

Abstract
Background
Exposure to chronic psychosocial stress is a risk factor for atherosclerosis‐based cardiovascular disease. We previously demonstrated the increased expressions of cathepsin S (CatS) in atherosclerotic lesions. Whether CatS participates directly in stress‐related neointimal hyperplasia has been unknown.
Methods and Results
Male wild‐type and CatS‐deficient mice that underwent carotid ligation injury were subjected to chronic immobilization stress for morphological and biochemical studies at specific times. On day 14 after stress/surgery, stress enhanced the neointima formation. At the early time points, the stressed mice had increased plaque elastin disruption, cell proliferation, macrophage accumulation, mRNA and/or protein levels of vascular cell adhesion molecule‐1, angiotensin II type 1 receptor, monocyte chemoattractant protein‐1, gp91phox, stromal cell–derived factor‐1, C‐X‐C chemokine receptor‐4, toll‐like receptor‐2, toll‐like receptor‐4, SC35, galectin‐3, and CatS as well as targeted intracellular proliferating‐related molecules (mammalian target of rapamycin, phosphorylated protein kinase B, and p‐glycogen synthase kinase‐3α/β). Stress also increased the plaque matrix metalloproteinase‐9 and matrix metalloproteinase‐2 mRNA expressions and activities and aorta‐derived smooth muscle cell migration and proliferation. The genetic or pharmacological inhibition of CatS by its specific inhibitor (Z‐FL‐COCHO) ameliorated the stressed arterial targeted molecular and morphological changes and stressed aorta‐derived smooth muscle cell migration. Both the genetic and pharmacological interventions had no effect on increased blood pressure in stressed mice.
Conclusions
These results demonstrate an essential role of CatS in chronic stress–related neointimal hyperplasia in response to injury, possibly via the reduction of toll‐like receptor‐2/toll‐like receptor‐4–mediated inflammation, immune action, and smooth muscle cell proliferation, suggesting that CatS will be a novel therapeutic target for stress‐related atherosclerosis‐based cardiovascular disease.
Keywords: hyperplasia, hypertension, protease, stress, vascular disease
Subject Categories: Atherosclerosis
Clinical Perspective
What Is New?
We studied chronic stress–accelerated cathepsin S (CatS) expression in failing mouse carotid vasculature in response to injury.
CatS gene absence attenuates chronic stress–related oxidative stress production, inflammatory actions, proteolysis, and vascular smooth muscle cell migration/proliferation and ameliorates positive remodeling without affecting blood pressure.
Pharmacological CatS inhibition mimics CatS deletion‐mediated vasculoprotective actions.
What Are the Clinical Implications?
This study provides direct evidence of the role of CatS in stress‐related vascular remodeling in response to injury.
CatS will be a novel therapeutic target for controlling chronic psychological stress–related vascular remodeling and restenosis after intravascular interventions.
Introduction
Chronic psychological stress (CPS) in modern lifestyles is closely linked to the incidence of vascular aging, metabolic syndrome, thromboembolism, and cardiovascular disease (CVD).1, 2, 3, 4 Most experimental and clinical studies that investigated the impact of CPS on the cardiovascular system focused on activated sympathoadrenomedullary and hypothalamic‐pituitary‐adrenocortical axes.5, 6, 7 Comprehensive review articles have highlighted the close relationship between CPS and atherosclerosis‐based CVD.7, 8 Social stress upregulates inflammatory gene expressions in the leukocyte transcription through a β‐adrenergic induction of myelopoiesis.9 It was also reported that hematopoietic stem cell proliferation was sensitive to various pathological stressors.5 Recent studies from our laboratories demonstrated that CPS accelerated inflammation and reduced the vascular regenerative capacity in mice.10 However, the precise mechanisms involved in CPS‐related remodeling and restenosis remain largely unknown.
Cathepsins, which are members of the cysteine protease family, are primary intracellular proteases that function in the terminal protein degradation in lysosomes and the protein processing in other intracellular organelles, such as hormone secretory granules.11, 12, 13 Cathepsins were also recently demonstrated to play an important role in the remodeling of extracellular matrix proteins (eg, elastin, collagens, and proteoglycans), and cathepsins are involved in the initiation and progression of atherosclerotic CVD.14, 15, 16 Emerging evidence indicates that biological oxidative and inflammatory stresses resulted in increases in the expression of several proteases, including members of the cysteinyl cathepsins (ie, cathepsins L and K), which then modulate vascular smooth muscle cell (SMC) events (ie, migration, invasion, apoptosis, and proliferation) in cardiovascular inflammatory and metabolic disorders.
Cathepsin S (CatS) is one of the most potent mammalian elastases and collagenases. Several experimental and clinical investigations have obtained several important findings that contribute to our understanding of a pivotal role of CatS in tissue regeneration and CVD.16 For example, CatS‐mediated fibroblast transdifferentiation contributed to cardiac remodeling in response to ischemic stress.17 CatS activity controls neovascularization via the modulation of insulin receptor substrate‐2/peroxisome proliferator‐activated receptor‐γ axis activation in response to hypoxic stress, both in vivo and in vitro.18 Genetic and pharmacological interventions targeted toward CatS ameliorated cardiovascular remodeling and aneurysm formation in animal models.17, 19 Interestingly, recent studies reported that the stressed atherosclerotic plaques contain high levels of CatS/K compared with nonstressed control plaques.20, 21
The primary aim of our present study was thus to investigate the role(s) of CatS in chronic stress–related neointimal hyperplasia in a mouse carotid artery injury model, with a special focus on the CatS‐mediated modulation of inflammation and oxidative stress.
Methods
The authors declare that all supporting data are available with the article.
Animals
The animal study protocol (No. 27304) was approved by the Institutional Animal Care and Use Committee of Nagoya University (Nagoya, Japan). All animal laboratory studies were conducted in accord with the guidelines on animal care of the Nagoya University Graduate School of Medicine. The male CatS‐deficiency mice (knockout, CatS−/− 22) and wild‐type (CatS+/+) littermates (C57BL/6J) used in this experiment were 8 weeks old and weighed between 21 and 25 g. All mice were maintained in a 22°C room with a 12‐hour light/dark cycle and received drinking water ad libitum.
Mouse Carotid Artery Ligation and Restraint Stress Model
Mouse carotid artery ligation mode was developed, as previously described.23 In brief, the right common carotid artery was dissected and ligated just proximal to its bifurcation with a 5 to 0 silk ligature. We randomly divided mice (n=63) that underwent right carotid artery ligation (Figure 1A) into a nonstressed group and a stressed group. Nonstressed control mice were permitted contact with each other and left undisturbed, whereas the stressed mice received a 4‐hour session in an immobilization stress tube (catalog No.551‐BSRR; Natsume Seisakusho, Tokyo, Japan) once daily (between 9:00 am and 1:00 pm) for 7 days per week for 2 weeks, as described.21 For the evaluation of the role of CatS in stress‐related experimental neointimal hyperplasia, CatS+/+ and CatS−/− mice (n=33 for each group) that had undergone ligation surgery were randomly subjected to stress, as described above for 2 weeks. For a separate specific CatS inhibitor (CatS‐I) experiment, CatS+/+ mice (n=64) that had undergone ligation surgery were randomly assigned to 1 of 2 groups and administered (by oral gavage) vehicle (0.5% carboxymethylcellulose; CatS‐I [−]) or a CatS‐I known as Z‐FL‐COCHO for the CatS‐I (+) mice, 5 mg/kg per day (Calbiochem, San Diego, CA), twice daily from 2 days before the injury to 14 days after the injury and under continued daily 4‐hour immobilization stress. The blood pressure of each mouse was measured by the tail‐cuff method, as described.5
Figure 1.
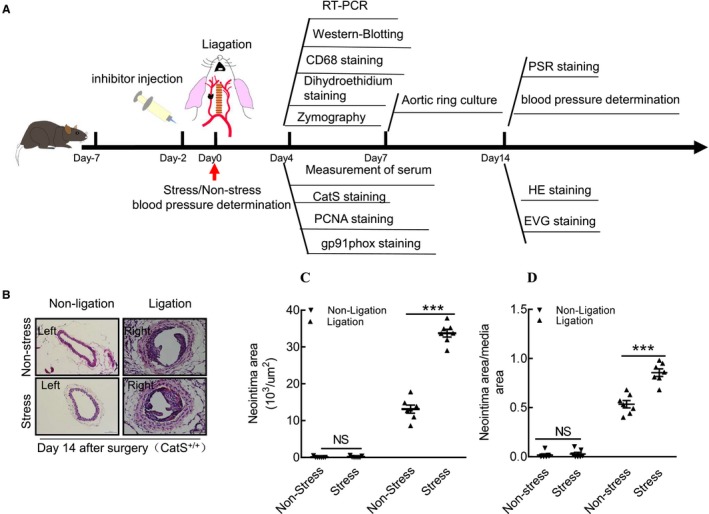
Chronic stress accelerated injury‐related neointimal formation in cathepsin S wild‐type (CatS+/+) mice. A, Schematic diagram of mouse right carotid ligation surgery and sampling procedures at the indicated time points. Dihydroethidium was used for oxidative stress staining. B, Representative hematoxylin and eosin (HE) staining images of right and left carotid arteries of the nonstressed and stressed groups. C and D, Quantitative data showing the neointimal areas and the ratio of neointima area/media area in injured arteries of both experimental groups. Bar=100 μm. Results are mean±SEM (n=7). EVG indicates Elastica van Gieson (for elastin staining); NS, no significance; PCNA, proliferating cell nuclear antigen (for proliferation staining); PSR, picrosirius red (for collagen staining); RT‐PCR, real‐time polymerase reaction chain. ***P<0.01 vs nonstressed group by Student t test.
Sample Collections
At the indicated time points after surgery and stress, mice were fasted 10 hours and received stress for 2 hours. At 1 hour after being released from the stress tube, the mice were then given an IP injection with an overdose of sodium pentobarbital (50 mg/kg; Dainippon Pharmaceutical, Osaka, Japan). The mice were then perfused with PBS under physiological pressure, and both carotid arteries were isolated. For the pathological evaluation, the arteries at day 14 after injury were fixed with 4% paraformaldehyde for 16 hours (4°C) and subsequently embedded in OCT (optimal cutting temperature) compound (Sakura Finetechnical, Tokyo, Japan) and stored at −30°C. For the biological analysis, the arteries at days 4 and 7 after injury were kept in RNAlater solution (for the gene assay) or liquid nitrogen (for the targeted protein evaluation).
