

Abstract
Background
Homoarginine (hArg) has been shown to be cardioprotective in a model of ischemic heart failure; however, the mechanism remains unknown. hArg can inhibit tissue‐nonspecific alkaline phosphatase (TNAP), an enzyme that promotes vascular calcification. We hypothesized that hArg will exert beneficial effects by reducing calcification in a mouse model of coronary artery disease associated with TNAP overexpression and hypercholesterolemia.
Methods and Results
TNAP was overexpressed in the endothelium in mice homozygous for a low‐density lipoprotein receptor mutation (wicked high cholesterol [WHC] allele). WHC and WHC–endothelial TNAP mice received placebo or hArg supplementation (14 mg/L in drinking water) starting at 6 weeks of age simultaneously with an atherogenic diet. Outcomes were compared between the groups after 4 to 5 weeks on treatment. Experiments were performed in males, which presented a study limitation. As expected, WHC–endothelial TNAP mice on the placebo had increased mortality (median survival 27 days, P<0.0001), increased coronary calcium and lipids (P<0.01), increased left ventricular end‐diastolic diameter (P<0.0001), reduced ejection fraction (P<0.05), and increased myocardial fibrosis (P<0.0001) compared with WHC mice. Contrary to our hypothesis, hArg neither inhibited TNAP activity in vivo nor reduced coronary artery calcification and atherosclerosis in WHC–endothelial TNAP mice; however, compared with the placebo, hArg prevented left ventricular dilatation (P<0.01), preserved ejection fraction (P<0.05), and reduced myocardial fibrosis (P<0.001).
Conclusions
The beneficial effect of hArg supplementation in the setting of calcified coronary artery disease is likely due to its direct protective actions on the myocardial response to the ischemic injury and not to the inhibition of TNAP activity and calcification.
Keywords: cardiac remodeling, coronary artery disease, coronary calcium, homoarginine
Subject Categories: Remodeling, Heart Failure
Clinical Perspective
What Is New?
In various epidemiologic studies, low homoarginine plasma levels have been found to be associated with adverse outcomes in multiple cardiovascular, metabolic, and renal pathologies; however, the mechanism explaining this is not known.
Our findings demonstrate that dietary homoarginine supplementation improved outcomes in a mouse model of coronary atherosclerosis and heart failure, most probably due to protection from myocardial remodeling.
What Are the Clinical Implications?
Our results imply that patients with heart failure might be a patient population with especially high chances to benefit from homoarginine supplementation—a well‐tolerated and nonharmful treatment that can be further tested in clinical studies.
Introduction
l‐Homoarginine (hArg) is a nonproteinogenic amino acid homologue of l‐arginine that has recently been proposed as an endogenous protective cardiovascular and metabolic factor.1 hArg is present in human plasma at levels of 1 to 2 μmol/L2 and is thought to be produced from arginine and l‐lysine by the mitochondrial enzyme arginine:glycine amidinotransferase, which is primarily expressed in the kidneys, liver, and pancreas.3, 4 hArg is also found in certain plants; however, the relative contribution of dietary hArg consumption to circulating hArg levels is still unclear.5, 6 There are 3 known pathways for enzymatic homoarginine catabolism—conversion to homocitrulline and nitric oxide by nitric oxide synthase (NOS), to lysine by arginase, and to 6‐guanidino‐2‐oxocaproic acid (GOCA) by alanine:glyoxylate aminotransferase 2 (AGXT2).7 hArg is also excreted by kidneys in unchanged form.8
Multiple epidemiological studies have demonstrated an association of low circulating hArg levels with adverse outcome and increased mortality in cardiovascular, metabolic, and renal diseases. hArg levels decline with advancement of chronic kidney disease and are inversely associated with the risk of progression to dialysis and mortality.9, 10 Low hArg concentration in plasma is also associated with an increased risk of stroke and cardiovascular mortality.11, 12, 13 In patients with lower extremity arterial disease, low hArg/ADMA ratio correlated with a higher cardiovascular mortality and higher incidence of cardiovascular events.14 In a cohort of participants with median age of 43 years, higher hArg was associated with a lower rate of major adverse cardiovascular events and lower all‐cause mortality.15
Several animal studies have tested whether hArg has direct cardiovascular or metabolic protective effects. Indeed, hArg supplementation has improved outcomes in mice with low and normal circulating hArg levels subjected to experimental stroke and helped to preserve cardiac function in a murine model of post–myocardial infarction heart failure.12, 16 hArg supplementation also rescued impaired cardiac contractile function in arginine:glycine amidinotransferase knockout mice.17 With respect to metabolic changes, hArg supplementation ameliorated blood glucose levels in mice on a high‐fat diet.18 Taken together, these studies suggest that at least in some settings hArg may have direct protective properties in addition to being merely a marker of favorable outcomes.
The possible mechanisms of the hypothesized direct protective effects of hArg are unclear. hArg is a weak substrate of nitric oxide synthase and a weak competitive inhibitor of l‐arginine catabolism by arginase.19, 20 However, it is unlikely that small changes in circulating hArg levels, which are associated with adverse outcomes, would affect the l‐arginine/nitric oxide pathway, taking into account that the plasma concentration of hArg is about 20 times lower than that of arginine.20, 21
Since the initial description of hArg as an noncompetitive inhibitor of tissue‐nonspecific alkaline phosphatase (TNAP) by Rufo and Fishman,22 several studies have shown that hArg can inhibit TNAP in vitro at low millimolar concentrations.23, 24, 25 Upregulation of TNAP expression in the endothelium leads to increased vascular calcification26 and is associated with accelerated coronary atherosclerosis and mortality in a mouse model of familial hypercholesterolemia,24 suggesting that at least some of the protective effects of hArg may be mediated by its inhibitory action on TNAP in coronary arteries. To test this hypothesis, we evaluated the effects of hArg supplementation in our recently developed mouse model, in which coronary atherosclerosis was induced by a combination of factors: a mutation in the LDL receptor gene (the wicked high cholesterol allele, WHC27), increased vascular calcification due to endothelium‐specific transgenic overexpression of TNAP (eTNAP), and an atherogenic diet.24
Surprisingly, and contrary to our hypothesis, hArg did not influence TNAP activity in vivo, coronary calcification, or atherosclerosis but, rather, protected mice from myocardial remodeling in this model of ischemic heart disease.
Methods
The authors declare that all supporting data are available within the article. Unprocessed data (echocardiographic and histologic images) are available from the corresponding authors on request.
Ethics Statement
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of New York Institute of Technology College of Osteopathic Medicine (Old Westbury, NY) and complied with the National Institutes of Health guidelines for humane treatment of laboratory animals. Mice were euthanized by exsanguination under 5% isoflurane anesthesia before terminal experiments.
