

Abstract
Bacterial adhesins mediate adhesion to substrates and biofilm formation. Adhesins of the LPXTG family are posttranslationally processed by the cell membrane–localized peptidase sortase A (SrtA), which cleaves the LPXTG motif. This generates a short C-terminal peptide (C-pep) that remains in the cell membrane while the mature adhesion is incorporated into the cell wall. Genes encoding adhesins of the oral bacterium Streptococcus gordonii were differentially expressed depending on whether the bacteria were isolated from saliva or dental plaque and appeared to be coordinately regulated. Deletion of sspA and sspB (sspAB), both of which encode LPXTG-containing adhesins, unexpectedly enhanced adhesion and biofilm formation. C-peps produced from a model LPXTG-containing adhesin localized to the cell membrane and bound to and inhibited the intramembrane sensor histidine kinase SGO_1180, thus preventing activation of the cognate response regulator, SGO_1181. The absence of SspAB C-peps induced the expression of the scaCBA operon encoding the lipoprotein adhesin ScaA, which was sufficient to preserve and even enhanced biofilm formation. This C-pep–driven regulatory circuit also exists in pathogenic streptococci and is likely conserved among Gram-positive bacteria. This quality control mechanism ensures that the bacteria can form biofilms under diverse environmental conditions and may play a role in optimizing adhesion and biofilm formation.
Introduction
Surface-attached microbial communities (biofilms) are involved in about 80% of all human bacterial infections (1–3). Adhesion to a surface is necessary for the survival of many Gram-positive bacteria (4–6) and is mediated by an array of cell-surface adhesive proteins (adhesins), including the ubiquitous LPXTG-motif protein family (7–9). In the Gram-positive oral bacterium Streptococcus gordonii, we previously reported that transcripts encoding the highly homologous LPXTG adhesins SspA and SspB (SspAB) are more highly expressed in planktonic and free-growing cells than in sessile cells adhering to saliva-coated hydroxyapatite in vitro (10). In this in vitro tooth surface model, mutation of sspAB does not prevent biofilm formation; instead, it causes sessile, planktonic, and free-growing S. gordonii to increase the expression of genes encoding several other adhesins (10), suggesting compensation through the coordinated regulation of other key adhesins to maintain biofilm formation.
Biofilm formation in the oral environment (4, 5) appears to require that LPXTG adhesins are correctly processed, presented, and covalently linked to the cell wall (11). Posttranslational sorting and processing of LPXTG proteins involves two enzymatic cleavages and produces three functional peptides: (i) an N-terminal signal peptide, which targets the polypeptide for secretion across the cell membrane and is cleaved by the signal peptidase (12); (ii) an adhesin domain(s) that mediates adhesion to surfaces; and (iii) a C-terminal peptide (C-pep), which is cleaved from the polypeptide between the threonine and glycine residues of the LPXTG motif by the cell membrane–anchored peptidase sortase A (SrtA) and is hypothesized to be retained within the cell membrane (12–14). The signal peptide targets the protein for export, and the LPXTG motif and its cleavage to produce the C-pep are required for SrtA-mediated linkage of the processed adhesin to the cell wall (13, 14). S. gordonii srtA mutants fail to anchor LPXTG adhesins (SspA, SspB, SGO_0707, SGO_1487, CshA, and CshB) to the cell wall, resulting in the release of these adhesins into the medium (15, 16). Cell adhesion and biofilm formation are disrupted even though the expression of most genes encoding putative adhesins increases in the mutants (16). Mutating genes encoding adhesins generally causes a loss of adhesion (17), but disruption of the highly homologous S. gordonii adhesins SspAB enhances adhesion (18).
Here, we describe a quality control mechanism that monitors the processing of LPXTG adhesins by SrtA. The C-peps generated by SrtA-mediated cleavage of adhesions repressed a previously uncharacterized intramembrane two-component system (TCS). Mutations that prevented the generation of C-peps de-repressed the TCS, resulting in increased expression of other adhesins that compensated for the loss of LXPTG adhesins.
Results
S. gordonii LPXTG-containing adhesins are differentially expressed in planktonic and plaque environments
In S. gordonii, many of the 26 LPXTG family proteins are adhesins (15). In biofilms formed on saliva-coated tooth surfaces in vitro, the abundance of S. gordonii sspA transcripts are higher in unattached cells than in cells attached to surfaces (10). To determine whether sessile and planktonic cells show differences in expression of LPXTG adhesins in the human oral environment, we measured the expression of several S. gordonii LPXTG adhesins from five individuals on two to three successive days. Expression of S. gordonii adhesins differed markedly between saliva and plaque within each subject, with some adhesins being more abundant in planktonic cells (saliva) and some more abundant in sessile cells (plaque), and the patterns appeared to be consistent from day to day (fig. S1). Notably, S. gordonii sspA mRNA expression was higher in cells recovered from saliva than in cells recovered from plaque, consistent with data from our in vitro model of tooth surfaces (10). Adhesin expression thus appeared to depend upon the salivary planktonic or sessile dental plaque niche that S. gordonii occupied.
S. gordonii compensates for loss of LPXTG adhesins
Because adhesin expression appears to be regulated in the oral environment when other adhesins are mutated (10), we determined whether biofilm phenotypes changed in response to deletion of the LPXTG-motif adhesins SspAB, SGO_0707, and PavB (also called SGO_1182, 61% identity with S. pneumoniae PavB). We generated single gene deletions of sspAB, SGO_0707, and pavB in the S. gordonii wild-type strain DL1 using a marker-free, in-frame mutation system (19). Growth rates of the adhesin mutants and the wild-type strain were similar in planktonic cultures (fig 2A–C). We tested these deletion strains for in vitro biofilm formation on uncoated and saliva-coated plastic surfaces to simulate the oral environment. When formed on saliva-coated surfaces, biofilms of the ΔSspAB, ΔSGO_0707 (Fig. 1A) and ΔPavB (fig. S3 strains showed increased biomass compared to wild type. Without saliva, the ΔSspAB strain showed lower biofilm biomass than the wild-type strain (Fig. 1A), whereas, the ΔSGO_0707 strain showed increased biofilm biomass in the absence of saliva (Fig. 1A). The magnitude of the biomass increase differed among the deletion mutants and was sensitive to the substrate.
Fig. 2. Co-localization of LPXTG-motif C-peps with the cell membrane.
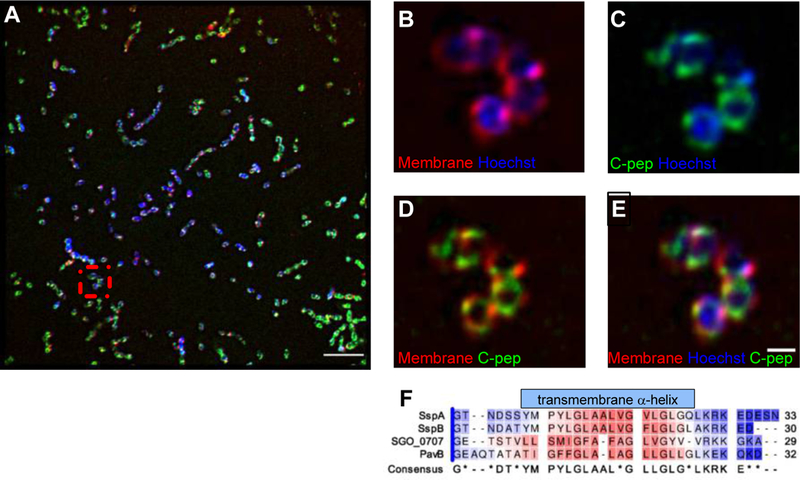
(A) Representative structured illumination microscopy (SIM) showing a field of S. gordonii cells fluorescently to show the cell membrane (FM® 4–46FX, red), chromosome (Hoeschst 33342, blue), and the Venus-tagged SGO_0707 C-pep (green). Scale bar, 5 μm. (B–E) Higher magnification views of the boxed area in (A) showing overlays of the indicated stains. Scale bar, 2 μm. n= 3 independent experiments. (F) ClustalW alignment of SspA, SspB, SGO_0707, and PavB C-peps showing the conserved hydrophobic transmembrane α-helix. Color coding indicates amino acid hydrophobicity intensity based on the Kyte-Doolittle Hydrophobicity Scale (red, hydrophobic; blue, hydrophilic).