Gene Expression Assay
Total RNA was extracted from the left and right carotid arteries of mice with the use of an RNeasy Fibrous Tissue Mini‐Kit (Qiagen, Hilden, Germany) and subjected to reverse transcription.23 The resulting cDNA was subjected to a quantitative real‐time polymerase reaction chain (PCR) analysis with primers specific for angiotensin II receptor 1 (ATR1), matrix metalloproteinase (MMP)‐9, MMP‐2, CatS, cathepsin L (CatL), intercellular adhesion molecule‐1 (ICAM‐1), vascular cell adhesion molecule‐1 (VCAM‐1), monocyte chemoattractant protein‐1 (MCP‐1), p22phox, p47phox, p67phox, gp91phox, nicotinamide‐adenine dinucleotide phosphate, reduced form (NADPH), oxidase 1, stromal cell–derived factor‐1 (SDF‐1α), C‐X‐C chemokine receptor‐4 (CXCR4), toll‐like receptor‐4 (TLR‐4), TLR‐2, tumor necrosis factor‐α (TNF‐α), and interleukin‐1β (IL‐1β) with the use of an ABI 7300 Real‐Time PCR System (Applied Biosystems, Foster City, CA). The transcription of targeted RNAs was normalized to GAPDH mRNA levels. The primer sequences are listed in the Table.
Table 1.
Primer Sequences for Mice Used for Quantitative Real‐Time PCR
| Genes | Forward Primers | Reverse Primers |
|---|---|---|
| p22phox | AACTACCTGGAGCCAGTTGAG | AATTAGGAGGTGGTGGAATATCGG |
| gp91phox | ACTTTCCATAAGATGGTAGCTTGG | GCATTCACACACCACTCAACG |
| p47phox | CTGAGGGTGAAGCCATTGAGG | GCCGGTGATATCCCCTTTCC |
| p67phox | AACTACCTGGAGCCAGTTGAG | AATTAGGAGGTGGTGGAATATCGG |
| NOX1 | TTGGCACAGTCAGTGAGGATG | AGATTTCAAGATGGAAGCAAAGGG |
| ICAM‐1 | CCCCGCAGGTCCAATTC | CCAGAGCGGCAGAGCAA |
| VCAM‐1 | ACAAAACGATCGCTCAAATCG | GGTGACTCGCAGCCCGTA |
| MCP‐1 | GCCCCACTCACCTGCTGCTACT | CCTGCTGCTGGTGATCCTCTTGT |
| SDF‐1α | CAGAGCCAACGTCAAGCATC | GTTCTTCAGCCGTGCAACAA |
| CXCR4 | CCACCCAGGACAGTGTGACTCTAA | GATGGGATTTCTGTATGAGGATT |
| TLR‐2 | AAGAAGCTGGCATTCCGAGGC | CGTCTGACTCCGAGGGGTTGA |
| TLR‐4 | AGTGGGTCAAGGAACAGAAGCA | CTTTACCAGCTCATTTCTCACC |
| TNF‐α | AGGCTGCCCCGACTACGT | GACTTTCTCCTGGTATGAGATAGCAAA |
| IL‐1β | TGCCACCTTTTGACAGTGATG | ATGTGCTGCTGCGAGATTTG |
| MMP‐9 | CCAGACGCTCTTCGA GAACC | GTTATAGAAGTGGCGGTTGT |
| ATR1α | TTTCCAGATCAAGTGCATTTTGA | AGAGTTAAGGGCCATTTTGCTTT |
| MMP‐2 | CCCCATGAAGCCTTGTTTACC | TTGTAGGAGGTGCCCTGGAA |
| CatL | GGCAACCCGATGCGC | TGTGTGACTCCTGTGAAGAACCA |
| CatS | GTGGCCACTA AAGGGCCTG | ACCGCTTTTGTAGAAGAAGAAGGAG |
| GAPDH | ATGTGTCCGTCGTGGATCTGA | ATGCCTGCTTCACCACCTTCT |
ATR1α indicates angiotensin receptor 1α; CatL, cathepsin L; CatS, cathepsin S; CXCR4, C‐X‐C motif chemokine receptor‐4; ICAM‐1, intercellular adhesion molecule‐1; IL‐1β, interleukin‐1β; MCP‐1, monocyte chemoattractant protein‐1; MMP, matrix metalloproteinase; NOX1, nicotinamide‐adenine dinucleotide phosphate, reduced form, oxidase 1; PCR, polymerase chain reaction; SDF‐1α, stromal cell–derived factor‐1α; TLR, toll‐like receptor; TNF‐α, tumor necrosis factor‐α; VCAM‐1, vascular cell adhesion molecule‐1.
Histological Characterization of the Carotid Arteries
The cross‐cryosections (5 μm) at 2 mm proximal to the ligated site were prepared and then stained with hematoxylin and eosin. The perimeters of the external elastic lamina, the internal elastic lamina, and the lumen were obtained by tracing the contours on digitized images. We measured the neointimal area by subtracting the lumen area from the area defined by the internal elastic lamina, and we calculated the medial area by subtracting the area defined by the internal elastic lamina from the area defined by the external elastic lamina.23
For the histological characterization of the plaques, we subjected corresponding sections of the arterial tissues at day 14 after surgery and stress to picrosirius red staining for collagen contents and Elastica van Gieson staining for elastin degradation. For the immunohistochemistry, corresponding sections of the arterial tissues at day 4 after surgery and stress were incubated with rat monoclonal antibodies against macrophages (CD68, 1:40; Chemicon International, Temecula, CA), mouse monoclonal antibodies against proliferating cell nuclear antigen (PCNA; catalog No. NA03; Merck Millipore, Darmstadt, Germany), or CatS (6686‐100; BioVision, Milpitas, CA), respectively. After 2 washes with PBS, the sections were then reacted with the secondary antibodies against rabbit or mouse IgG (1:200; all from Vector Laboratories, Burlingame, CA) for 1 hour at room temperature and were visualized with an ABC or M.O.M. substrate kit, respectively (Vector Laboratories) in accord with the manufacturer's instructions. For the evaluation of oxidative stress production, fresh‐frozen sections (5‐μm thick) were treated for 30 minutes at 37°C with dihydroethidium (1 mmol/L; Sigma‐Aldrich, St. Louis, MO). We also performed immunofluorescence with gp91phox antibody to show the cell source of the oxidative stress production in the vascular wall.
We analyzed images of sections stained for collagen content, elastin disruption, dihydroethidium positivity, PCNA+, CatS+, and CD68+ with the use of ImagePro software (BZ9000 Analysis; Keyence, Japan) under ×200 magnification. A total of 5 to 7 cross‐sections of vessels in each carotid artery were quantified and averaged for each animal. The results are reported as the percentage of the intima area that contained lesions.
Gelatin Zymography
For gelatin zymography, 20 μg of carotid protein extract was added to SDS sample buffer without reducing agent and loaded onto a 10% SDS–polyacrylamide gel containing 1 mg/mL gelatin, as described.20 After electrophoresis, the gels were washed twice with 2.5% Triton X‐100 (v/v) and then incubated in reaction buffer (0.15 mol/L NaCl, 0.02% NaN3, 50 mmol/L Tris‐HCl, and 10 mmol/L CaCl2, pH 7.4) at 37°C overnight. After staining with Coomassie Brilliant Blue, the gels were washed with destaining buffer for 30 minutes. Digestion bands were analyzed by an image analyzer software program (NIH Image 1.62).
Western Blot Analysis
The carotid artery was homogenized in lysis buffer (150 mmol/L NaCl, 1 mmol/L EDTA, Triton X‐100, 20 mmol/L Tris‐Cl, 1% SDS, 1% Na‐deoxycholate, and fresh 1× protease inhibitor cocktail; pH 7.4). The protein concentration was measured with a DC protein assay kit (Bio‐Rad Laboratories, Hercules, CA). The same amounts of protein (40 μg) were loaded and separated by SDS‐PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis). The membranes were incubated overnight with primary antibodies against protein kinase B (Akt; 2967), phosphorylated Akt (p‐Akt)s473 (4060), mammalian target of rapamycin (mTOR; 4517), phosphorylated mTOR (p‐mTOR)s2448 (2971), GSK (phosphoglycogene synthesis kinase)3α/β (5676), phosphorylated GSK3α/β (p‐GSK3α/β)s21/9 (9331), p38 mitogen‐activated protein kinase (p38MAPK; 9212), phosphorylated p38MAPK (p‐p38MAPK)t180/t182 (4511), extracellular signal‐regulated kinase (Erk) 1/2 (9107), and phosphorylated Erk (p‐Erk)t202/t204 (4377) (Cell Signaling Technology, Beverly, MA; 1:1000); ATR1 (sc‐1173), p22phox (sc‐11712), and GAPDH (sc‐20357) (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500); gp91phox (clone 53; BD Transduction Laboratories, San Jose, CA; 1:1000); galectin‐3 (1:1000, ab76245; Abcam, Cambridge, MA); SC35 (1:1000, clone αSC35; BD Pharmingen, San Diego, CA); and p16INK4A (1:1000, catalog No. 10883‐1‐AP; Proteintech, Chicago, IL). Then, they were treated with the related secondary antibodies at a 1:10 000 to 1:15 000 dilution. The Amersham ECL Prime Western Blotting Detection kit (GE Healthcare, Freiburg, Germany) was used for the evaluation of targeted proteins. Targeted protein levels quantitated from Western blots were normalized by loading internal controls.
Assay of NADPH Oxidase‐Dependent Superoxide Production
Superoxide production by total homogenates of the fresh aortic tissue was measured with the use of a lucigenin‐based enhanced chemiluminescence assay, as described.24 A low lucigenin concentration (5 μmol/L) was used to minimize artifactual superoxide production attributable to oxidation‐reduction cycling. In brief, 100 μg of homogenate protein diluted in 1 mL of lysis buffer (20 mmol/L Tris‐HCl, 150 mmol/L NaCl, 1 mmol EDTA, 1 mmol/L EGTA, and 1% Triton X‐100; pH 7.5) was transferred to an assay tube, and NADPH and dark‐adapted lucigenin were added to final concentrations of 100 and 5 μmol/L, respectively, immediately before the measurement of chemiluminescence. The chemiluminescence signal was sampled every minute for 12 minutes with a tube luminometer (20/20; Tuner Designs), and the respective background counts were subtracted from the experimental values. All the assays were performed in triplicate.