Experimental Animals
Hprt ALPL mice, designed to overexpress human ALPL gene encoding TNAP in a Cre/lox‐dependent manner, were previously described.28 The ALPL coding sequence with a floxed “stop” cassette was integrated into the X‐linked hypoxanthine phosphoribosyltransferase (Hprt) locus.29 These mice were backcrossed to the C57BL/6‐Ldlr whc strain, which has carried a familial hypercholesterolemia mutation in the low‐density lipoprotein receptor (Ldlr) gene for 2 generations (WHC allele, The Jackson Laboratory, Bar Harbor, ME; Stock No. 005061). Likewise, B6‐Tie2cre mice, which expresses Cre recombinase under control of the Tie2 gene promoter30 (The Jackson Laboratory, 008863–B6.Cg‐Tg(Tek‐cre)1Ywa/J), were crossed to C57BL/6‐Ldlr whc for 2 generations. Male mice of 2 genotypes (Ldlr whc ;Hprt ALPL ;Tie2cre Tg/+ and Ldlr whc ;Hprt ALPL ;Tie2cre +/+) were produced by intercrossing homozygous Ldlr whc ;Hprt ALPL females with Ldlr whc ;Tie2cre tg/+ males as previously described.24 Female mice were not used in this study because of the limitations imposed by random X chromosome inactivation,31 which was expected to result in a mosaic expression of ALPL from the X‐linked Hprt locus. Mice overexpressing TNAP in a Tie2cre‐specific manner in endothelial cells are referred to as WHC‐eTNAP, and their control littermates WHC. It is important to note that Tie2cre promoter is also active in macrophages and other myeloid cells, and it is plausible that TNAP‐expressing macrophages contribute to vascular calcification in WHC‐eTNAP mice. Animals were fed Paigen diet containing 1.25% cholesterol and 0.5% cholate (TD.02028) starting at 6 weeks of age. Data were collected at baseline and 4 to 5 weeks after induction of atherosclerosis by an atherogenic diet, which corresponded to the median survival age of WHC‐eTNAP mice. Sixty‐five mice were enrolled in the study, of which 26 were WHC and 39 WHC‐eTNAP. Of these, 16 WHC mice were assigned to the placebo treatment, and 10 WHC mice to the hArg arm. Twenty‐three WHC‐eTNAP were treated with placebo and 16 with hArg supplementation.
Supplementation With l‐Homoarginine
l‐Homoarginine was obtained from Sigma (St. Louis, MO; H1007) and dissolved at 14 mg/L in drinking tap water. Supplemented water was provided ad libitum and refreshed weekly. The same regimen has been used by other groups and resulted in ≈3‐fold increase in plasma l‐homoarginine levels after 4 weeks.16, 17, 18 In humans the supplementation was performed with 125 mg l‐homoarginine/day and after 4 weeks led to ≈7‐fold increase in systemic l‐homoarginine levels.20, 32 Both regimens (in humans and in mice) resulted in the addition of ≈2 mg hArg/kg body weight.
Echocardiography
A Vivid 7 ultrasound instrument (GE Healthcare, Port Washington, NY) equipped with an i13L transducer (recommended for cardiac studies in rodents, 5.9‐14.1 MHz) was used in all experiments. Mice were anesthetized with 0.8% to 1.5% isoflurane, and a mid–left ventricular (LV) short‐axis view at the level of the papillary muscles was obtained in B‐mode and recorded in M‐mode. LV diameter, LV posterior, and septal wall thicknesses in systole and diastole as well as time intervals were measured and averaged over 3 cardiac cycles using EchoPAC PC software (GE Healthcare, Port Washington, NY). Heart rate, fractional shortening, ejection fraction, cardiac output, and LV mass were calculated using standard equations for rodents (as recommended by VisualSonics, Bothell, WA). Matched baseline and final echo data were available for 9 mice in the WHC+placebo group, 9 in the WHC+hArg group, 5 in WHC‐eTNAP+placebo group, and 6 in the WHC‐eTNAP+hArg group.
Collection of Plasma and Tissues
Heparin plasma was prepared from venous blood collected from the right ventricle of anesthetized mice. Mice were fasted at least 5 hours before blood collection. Following blood collection, whole‐body perfusion with 10% formalin was performed, and tissues were stored in 10% formalin until dissection.
Blood Chemistry
Alkaline phosphatase, total cholesterol, triglycerides, inorganic phosphate, and calcium were determined using specific reagents (Pointe Scientific, Canton, MI). Lipemic plasma samples were cleared with an equal volume of StatSpin LipoClear reagent (Beckman Coulter, Brea, CA) before inorganic phosphate and calcium determination. All samples were analyzed using manual protocols and a plate reader (BioTek Instruments, Winooski, VT). Measurements of l‐homoarginine were conducted by liquid chromatography–tandem mass spectrometry according to a previously described procedure.33, 34 Pyridoxal phosphate was measured by liquid chromatography–tandem mass spectrometry according to the method adapted from Roelofsen‐de Beer et al.35
Histology
Tissues were embedded in Optimal Cutting Temperature compound and cryosectioned at 10 μm. Serial sections were mounted on replica slides. For the aortic root samples, we collected 72 consecutive sections on 8 slides. For the midventricular slices of the heart, 36 sections were mounted on 6 slides.
To detect calcium, sections were rehydrated in water, stained in 2% alizarin red S (CI 58005) solution, pH 4.2, for 3 minutes, rinsed in distilled water followed by 3 changes of phosphate‐buffered saline, pH 7.4. Slides were then air‐dried and mounted in Clearium Mounting Medium (Leica, Wetzlar, Germany). Another set of slides was rehydrated in water, rinsed in 60% isopropanol, and stained in 0.2% oil red O (CI 26125) solution in 60% isopropanol for 15 minutes for the detection of lipids. Following oil red O staining, slides were rinsed in 60% isopropanol and 3 changes of tap water and mounted in glycerin jelly mounting medium (Poly Scientific, Bay Shore, NY). Picrosirius red staining was used to detect collagen according to a published protocol for cryosections.36
Tissues for histological analyses were extracted from 6 mice in the WHC+placebo group, 6 mice in the WHC+hArg group, 5 mice in WHC‐eTNAP+placebo group, and 6 mice in the WHC‐eTNAP+hArg group.
Morphometric Analysis
Morphometric analysis of alizarin red, oil red O, and picrosirius red staining was performed using ImageJ.37 Regions of interest representing the left anterior descending coronary artery (LAD), the septal artery, the aortic sinus, and the left ventricle were manually outlined. Areas outside of the regions of interest were cleared, and the resulting images were sampled for colors representing positively stained areas by visual inspection using the “adjust color threshold” function of ImageJ. The areas above the set color threshold were defined as positive regions of interest and recorded. Aortic root sections (6‐8 per sample) were used to measure calcium and lipids in the septal artery and plaques in the aortic root. Mid‐LV sections at the level of papillary muscles (4‐6 per sample) were used to measure calcium and lipids in the LAD. Mid‐LV sections were also used to measure picrosirius red–positive regions representing collagen. Picrosirius red–positive areas were quantified over the entire LV cross section and expressed as percentage of area.