Fig. 1. The roles of substratum and LPXTG-adhesin C-peps in biofilm biomass.
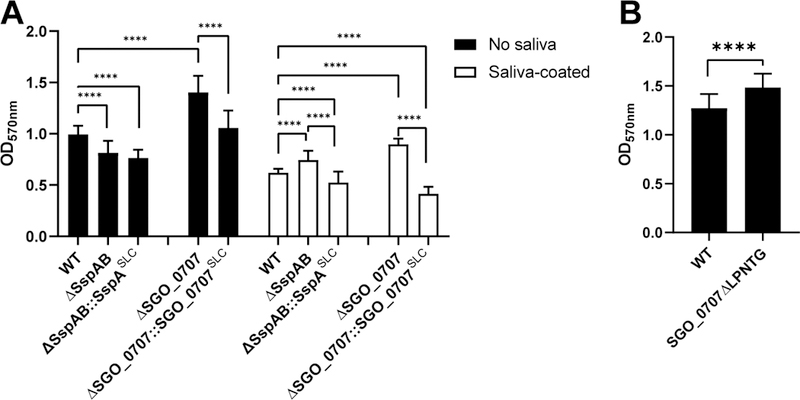
(A) Quantification of biofilm formation by the indicated S. gordonii mutants in wells that were uncoated (filled bars) or saliva-coated (unfilled bars). Wild-type (WT) cells were compared to mutants lacking LPXTG adhesins SspA and SspB (ΔSspAB) or SGO_0707 (ΔSGO_0707) and to each deletion mutant expressing the SLC form of the deleted adhesin, which contains the engineered polypeptide: signal peptide, LPXTG motif, and C-pep (adhesin domain missing). Each substrata dataset was analyzed separately using one-way ANOVA with Tukey’s multiple comparisons test. (B) Quantification of biofilm formation by mutant S. gordonii in which the SGO_0707 C-pep production was eliminated by deleting the SrtA cleavage site, LPNTG, from the endogenous sgo_0707 locus (SGO_0707ΔLPNTG). Data were analyzed using an unpaired, two-tailed Student’s t-test. All data represent the means ± SD of at least three independent experiments, with at least three technical replicates for each strain per experiment. Significant differences are denoted by asterisk(s). * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
The release of C-peps affects biofilm formation
The increased biomass of biofilms in S. gordonii lacking SspAB could be explained by increased production of other adhesins (10). Further, given that LPXTG-motif genes and proteins are globally increased when srtA is deleted (16), we proposed that SrtA activity produces a repressive signal during LPXTG processing (12). To determine whether the nascent adhesin polypeptide contained a repressive signal, we constructed new strains that lacked specific functional domains. Strains missing only the adhesin domain produce a polypeptide in which the secretion signal peptide (S) is directly coupled to the LPXTG motif (L) and the C-pep (C). Alleles in which we deleted the adhesin domains of SspAB, SGO_0707, or PavB are referred to as SspASLC, SGO_0707SLC, and PavBSLC, respectively. Because sspA and sspB have high genetic homology, the sspA and sspB alleles were replaced with a sequence that only produced the SspASLC polypeptide; the respective SLC alleles were used for SGO_0707 and PavB. The resultant strains, a SspAB deletion mutant expressing SspASLC (ΔSspAB::SspASLC) and a SGO_0707 deletion mutant expressing SGO_0707SLC (ΔSGO_0707::SGO_0707SLC), formed biofilms with 16% and 33% less biomass, respectively, on saliva than did wild-type cells (Fig. 1A). In the absence of saliva, ΔSspAB::SspASLC biofilms mimicked ΔSspAB and formed 23% less biomass than wild type, whereas, ΔSGO_0707::SGO_0707SLC and ΔPavB::PavBSLC formed biofilms that were equal in biomass to wild type (Fig. 1A and fig. S3). Biofilms of the ΔSspAB::SspASLC, ΔSGO_0707::SGO_0707SLC, and ΔPavB::PavBSLC constructs did not show the characteristic enhanced biomass observed in the respective complete adhesin deletion strains, indicating that the repressive signal was not contained within the functional adhesin domain. To determine whether the LPXTG motif was required for repression, we deleted the LPNTG motif from SGO_0707 (SGO_0707ΔLPNTG), creating a protein in which the signal peptide and adhesin domain were connected directly to the C-pep. Without saliva, SGO_0707ΔLPNTG biofilms showed enhanced biomass similar to the ΔSGO_0707 strain (Fig. 1B). Thus, cleavage of the LPXTG motif by SrtA and release of C-pep in the cell membrane was required to repress compensatory adhesin production that enhanced biofilm formation.
C-peps localize to the streptococcal plasma membrane
C-peps are hydrophobic peptides of 29–34 amino acids, sufficient to span the lipid bilayer; glycine residues may form intrahelical salt bridges that stabilize the transmembrane helix (12, 14, 20–22). To model the localization of SGO_0707 C-peps within the streptococcal cell, we inserted the Venus yellow fluorescent protein (YFP) into the SGO_0707 locus (23) between the penultimate and stop codon of sgo_0707. High genetic homology made it not feasible to perform this insertion with sspA and sspB. If properly intercalated in the cell membrane, the SrtA-processed SGO_0707 C-pep-YFP should be juxtaposed or overlapping with the lipid bilayer. Using structured-illumination microscopy (SIM), we found that C-pep-YFP appeared to colocalize with a membrane marker and formed a punctate halo around the chromosome (Fig. 2, A to E, fig. S4, A to D, and movies S1 and S2), consistent with streptococcal cell architecture (16). Thus, SGO_0707 C-pep (Fig. 2F), and likely the structurally similar SspA, SspB, and PavB C-peps (12), appeared to localize within the S. gordonii lipid membrane as observed at sub-visible light resolution facilitated by SIM.
SGO_1180 and SGO_1181 constitute a two-component system that controls biofilm formation
Because C-peps are localized within the membrane (Fig. 2), we reasoned that C-peps could be “sensed” by a two-component system (TCS). TCSs consist of a sensor histidine kinase (HK) and a response regulator (RR). Most HKs are transmembrane receptors that phosphorylate intracellular RRs that often act as transcription factors when activated by phosphorylation. Most TCSs detect intra- or extracellular stimuli (24) but some detect signals that originate in the membrane (25, 26). Intramembrane sensor kinases (IM-HKs) are characterized by an intramembrane sensor domain (25) formed by at least two transmembrane helix regions connected by a linker(s) of less than 25 amino acids (25). By querying the MIST2 database (http://mistdb.com), we identified 16 known or potential sensor HKs (table S1). One potential sensor HK, SGO_1180, has characteristics of an intramembrane sensor. It lacks predicted extracellular or intracellular sensor or ligand-binding domains and has an extracellular linker of only nine or ten amino acids connecting the two transmembrane helices (table S1). The sgo_1180 gene shares an overlapping translational reading frame with a gene encoding a predicted DNA-binding RR, sgo_1181, and is located downstream of pavB (sgo_1182) (27).