Plasma Corticosterone Level Analysis
The levels of mouse plasma corticosterone were examined at a commercial laboratory (SRL, Tokyo, Japan).10
Preparation of Explants and Migration Assay
For aorta‐ring cultures, the aortae were removed and opened out, and the endothelium was removed by gentle abrasion, as described.23 After the aorta was cut into 1‐×1‐mm explants, the explants were individually plated with the lumen side down into collagen type 1–coated 24‐well plates and cultured in 500 μL of DMEM (Dulbecco’s Modified Eagle Medium) containing transferrin, insulin, 0.1% BSA (bovine serum albumin), and platelet‐derived growth factor BB (50 ng/mL; PeproTech, London, UK). At the indicated time points, we performed a quantitative analysis of SMC sprouts at the edge of the explants using a BZ‐X700 microscope and a BZ‐X Analyzer (Keyence) under ×200 magnification. The SMC migratory ability is expressed as the sprouted total cell numbers and areas (average of 5–7 explants for each animal).
Statistical Analysis
Data are expressed as the mean±SEM. We performed a 1‐way ANOVA for comparisons of ≥3 groups, followed by Tukey's post hoc test or by the Student t test (for comparisons of 2 groups) with SPSS software, version 19.0 (SPSS, Chicago, IL). P<0.01 was considered significant. After testing the data distribution status, the data were subjected to the statistical analysis. If the homogeneity of variance assumption was violated, the nonparametric Kruskal‐Wallis test was used instead. All morphological analyses were evaluated by 2 observers in a blinded manner, and the values they obtained were averaged by 2 observers.
Results
Chronic Stress Accelerated the Injury‐Related Neointimal Formation in CatS+/+ Mice
Representative images of the nonstressed and stressed carotids for both left and right arteries of CatS+/+ mice are provided in Figure 1B. We assessed the impact of chronic stress on injury‐related neointimal information by measuring the neointima area and the ratio of the neointima area/the media area in cross‐sections of the carotid. The results demonstrated that the chronic stress significantly enhanced the area of the intima compared with the nonstressed controls (Figure 1C). The stress also significantly accelerated the ratio of the neointima area/the media area in the lesions compared with the corresponding controls (Figure 1D). However, stress exhibited no effect on neointima formation in the uninjured carotid arteries compared with the corresponding control arteries (Figure 1B through 1D).
The immunostaining revealed a marked and significant increase in PCNA+ cells in the injured tissues of the stressed mice (Figure 2A and 2B). The Elastica van Gieson staining revealed that the degradation of the elastic in the carotid of the stressed mice was significantly more serious than that in the nonstressed mice (Figure 2C and 2D). The picrosirius red staining showed a significant reduction of the collagen content in the stressed mice compared with the nonstressed mice (Figure 2E and 2F). The stressed aortae had increased sprouted SMC numbers and sprouted areas at days 1, 4, and 7 after culture (Figure 3A through 3C). In addition, the levels of plasma corticosterone were higher in the stressed mice than those of the unstressed mice (141.0±15.7 versus 97.6±11.4 ng/mL; P<0.01). The stressed mice had significantly increased blood pressure compared with the nonstressed mice (120.8±2.6 versus 92.5±2.3 mmHg; P<0.01).
Figure 2.
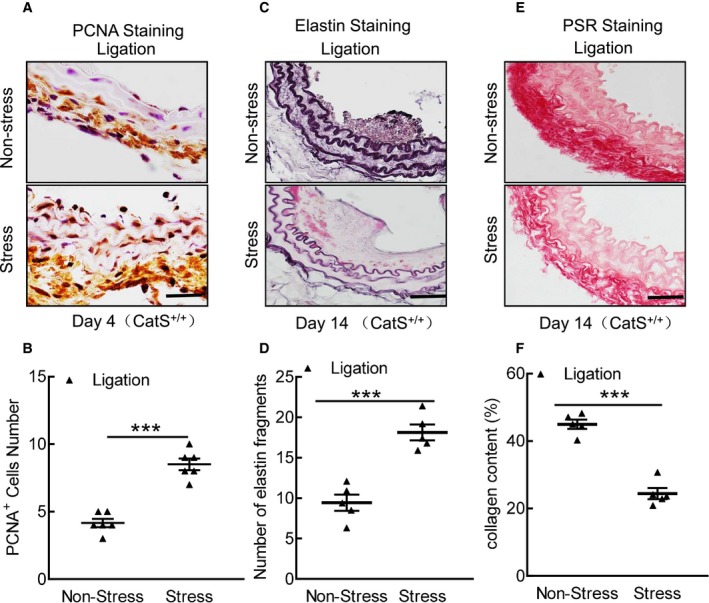
Stress accelerates cell proliferation and media elastic laminal degradation and collagen in cathepsin S wild‐type (CatS+/+) mice. A and B, Representative proliferating cell nuclear antigen (PCNA) immunostaining images of media smooth muscle cell proliferation and combined quantitative data for PCNA + cells. C through F, Representative images and quantitative data for the number of broken elastin fragments (Elastica van Gieson) and collagen content (picrosirius red [PSR]). Bar=100 μm. The results are mean±SEM (n=5–6). ***P<0.001 vs CatS+/+ group by Student t test.
Figure 3.
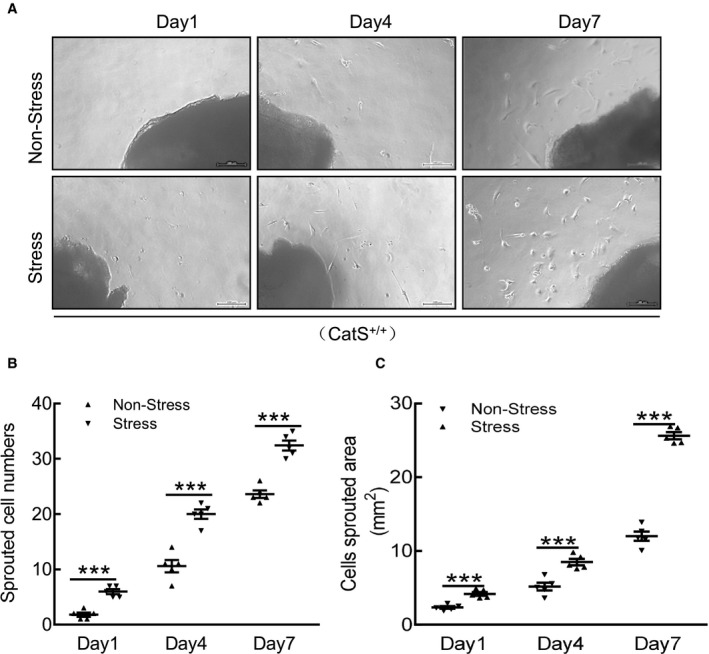
Stress accelerates cathepsin S wild‐type (CatS+/+) aorta‐derived smooth muscle cell migration. A, Representative images and quantitative data for the sprouted cell numbers and cell sprouted areas at the indicated time points. B and C, Quantitative data showing the sprouted cell numbers and cell sprouted areas of the 2 experimental groups at the indicated time points. Bar=100 μm. Results are mean±SEM (n=5). ***P<0.001 vs corresponding nonstressed groups by 1‐way ANOVA, followed by Tukey post hoc tests.
The Impact of the Stress on Inflammation, Oxidative Stress, and Proteolysis
At day 4 after ligation and stress, the expressions of the inflammation‐ and oxidative stress–related genes (ie, p22phox, gp91phox, p47phox, p67phox, NADPH oxidase 1, ICAM‐1, VCAM‐1, MCP‐1, SDF‐1α, CXCR4, TLR‐2, TLR‐4, TNF‐α, and IL‐1β) in the carotid of CatS+/+ mice were analyzed by quantitative real‐time PCR. The results demonstrated that all these parameters were significantly elevated in the stressed mice compared with the nonstressed mice (Figure 4A through 4D). We also observed that the mRNA expressions of ATR1, MMP‐2, MMP‐9, CatL, and CatS were significantly elevated in the stressed mice compared with the nonstressed mice (Figure 4E and 4F). Next, we performed immunostaining to identify the cell sources of CatS in injured arteries. As shown in Figure 5A, the positive staining signaling was pronounced in the media and neointima (thin) of injured vessel from CatS+/+ mice on day 4, whereas no signaling was detected in the whole arterial walls of CatS−/− mice (Figure 5B). CatS staining of the aorta cross‐sections taken on day 4 after ex vivo cultured aortae of CatS+/+ mice had increased levels of CatS protein (Figure 5C).
Figure 4.
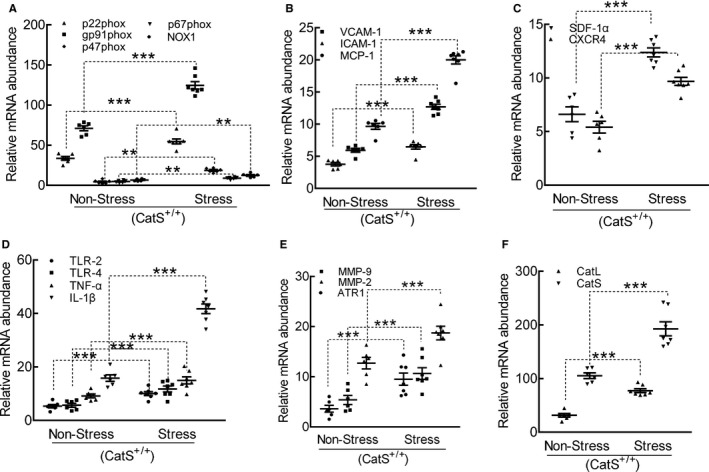
Stress produced a harmful change in the targeted oxidative stress–, inflammation‐, and proteolysis‐related gene expressions in the carotid arteries of cathepsin S wild‐type (CatS+/+) mice. A through F, Quantitative polymerase chain reaction data show the levels of p22phox, p47phox, p67phox, nicotinamide‐adenine dinucleotide phosphate, reduced form, oxidase 1 (NOX‐1), gp91phox, intercellular adhesion molecule‐1 (ICAM‐1), vascular cell adhesion molecule‐1 (VCAM‐1), monocyte chemoattractant protein‐1 (MCP‐1), stromal cell–derived factor‐1α (SDF‐1α), C‐X‐C chemokine receptor‐4 (CXCR‐4), toll‐like receptor (TLR)‐2, TLR‐4, tumor necrosis factor‐α (TNF‐α), interleukin‐1β (IL‐1β), angiotensin II receptor 1α (ATR1α), matrix metalloproteinase (MMP)‐9, MMP‐2, cathepsin L (CatL), and CatS mRNAs. Results are mean±SEM (n=5–7). ***P<0.001 vs nonstressed group by 1‐way ANOVA, followed by Tukey post hoc tests.
