j.bmc.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Maini R.; Chowdhury S. R.; Dedkova L. M.; Roy B.; Daskalova S. M.; Paul R.; Chen S.; Hecht S. M. Protein synthesis with ribosomes selected for the incorporation of β-amino acids. Biochemistry 2015, 54, 3694–3706. 10.1021/acs.biochem.5b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo Czekster C.; Robertson W. E.; Walker A. S.; Söll D.; Schepartz A. In vivo biosynthesis of a β-amino acid-containing protein. J. Am. Chem. Soc. 2016, 138, 5194–5197. 10.1021/jacs.6b01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini R.; Dedkova L. M.; Paul R.; Madathil M. M.; Chowdhury S. R.; Chen S.; Hecht S. M. Ribosome-mediated incorporation of dipeptides and dipeptide analogues into proteins in vitro. J. Am. Chem. Soc. 2015, 137, 11206–11209. 10.1021/jacs.5b03135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Ji X.; Gao M.; Dedkova L. M.; Hecht S. M. In cellulo synthesis of proteins containing a fluorescent oxazole amino acid. J. Am. Chem. Soc. 2019, 141, 5597–5601. 10.1021/jacs.8b12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny A. O.; Hartman M. C. T.; Szostak J. W. Ribosomal synthesis of N-methyl peptides. J. Am. Chem. Soc. 2008, 130, 6131–6136. 10.1021/ja710016v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova L. M.; Fahmi N. E.; Golovine S. Y.; Hecht S. M. Enhanced d-amino acid incorporation into protein by modified ribosomes. J. Am. Chem. Soc. 2003, 125, 6616–6617. 10.1021/ja035141q. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Murakami H.; Suga H. Initiating translation with d-amino acids. RNA 2008, 14, 1390–1398. 10.1261/rna.1020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A.; Murakami H.; Higashimura E.; Suga H. Synthesis of polyester by means of genetic code reprogramming. Chem. Biol. 2007, 14, 1315–1322. 10.1016/j.chembiol.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Fujino T.; Goto Y.; Suga H.; Murakami H. Ribosomal synthesis of peptides with multiple β-amino acids. J. Am. Chem. Soc. 2016, 138, 1962–1969. 10.1021/jacs.5b12482. [DOI] [PubMed] [Google Scholar]
- Katoh T.; Suga H. Ribosomal incorporation of consecutive β-amino acids. J. Am. Chem. Soc. 2018, 140, 12159–12167. 10.1021/jacs.8b07247. [DOI] [PubMed] [Google Scholar]
- Wang P. S.; Schepartz A. β-peptide bundles: Design. Build. Analyze. Biosynthesize.. Chem. Commun. 2016, 52, 7420–7432. 10.1039/C6CC01546H. [DOI] [PubMed] [Google Scholar]
- Checco J. W.; Gellman S. H. Targeting recognition surfaces on natural proteins with peptidic foldamers. Curr. Opin. Struct. Biol. 2016, 39, 96–105. 10.1016/j.sbi.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalvo G.; Waegele M. M.; Shandler S.; Gai F.; DeGrado W. F. Infrared signature and folding dynamics of a helical β-peptide. J. Am. Chem. Soc. 2010, 132, 5616–8. 10.1021/ja100459a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach D.; Gardiner J. Beta-peptidic peptidomimetics. Acc. Chem. Res. 2008, 41, 1366–1375. 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- García J. M.; García F. C.; Serna F.; de la Peña J. L. High-performance aromatic polyamides. Prog. Polym. Sci. 2010, 35, 623–686. 10.1016/j.progpolymsci.2009.09.002. [DOI] [Google Scholar]
- Tanner D.; Fitzgerald J. A.; Phillips B. R. The kevlar story - an advanced materials case study. Angew. Chem., Int. Ed. Engl. 1989, 28, 649–654. 10.1002/anie.198906491. [DOI] [Google Scholar]
- Baumann S.; Herrmann J.; Raju R.; Steinmetz H.; Mohr K. I.; Háttel S.; Harmrolfs K.; Stadler M.; Máller R. Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew. Chem., Int. Ed. Engl. 2014, 53, 14605–14609. 10.1002/anie.201409964. [DOI] [PubMed] [Google Scholar]
- Saraogi I.; Incarvito C. D.; Hamilton A. D. Controlling curvature in a family of oligoamide α-helix mimetics. Angew. Chem., Int. Ed. 2008, 47, 9691–9694. 10.1002/anie.200803778. [DOI] [PubMed] [Google Scholar]
- Meisel J. W.; Hu C. T.; Hamilton A. D. Mimicry of a β-hairpin turn by a nonpeptidic laterally flexible foldamer. Org. Lett. 2018, 20, 3879–3882. 10.1021/acs.orglett.8b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S.; Kauffmann B.; Ferrand Y.; Huc I. Selective encapsulation of disaccharide xylobiose by an aromatic foldamer helical capsule. Angew. Chem., Int. Ed. 2018, 57, 13542–13546. 10.1002/anie.201808370. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Kwon S.; Dawson S. J.; Mandal P. K.; Suga H.; Huc I. Ribosomal synthesis and folding of peptide-helical aromatic foldamer hybrids. Nat. Chem. 2018, 10, 405–412. 10.1038/s41557-018-0007-x. [DOI] [PubMed] [Google Scholar]