We confirmed that pavB, sgo_1180, and sgo_1181 were co-transcribed (fig. S5A) using RT-PCR (fig. S5B–C). The predicted protein products, SGO_1180, SGO_1181, show high sequence homology to the HK and RR of the S. pneumoniae TCS TCS08 (76 and 83% identity, respectively) and are conserved among streptococci (28, 29). The HK and RR of the SaeRS TCS, regulators of adhesins and virulence factors in staphylococci that have similar protein structures, show 31 and 47% identity, respectively to SGO_1180 and SGO_1181 (30, 31).
To test if C-peps repress adhesin production through an intramembrane TCS, we deleted sgo_1180 and sgo_1181 alone and in combination with sspAB, sgo_0707, or pavB. Whereas the deletion of single adhesin genes in the wild-type background resulted in biofilms of increased biomass (Fig 1A), the deletion of sgo_1180 and sgo_1181 (ΔSGO_1180–81) in wild type reduced biofilm formation in both saliva-coated (25%) and non-coated (6%) wells (Fig. 3A). Deleting individual adhesins in the TCS mutant background (ΔSspABΔSGO_1180–81, ΔSGO_0707ΔSGO_1180–81, and ΔPavBΔSGO_1180–81) prevented the increase in biofilm mass conferred by deletion of the adhesin alone on saliva-coated or uncoated surfaces (Fig. 3A, fig. S3). We then deleted sgo_1180 and sgo_1181 in the ΔSspAB::SspASLC and ΔSGO_0707::SGO_0707SLC strains, creating ΔSspAB::SspASLCΔSGO_1180–81 and ΔSGO_0707::SGO_0707SLCΔSGO_1180–81. Biofilm formation was similarly reduced in all strains with a ΔSGO_1180–81 background (Fig. 3A). The SGO_1180–1181 TCS was therefore essential for repressing the enhanced biofilm response in the absence of an LPXTG adhesin.
Fig. 3. The SGO_1180 and SGO_1181 two-component system is required for enhanced biofilm formation in the absence of SspAB C-peps.
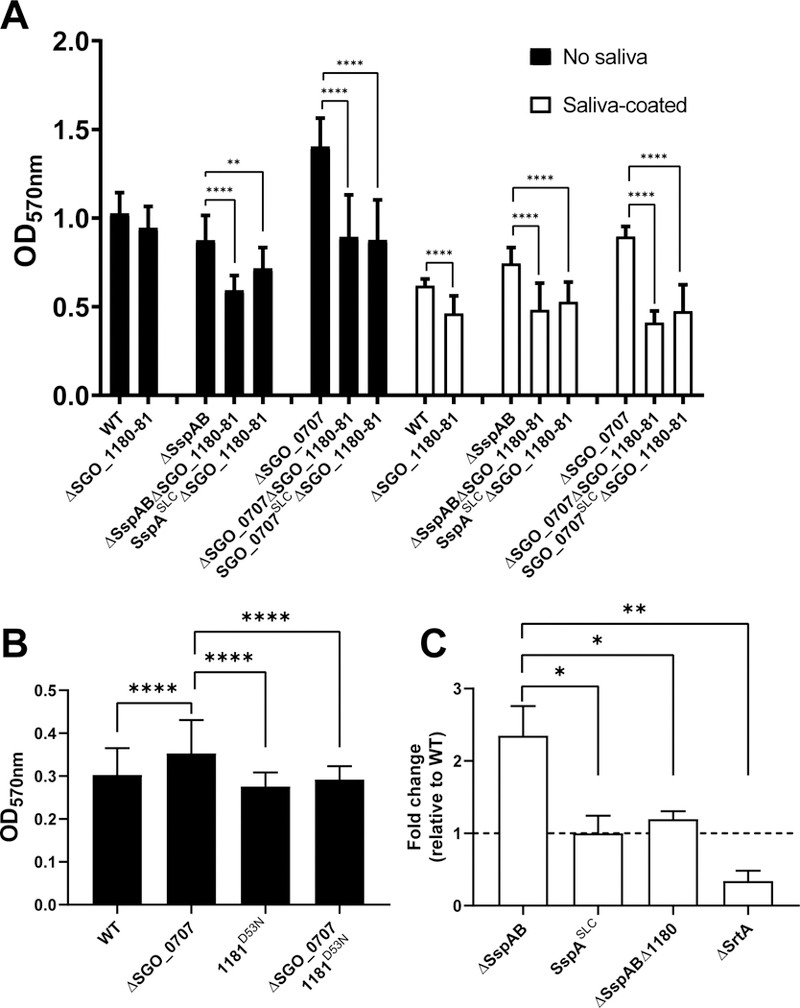
(A) Quantification of biofilm formation by S. gordonii lacking SGO_1180 and SGO_1181 (Δ1180–81) on uncoated (filled bars) or saliva-coated (unfilled bars) wells relative to WT. Biofilm formation was quantified in Δ1180–81 mutants in combination with deletion of SspAB (ΔSspAB Δ1180–81), with replacement of SspAB with SspASLC (SspASLC Δ1180–81), with deletion of SGO_0707 (ΔSGO_0707 Δ1180–81), or with replacement of SGO_0707 with SGO_0707SLC (SGO_0707SLC Δ1180–81). Data represent the means ± SD of at least three independent experiments with at least three technical replicates per experiment. (B) Quantification of biofilm formation in SGO_1181 D53N point mutants in the ΔSGO_0707 background relative to WT. Data represent the means ± SD of at least three independent experiments with at least three technical replicates per experiment. (C) Quantification of the competitiveness of S. gordonii mutants lacking SspAB adhesins, SGO_1180, or SrtA in ex vivo multi-species dental plaque biofilms. S. gordonii were added to supragingival multi-species plaque biofilms and cultured overnight. The amount of each strain that colonized the plaque ex vivo community was quantified using qPCR. Data represent the means ± SD of N ≥ 2 experiments with at least duplicate biofilms. Horizontal hatched line indicates WT S. gordonii. Each dataset was analyzed using one-way ANOVA with Tukey’s multiple comparisons test. Significant differences are denoted by asterisk(s). * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
To transduce signal, sensor HKs phosphorylate cognate RRs at a conserved aspartic acid residue, activating the effector function of the RR (32). SGO_1181 is similar to the RR OmpR, which binds to DNA and acts as a transcriptional regulator, so we predicted that the kinase activity of SGO_1180 towards SGO_1181 would be inhibited by C-peps. To test this prediction, we created a non-phosphorylatable mutant form of SGO_1181 (1181D53N) in the wild-type and ΔSGO_0707 backgrounds. As expected, ΔSGO_0707 formed biofilms of 16% greater biomass than wild-type cells in uncoated wells, whereas 1181D53N and 1181D53NΔSGO_0707 biofilms had 10% less biomass than wild-type biofilms (Fig. 3B). In the absence of a C-pep from SGO_0707, therefore, SGO_1181 phosphorylation was required to de-repress the enhanced biofilm response.