Figure 5.
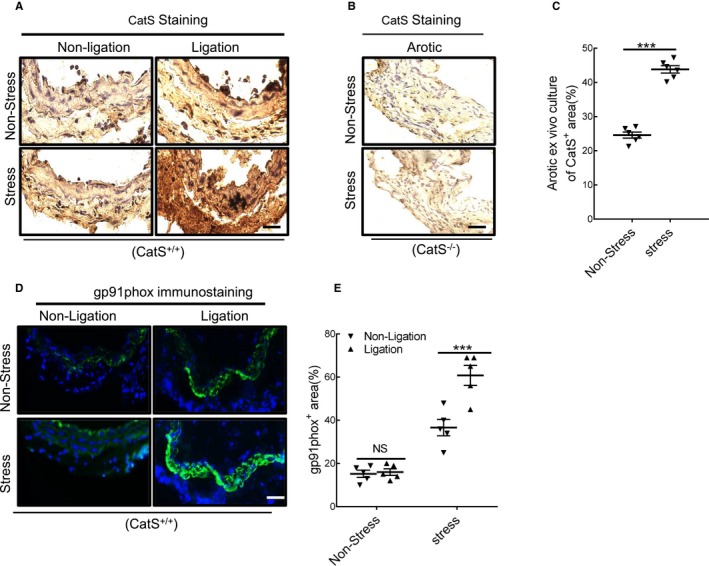
Immunostaining showed the cell sources of cathepsin S (CatS) in injured arteries. A and B, Representative images of CatS staining that show the positive staining signaling was pronounced in the media and neointima (thin) of injured vessels from wild‐type CatS (CatS+/+) mice on day 4 after surgery, whereas no signaling was detected in the whole arterial walls of CatS‐deficient (CatS−/−) mice. C, CatS staining of cross‐sections taken on day 4 after ex vivo cultured aortae of CatS+/+ mice received stress. D and E, Representative images and quantitative data show the gp91phox+ staining area. Results are mean±SEM (n=5–7). NS indicates no significance. ***P<0.001 vs nonstressed group by 1‐way ANOVA, followed by Tukey post hoc tests.
Immunofluorescence revealed that the stressed arteries had dramatically increased the levels of gp91phox protein than those in the nonstressed mice (Figure 5D and 5E). The dihydroethidium staining showed that chronic stress enhanced the oxidative stress production (Figure 6A and 6B). As anticipated, stress also significantly increased NADPH oxidase activity in the injured arterials than that of the nonstressed control arterials (189.3±11.6 versus 123.3±7.5 RLU (relative luminescence units)/mg protein; P<0.01). Similarly, the stressed lesions had accelerated macrophage infiltration (Figure 6C and 6D). The gelatin zymography revealed that stressed lesions had increased MMP‐9 and MMP‐2 activities (Figure 6E through 6G). As expected, the quantitative data of immunoblots revealed that the injured arterial tissues had increased levels of targeted oxidative stress–, inflammation‐, protein synthesis–, and cytoprotection‐related proteins (ie, gp91phox, p22phox, galectin‐3, ATR1, p16INK4A, p‐Akt, p‐mTOR, p‐Erk1/2, p‐p38MAPK, and p‐GSK3α/β; Figure 7A through 7C). However, there was no difference in the levels of the targeted molecules in uninjured vessels of CatS+/+ mice (Figures 6 and 7).
Figure 6.
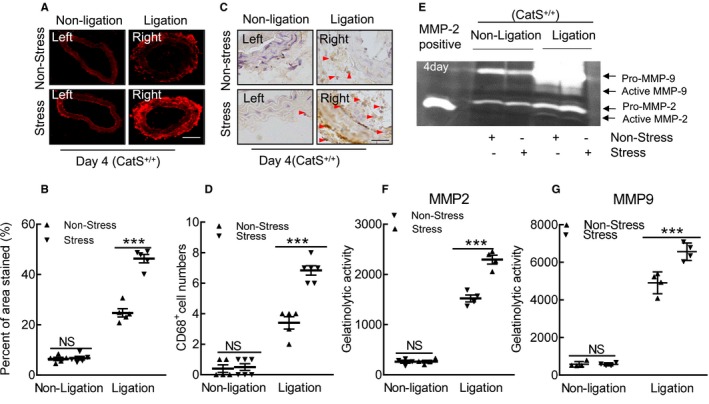
Stress accelerated the oxidative stress production and macrophage infiltration in response to injury at day 4. A and B, Representative images of dihydroethidium staining and quantitative data for positive staining area of the injured arterial tissues. C and D, Representative images and quantitative data for CD68+ cell numbers (arrowheads). E through G, Representative images of gelatin zymography and combined quantitative data for gelatinolytic activities of matrix metalloproteinase (MMP)‐2 and MMP‐9 in the nonstressed and stressed groups. Bar=100 μm. Results are mean±SEM (n=4–6). CatS+/+ indicates cathepsin S wild type; NS, no significance. ***P<0.001 vs nonstressed group by 1‐way ANOVA, followed by Tukey post hoc tests.
Figure 7.
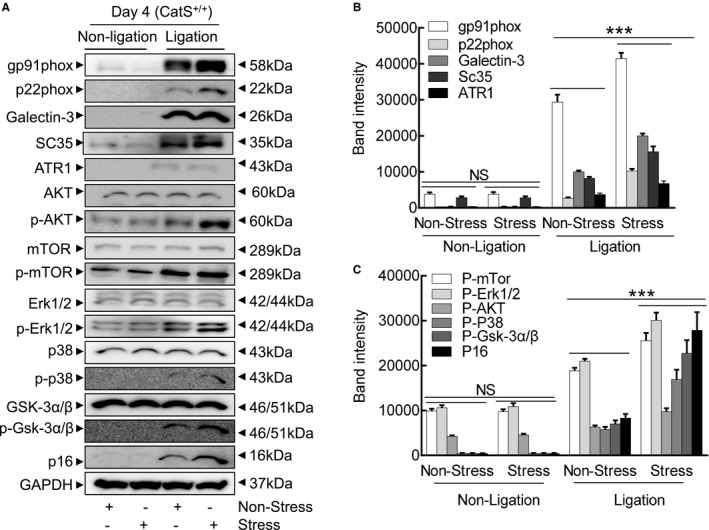
Stress altered the levels of targeted protein in the injured arteries. A through C, Representative images and combined quantitative data for the levels of targeted proteins. Results are mean±SEM (n=5–7). AKT indicates protein kinase B; ATR1, angiotensin II receptor 1; CatS+/+, cathepsin S wild type; Erk, extracellular signal‐regulated kinase; GSK, phosphoglycogene synthesis kinase; mTOR, mammalian target of rapamycin; NS, no significance; p‐AKT, phosphorylated AKT; p‐Erk, phosphorylated Erk; p‐mTOR, phosphorylated mTOR. ***P<0.001 vs the relative controls by 1‐way ANOVA, followed by Tukey post hoc tests.
CatS−/− Mitigated the Stress‐Related Neointimal Formation in Mice That Received Stress
As shown in Figure 8A and 8B, significantly less intimal lesion formation was observed in the injured carotid arterials of the stressed CatS−/− mice on day 14 compared with the stressed CatS+/+ mice, but no significant difference in media thickness was observed between the stressed mice of both genotypes (data not shown). Therefore, the ratio of intimal/medial area was significantly higher in the stressed CatS+/+ mice (Figure 8C). As anticipated, we observed a marked and significant reduction in PCNA+ cells in the injured tissues of the stressed CatS−/− mice (Figure 9A and 9B). Our quantitative morphological analysis revealed that CatS deletion significantly mitigated vascular collagen and elastic lamina metabolic degradation (Figure 9C through 9F). The stressed CatS−/− aortae had impaired sprouted SMC numbers and sprouted areas at day 7 after culture (Figure 10A through 10C). However, CatS deficiency did not significantly affect the increased blood pressure (118.8±2.7 versus 122.0±2.2 mmHg; P>0.05) and plasma corticosterone levels (129.8±28.3 versus 135.8±11.3 ng/mL; P>0.05) in the stressed mice. In addition, it is consistent with our previous study that CatS deletion also markedly improved neointima hyperplasia in mice that received ligation injury without chronic stress (Figure 10D through 10F).
Figure 8.
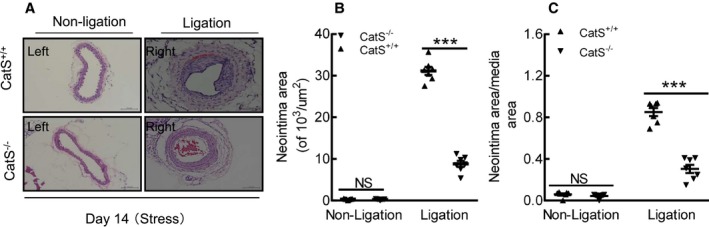
Cathepsin S deficiency (CatS−/−) alleviated injury‐induced neointimal formation in the mice subjected to chronic stress. A, Representative hematoxylin and eosin staining images of right and left carotid arteries of wild‐type (CatS+/+) and CatS−/− stressed mice. B and C, Quantitative data showing the neointimal areas and the ratio of neointima area/media area in injured arteries of the 2 experimental groups. Bar=100 μm. The results are mean±SEM (n=5–7). NS indicates no significance. ***P<0.01 vs CatS+/+ group by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
Figure 9.