Statistical Analyses
Statistical analyses were performed in GraphPad Prism 8 (GraphPad Software, San Diego, CA). Survival fractions and curves were compared using Chi‐square and Gehan‐Breslow‐Wilcoxon tests, respectively. All other parameters were first tested for equality of variances using the Brown‐Forsythe method. The parametric variables were then compared using ANOVA followed by Holm‐Sidak multiple comparisons of prespecified groups. The nonparametric variables were analyzed using a Kruskal‐Wallis test followed by prespecified Dunn multiple comparisons. The prespecified primary comparisons were formulated to test (1) the effect of TNAP on the final outcomes by comparing the WHC+placebo and WHC‐eTNAP+placebo groups, (2) the effect of hArg in the absence of coronary artery disease (CAD) by comparing the WHC+placebo and WHC+hArg groups, and (3) the effect of hArg in mice with CAD by comparing the WHC‐eTNAP+placebo and WHC‐eTNAP+hArg groups. The percentage change from baseline was calculated for all reported echocardiographic parameters and compared between the groups by ANOVA or Kruskal‐Wallis methods as described above. Secondary analyses were performed to compare body weight and plasma parameters (inorganic phosphate, calcium, total cholesterol, triglycerides) between the WHC and WHC‐eTNAP groups at baseline and to determine the effect of an atherogenic diet on these measures in each group (WHC, WHC‐eTNAP). In addition, a linear regression was calculated to determine the effect of the duration of an atherogenic diet on plasma lipids in WHC‐eTNAP mice (the placebo group). Statistical significance was accepted at P<0.05. Data are reported as mean±SD.
Results
Homoarginine Supplementation and Survival in a CAD Model
WHC‐eTNAP mice had significantly reduced survival under the atherogenic conditions of our experiment compared with WHC mice (21.7% versus 93.8%, P<0.0001, Table 1 and Figure 1A) as was expected.24 The median survival in the WHC‐eTNAP+placebo group was 27 days. The WHC‐eTNAP+hArg group showed a 37.5% survival under conditions of our study with the median survival of 33 days (Table 1 and Figure 1A). To verify if our supplementation protocol indeed increased hArg levels, we measured hArg concentrations in plasma. Plasma levels of hArg were increased ≈3‐fold in hArg‐supplemented mice compared with mice given the placebo treatment in both comparisons, with and without TNAP overexpression (Figure 1B, P<0.001).
Table 1.
Survival
| WHC+Placebo | WHC+hArg | WHC‐eTNAP+Placebo | WHC‐eTNAP+hArg | |
|---|---|---|---|---|
| N | 16 | 10 | 23 | 16 |
| Survival | 93.75% | 90.00% | 21.74% a | 37.50% |
| Median survival, d | >42b | >42b | 27 | 33c |
eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; WHC, wicked high‐cholesterol allele.
P<0.0001 vs WHC+placebo by Chi‐squared test.
Undetermined.
Not significant (P=0.10 vs WHC‐eTNAP+placebo by Gehan‐Breslow‐Wilcoxon test).
Figure 1.
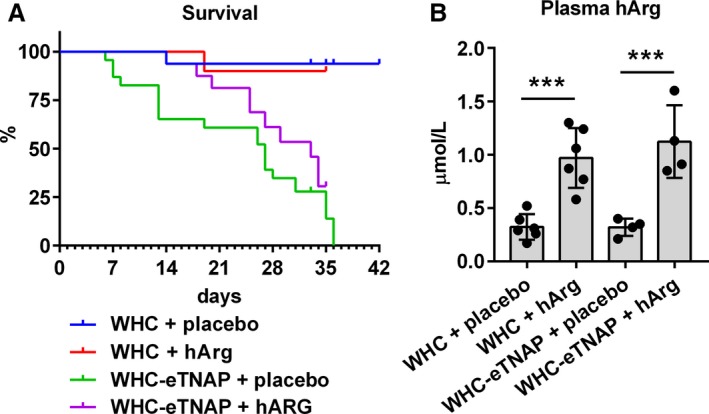
Survival data and plasma homoarginine (hArg) levels. A, Survival was monitored over 6 weeks starting at 6 weeks of age. B, Homoarginine was measured in heparin plasma in a subset of mice, ***P<0.001 vs placebo. eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; WHC, wicked high‐cholesterol allele.
Homoarginine Supplementation Did Not Influence Alkaline Phosphatase Activity
Homoarginine supplementation influenced neither alkaline phosphatase activity in plasma (Figure 2A) nor systemic levels of pyridoxal phosphate, which can be regarded as a marker of changes in alkaline phosphatase activity in vivo38 (Figure 2B).
Figure 2.

Effect of hArg on alkaline phosphatase activity and the levels of endogenous TNAP substrate pyridoxal phosphate (PLP). A, Alkaline phosphatase activity was measured in heparin plasma in a subset of mice, **P<0.01. B, Plasma levels of PLP, an endogenous substrate of TNAP, *P<0.05. eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; TNAP, tissue‐nonspecific alkaline phosphatase; WHC, wicked high‐cholesterol allele.
Homoarginine Supplementation, Body Weight, and Plasma Parameters in Mice With CAD
Plasma inorganic phosphate was elevated in the WHC+placebo group compared with baseline (P<0.001, Figure 3A), whereas in the WHC+hArg group this effect was insignificant. Plasma calcium appeared to be suppressed in mice on an atherogenic diet, but this reduction was insignificant in 2 prespecified comparisons (WHC+placebo versus WHC baseline and WHC‐eTNAP+placebo versus WHC‐eTNAP baseline, Figure 3B). There were no differences in plasma inorganic phosphate or calcium between the placebo‐ and hArg‐treated groups (Figure 3A and 3B).
Figure 3.