As a resident of dental plaque, S. gordonii exists in complex multi-species biofilms. To understand whether this C-pep signal and response circuitry regulates fitness of S. gordonii in the absence of sspAB, we isolated and cultured supragingival plaque from healthy donors and then inoculated these multi-species plaque biofilms with genetically-marked S. gordonii strains at low cell density. After overnight culture, we quantified the relative amount of each S. gordonii strain present in the plaques by qPCR. Like mono-species biofilms, multi-species biofilms supported >2-fold more ΔSspAB than wild-type cells (Fig. 3C). Further, the abundance of ΔSspAB::SspASLC and ΔSspABΔ1180 strains was similar to wild type (Fig. 3C). Relative to wild type, the amount of the ΔSrtA strain was greatly reduced in the multi-species biofilm (Fig. 3C), which is consistent with our previous reports showing that LPXTG adhesins are released into the medium (rather than covalently linked to the cell wall) and biofilm formation is dramatically reduced in the absence of SrtA (15, 16). These results also indicate that S. gordonii shows fitness in colonizing a multi-species ex vivo biofilm in the absence of the major SspAB adhesins, which had been suggested to be crucial for survival in plaque (33). We conclude that the TCS regulatory circuit coordinates the expression of adhesins with biofilm formation and that fitness to colonize a multi-species biofilm is established through SspAB C-pep–mediated repression of the SGO_1180-1181 TCS.
SspA C-pep and the SGO_1180-1181 TCS control scaCBA expression
To learn whether transcriptional activation of alternative adhesins explains the increased biomass of biofilms (10) and S. gordonii fitness in multi-species biofilms, we performed next-generation RNA sequencing on biofilms of various strains: wild type, ΔSspAB, ΔSspAB::SspASLC, ΔSGO1180–81, ΔSspABΔSGO_1180–81, and ΔSspABΔSGO_1180–81::SspASLC. Sessile biofilm cells were recovered after 4 hours of growth on uncoated plastic dishes. In pairwise comparison to wild type, only three transcripts were significantly increased (~5-fold) in ΔSspAB sessile biofilms: scaC, scaB, and scaA of the adhesion-associated scaCBA operon (table S2) (34, 35). In ΔSspAB biofilms, other adhesin transcripts including pavB, sgo_2004, sgo_2005, sgo_0707, cshA, sgo_1487, and sgo_0890 were comparable to wild type biofilms. Likewise, expression of transcripts encoding glycosyltransferases, which can contribute to biofilm matrices (36, 37), were similar in all strains tested (table S2). Changes in adhesin gene expression before or after 4 hours of growth were not reflected in our single time-point analysis.
SGO_1180–81 TCS was required for the increase in scaCBA expression because scaCBA was not up-regulated in strains lacking SGO_1180 and SGO_1181 (table S2). scaA mRNA abundance was confirmed to increase 5.5 ± 1.1-fold in ΔSspAB biofilms relative to wild type, whereas mRNA abundance in the ΔSGO_1180–81 biofilms was similar to wild-type using RT-qPCR (table S2). Using RNA isolated from biofilms grown on plastic, transcription of scaA in ΔSGO_0707 and ΔPavB mutants was similar to wild type as determined by RT-qPCR. In free-growing (no adhesion substrate) ΔSspAB cells, scaA expression was 4.7 ± 0.3-fold greater than in wild-type cells. Expression of scaA in ΔSspAB::SspASLC, ΔSGO_1180–81 mutants, and wild type was similar in free-growing cells (table S3) and sessile biofilms (table S2). Thus, SGO_1180–81 likely transcriptionally activates the adhesion-related operon scaCBA to stimulate production of alternative adhesins when SspAB C-peps are missing.
To determine whether the ScaA adhesin enhances the biomass of ΔSspAB biofilms, we tested mutants in both plastic and saliva-coated wells. The scaA mutant strain (scaA::tetM) formed biofilms of 47% less biomass in the presence of saliva and 60% less biomass in the absence of saliva (fig. S5). When scaA was inactivated in the ΔSspAB background (scaA::tetMΔSspAB), biofilm biomass was only 48% of wild type in the presence of saliva and 8% in the absence of saliva (fig. S5). Because the enhanced biomass in ΔSspAB biofilms was independent of changes in glycosyltransferase expression based upon our RNAseq data (table S2), increased expression of scaA adhesin gene best explains the enhanced biomass of ΔSspAB biofilms. Further, ScaA-dependent enhanced biofilms appear to strictly depend on SGO_1180–1181 because the sca operon regulator, scaR, is not co-regulated with scaA (table S2) (41).
SGO_0707 C-pep interacts directly with SGO_1180
We next asked if C-peps interact directly with SGO_1180 in the lipid bilayer of S. gordonii cells using bimolecular fluorescence complementation (BiFC) (23, 42, 43). Again, due to the high nucleotide sequence homology between sspA and sspB, we focused on sgo_0707 for genetic manipulations. The C-pep of SGO_0707 and the transmembrane regions of SGO_1180 (TM1 or TM2) were each chromosomally tagged with reciprocal halves (N- or C-fragments) of the Venus-YFP (V) protein and expressed in wild-type cells. All possible combinations of YFP fragment–labeled SGO_0707-C-pep and SGO_1180 TMs were constructed (42). Single N- or C-fragment–labeled strains emitted fluorescence equivalent to unlabeled wild-type cells. Only two of the four strains carrying complete BiFC combinations (Fig. 4A) emitted fluorescence signals above controls. Strains SGO_0707-V-N::SGO_1180-V-C-TM1 and SGO_0707-V-C::SGO_1180-V-N-TM2 emitted 25-fold and 8-fold more fluorescence than their respective single-labeled SGO_1180 BiFC strains, respectively (Fig. 4B). This suggested that the SGO_0707 C-pep (Fig. 4A) interacted with, or was at least localized in very close proximity to, the transmembrane domains of SGO_1180 within the streptococcal cell membrane. Furthermore, our observation that the C-pep interacted with SGO_1180 to reconstitute fluorescence emission only when the YFP fragments were in two of the possible orientations implies that interaction with the C-pep was restricted to specific faces of the TM helices of SGO_1180 (44).
Fig. 4. LPXTG-adhesin C-peps interact with SGO_1180.