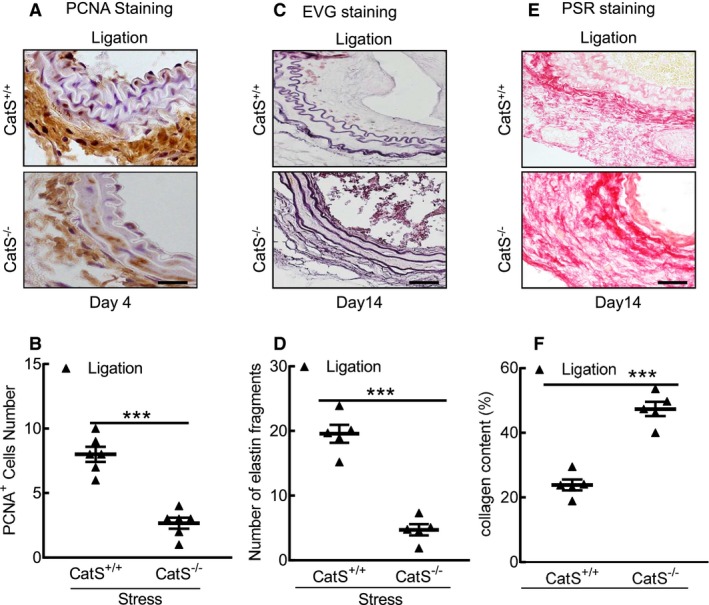
Cathepsin S deficiency (CatS−/−) mitigated cell proliferation and elastin degradation. A and B, Representative proliferating cell nuclear antigen (PCNA) immunostaining images of media smooth muscle cell proliferation and combined quantitative data for PCNA + cells. C through F, Representative images and quantitative data for the number of broken elastin fragments (Elastica van Gieson [EVG]) and collagen content (picrosirius red [PSR]). Bar=100 μm. Results are mean±SEM (n=5–6). ***P<0.001 vs cathepsin S wild type (CatS+/+) by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
Figure 10.
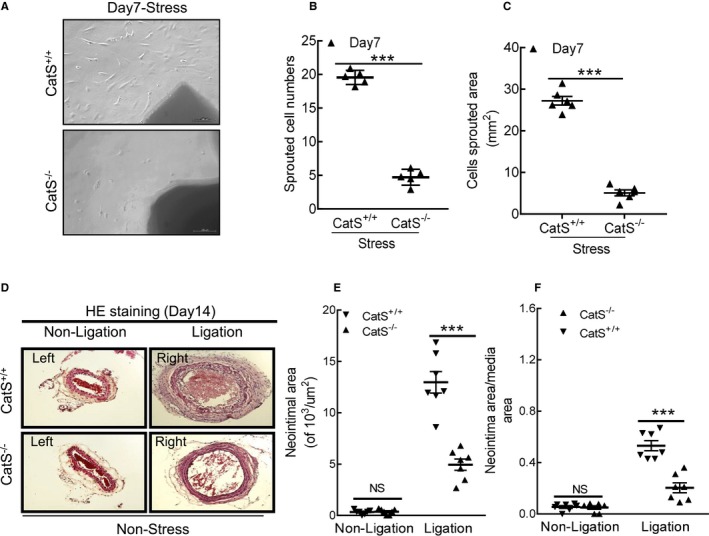
Cathepsin S deficiency (CatS−/−) impaired aorta‐derived smooth muscle migration in the stressed mice. A through C, Representative images and quantitative data showing the sprouted cell numbers and sprouted areas. D through F, Representative images and quantitative data showed that CatS−/− reduced the neointimal areas and the ratio of neointima area/media area in injured arteries of the mice even without chronic stress. Bar=100 μm. The results are mean±SEM (n=6–7). CatS+/+ indicates cathepsin S wild type; HE, hematoxylin and eosin; NS, no significance. ***P<0.001 vs corresponding control group by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
CatS−/− Suppressed Inflammation, Oxidative Stress, and Proteolysis in Response to Stress and Injury
As shown in Figure 11, the quantitative PCR data revealed that the levels of all of the targeted inflammatory (ICAM‐1, VCAM‐1, MCP‐1, SDF‐1α, CXCR4, TLR‐2, TLR‐4, TNF‐α, and IL‐1β) and oxidative stress–related (p22phox, p47phox, p67phox, gp91phox, and NADPH oxidase 1) genes were lower in the stressed CatS−/− mice compared with the control mice (Figure 11A through 11D). We also observed that the stressed CatS−/− mice had decreased levels of ATR1, MMP‐2, MMP‐9, CatL, and CatS genes in the injured arteries (Figure 11E and 11F). This was consistent with the PCR findings that CatS−/− reduced NADPH oxidase activity (148.1±11.8 versus 202.8±15.2 RLU/mg protein) and the oxidative stress production (Figure 12A and 12B). Similarly, CatS−/− significantly mitigated macrophage infiltration (Figure 12C and 12D).
Figure 11.
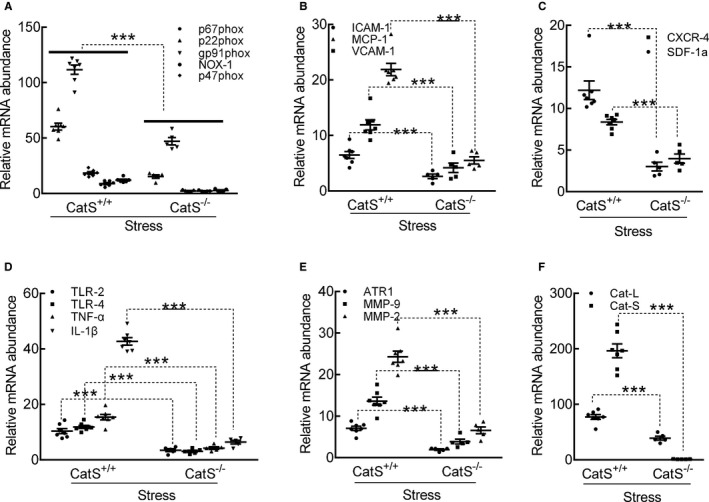
Cathepsin S deficiency (CatS−/−) mitigated the expressions of targeted oxidative stress–, inflammation‐, and proteolysis‐related genes in the carotid arteries of the stressed mice at day 4 after surgery. A through F, Quantitative polymerase chain reaction data show the levels of p22phox, gp91phox, p47phox, p67phox, nicotinamide‐adenine dinucleotide phosphate, reduced form, oxidase 1 (NOX‐1), intercellular adhesion molecule‐1 (ICAM‐1), vascular cell adhesion molecule‐1 (VCAM‐1), monocyte chemoattractant protein‐1 (MCP‐1), stromal cell–derived factor‐1α (SDF‐1α), C‐X‐C chemokine receptor‐4 (CXCR‐4), toll‐like receptor (TLR)‐2, TLR‐4, tumor necrosis factor (TNF)‐α, interleukin‐1β (IL‐1β), angiotensin II receptor 1α (ATR1α), matrix metalloproteinase (MMP)‐9, MMP‐2, cathepsin L (CatL), and CatS mRNAs. Results are mean±SEM (n=5–7). CatS+/+ indicates CatS wild type. ***P<0.001 vs corresponding CatS+/+ by 1‐way ANOVA, followed by Tukey post hoc tests.
Figure 12.
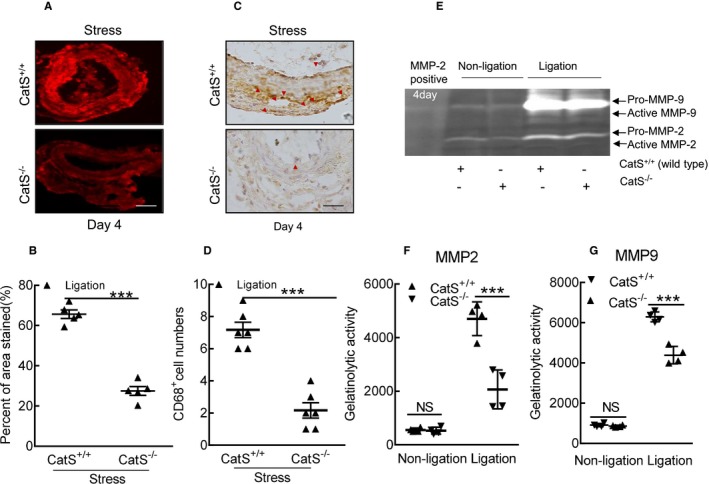
Cathepsin S deficiency (CatS−/−) reduced the oxidative stress production and macrophage infiltration in the injured arteries of stressed mice at day 4 after surgery. A and B, Representative images and quantitative data for the dihydroethidium staining area of the injured carotid arteries of stressed CatS wild type (CatS+/+) and CatS−/− mice. C and D, Representative images and quantitative data of the numbers of CD68+ macrophages. E through G, Representative images of gelatin zymography and combined quantitative data for the gelatinolytic activities of matrix metalloproteinase (MMP)‐2 and MMP‐9 in the carotid arteries of stressed CatS+/+ and CatS−/− mice. Bar=100 μm. Results are mean±SEM (n=4–6). NS indicates no significance. ***P<0.001 vs corresponding CatS+/+ by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
The gelatin zymography revealed that the CatS−/− lesions had much less gelatinolytic activities of MMP‐9 and MMP‐2 compared with CatS+/+ lesions (Figure 12E through 12G). As expected, the quantitative data of immunoblots revealed that CatS−/− resulted in decreases in the levels of targeted proteins (gp91phox, p22phox, galectin‐3, ATR1, p16, p‐Akt, p‐mTOR, p‐Erk1/2, p‐p38MAPK, and p‐GSK3α/β) (Figure 13A through 13C).
Figure 13.
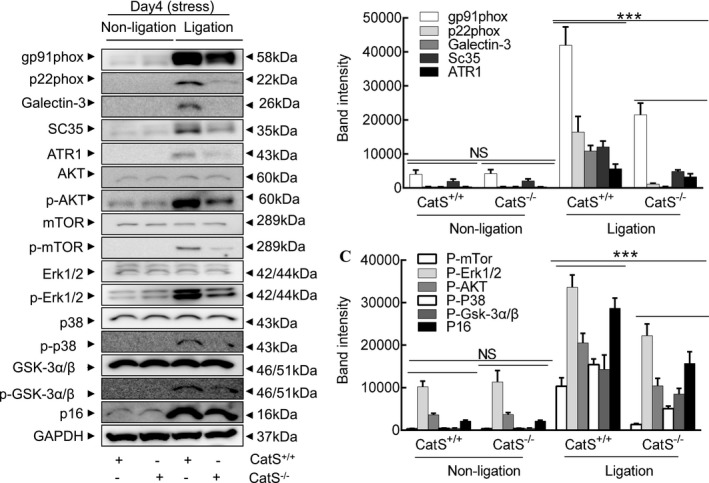
Cathepsin S (CatS) deletion improved targeted molecule changes in the injured arteries of the stressed mice. A through C, Representative images and combined quantitative data for the levels of targeted molecule proteins. The results are mean±SEM (n=5–7). AKT indicates protein kinase B; ATR1, angiotensin II receptor 1; CatS+/+, CatS wild type; CatS−/−, CatS deficient; Erk, extracellular signal‐regulated kinase; GSK, phosphoglycogene synthesis kinase; mTOR, mammalian target of rapamycin; NS, no significance; p‐AKT, phosphorylated AKT; p‐Erk, phosphorylated Erk; p‐GSK, phosphorylated GSK; p‐mTOR, phosphorylated mTOR. ***P<0.001 vs CatS+/+ by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
Pharmacological CatS Inhibition Ameliorated Stress‐ and Injury‐Related Neointimal Formation in Mice That Received Injury
Similar to the case with CatS genetic deletion, pharmacological inhibition with a CatS‐specific inhibitor demonstrated that the neointima area and the ratio of the neointima area/the media area were significantly lower in the injured arterial tissues of stressed CatS‐I (+) mice than those of control CatS‐I (−) mice (Figure 14A through 14C). Moreover, the CatS‐I (+) aortae had impaired sprouted SMC numbers and sprouted areas at day 7 after culture (Figure 15A through 15C). In addition, we also observed that CatS inhibition markedly improved neointima hyperplasia in mice that received ligation injury without chronic stress (Figure 15D through 15F).