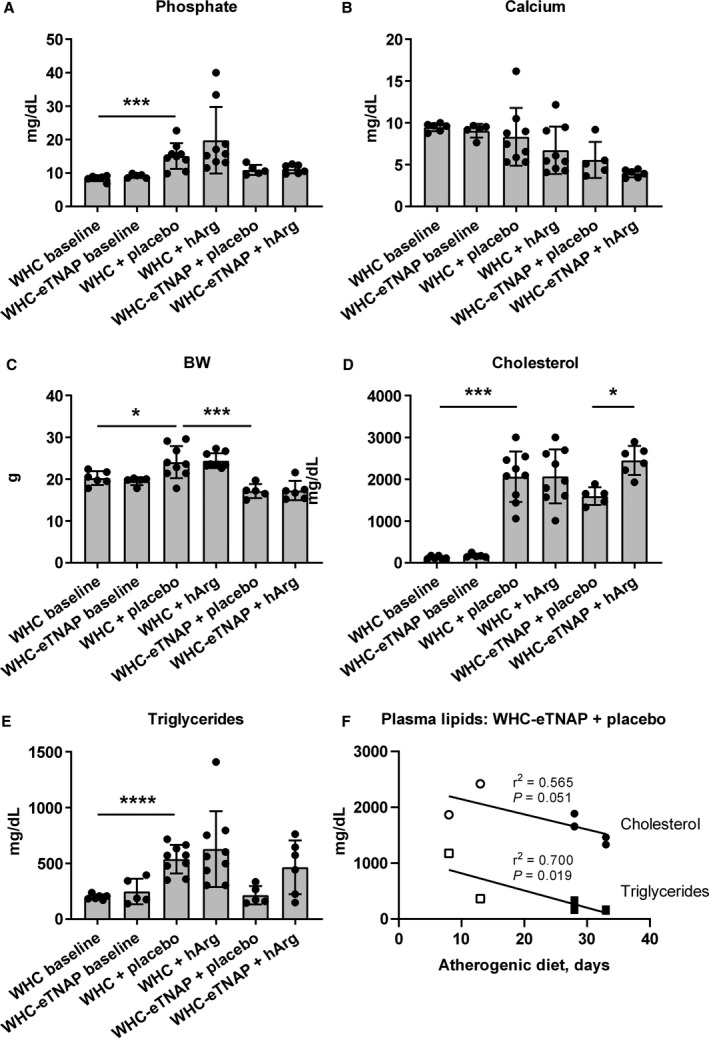
Effects of TNAP overexpression, an atherogenic diet, and hArg supplementation on plasma inorganic phosphate, calcium, body weight, and plasma lipids (cholesterol and triglycerides). A, Plasma inorganic phosphate, ***P<0.001. B, Plasma calcium. C, Body weight (BW), *P<0.05, ***P<0.001. D, Plasma total cholesterol, *P<0.05, ***P<0.001. E, Plasma triglycerides, ****P<0.0001. F, Linear regression analysis of the correlation between the duration of an atherogenic diet and plasma lipids in WHC‐eTNAP mice; open symbols represent early time points added to construct the correlation curves. BW indicates body weight; eTNAP, overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; TNAP, tissue‐nonspecific alkaline phosphatase; WHC, wicked high‐cholesterol allele.
There was a significant increase in body weight (BW) in the WHC+placebo group as compared with baseline (P<0.05). WHC‐eTNAP+placebo mice were significantly lighter than WHC+placebo mice at the end of the study (P<0.001, Figure 3C). Compared with baseline, plasma cholesterol was higher in the WHC+placebo (P<0.001) group but not in the WHC‐eTNAP+placebo group (Figure 3D). There was an ≈50% increase in total plasma cholesterol in WHC‐eTNAP mice receiving hArg compared with placebo (P<0.05, Figure 3D). Similarly to cholesterol, triglycerides were elevated in the WHC+placebo group (P<0.0001) but not in the WHC‐eTNAP+ placebo group compared with baseline (Figure 3E). The effects of hArg on triglycerides were not significant in either the WHC or the WHC‐eTNAP group compared with the corresponding placebo groups (Figure 3E). Further examination of plasma lipids in WHC‐eTNAP+placebo mice with the addition of 2 samples collected at the earlier time points (8 and 13 days after initiation of an atherogenic diet, Figure 3F, open symbols) showed a gradual decline in plasma triglycerides (P<0.05) and a nonsignificant trend for the reduction of cholesterol in WHC‐eTNAP+placebo mice over time (P=0.051, linear regression, Figure 3F).
Homoarginine Supplementation Did Not Influence Calcification and Lipid Deposition in Coronary Arteries or Aortic Root
Consistent with our previous observations,24 endothelial TNAP overexpression resulted in increased calcification of the LAD (P<0.001) and atherosclerotic plaques in the aortic root in WHC‐eTNAP mice (P<0.05, Figure 4). Contrary to our hypothesis, however, hArg supplementation led to no significant changes in calcium in any of the 3 vascular segments tested (Figure 4).
Figure 4.
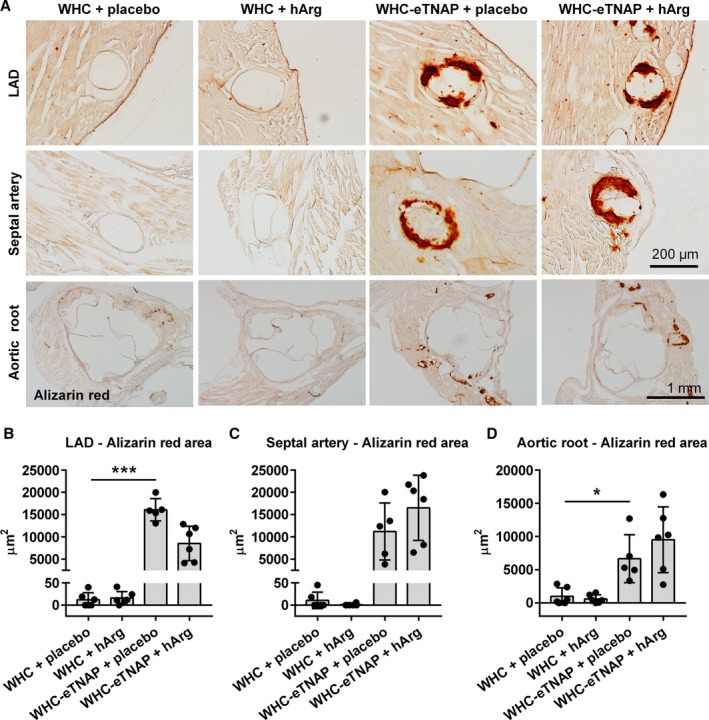
Alizarin red staining for calcium. A, Left anterior descending artery (LAD, top panels), proximal segment of the septal artery (middle panels), and the aortic root (bottom panels). B through D, Morphometric quantification of alizarin red positive area in the LAD (B), septal artery (C), and the aortic root (D). *P<0.05, ***P<0.001. eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; LAD, left anterior descending artery; WHC, wicked high‐cholesterol allele.
TNAP overexpression resulted in an increase in coronary lipid deposition (LAD, P<0.001, Figure 5B) and a decrease in the size of atherosclerotic plaques in the aortic root of WHC‐eTNAP mice compared with WHC (P<0.05, Figure 5D). hArg supplementation did not influence the amount of lipids deposited in the LAD, the septal branch of coronary arteries, or the aortic root as quantified by oil red O staining (Figure 5).
Figure 5.