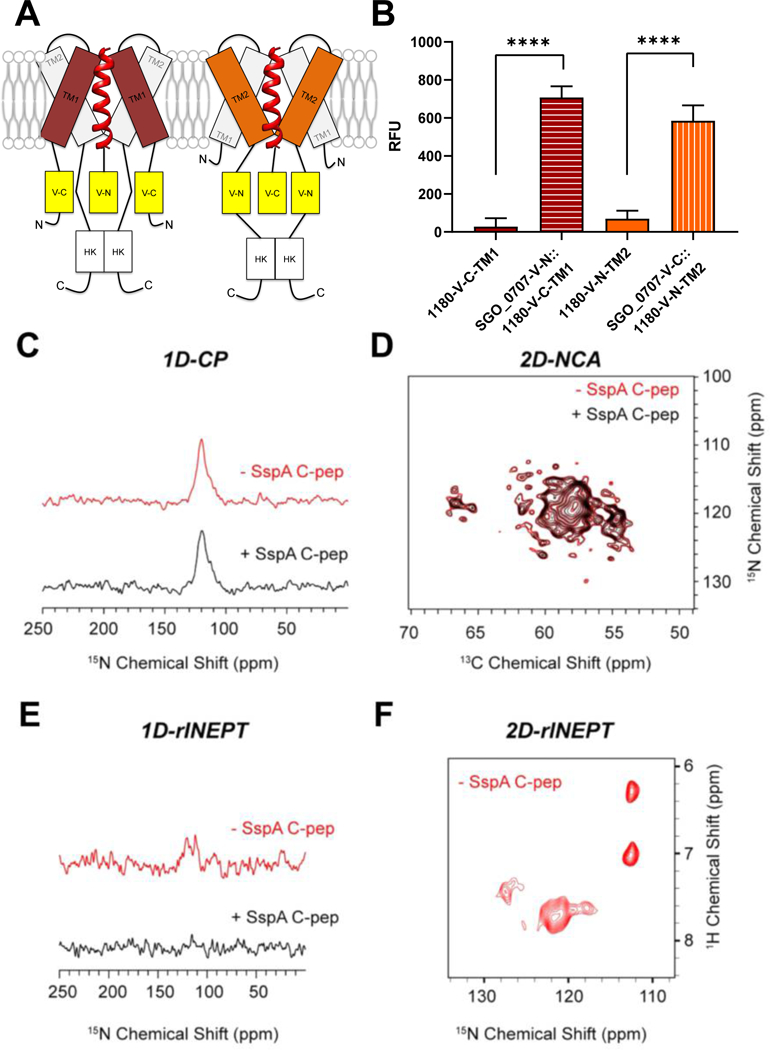
(A) Schematic of the interaction between SGO_0707 C-pep (red helical ribbon) and transmembrane domains (TM1 and TM2) of SGO_1180. The N- or C-terminal (N or C) domain of Venus YFP was inserted in-frame between the penultimate and stop codon of sgo_0707 and on the cytoplasmic side of TM1 or TM2 of sgo_1180. All four combinations of the constructs that could reconstitute active YFP were tested in cells, but only the two that produced fluorescence are shown in the diagram. (B) Quantification of fluorescence emission from the single and dual-labeled bifluorescent strains shown in (A). Fluorescence was only detected when the N-terminal domain of YFP fused to the SGO_0707 C-pep (SGO_0707-V-N) was combined with the C-terminal domain of YFP fused to TM1 of SGO_1180 (1180-V-C-TM1) or when the C-terminal domain of YFP fused to the SGO_0707 C-pep (SGO_0707-V-C) was combined with the N-terminal domain of YFP fused to TM2 of SGO_1180 (1180-V-C-TM2). RFU, relative fluorescence units, fluorescence units divided by the culture optical density at λ = 600 nm. Data represents the mean ± SD of three independent experiments with three technical replicates per experiment. Data were analyzed using an unpaired Student’s t-test. Significant differences between strains are denoted by asterisk(s): * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001. (C-F) Magic-angle spinning (MAS) NMR spectroscopy of the SspA C-pep and SGO_1180 (1:1) in artificial lipid membranes. The 1D cross-polarization (CP) spectrum to detect the TM helical domains (C), the overlay of 2D-NCA spectra of free and SspA C-pep–bound SGO_1180 (D), the refocused INEPT (rINEPT) of free and SspA C-pep–bound SGO_1180 to capture the dynamic residues of SGO_1180, and the 2D-rINEPT spectrum of SGO_1180 alone to detect the dynamic residues. Red, SGO_1180 alone. Black, SGO_1180 + SspA C-pep (1:1).
C-pep docking could change the conformation of SGO_1180, interfering with the ability of SGO_1180 to undergo autophosphorylation or transphosphorylate SGO_1181, or both. HKs such as SGO_1180 have three domains critical to signal transduction: (1) Histidine kinases, Adenylate cyclases, Methyl accepting proteins and Phosphatases (HAMP); (2) dimerization and histidine phosphotransfer (DHp); and (3) Catalytic ATP-binding (CA). To analyze changes in conformation, purified recombinant SspA C-pep and SGO_1180 were combined in 1,2-dimyristoyl-sn-glycero-3-phosphorylcholine (DMPC) vesicles (45) and analyzed by magic-angle spinning (MAS) NMR spectroscopy. To detect rigid TM helical domains in the kinase, we acquired a 1D cross-polarization (CP) spectrum. The envelope of the TM domain residues resonated between 100 and 150 ppm (Fig. 4C). By capturing the 2D-N to Cα (NCA) spectrum (46), the free and SspA C-pep–bound forms were found to overlay almost entirely in fingerprint resonances (N-Cα correlations) of SGO_1180 (Fig. 4D). We then visualized the dynamic residues of SGO_1180 using a refocused INEPT (rINEPT) experiment (47). In the absence of SspA C-pep, we observed a few peaks in the 100–150 ppm region (Fig. 4E), corresponding to the most mobile regions of the HK. Several of these dynamic residues were also detectable in the 2D region of the rINEPT spectrum (Fig. 4F). In the presence of SspA C-pep, these dynamic residues were no longer detectable (Fig. 4E), indicating that these residues became immobilized due to intermolecular interactions. These NMR spectra in lipid membranes showed significant rigidification of the most dynamic region of SGO_1180 when physically interacting with SspA C-pep.
Conformational integrity of SGO_1180 is critical for sensing C-peps
Given that intramembrane C-peps directly interact with and negatively regulate SGO_1180, the transmembrane domains of SGO_1180 may contain tertiary structural determinants that respond to the absence of C-peps with downstream signaling. In the intramembrane HK SaeS of Staphylococcus aureus strain Newman, for example, the substitution mutation L18P converts the transmembrane domain alpha-helix to a beta-sheet and constitutively activates the kinase (48). Based on the high structural similarity to SaeS, the alpha-helical transmembrane structure of SGO_1180 was hypothesized to be critical for “sensing” C-peps. We substituted proline for the conserved lysine in the first transmembrane helix of SGO_1180 (Lys16), creating SGO_1180L16P (Fig. 5A). The 1180L16P strain produced a biofilm of 27% greater biomass than wild type (Fig. 5B). Hence, the native conformation of the first SGO_1180 transmembrane loop is essential for proper repression of biofilm formation (32). Expression of scaA was similar in 1180L16P and wild-type strains; suggesting that SGO_1180-mediated phosphorylation of SGO_1181 alone may be insufficient for induction of scaA.
Fig. 5. SGO_1180 conformational integrity and functional conservation across species.