Figure 14.
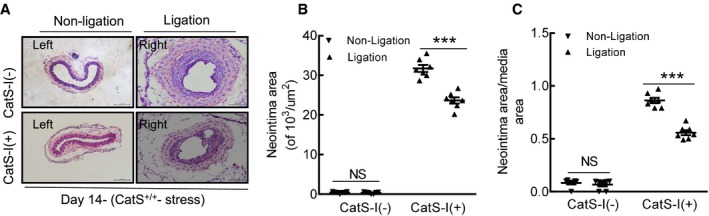
Cathepsin S inhibitor (CatS‐I) alleviated injury‐induced neointimal formation at day 14 after surgery. A, Representative hematoxylin and eosin staining images of the right and left carotid arteries of stressed CatS wild type (CatS+/+) mice treated with CatS‐I (CatS‐I [+]) or without CatS‐I (CatS‐I [−]). B and C, Quantitative data showing the neointimal areas and the ratio of neointima area/media area in injured arteries of the 2 experimental groups. Bar=100 μm. Results are mean±SEM (n=7). NS indicates no significance. ***P<0.001 vs CatS‐I (−) group by Student t test.
Figure 15.
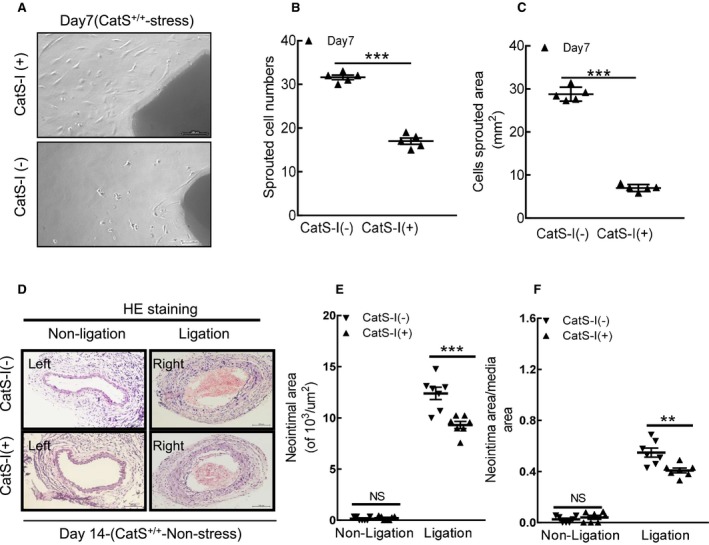
Cathepsin S inhibition (CatS‐I) impaired the aorta‐derived smooth muscle cell migration in stressed CatS wild‐type (CatS+/+) mice at day 7 after surgery. A through C, Representative images and quantitative data for sprouted cell numbers and sprouted areas at day 7 after culture. D through F, Representative images and quantitative data revealed that CatS‐I also suppressed the neointimal areas and the ratio of neointima area/media area in injured arteries of the mice without stress. Bar=100 μm. Results are mean±SEM (n=6–7). HE indicates hematoxylin and eosin; NS, no significance. ***P<0.001 vs without CatS‐I by Student t test.
CatS Inhibition Reduced the Stress‐ and Injury‐Related Inflammation, Oxidative Stress, and Proteolysis
Compared with the control, CatS inhibition suppressed the levels of targeted gene expressions (p22phox, p47phox, p67phox, gp91phox, ICAM‐1, VCAM‐1, MCP‐1, SDF‐1α, CXCR4, TLR‐2, TLR‐4, TNF‐α, IL‐1β, ATR1, MMP‐2, MMP‐9, CatL, and CatS) (Figure 16A through 16F). The dihydroethidium staining showed that CatS‐I (+) alleviated the oxidative stress production (Figure 17A and 17B). Similarly, CatS‐I (+) significantly mitigated the macrophage infiltration (Figure 17C and 17D).
Figure 16.
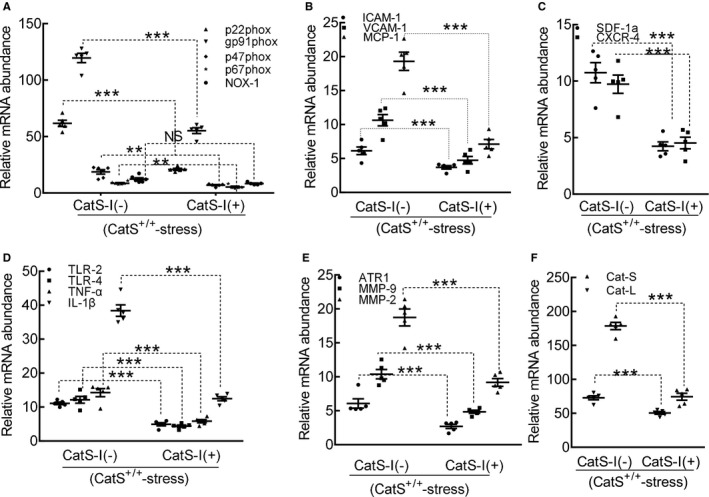
Cathepsin S inhibition (CatS‐I) mitigated the expressions of targeted oxidative stress–, inflammation‐, and proteolysis‐related genes in the carotid arteries of the stressed CatS wild‐type (CatS+/+) mice at day 4 after surgery. A through F, Quantitative polymerase chain reaction data show the levels of p22phox, gp91phox, p47phox, p67phox, nicotinamide‐adenine dinucleotide phosphate, reduced form, oxidase 1 (NOX‐1), intercellular adhesion molecule‐1 (ICAM‐1), vascular cell adhesion molecule‐1 (VCAM‐1), monocyte chemoattractant protein‐1 (MCP‐1), stromal cell–derived factor‐1α (SDF‐1α), C‐X‐C chemokine receptor‐4 (CXCR‐4), toll‐like receptor (TLR)‐2, TLR‐4, tumor necrosis factor (TNF)‐α, interleukin‐1β (IL‐1β), angiotensin II receptor 1α (ATR1α), matrix metalloproteinase (MMP)‐9, MMP‐2, cathepsin (CatL), and CatS mRNAs of mice with CatS‐I (CatS‐I [−]) and mice with CatS‐I (CatS‐I [+]). Results are mean±SEM (n=5–7). NS indicates no significance. **P<0.01/***P<0.001 vs corresponding CatS‐I (−) mice by 1‐way ANOVA, followed by Tukey post hoc tests.
Figure 17.
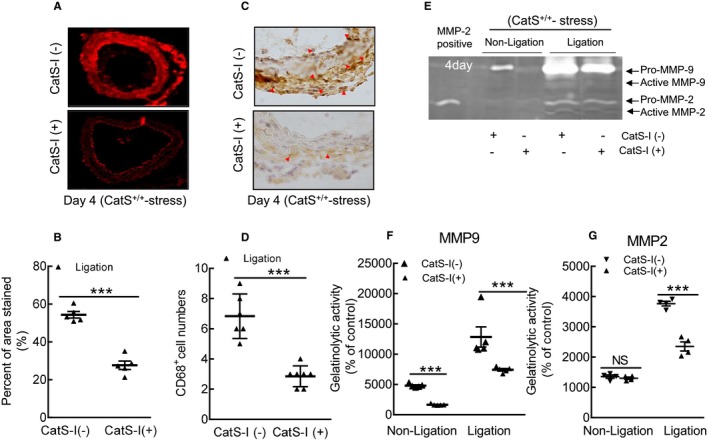
Cathepsin S inhibitor (CatS‐I) mitigated oxidative stress production and reduced the macrophage infiltration in injured arteries of stressed CatS wild‐type (CatS+/+) mice at day 4 after injury. A and B, Representative images and quantitative data for the dihydroethidium staining area of the injured carotid arteries of stressed mice without CatS‐I (CatS‐I [−]) and mice with CatS‐I (CatS‐I [+]). C and D, Representative images and quantitative data of the numbers of CD68+ macrophages. E through G, Representative images of gelatin zymography and combined quantitative data for the gelatinolytic activities of matrix metalloproteinase (MMP)‐2 and MMP‐9 in the carotid arteries of stressed CatS‐I (−) and CatS‐I (+) mice. Bar=100 μm. Results are mean±SEM (n=4–7). NS indicates no significance. ***P<0.001 vs corresponding CatS‐I (−) group by 1‐way ANOVA, followed by Tukey post hoc tests or Student t test.
The gelatin zymography revealed that CatS‐I (+) had mitigated the gelatinolytic activities of MMP‐9 and MMP‐2 (Figure 17E through 17G). As expected, CatS inhibition reduced the levels of targeted gp91phox, p22phox, galectin‐3, ATR1, p16 p‐Akt, p‐mTOR, p‐Erk1/2, p‐p38MAPK, and p‐GSK3α/β proteins (Figure 18A through 18C). Moreover, the pharmacological CatS inhibition also showed no influence on the increased blood pressure (116.7±6.6 versus 119.7±5.4 mmHg; P>0.05) and plasma corticosterone levels (135.3±19.3 versus 126.5±12.9 ng/mL; P>0.05) in the stressed mice.
Figure 18.