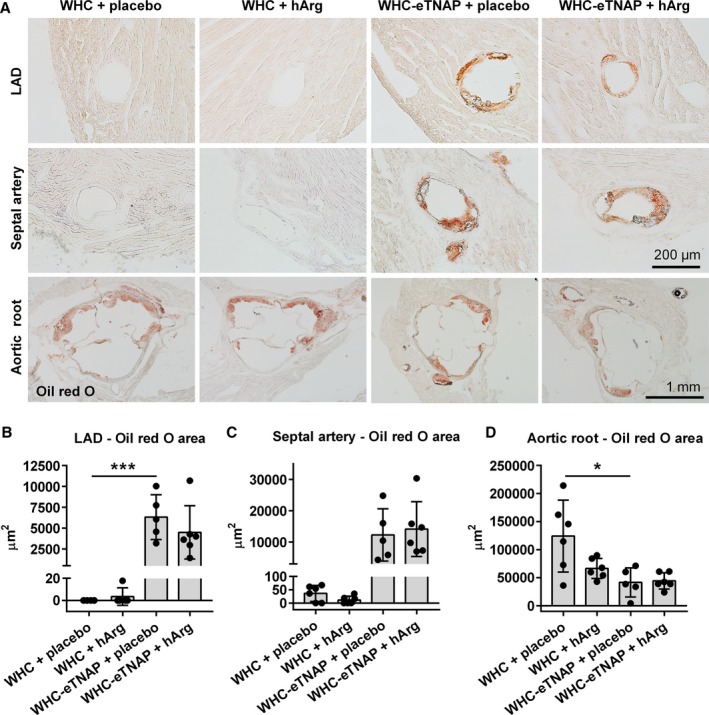
Oil red O staining for lipids. A, Left anterior descending artery (LAD, top panels), proximal segment of the septal artery (middle panels), and the aortic root (bottom panels). B through D, Morphometric quantification of oil red O—positive areas in the LAD (B), septal artery (C), and the aortic root (D), *P<0.05, ***P<0.001. eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; LAD, left anterior descending artery; WHC, wicked high‐cholesterol allele.
Protective Effects of Homoarginine on the Myocardium
Unable to detect an inhibitory effect of hArg on the calcification‐promoting activity of TNAP, we next investigated if the protective action of hArg in the setting of calcified CAD could be mediated by its effects on the myocardial structure and function.
WHC‐eTNAP mice were characterized by reduced BW (P<0.05), increased LV diameter (per BW, P<0.0001), LV mass index (LVmass/BW, P<0.05), and reduced LV fractional shortening (P<0.05) and ejection fraction (P<0.05) compared with WHC (Table 2). Supplementation of WHC‐eTNAP mice with hArg prevented LV dilatation (per BW, P<0.01), reduced LV hypertrophy (LVmass/BW, P<0.05), and preserved LV fractional shortening and ejection fraction (P<0.05, Table 2).
Table 2.
Echocardiography
| WHC+Placebo | WHC+hArg | WHC‐eTNAP+Placebo | WHC‐eTNAP+hArg | |
|---|---|---|---|---|
| N | 9 | 9 | 5 | 6 |
| Age, wk | ||||
| Baseline | 6.2±0.3 | 6.0±0.2 | 6.1±0.3 | 6.2±0.4 |
| Final | 11.2±0.5 | 11±0.2 | 10.3±0.4† | 10.7±0.6 |
| BW, g | ||||
| Baseline | 21.2±1.2 | 21.8±1.8 | 20.5±1.4 | 21.6±1.2 |
| Final | 24.1±3.9 | 24.4±1.7 | 17.1±1.7* | 17.3±2.3 |
| HR, bpm | ||||
| Baseline | 461±60 | 406±46 | 429±27 | 403±7 |
| Final | 431±62 | 461±44 | 325±84 | 371±128 |
| LVPWd/BW, mm/g | ||||
| Baseline | 0.036±0.007 | 0.033±0.007 | 0.032±0.004 | 0.033±0.006 |
| Final | 0.034±0.007 | 0.03±0.005 | 0.059±0.028 | 0.057±0.013 |
| IVSd/BW, mm/g | ||||
| Baseline | 0.036±0.003 | 0.034±0.007 | 0.041±0.011 | 0.036±0.005 |
| Final | 0.036±0.005 | 0.032±0.004 | 0.055±0.027 | 0.063±0.008 |
| LVIDd/BW, mm/g | ||||
| Baseline | 0.17±0.02 | 0.17±0.02 | 0.19±0.03 | 0.19±0.02 |
| Final | 0.15±0.02 | 0.16±0.01 | 0.25±0.07‡ | 0.18±0.05∥ |
| LV mass/BW, mg/g | ||||
| Baseline | 3.45±0.54 | 3.32±0.62 | 4.00±1.13 | 4.15±0.47 |
| Final | 3.6±0.75 | 3.26±0.44 | 8.04±3.7* | 4.92±0.47§ |
| FS, % | ||||
| Baseline | 37.2±10.9 | 29.4±6.3 | 30.8±11.6 | 30.2±9.2 |
| Final | 30.9±9.9 | 24.3±6 | 20.4±6.2* | 34.0±8.2§ |
| EF, % | ||||
| Baseline | 71.9±14.3 | 62.4±9.6 | 62.8±15 | 63±13.4 |
| Final | 63.9±13.8 | 54.3±10.4 | 46.8±12.5* | 69.2±12§ |
| CO/BW, μL/min per g | ||||
| Baseline | 0.71±0.17 | 0.60±0.12 | 0.76±0.17 | 0.85±0.21 |
| Final | 0.57±0.16 | 0.56±0.1 | 0.75±0.44 | 0.44±0.28 |
Data are expressed as mean±SD. BW indicates body weight; CO, cardiac output; EF, ejection fraction; eTNAP, overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; FS, fractional shortening; hArg, homoarginine; HR, heart rate; IVSd, interventricular septum thickness in diastole; LVIDd, left ventricular end diastolic diameter; LVmass, left ventricular mass; LVPWd, left ventricular posterior wall thickness in diastole; WHC, wicked high‐cholesterol allele.
*P<0.05, † P<0.01, ‡ P<0.01 vs WHC+placebo.
§ P<0.05, ∥ P<0.01 vs WHC‐eTNAP+placebo.
Analyzing echocardiographic changes from baseline, we found that WHC‐eTNAP+placebo mice experienced a 33% (0%, 66%) increase in the LV diameter (per BW), whereas WHC‐eTNAP mice treated with hArg showed a small −8% (−14%, −3%) reduction in the LV diameter (P<0.001, Figure 6A and 6B). hArg also reversed a negative change in fractional shortening from a −30% (−57%, −3%) reduction in the placebo group to a 19% (−21%, 58%) increase in hArg‐treated WHC‐eTNAP mice (P<0.05, Figure 6C). Likewise, hArg reversed the effect on the ejection fraction in WHC‐eTNAP mice from a −25% (−47%, −2%) reduction in the placebo group to a 13% (−15%, 41%) increase in the hArg group (P<0.05, Figure 6D).
Figure 6.