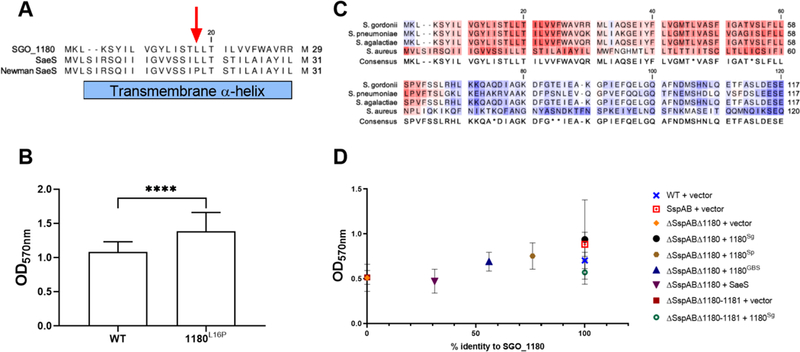
(A) ClustalW alignment of transmembrane helix 1 of SGO_1180, consensus S. aureus SaeS, and S. aureus Newman SaeS. The red arrow indicates the SaeS L18P mutation that constitutively activates signaling in the Newman SaeS. (B) Quantification of S. gordonii SGO_1180 L16P biofilm biomass in saliva-coated polystyrene wells. The L16P substitution was engineered into the endogenous sgo_1180 locus and corresponds to the L18P substitution in S. aureus Newman SaeS. Data represents the mean ± SD of three independent experiments with at least three technical replicates per experiment. Data were analyzed using an unpaired Student’s t-test. Significant differences between strains are denoted by asterisk(s): **** = p ≤ 0.0001. (C) Sequence alignment of the first 120 amino acids of SGO_1180 with homologs from S. pneumoniae, S. agalactiae, and S. aureus using ClustalW. (D) Functional complementation of S. gordonii SGO_1180 (Sg) with orthologs from S. pneumoniae (Sp), S. agalactiae (GBS), and S. aureus (SaeS). Biofilm formation in saliva-coated wells was quantified for ΔSspABΔ1180 S. gordonii expressing each of the homologs from an E. coli-streptococcal shuttle vector. Data represents the mean ± SD of at least three independent experiments with at least three technical replicates per experiment. Data were analyzed using Kruskal-Wallis with Dunn’s multiple comparisons test. Significant differences are denoted by asterisk(s). * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
SGO_1180 is functionally conserved among streptococci
We next determined the extent of structural and functional conservation of SGO_1180. Using NCBI BLASTP, we identified proteins orthologous to the first 120 amino acids of SGO_1180, which contains both the transmembrane and HAMP domains. With the notable exception of Group A Streptococcus (S. pyogenes) and S. mutans, SGO_1180 orthologs highly conserved at the level of amino acid sequence were present throughout the Streptococci (fig. S6). SGO_1180 orthologs were also identified among related genera, including enterococci and Listeria (fig. S7). Given that enhanced biofilms in ΔSspAB strains depended on a functional SGO_1180–81 TCS, we hypothesized that SGO_1180 orthologs function similarly across species.
To test this hypothesis, we determined if S. aureus SaeS (designated here as 1180SaeS) or potential orthologs from S. pneumoniae (TCS08HK, designated here as 1180Sp) or S. agalactiae (designated here as 1180GBS complemented S. gordonii ΔSGO_1180ΔSspAB. The first 120 amino acids of 1180Sp and 1180GBS are 69% and 48% identical to SGO_1180, respectively (Fig. 5C). The structurally related intramembrane HK, SaeS, has 22% amino acid identity with the first 120 amino acids of SGO_1180. We then tested whether each possible ortholog could restore enhanced biofilm biomass in S. gordonii ΔSspABΔSGO_1180 grown in saliva-coated microtiter wells (Fig. 5D). As expected, 1180Sg fully complemented the enhanced biofilm of the ΔSspAB strain (100%). The biofilm biomasses of the strains complemented with 1180Sp or 1180GBS were greater than those expressing vector alone. Further, complementation of ΔSspABΔSGO_1180–1181 with 1180Sg did not restore biofilm biomass, indicating that the presence of both the HK 1180 and RR 1181 are necessary for the compensatory biofilm response in the absence of SspA C-pep.
These data strongly suggest that SGO_1180 and the predicted streptococcal orthologs are functionally orthologous. In contrast, S. aureus SaeS does not appear to functionally complement SGO_1180 within the parameters of this assay. Despite structural conservation, SGO_1180 and SaeS appear to be functionally dissimilar intramembrane HKs. Nonetheless, the SGO_1180–81 TCS and highly conserved pan-streptococcal orthologs, however, are strongly suggested to regulate the production of alternative adhesins and sustain biofilm formation in the absence of C-pep–containing LPXTG adhesins.
Discussion
Adhesins are important for bacterial attachment and establishment of biofilms on abiotic and biotic surfaces and for intercellular communication. The inability to properly produce adhesins may lead to niche extinction. Using the oral commensal bacterium S. gordonii as a model organism, we showed that the expression of adhesin genes differed in samples taken simultaneously from the dental plaque and saliva from the same individuals on different days, indicating that adhesin expression varies within the host environment. SrtA, which is ubiquitous in Gram-positive bacteria (12), processes cell wall–anchored LPXTG-containing adhesins, generally large proteins or glycoproteins that are energetically expensive to produce. Monitoring for proper sorting would conserve energy in all organisms expressing this regulatory circuit. We identified an intramembrane peptide-protein sensory circuit that monitors the SrtA processing fidelity of LPXTG adhesins that maintains the organism’s ability to form biofilms even when processing is compromised.
We present here the characterization of a previously unrecognized intramembrane signaling mechanism that monitors LPXTG adhesin processing by SrtA to sustain biofilm formation. The intramembrane C-pep signals are structurally conserved, short, single-transmembrane peptides (Fig. 2F), whereas the functional adhesive domains of LPXTG adhesins are structurally divergent (12). We speculate that differences in primary sequence could confer signaling specificity to C-peps, and more experiments are needed to decipher whether or not there is a specific C-pep code for inhibition of SGO_1180, given that scaA was not induced when SGO_0707 or PavB was deleted or SGO_1180 was mutated (1180L16P). We cannot exclude that additional unknown proteins or other transcriptional programs may be at work. We do not yet know how these C-pep signals enable the 26 LPXTG family proteins to work collectively to repress or activate the production of alternative adhesins through SGO_1180. We speculate that C-pep sequences may cluster among groups of adhesins and be distinguished by SGO_1180 to induce different patterns of transcriptional adhesin gene responses. How this mechanism might influence the function of a particular adhesin, perhaps during adaptation to an ecological niche such as the oral cavity or during establishment as a pathobiont remains unknown but is under active investigation. As a regulator of biofilm formation, however, we consider SGO_1180 and orthologs to be potential targets for the development of anti-infective therapies.
Indeed, this mechanism for monitoring LPXTG adhesin processing is likely to be present in many Gram-positive bacteria because SGO_1180–81 orthologs were identified in related genera, and the sensing function of SGO_1180 is likely to be conserved based on sequence homology. Although not exhaustive, our functional complementation of SGO_1180 with orthologs from S. pneumoniae and S. agalactiae (Fig. 5D) supports the assertion that SGO_1180 orthologs in streptococcal species also monitor sorting of LPXTG-containing proteins. The monitoring of SrtA processing fidelity by TCS signaling in response to C-peps also appears conserved among several LPXTG adhesins.
The HK SGO_1180 docks with the SspAB C-peps. In the absence of C-peps, SGO_1180-mediated activation of SGO_1181 likely transcriptionally activates compensatory expression of the adhesion-related operon, scaCBA, resulting in production of the alternative adhesin, ScaA (10). Based on homology to other RRs of the OmpR family, a potential SGO_1181 binding site overlaps the −10 and −35 promoter elements (ATTAACTTGACTTAATTTTTATGTTAAGGTATATTAA) in the scaCBA promoter (49–51).
Increased biofilm biomass in response to the loss of SspAB depended on the lipoprotein adhesin ScaA, which mediates co-aggregation with Actinomyces naeslundii (9) and binding to oral glycoproteins and the salivary film (52). Increased ScaA may directly promote adhesion, therefore, to the substrate or neighboring cells. S. gordonii biofilm formation associated with scaA induction was enhanced (Fig. 1A) and may contribute to increased fitness in a saliva-based multi-species biofilm (Fig. 3C). Whereas scaCBA induction is not seen in ΔSGO_0707 and ΔPavB strains, other adhesins may be induced to rescue biofilm formation. Furthermore, additional transcriptional regulators may indirectly control biofilm formation (53, 54) because the transcriptome data suggested altered expression patterns of known and predicted transcriptional regulators.