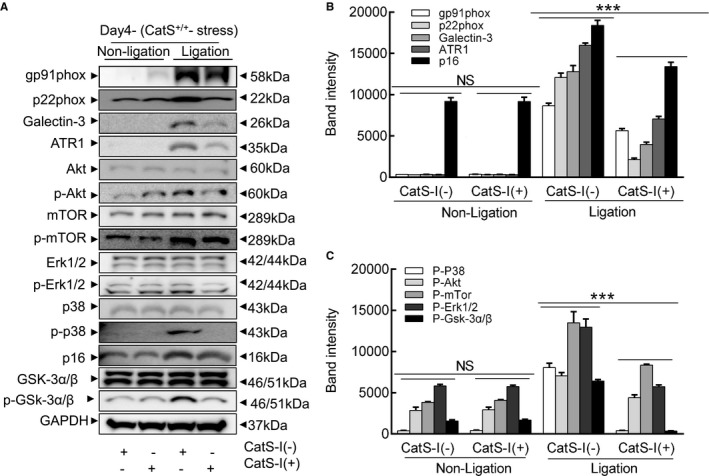
Cathepsin S inhibition (CatS‐I) mitigates the stress‐related harmful targeted protein changes in the injured arteries of CatS wild‐type (CatS+/+) mice at day 4 after injury. A through C, Representative images and quantitative data showing the levels of targeted proteins. Results are mean±SEM (n=5–7). Akt indicates protein kinase B; ATR1, angiotensin II receptor 1; CatS‐I (+), with CatS‐I; CatS‐I (−), without CatS‐I; Erk, extracellular signal‐regulated kinase; GSK, phosphoglycogene synthesis kinase; mTOR, mammalian target of rapamycin; NS, no significance; p‐Akt, phosphorylated Akt; p‐Erk, phosphorylated Erk; p‐GSK, phosphorylated GSK; p‐mTOR, phosphorylated mTOR. ***P<0.001 vs corresponding CatS‐I (−) mice by 1‐way ANOVA, followed by Tukey post hoc tests.
Discussion
Chronic stress–related vascular positive remodeling is the leading cause of restenosis and cardiovascular events in patients with CVD.1, 2 Identifying novel targets to suppress endovascular treatment–related maladaptive vascular negative remodeling will contribute to therapeutic strategies to preempt restenosis in patients with atherosclerotic CVD and CPS. The important traditional and nontraditional roles of lysosomal CatS in various physiological and pathological conditions have been revealed by experimental and clinical studies.14, 15, 16, 17 Although those investigations uncovered proteolysis‐dependent and proteolysis‐independent mechanisms underlying ischemia‐induced neovascularization and aneurysm formation,17, 25 a limited number of experimental studies reported that the atherosclerotic plaques of chronically stressed mice had increased CatS mRNA and protein among apolipoprotein E–deficient mice fed a high‐fat diet.20, 21 To the best of our knowledge, the present study is the first to report that that genetic and pharmacological interventions targeted toward CatS confer vascular protection against chronic stress with mechanical injury.
Experimental and clinical evidence indicates that oxidative stress plays a critical role in endothelial cell injury and cytokine/chemokine secretions, leading to atherosclerotic lesion formation.21, 26, 27 Our present findings demonstrated that the stressed lesions had increased levels of p22phox and gp91phox genes and proteins as well as oxidative stress production. The genes gp91phox and p22phox are important membrane subunits of NAD(P)H oxidase.27 NADPH oxidases are the main source of reactive oxygen species, and it has been demonstrated that genetic and pharmacological inhibitions of NADPH oxidase subunits ameliorated the development of atherosclerotic plaque growth in apolipoprotein E–deficient mouse fed a high‐fat diet.26, 27 Because the stressed mice had increased neointima hyperplasia under our experimental conditions, we propose that the enhancement of stress‐induced oxidative stress may contribute to vascular remodeling in mice that had been subjected to injury. A significant finding of our present work is that CatS deletion as well as its pharmacological inhibition mitigated vascular changes, accompanied by the reduction of gp91phox and p22phox expression and oxidative stress production in the arterial lesions of stressed mice. Our findings thus provide evidence that the vasculoprotective actions of CatS inhibition by genetic and pharmacological approaches occur, at least in part, through the modulation of NADPH oxidase expression and activity.
Clinical and experimental evidence has revealed that CPS can enhance the inflammatory process in different tissues (eg, adipose and vascular tissues).4, 5, 6 Our present findings confirmed that stress increased the macrophage accumulation in the plaques compared with the nonstressed mice. The stressed lesions had increased inflammatory gene and/or protein expressions (ie, TLR‐2, TLR‐4, IL‐1β, TNF‐α, ICAM‐1, VCAM‐1, MCP‐1, SDF‐1, CXCR4, and galectin‐3). Proinflammatory effects of these molecules on the process of vascular remodeling and atherosclerotic plaque growth have been sufficiently demonstrated by studies from our group and other groups.19, 20, 21 Thus, the chronic stress promoted the development of injury‐induced harmful vascular changes by enhancing inflammatory actions. Our results show that genetic and pharmacological interventions targeted toward CatS ameliorated stressed arterial inflammatory actions in response to injury. Taken together, our findings indicate the ability of CatS inhibition to mitigate macrophage activation, and they suggest that the production of inflammatory cytokine exerts salutary effects on injured vasculature by TLR‐2/TLR‐4 signaling inactivation, thereby improving morphological alterations under stress conditions.
Because MMP‐2 and MMP‐9 are expressed and secreted mainly by inflammatory cells (ie, macrophages and leukocytes) in atherosclerotic plaques,28 this notion is further supported by our observation that CatS inhibition ameliorated MMP‐2 and MMP‐9 gene expressions and their activities in the stressed arteries with injury. The mice that received 2‐week immobilized stress resulted in an increase in blood pressure and plasma corticosterone compared with the control mice; this effect was not influenced by CatS inhibition. This result supports our hypothesis that CatS‐absence–mediated vasculoprotective effects are not attributable to a reduction in blood pressure and plasma corticosterone but rather to the antioxidative and anti‐inflammatory actions under the experimental conditions used herein. Further investigation is needed to resolve this issue.
The ability of chronic stress to increase CatS expression and activity probably contributed to repair under our experimental conditions. In agreement with an animal study in which CatS deletion reduced atherosclerotic lesion formation in apolipoprotein E–deficient mice, we observed that neointimal formation after injury was mitigated by our modifications of CatS activity. Early in the formation of the thickened intima, as in neointimal lesions and atherosclerotic lesions, vascular medial SMCs must migrate from the tunica media into the developing intima.23 The results of our aorta‐ring culture assay show that CatS−/− reduced the numbers of sprouted SMCs and areas from aortic explants. Previous data obtained in in vitro experiments demonstrated that CatS on the cell surface with integrin ανβ3 modulated SMC invasiveness across the collagen/gelatin gel barrier.29 It was reported that patients with coronary artery disease and restenosis after intravascular therapy had higher levels of plasma CatS compared with patients without restenosis.30 Herein, we have shown that genetic and pharmacological inhibition of CatS ameliorated the chronic stress– and injury‐induced collagen and elastin degradation and intimal hyperplasia. Thus, the upregulation of CatS expression and activity could represent a common proteolysis‐related mechanism in CPS‐related remodeling in CatS+/+ mice under our experimental conditions. In addition, accumulating evidence indicates that oxidative stress and inflammatory cytokines modulated CatS expression and activity in vascular cells (SMCs and endothelial cells) and inflammatory macrophages.22, 30, 31 Previous studies from our group and other groups reported close interactions between ATR1 and the TLR‐2/TLR‐4 axis in the regulation of the MMP‐2/MMP‐9 and CatS/K expression in macrophages and endothelial cells by several intracellular signaling pathways.28, 32 Collectively, these findings raise the possibility that CPS can accelerate neointima hyperplasia in response to injury via the enhancement of CatS‐dependent proteolytic mechanisms that may be mediated by ATR1 and TLR‐2/TLR‐4 signaling activations.
A recent comprehensive review article highlighted the roles of a CatS and its endogenous inhibitor (cystatin C) system in proliferative diseases.16 It is notable that CatS seems to be of particular importance for SMC proliferation in the vascular repair process. It was reported that the atherosclerotic plaques of CatL−/− mice contained fewer Ki67+ proliferating cells compared with CatL+/+ mice.33 Our present results show that the lesions of the stressed CatS−/− mice and CatS‐I–treated CatS+/+ mice had lower PCNA+ proliferating cells compared with the control mice. Because stress induces CatS activation, we favor the hypothesis that CPS accelerates neointimal hyperplasia in response to injury through its ability to activate elevated CatS‐related SMC proliferative activity. Akt‐mTOR and Akt‐Erk1/2 signaling has been widely accepted as an initiator of protein synthesis and cell growth. Our observations indicate that stress‐related elevations of p‐Akt, p‐p38MAPK, p‐Erk1/2, p‐GSK3α/β, and p‐mTOR proteins in the lesions were rectified by the negative modifications of CatS activity. Our results also showed that the stressed lesions had elevated levels of TLR‐2 and TLR‐4 genes; these changes were also rectified by CatS inhibition. Several lines of evaluation revealed that TLR2 and TLR4 are involved in vascular SMC protein synthesis and proliferation via Akt‐mTOR and Akt‐Erk1/2 signaling activation.23 The TLR‐4/MyD88 axis has been shown to modulate the injury‐induced vascular repair process.34 Thus, both therapeutic interventions of CatS activity may rectify the alterations in the expressions of TLR‐2 and TLR‐4 genes, resulting in Akt‐mTOR and Akt‐Erk1/2 signaling overactivation, contributing to the mitigation of vascular repair in mice under our experimental conditions.
As known, the dihydroethidium assay to measure reactive oxygen species is a nonspecific assay. Study limitations should be considered. One potential limitation of the present study is that we could not evaluate the levels of additional oxidative stress biomarker. We also did not perform complimentary assays (ie, cytochrome c, Amplex Red, or dihydroethidium high‐performance liquid chromatography) to support our findings. Finally, we have no data to show chronic stress mediated the close talk between p38MAPK signaling activation and oxidative stress production.
Conclusions
The expressions of cysteinyl CatS gene and protein increase in the injured carotid arteries of the mice under CPS. CatS deletion alleviates chronic stress–related vascular overrepair in response to injury in mice. The pharmacological inhibition of CatS mimics the vascular protective effects of genetic CatS deletion. It seems that a selective CatS‐I may have potential utility in the treatment or control of restenosis after intravascular intervention therapies in atherosclerotic CVD.