- Schepartz A. Foldamers wave to the ribosome. Nat. Chem. 2018, 10, 377–379. 10.1038/s41557-018-0036-5. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Suga H. Flexizymes as a tRNA acylation tool facilitating genetic code reprogramming. Methods Mol. Biol. 2012, 848, 465–478. 10.1007/978-1-61779-545-9_29. [DOI] [PubMed] [Google Scholar]
- Murakami H.; Saito H.; Suga H. A versatile tRNA aminoacylation catalyst based on RNA. Chem. Biol. 2003, 10, 655–662. 10.1016/S1074-5521(03)00145-5. [DOI] [PubMed] [Google Scholar]
- McMurry J. L.; Chang M. C. Y. Fluorothreonyl-tRNA deacylase prevents mistranslation in the organofluorine producer Streptomyces cattleya. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11920–11925. 10.1073/pnas.1711482114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H.; Ohta A.; Ashigai H.; Suga H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- Nawrot B.; Sprinzl M. Aminoacyl-tRNA analogues; synthesis, purification and properties of 3-anthraniloyl oligoribonucleotides. Nucleosides Nucleotides 1998, 17, 815–829. 10.1080/07328319808004677. [DOI] [PubMed] [Google Scholar]
- Mortimer S. A.; Weeks K. M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 2007, 129, 4144–4145. 10.1021/ja0704028. [DOI] [PubMed] [Google Scholar]
- Drossman H.; Johnson H.; Mill T. Structure activity relationships for environmental processes 1: Hydrolysis of esters and carbamates. Chemosphere 1988, 17, 1509–1530. 10.1016/0045-6535(88)90204-4. [DOI] [Google Scholar]
- Xiao H.; Murakami H.; Suga H.; Ferré-D’Amaré A. R. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 2008, 454, 358–361. 10.1038/nature07033. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Lawson K. R.; Perkins J.; Urch C. J. Aromatic interactions. J. Chem. Soc. Perkins Trans. 2 2001, 2, 651–669. 10.1039/b008495f. [DOI] [Google Scholar]
- Meyer E. A.; Castellano R. K.; Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Kawakami T.; Ogawa K.; Hatta T.; Goshima N.; Natsume T. Directed evolution of a cyclized peptoid-peptide chimera against a cell-free expressed protein and proteomic profiling of the interacting proteins to create a protein-protein interaction inhibitor. ACS Chem. Biol. 2016, 11, 1569–1577. 10.1021/acschembio.5b01014. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Ohta A.; Sako Y.; Yamagishi Y.; Murakami H.; Suga H. Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol. 2008, 3, 120–129. 10.1021/cb700233t. [DOI] [PubMed] [Google Scholar]
- Gualerzi C. O.; Pon C. L. Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell. Mol. Life Sci. 2015, 72, 4341–4367. 10.1007/s00018-015-2010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Katoh T.; Suga H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. 10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]
- Staunton J.; Weissman K. J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001, 18, 380–416. 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- Dutta S.; Whicher J. R.; Hansen D. A.; Hale W. A.; Chemler J. A.; Congdon G. R.; Narayan A. R. H.; Håkansson K.; Sherman D. H.; Smith J. L.; Skiniotis G. Structure of a modular polyketide synthase. Nature 2014, 510, 512–517. 10.1038/nature13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins T.; Liu Y.-C.; Cane D. E.; Khosla C. Structure and mechanism of assembly line polyketide synthases. Curr. Opin. Struct. Biol. 2016, 41, 10–18. 10.1016/j.sbi.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Sanchez C.; Shen B. Hybrid peptide-polyketide natural products: biosynthesis and prospects toward engineering novel molecules. Metab. Eng. 2001, 3, 78–95. 10.1006/mben.2000.0171. [DOI] [PubMed] [Google Scholar]
- Silakowski B.; Nordsiek G.; Kunze B.; Blöcker H.; Máller R. Novel features in a combined polyketide synthase/non-ribosomal peptide synthetase: the myxalamid biosynthetic gene cluster of the myxobacterium Stigmatella aurantiaca Sga151. Chem. Biol. 2001, 8, 59–69. 10.1016/S1074-5521(00)00056-9. [DOI] [PubMed] [Google Scholar]
- Walsh C. T. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 2004, 303, 1805–1810. 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- Fujino T.; Kondo T.; Suga H.; Murakami H.. Exploring of minimal RNA substrate of flexizymes. ChemBioChem. 2019, in press. 10.1002/cbic.201900150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.