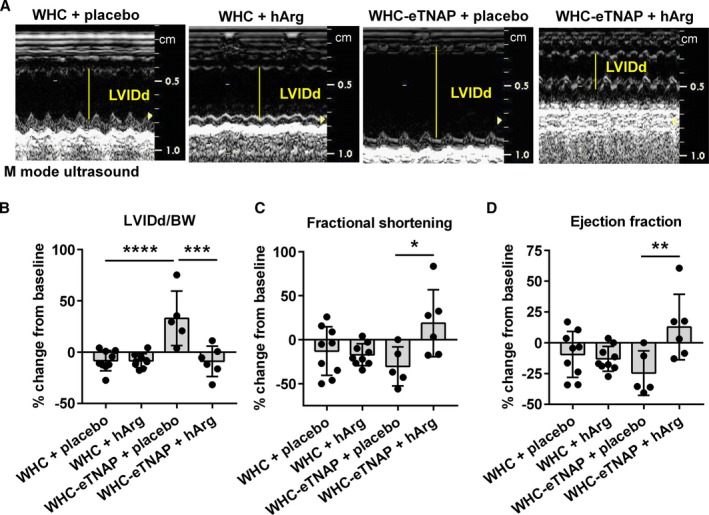
Echocardiographic data. A, M‐mode echocardiographic images of the left. B, LVIDd normalized to body weight, change from baseline. C, Fractional shortening, change from baseline. D, Ejection fraction, change from baseline. *P<0.5, **P<0.01, ***P<0.001, ****P<0.0001. BW indicates body weight; eTNAP, overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; LVIDd, left ventricular internal diameter in diastole (left ventricular end‐diastolic diameter); WHC, wicked high‐cholesterol allele.
LV collagen content (measured by picrosirius red staining) was increased in WHC‐eTNAP+placebo mice compared with WHC+placebo (P<0.0001, Figure 7). Treatment of WHC‐eTNAP mice with hArg reduced myocardial collagen deposition compared with the placebo treated group (P<0.01, Figure 7).
Figure 7.
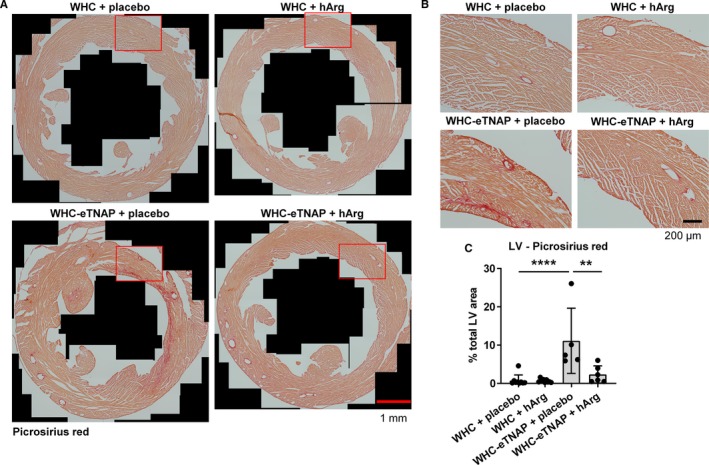
Picrosirius red staining for collagen. A, Representative images of picrosirius red–stained sections through the left ventricle (LV) of the heart. B, Representative high‐resolution images corresponding to the insets in (A). C, Morphometric quantification of collagen (percentage picrosirius red–positive area relative to the total LV area), **P<0.01, ****P<0.0001. eTNAP indicates overexpression of tissue‐nonspecific alkaline phosphatase in the endothelium; hArg, homoarginine; WHC, wicked high‐cholesterol allele.
Discussion
Contrary to our mechanistic hypothesis regarding the effects of hArg on TNAP activity and coronary calcification as a possible explanation for cardioprotective properties of this molecule in vivo, we found no evidence that hArg inhibits TNAP activity or reduces coronary calcification or atherosclerosis (Figures 1, 2 through 3). Interestingly, however, hArg treatment was associated with protection from LV dilatation on the background of severe dyslipidemia and coronary artery disease (Figures 3, 6, and 7, Table 2) and a modest improvement in survival (Figure 1 and Table 1).
Low hArg levels predict an adverse outcome in multiple cardiovascular, metabolic, and renal pathologies; however, it is still not entirely clear whether hArg plays a direct protective role in at least some of those pathologies. Our finding that dietary hArg supplementation improved outcomes in an experimental model of coronary atherosclerosis and heart failure supports the hypothesis that hArg is more than just a marker and can have direct protective cardiovascular effects. This observation is in line with the previous studies, demonstrating direct protective effects of homoarginine in experimental cardiovascular and metabolic models.12, 16, 17, 18
We hypothesized that protection might be mediated through the hArg effect on calcification, based on its ability to inhibit TNAP activity in vitro.23, 24 At least in our experimental model we could not detect any inhibitory effect of hArg on TNAP activity or vascular calcification in vivo, as evidenced by a lack of changes in alkaline phosphatase activity and pyridoxal phosphate levels in plasma and a lack of protective effect of hArg on coronary calcification. Overall, our findings in this model of coronary artery disease suggest that hArg improved the disease course through a mechanism that is independent of vascular calcification. A lack of hArg effects on vascular calcification in our study is consistent with the report by Alesutan and colleagues, who observed augmented vascular calcification in response to hArg treatment in hyperphosphatemic klotho‐hypomorphic mice, in mice with renal failure, and in mice after vitamin D3 overload.39 A lack of association between low plasma hArg and coronary calcification was also observed in the population‐based Dallas Heart Study.15 On the other hand, it was shown that l‐lysine, a hArg precursor, prevented arterial calcification in adenine‐induced uremic rats, whereas hArg inhibited precipitation of minerals in vitro in a solution of supersaturated calcium/phosphate.40 However, the same study by Shimomura also suggested several potential hArg‐independent mechanisms for l‐lysine to affect vascular calcification, and the physiologic relevance of the effect of hArg on precipitation of calcium phosphate in vitro is currently unclear.
Our study did not show any protective effects of hArg on coronary atherosclerosis in WHC‐eTNAP mice on the Paigen diet. This finding is consistent with the lack of association between low hArg levels and aortic plaques in the Dallas Heart Study15 and suggests that hArg might not have a major effect on atherosclerosis progression. The effect of hArg on coronary atherosclerosis in WHC‐eTNAP mice might have been masked by the restoration of hypercholesterolemia to the levels observed in WHC mice. It also must be noted that our model, although it recapitulates the genetics and lipid abnormalities of familial hypercholesterolemia,41, 42, 43 is not the classical experimental model of atherosclerosis. We therefore suggest that the effect of hArg on atherosclerosis progression will need to be systematically addressed in models that are more appropriate for this study question, such as apoE knockout and Ldlr knockout mice.
The key finding of our study is that hArg mediated protection from LV dilatation and preserved LV ejection fraction. Our data are consistent with the recent reports that hArg supplementation improved cardiac contractile function in a murine model of post–myocardial infarction heart failure.16 Furthermore, impaired cardiac contractile reserve in arginine:glycine amidinotransferase knockout mice with low creatine and hArg levels was rescued not by creatine supplementation but by hArg supplementation.17 Bahls and colleagues recently reported a correlation between low plasma hArg and decreased LV function in a large cohort of 3113 subjects, suggesting that the protective effects of hArg on myocardial contractility that were observed in animal models might also be relevant for humans.44 Our characterizations of the positive effects of hArg on LV geometry are also consistent with the reported association between hArg and LV dilatation.44 The mechanisms underlying this direct effect of hArg on LV remodeling are still unclear and need to be addressed in future studies.
The study presented here is limited to male mice and was conducted at extremely high levels of dyslipidemia as well as artificially increased levels of plasma alkaline phosphatase. The study was focused on the heart and has not yet considered the effect of hArg on other organ systems under conditions of coronary artery disease and/or heart failure.