Author Contributions
Dr Wang researched biological and histological data and wrote the first draft of the manuscript. Drs Meng, Hu, and Takeshi researched the morphological data and assisted with the carotid artery injury mouse models. Drs Piao, Inoue, Xu, Yu, and Wu researched the real‐time polymerase chain reaction data and mouse genotyping. Dr Shi reviewed the manuscript, contributed to the discussion, and provided the transgenic mice. Drs Unno, Murohara, and Kuzuya edited the manuscript or/and planned the study. Dr Cheng designed the study and handled the funding and supervision.
Sources of Funding
This work was supported, in part, by grants from the National Natural Science Foundation of China (Nos. 81560240, 81460082, 81660240, and 81770485) and by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Nos. 15H04801 and 15H04802).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011994 DOI: 10.1161/JAHA.119.011994.)
References
- 1. Roepke SK, Allison M, Von Kanel R, Mausbach BT, Chattillion EA, Harmell AL, Patterson TL, Dimsdale JE, Mills PJ, Ziegler MG, Ancoli‐Israel S, Grant I. Relationship between chronic stress and carotid intima‐media thickness (IMT) in elderly Alzheimer's disease caregivers. Stress. 2012;15:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosengren A, Hawken S, Ounpuu S, Sliwa K, Zubaid M, Almahmeed WA, Blackett KN, Sitthi‐amorn C, Sato H, Yusuf S; INTERHEART investigators. Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): case‐control study. Lancet. 2004;364:953–962. [DOI] [PubMed] [Google Scholar]
- 3. Chandola T, Brunner E, Marmot M. Chronic stress at work and the metabolic syndrome: prospective study. BMJ. 2006;332:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uchida Y, Takeshita K, Yamamoto K, Kikuchi R, Nakayama T, Nomura M, Cheng XW, Egashira K, Matsushita T, Nakamura H, Murohara T. Stress augments insulin resistance and prothrombotic state: role of visceral adipose‐derived monocyte chemoattractant protein‐1. Diabetes. 2012;61:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu E, Hu L, Wu H, Piao L, Zhao G, Inoue A, Kim W, Yu C, Xu W, Bando YK, Li X, Lei Y, Hao CN, Takeshita K, Kim WS, Okumura K, Murohara T, Kuzuya M, Cheng XW. Dipeptidyl peptidase‐4 regulates hematopoietic stem cell activation in response to chronic stress. J Am Heart Assoc. 2017;6:006394 DOI: 10.1161/JAHA.117.006394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223:69–77. [DOI] [PubMed] [Google Scholar]
- 8. Steptoe A, Kivimaki M. Stress and cardiovascular disease. Nat Rev Cardiol. 2012;9:360–370. [DOI] [PubMed] [Google Scholar]
- 9. Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW. Social stress up‐regulates inflammatory gene expression in the leukocyte transcriptome via beta‐adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. 2013;110:16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piao L, Zhao G, Zhu E, Inoue A, Shibata R, Lei Y, Hu L, Yu C, Yang G, Wu H, Xu W, Okumura K, Ouchi N, Murohara T, Kuzuya M, Cheng XW. Chronic psychological stress accelerates vascular senescence and impairs ischemia‐induced neovascularization: the role of dipeptidyl peptidase‐4/glucagon‐like peptide‐1‐adiponectin axis. J Am Heart Assoc. 2017;6:e006421 DOI: 10.1161/JAHA.117.006421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedrichs B, Tepel C, Reinheckel T, Deussing J, von Figura K, Herzog V, Peters C, Saftig P, Brix K. Thyroid functions of mouse cathepsins B, K, and L. J Clin Invest. 2003;111:1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation. 2012;125:1551–1562. [DOI] [PubMed] [Google Scholar]
- 13. Cheng XW, Huang Z, Kuzuya M, Okumura K, Murohara T. Cysteine protease cathepsins in atherosclerosis‐based vascular disease and its complications. Hypertension. 2011;58:978–986. [DOI] [PubMed] [Google Scholar]
- 14. Guo R, Hua Y, Rogers O, Brown TE, Ren J, Nair S. Cathepsin K knockout protects against cardiac dysfunction in diabetic mice. Sci Rep. 2017;7:8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Folkesson M, Vorkapic E, Gulbins E, Japtok L, Kleuser B, Welander M, Lanne T, Wagsater D. Inflammatory cells, ceramides, and expression of proteases in perivascular adipose tissue adjacent to human abdominal aortic aneurysms. J Vasc Surg. 2017;65:1171–1179.e1171. [DOI] [PubMed] [Google Scholar]
- 16. Wu H, Du Q, Dai Q, Ge J, Cheng X. Cysteine protease cathepsins in atherosclerotic cardiovascular diseases. J Atheroscler Thromb. 2018;25:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen H, Wang J, Xiang MX, Lin Y, He A, Jin CN, Guan J, Sukhova GK, Libby P, Wang JA, Shi GP. Cathepsin S‐mediated fibroblast trans‐differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2013;100:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Cheng XW, Hu L, Wu H, Guo P, Hao CN, Jiang H, Zhu E, Huang Z, Inoue A, Sasaki T, Du Q, Takeshita K, Okumura K, Murohara T, Kuzuya M. Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol. 2015;183:198–208. [DOI] [PubMed] [Google Scholar]
- 19. Qin Y, Cao X, Guo J, Zhang Y, Pan L, Zhang H, Li H, Tang C, Du J, Shi GP. Deficiency of cathepsin S attenuates angiotensin II‐induced abdominal aortic aneurysm formation in apolipoprotein E‐deficient mice. Cardiovasc Res. 2012;96:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang G, Lei Y, Inoue A, Piao L, Hu L, Jiang H, Sasaki T, Wu H, Xu W, Yu C, Zhao G, Ogasawara S, Okumura K, Kuzuya M, Cheng XW. Exenatide mitigated diet‐induced vascular aging and atherosclerotic plaque growth in ApoE‐deficient mice under chronic stress. Atherosclerosis. 2017;264:1–10. [DOI] [PubMed] [Google Scholar]
- 21. Lei Y, Yang G, Hu L, Piao L, Inoue A, Jiang H, Sasaki T, Zhao G, Yisireyili M, Yu C, Xu W, Takeshita K, Okumura K, Kuzuya M, Cheng XW; FAHA . Increased dipeptidyl peptidase‐4 accelerates diet‐related vascular aging and atherosclerosis in apoe‐deficient mice under chronic stress. Int J Cardiol. 2017;243:413–420. [DOI] [PubMed] [Google Scholar]
- 22. Shi GP, Sukhova GK, Kuzuya M, Ye Q, Du J, Zhang Y, Pan JH, Lu ML, Cheng XW, Iguchi A, Perrey S, Lee AM, Chapman HA, Libby P. Deficiency of the cysteine protease cathepsin S impairs microvessel growth. Circ Res. 2003;92:493–500. [DOI] [PubMed] [Google Scholar]
- 23. Hu L, Cheng XW, Song H, Inoue A, Jiang H, Li X, Shi GP, Kozawa E, Okumura K, Kuzuya M. Cathepsin K activity controls injury‐related vascular repair in mice. Hypertension. 2014;63:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng XW, Murohara T, Kuzuya M, Izawa H, Sasaki T, Obata K, Nagata K, Nishizawa T, Kobayashi M, Yamada T, Kim W, Sato K, Shi GP, Okumura K, Yokota M. Superoxide‐dependent cathepsin activation is associated with hypertensive myocardial remodeling and represents a target for angiotensin II type 1 receptor blocker treatment. Am J Pathol. 2008;173:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y, Wu H, Takeshita K, Li X, Huang Z, Song H, Asai M, Hao CN, Unno K, Koike T, Oshida Y, Okumura K, Murohara T, Kuzuya M. Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun. 2014;5:3838. [DOI] [PubMed] [Google Scholar]
- 26. Lozhkin A, Vendrov AE, Pan H, Wickline SA, Madamanchi NR, Runge MS. Nadph oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J Mol Cell Cardiol. 2017;102:10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Juni RP, Duckers HJ, Vanhoutte PM, Virmani R, Moens AL. Oxidative stress and pathological changes after coronary artery interventions. J Am Coll Cardiol. 2013;61:1471–1481. [DOI] [PubMed] [Google Scholar]
- 28. Cheng XW, Song H, Sasaki T, Hu L, Inoue A, Bando YK, Shi GP, Kuzuya M, Okumura K, Murohara T. Angiotensin type 1 receptor blocker reduces intimal neovascularization and plaque growth in apolipoprotein e‐deficient mice. Hypertension. 2011;57:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng XW, Kuzuya M, Nakamura K, Di Q, Liu Z, Sasaki T, Kanda S, Jin H, Shi GP, Murohara T, Yokota M, Iguchi A. Localization of cysteine protease, cathepsin S, to the surface of vascular smooth muscle cells by association with integrin alphanubeta3. Am J Pathol. 2006;168:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arnlov J. Cathepsin s as a biomarker: where are we now and what are the future challenges? Biomark Med. 2012;6:9–11. [DOI] [PubMed] [Google Scholar]
- 31. Sasaki T, Kuzuya M, Nakamura K, Cheng XW, Hayashi T, Song H, Hu L, Okumura K, Murohara T, Iguchi A, Sato K. AT1 blockade attenuates atherosclerotic plaque destabilization accompanied by the suppression of cathepsin S activity in apoE‐deficient mice. Atherosclerosis. 2010;210:430–437. [DOI] [PubMed] [Google Scholar]
- 32. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M, Jerome WG, Ostrowski MC, Bromme D, Libby P, Tabas IA, Welch CL, Tall AR. Free cholesterol accumulation in macrophage membranes activates Toll‐like receptors and p38 mitogen‐activated protein kinase and induces cathepsin K. Circ Res. 2009;104:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun J, Sukhova GK, Zhang J, Chen H, Sjoberg S, Libby P, Xiang M, Wang J, Peters C, Reinheckel T, Shi GP. Cathepsin L activity is essential to elastase perfusion‐induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2011;31:2500–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saxena A, Rauch U, Berg KE, Andersson L, Hollender L, Carlsson AM, Gomez MF, Hultgardh‐Nilsson A, Nilsson J, Bjorkbacka H. The vascular repair process after injury of the carotid artery is regulated by IL‐1R1 and MyD88 signalling. Cardiovasc Res. 2011;91:350–357. [DOI] [PubMed] [Google Scholar]