In the cell membrane, SGO_0707 C-pep directly interacted within molecular distances of both predicted transmembrane domains of SGO_1180. The helical structure of the transmembrane domains was critical for C-pep repression through SGO_1180 because the L16P substitution blinded SGO_1180 to C-pep repression (Fig. 5C). Using solid-state NMR, we confirmed that SspA C-pep docked with SGO_1180 without accessory proteins or other factors (Fig. 4C–D) (55). In the presence of SspA C-pep, the soluble HAMP, DHp, and CA domains of SGO_1180 became rigid when intercalated in lipid vesicles. Thus, C-peps appear to inhibit the conformational rearrangement necessary for transmembrane signaling by the sensor HK (56). Hence, C-peps appear to be analogues of PilA, which confers autoregulation in Pseudomonas aeruginosa (57). The single conserved N-terminal domain of PilA interacts with and inhibits the kinase activity of the intramembrane HK PilS, controlling the abundance of PilA on the cell surface (57). Although the ligands for many other intramembrane HKs have not been identified, we show that C-peps are the ligands for SGO_1180.
Our studies provide strong evidence that C-peps cleaved from LPXTG-containing adhesins by SrtA act as signaling ligands within the cell membrane as shown for SspAB, SGO_0707 and PavB. We, therefore, propose a model (Fig. 6) in which C-peps cleaved by SrtA during processing of a structural adhesin are retained within the membrane and dock with the intramembrane sensor domains of the intramembrane HK SGO_1180. We speculate that SGO_1180 can bind to C-peps from multiple LPXTG-containing proteins and respond to their loss. Whether or not the absence of different C-peps elicits distinct transcriptional responses remains to be determined. Docking with C-pep prevents conformational changes of the cytoplasmic signal transduction domains of SGO_1180 (56) and informs the cell that the LPXTG adhesins have been properly sorted to the cell wall. In addition to missing LPXTG adhesins, C-pep abundance could also be reduced by other pre- or posttranscriptional regulatory processes. The absence of C-pep binding likely leads to phosphorylation of the cognate RR SGO_1181, which sustains biofilm-related expression of genes encoding alternative adhesins. Through this mechanism, S. gordonii appears to monitor sorting of LPXTG-containing proteins (15) and provides an efficient process for correction when needed. This regulatory mechanism constitutes a previously unknown control of the fidelity of SrtA-mediated processing in the life cycle of streptococci and perhaps other Gram-positive bacteria. LPXTG adhesins also appear to distinguish between different environmental surface cues, suggesting that optimization of adhesion and biofilm formation may involve this adhesin regulatory circuit.
Fig. 6. Model of the SspAB C-pep–SGO_1180–SGO_1181 regulatory circuit for monitoring successful processing of LPXTG-containing proteins by SrtA.
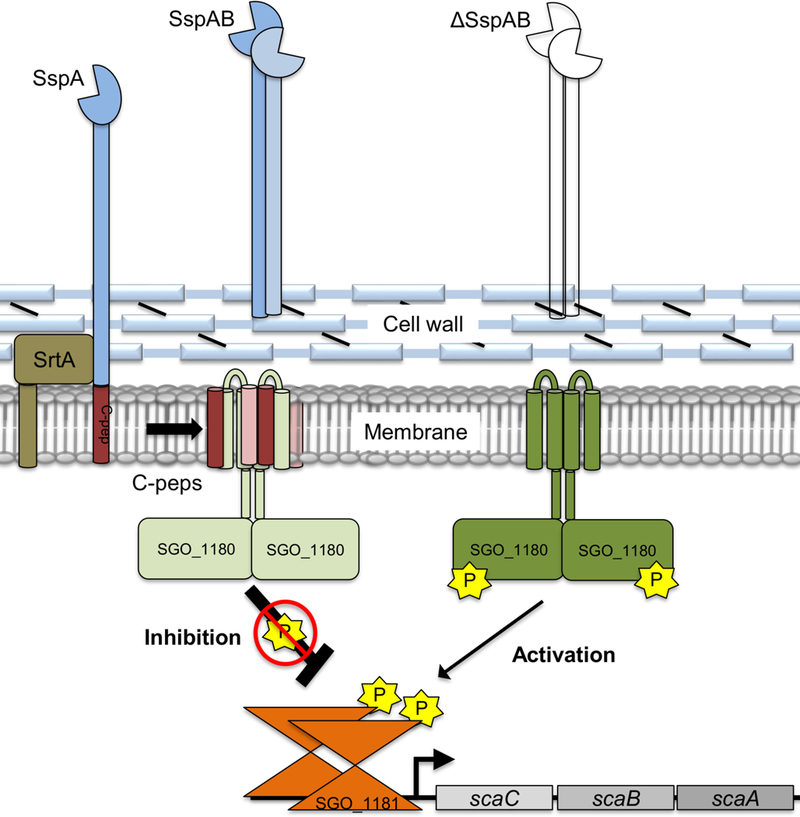
After cleavage of the LPXTG-motif by SrtA, the mature adhesin is anchored to the cell wall and the C-pep is retained in the cell membrane. Within the cell membrane, the C-peps bind to the transmembrane domains of the HK SGO_1180, thereby negatively regulating the phosphorylation cascade and preventing compensatory gene expression. In the absence of one or more C-peps, kinase activity of SGO_1180 is increased and induces compensatory gene expression through SGO_1181 to maintain biofilm formation. This circuit indirectly monitors proper placement of LPXTG-containing proteins on the cell wall.
Materials and Methods
Bacterial strains and media
Todd Hewitt (TH) broth (Difco) was used for routine culturing of S. gordonii DL1 and isolation and culture of mutant strains. When required for mutant isolation, agar was used to solidify TH broth. Brain Heart Infusion (BHI) broth (Difco) was used for biofilm biomass experiments. All experiments were incubated at 37°C with 5% CO2. To construct in-frame deletions, strains were cultured at 37°C on TH agar in an anaerobic mixed-gas chamber (Coy Laboratory Products). E. coli was cultured at 37°C in LB medium (Difco) or BHI. Antibiotic concentrations in TH and BHI media were used as follows: for S. gordonii, 5 μg/mL erythromycin (THerm) and 700 μg/mL spectinomycin (THspec); and for E. coli, LB medium was used with 75 μg/mL spectinomycin (LBspec) or 100 μg/mL ampicillin.
Saliva and plaque collection
Whole unstimulated saliva and supragingival plaque samples were collected concurrently from 5 healthy adult volunteers on at least two occasions. Volunteers were asked to refrain from brushing their teeth for 24 hours before each sample collection. Saliva was collected by expectoration into tubes on ice, and 20 mL from each donor was centrifuged for 10 minutes at 4°C at 1000 × g (Beckman SX4750 rotor) to remove large debris and bacterial aggregates. The supernatants (~15 mL) were collected, and the planktonic bacteria were pelleted by centrifugation at 13000 × g for 1 minute at 4°C and washed once with pre-chilled sterile PBS. Supragingival plaque was obtained by scraping from the buccal surface of first and second maxillary molars using sterile micropipette tips. The plaque samples were immediately transferred to 1.7 mL micro-centrifuge tubes containing 1 mL of pre-chilled sterile PBS, pelleted by centrifugation at 13000 × g (Eppendorf 5415D) for 1 minute at 4°C, and washed once with pre-chilled sterile PBS. Sessile plaque and planktonic salivary bacterial community samples were stored at −80°C in 500 μL of TRIzol® Reagent (Ambion®) until RNA isolation. Total RNA was purified using the Direct-Zol RNA MiniPrep kit (Zymo Research) according to the manufacturer’s protocol. RT-qPCR was performed as described below, and relative expression of adhesin genes was calculated using the ΔΔCt method.