In summary, our study further supports the hypothesis that hArg not only is a risk assessment marker but also has direct protective cardiovascular and metabolic effects. Specifically, we observed direct protective effects of hArg on myocardial remodeling. Daily oral supplementation with 125 mg hArg in capsules increased its plasma levels severalfold in healthy volunteers without causing any detectable side effects.20, 32 This identifies hArg supplementation as a well‐tolerated and nonharmful treatment that can be further tested in phase 2 and phase 3 clinical studies for improvement of cardiovascular outcome in patients at risk. Based on our results and the results by others, patients with heart failure might be a patient population with especially high chances to benefit from hArg supplementation.
Sources of Funding
This work was supported in part by National Institutes of Health grants R01 DE012889 (to Millan) and R56 HL131547 (to Savinova) and by funding from the German Heart Foundation/German Foundation of Heart Research F/24/17 (to Rodionov).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e012486 DOI: 10.1161/JAHA.119.012486.)
Contributor Information
Roman N. Rodionov, Email: roman.rodionov@uniklinikum-dresden.de.
Olga V. Savinova, Email: osavinov@nyit.edu.
References
- 1. Tsikas D, Wu G. Homoarginine, arginine, and relatives: analysis, metabolism, transport, physiology, and pathology. Amino Acids. 2015;47:1697–1702. [DOI] [PubMed] [Google Scholar]
- 2. May M, Kayacelebi AA, Batkai S, Jordan J, Tsikas D, Engeli S. Plasma and tissue homoarginine concentrations in healthy and obese humans. Amino Acids. 2015;47:1847–1852. [DOI] [PubMed] [Google Scholar]
- 3. Braissant O, Henry H, Villard AM, Speer O, Wallimann T, Bachmann C. Creatine synthesis and transport during rat embryogenesis: spatiotemporal expression of AGAT, GAMT and CT1. BMC Dev Biol. 2005;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsikas D, Bollenbach A, Hanff E, Kayacelebi AA. Asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA) and homoarginine (hArg): the ADMA, SDMA and hArg paradoxes. Cardiovasc Diabetol. 2018;17:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sacristan M, Varela A, Pedrosa MM, Burbano C, Cuadrado C, Legaz ME, Muzquiz M. Determination of β‐N‐oxalyl‐l‐α,β‐diaminopropionic acid and homoarginine in Lathyrus sativus and Lathyrus cicera by capillary zone electrophoresis. J Sci Food Agric. 2015;95:1414–1420. [DOI] [PubMed] [Google Scholar]
- 6. Onar AN, Erdogan BY, Ayan I, Acar Z. Homoarginine, β‐ODAP, and asparagine contents of grass pea landraces cultivated in Turkey. Food Chem. 2014;143:277–281. [DOI] [PubMed] [Google Scholar]
- 7. Rodionov RN, Oppici E, Martens‐Lobenhoffer J, Jarzebska N, Brilloff S, Burdin D, Demyanov A, Kolouschek A, Leiper J, Maas R, Cellini B, Weiss N, Bode‐Boger SM. A novel pathway for metabolism of the cardiovascular risk factor homoarginine by alanine:glyoxylate aminotransferase 2. Sci Rep. 2016;6:35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frenay AR, Kayacelebi AA, Beckmann B, Soedamah‐Muhtu SS, de Borst MH, van den Berg E, van Goor H, Bakker SJ, Tsikas D. High urinary homoarginine excretion is associated with low rates of all‐cause mortality and graft failure in renal transplant recipients. Amino Acids. 2015;47:1827–1836. [DOI] [PubMed] [Google Scholar]
- 9. Ravani P, Maas R, Malberti F, Pecchini P, Mieth M, Quinn R, Tripepi G, Mallamaci F, Zoccali C. Homoarginine and mortality in pre‐dialysis chronic kidney disease (CKD) patients. PLoS One. 2013;8:e72694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martens‐Lobenhoffer J, Emrich IE, Zawada AM, Fliser D, Wagenpfeil S, Heine GH, Bode‐Boger SM. l‐Homoarginine and its AGXT2‐metabolite GOCA in chronic kidney disease as markers for clinical status and prognosis. Amino Acids. 2018;50:1347–1356. [DOI] [PubMed] [Google Scholar]
- 11. Marz W, Meinitzer A, Drechsler C, Pilz S, Krane V, Kleber ME, Fischer J, Winkelmann BR, Bohm BO, Ritz E, Wanner C. Homoarginine, cardiovascular risk, and mortality. Circulation. 2010;122:967–975. [DOI] [PubMed] [Google Scholar]
- 12. Choe CU, Atzler D, Wild PS, Carter AM, Böger RH, Ojeda F, Simova O, Stockebrand M, Lackner K, Nabuurs C, Marescau B, Streichert T, Müller C, Lüneburg N, De Deyn PP, Benndorf RA, Baldus S, Gerloff C, Blankenberg S, Heerschap A, Grant PJ, Magnus T, Zeller T, Isbrandt D, Schwedhelm E. Homoarginine levels are regulated by L‐arginine:glycine amidinotransferase and affect stroke outcome: results from human and murine studies. Circulation. 2013;128:1451–1461. [DOI] [PubMed] [Google Scholar]
- 13. Pilz S, Tomaschitz A, Meinitzer A, Drechsler C, Ritz E, Krane V, Wanner C, Bohm BO, Marz W. Low serum homoarginine is a novel risk factor for fatal strokes in patients undergoing coronary angiography. Stroke. 2011;42:1132–1134. [DOI] [PubMed] [Google Scholar]
- 14. Jud P, Hafner F, Verheyen N, Meinitzer A, Gary T, Brodmann M, Seinost G, Hackl G. Homoarginine/ADMA ratio and homoarginine/SDMA ratio as independent predictors of cardiovascular mortality and cardiovascular events in lower extremity arterial disease. Sci Rep. 2018;8:14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Atzler D, Gore MO, Ayers CR, Choe CU, Boger RH, de Lemos JA, McGuire DK, Schwedhelm E. Homoarginine and cardiovascular outcome in the population‐based Dallas Heart Study. Arterioscler Thromb Vasc Biol. 2014;34:2501–2507. [DOI] [PubMed] [Google Scholar]
- 16. Atzler D, McAndrew DJ, Cordts K, Schneider JE, Zervou S, Schwedhelm E, Neubauer S, Lygate CA. Dietary supplementation with homoarginine preserves cardiac function in a murine model of post‐myocardial infarction heart failure. Circulation. 2017;135:400–402. [DOI] [PubMed] [Google Scholar]
- 17. Faller KME, Atzler D, McAndrew DJ, Zervou S, Whittington HJ, Simon JN, Aksentijevic D, Ten Hove M, Choe CU, Isbrandt D, Casadei B, Schneider JE, Neubauer S, Lygate CA. Impaired cardiac contractile function in arginine:glycine amidinotransferase knockout mice devoid of creatine is rescued by homoarginine but not creatine. Cardiovasc Res. 2018;114:417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stockebrand M, Hornig S, Neu A, Atzler D, Cordts K, Böger RH, Isbrandt D, Schwedhelm E, Choe CU. Homoarginine supplementation improves blood glucose in diet‐induced obese mice. Amino Acids. 2015;47:1921–1929. [DOI] [PubMed] [Google Scholar]
- 19. Hecker M, Walsh DT, Vane JR. On the substrate specificity of nitric oxide synthase. FEBS Lett. 1991;294:221–224. [DOI] [PubMed] [Google Scholar]
- 20. Atzler D, Schonhoff M, Cordts K, Ortland I, Hoppe J, Hummel FC, Gerloff C, Jaehde U, Jagodzinski A, Boger RH, Choe CU, Schwedhelm E. Oral supplementation with l‐homoarginine in young volunteers. Br J Clin Pharmacol. 2016;82:1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Waardenburg DA, de Betue CT, Luiking YC, Engel M, Deutz NE. Plasma arginine and citrulline concentrations in critically ill children: strong relation with inflammation. Am J Clin Nutr. 2007;86:1438–1444. [DOI] [PubMed] [Google Scholar]
- 22. Rufo MB, Fishman WH. L‐Homoarginine, a specific inhibitor of liver‐type alkaline phosphatase, applied to the recognition of liver‐type enzyme activity in rat intestine. J Histochem Cytochem. 1972;20:336–343. [DOI] [PubMed] [Google Scholar]
- 23. Romanul FC, Bannister RG. Localized areas of high alkaline phosphatase activity in the terminal arterial tree. J Cell Biol. 1962;15:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romanelli F, Corbo A, Salehi M, Yadav MC, Salman S, Petrosian D, Rashidbaigi OJ, Chait J, Kuruvilla J, Plummer M, Radichev I, Margulies KB, Gerdes AM, Pinkerton AB, Millan JL, Savinov AY, Savinova OV. Overexpression of tissue‐nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS One. 2017;12:e0186426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Millan JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley‐VCH Verlag GmbH & Co, 2006; 1–322. [Google Scholar]
- 26. Savinov AY, Salehi M, Yadav MC, Radichev I, Millan JL, Savinova OV. Transgenic overexpression of tissue‐nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc. 2015;4:e002499 DOI: 10.1161/JAHA.115.002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Svenson KL, Ahituv N, Durgin RS, Savage H, Magnani PA, Foreman O, Paigen B, Peters LL. A new mouse mutant for the LDL receptor identified using ENU mutagenesis. J Lipid Res. 2008;49:2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Chhea TN, Sergienko EA, Kapoor K, Jackson MR, Hoylaerts MF, Pinkerton AB, O'Neill WC, Millan JL. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res. 2015;30:824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bronson SK, Plaehn EG, Kluckman KD, Hagaman JR, Maeda N, Smithies O. Single‐copy transgenic mice with chosen‐site integration. Proc Natl Acad Sci USA. 1996;93:9067–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2‐cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo. Dev Biol. 2001;230:230–242. [DOI] [PubMed] [Google Scholar]
- 31. Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, Smallwood PM, Erlanger B, Wheelan SJ, Nathans J. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron. 2014;81:103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schonhoff M, Weineck G, Hoppe J, Hornig S, Cordts K, Atzler D, Gerloff C, Boger R, Neu A, Schwedhelm E, Choe CU. Cognitive performance of 20 healthy humans supplemented with L‐homoarginine for 4 weeks. J Clin Neurosci. 2018;50:237–241. [DOI] [PubMed] [Google Scholar]
- 33. Martens‐Lobenhoffer J, Surdacki A, Bode‐Böger SM. Fast and precise quantification of L‐homoarginine in human plasma by HILIC‐isotope dilution‐MS–MS. Chromatographia. 2013;76:1755–1759. [Google Scholar]
- 34. Martens‐Lobenhoffer J, Bode‐Boger SM. Quantification of L‐arginine, asymmetric dimethylarginine and symmetric dimethylarginine in human plasma: a step improvement in precision by stable isotope dilution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;904:140–143. [DOI] [PubMed] [Google Scholar]
- 35. Roelofsen‐de Beer R, van Zelst BD, Wardle R, Kooij PG, de Rijke YB. Simultaneous measurement of whole blood vitamin B1 and vitamin B6 using LC‐ESI‐MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1063:67–73. [DOI] [PubMed] [Google Scholar]
- 36. Hadi AM, Mouchaers KT, Schalij I, Grunberg K, Meijer GA, Vonk‐Noordegraaf A, van der Laarse WJ, Belien JA. Rapid quantification of myocardial fibrosis: a new macro‐based automated analysis. Cell Oncol. 2011;34:343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non‐specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B‐6. Nat Genet. 1995;11:45–51. [DOI] [PubMed] [Google Scholar]
- 39. Alesutan I, Feger M, Tuffaha R, Castor T, Musculus K, Buehling SS, Heine CL, Kuro OM, Pieske B, Schmidt K, Tomaschitz A, Maerz W, Pilz S, Meinitzer A, Voelkl J, Lang F. Augmentation of phosphate‐induced osteo‐/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc Res. 2016;110:408–418. [DOI] [PubMed] [Google Scholar]
- 40. Shimomura A, Matsui I, Hamano T, Ishimoto T, Katou Y, Takehana K, Inoue K, Kusunoki Y, Mori D, Nakano C, Obi Y, Fujii N, Takabatake Y, Nakano T, Tsubakihara Y, Isaka Y, Rakugi H. Dietary L‐lysine prevents arterial calcification in adenine‐induced uremic rats. J Am Soc Nephrol. 2014;25:1954–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pecin I, Whittall R, Futema M, Sertic J, Reiner Z, Leigh SE, Humphries SE. Mutation detection in Croatian patients with familial hypercholesterolemia. Ann Hum Genet. 2013;77:22–30. [DOI] [PubMed] [Google Scholar]
- 42. Mozas P, Castillo S, Tejedor D, Reyes G, Alonso R, Franco M, Saenz P, Fuentes F, Almagro F, Mata P, Pocovi M. Molecular characterization of familial hypercholesterolemia in Spain: identification of 39 novel and 77 recurrent mutations in LDLR. Hum Mutat. 2004;24:187. [DOI] [PubMed] [Google Scholar]
- 43. Humphries SE, Cranston T, Allen M, Middleton‐Price H, Fernandez MC, Senior V, Hawe E, Iversen A, Wray R, Crook MA, Wierzbicki AS. Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J Mol Med. 2006;84:203–214. [DOI] [PubMed] [Google Scholar]
- 44. Bahls M, Atzler D, Markus MRP, Friedrich N, Boger RH, Volzke H, Felix SB, Schwedhelm E, Dorr M. Low‐circulating homoarginine is associated with dilatation and decreased function of the left ventricle in the general population. Biomolecules. 2018;8:E63. [DOI] [PMC free article] [PubMed] [Google Scholar]