For experiments testing biofilm formation in the presence of human saliva, stimulated whole saliva was collected from two to five donors on ice and pooled. Saliva and plaque collections were performed using a protocol that was reviewed and approved by the University of Minnesota Institutional Review Board.
DNA manipulations
Standard recombinant DNA techniques were employed as described by Sambrook et al. (58). Oligonucleotides (table S4) and synthetic DNA gBlocks were synthesized by Integrated DNA Technologies. All strains and plasmids used or created in this study are listed in tables S5 and S6, respectively. Plasmids were purified from E. coli using the QIAquick Spin Miniprep purification Kit (Qiagen). Chromosomal DNA was prepared as previously described (10). DNA was amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs). DNA restriction and modification enzymes were used according to the manufacturer’s directions (New England Biolabs). PCR products were purified using GenCatch Advanced PCR Extraction Kit (Epoch Life Sciences), and DNA was extracted from agarose gels using the QIAquick Gel Extraction Kit (Qiagen).
Construction of Streptococcus gordonii mutants
The principles of a markerless mutagenesis system, originally designed for use in S. mutans (19), were adapted to construct a S. gordonii–specific mutagenesis cassette, JHMD1. All bacterial strains are listed in Table S5. Briefly, the cassette uses erythromycin resistance for the first allelic exchange event. A mutated S. gordonii pheS sensitizes cells to 4-Cl-Phe providing identification of the final allelic exchange, and resolution of the in-frame mutation. The S. gordonii ldh promoter in the JHMD1 cassette drives expression of the selection markers. The cassette was constructed in silico and produced by GenScript USA, producing plasmid pJHMD1. To construct the single in-frame markerless gene deletions, the JHMD1 cassette and regions of homology (~300–500 bp) upstream and downstream of the region to be mutated were PCR-amplified from pJHMD1 and DL1 genomic DNA, respectively. The PCR primers were designed to delete >98% of the structural gene but maintain the native transcription regulatory regions, and start and stop codon sequences. The upstream and downstream regions were spliced to the JHMD1 cassette using Splice-Overlap Extension PCR (SOE-PCR) (19). The PCR product was transformed into wild type DL1 (10) and plated on selective THerm agar. The same regions of chromosomal homology were then spliced to each other using SOE-PCR, transformed into the erythromycin-resistant intermediary strain, and plated on selective TH4-Cl-Phe. The mutations were confirmed by diagnostic PCR and sequencing.
Double deletions were constructed progressively using DNA from a verified Ermr-intermediary. Next, the genomic DNA of a resolved, sequence-verified mutant was used as a template for PCR amplification. Each PCR product was transformed sequentially through positive and negative selection until the second mutation was achieved. Chromosomal nucleotide changes were constructed as above. The mutation of interest was incorporated into the upstream reverse (UpRev) oligonucleotide PCR primer and carried through until resolution of the mutation.
Construction of SspASLC, SGO_0707SLC, and PavBSLC strains
The nucleotide sequences of sspA and sspB are highly homologous, which complicates the construction of mutations in the individual genes. To create a strain devoid of the functional SspA and SspB adhesin domains, we first constructed the sspA promoter-signal peptide (S)-LPXTG (L)-C-pep (C) (SspASLC) nucleotide sequence using a two-step cloning process, creating pC-pep (see below). The pC-pep plasmid was used as PCR template to amplify the in-frame pro-C-pep region using the primer set JKR1-B/JKR1-H-JHMD1. To simultaneously delete sspB, we amplified a ~500 bp region starting at the nucleotide directly following the stop codon of sspB. Chromosomal DNA was used as a template for the primer set C-pepDnFor-JHMD1/sspBDnRev. The PCR products (SspASLC and SspB-Down) were purified and spliced by SOE-PCR to JHMD1. The product was then transformed into wild-type DL1 and plated on THerm agar. To remove the mutagenesis cassette, the same regions were amplified from the intermediary strain using the primer sets JKR1-B/JKR1-H (synthesized without restriction endonuclease sites) and SspAC-pepDnFor/sspBDnRev. The two purified PCR products were spliced together using SOE-PCR, transformed into the ErmR intermediary pro-C-pep strain, and plated on TH4-Cl-Phe agar. Resolution of the mutation created a strain that chromosomally encodes the SspA promoter, signal peptide (S), LPXTG-motif (L), and C-pep (C), with complete deletion of sspB and of the sspA adhesin domain coding sequence (strain SspASLC; ΔsspAB::sspASLC).
Construction of pro-C-pep plasmid
The C-pep plasmid (pC-pep) was constructed to model the sequence in the native genome but deleting most of the structural adhesin gene. Amplified by PCR, the construct contained, in order, the predicted promoter region of sspA, the signal sequence, the coding region for the first eight amino acids of the mature SspA, the four amino acids preceding the LPXTG motif, the LPXTG motif, the sequence encoding C-pep, and the stop codon. The oligonucleotides JKR1-B and JKR1-Sup introduced restriction enzyme cleavage sites for BamHI and SalI into the amplified promoter-signal peptide fragment. Similarly, the PCR primers JKR1-Sdown and JKR1-H introduced restriction enzyme cleavage sites for HindIII and SalI into the amplified fragment encoding LPXTG-C-pep. Each fragment was further cloned into pGEM®-T (Promega, Madison, WI) and sequenced. For genes without a homologous partner, sgo_0707 and pavB (sgo_1182), we deleted the nucleotide region between the signal peptide sequence and the LPXTG-motif using the protocol for whole gene deletion as above, creating strains SGO_0707SLC and PavBSLC.
Growth curves
Each strain was streaked on THAspec and three individual colonies were selected and cultured overnight. Cultures were diluted 1:100 into fresh THBspec and 200 μl of culture was aliquoted into triplicate wells of a sterile 96-well microtiter plate (Sarstedt). Each plate was sealed with a Breathe-Easy® (Sigma-Aldrich) membrane. Plates were incubated for 16 hours at 37 °C in a BioTek Synergy II reader. Every 30 minutes the plates were shaken for 10 seconds and the optical density (λ = 600nm) of each well was recorded. The data in Figure S2 represents the mean of all technical replicates for the three colonies of each strain.
Biofilm biomass assay
To test for biofilm formation in the absence of saliva as reported previously (16, 53), bacteria in BHI broth were grown at 37°C for 16 to 18 h in 5% CO2 in 96-well plastic plates (Corning). The plates were inverted and shaken to remove excess liquid growth medium, stain, and buffer. The retained biomass was stained for 15 min with 50 μl of 0.1% (w/v) crystal violet. The wells were then washed four times with sterile PBS. Excess PBS was removed by rapping the plate onto paper towels and air-drying for two minutes. To quantify sessile biomass, 200 μl of acidified ethanol (4% 1N HCl/EtOH) was added to each well. In each well, the ethanol and crystal violet were gently mixed, 125 μl of the extracted crystal violet was transferred to a new flat-bottom well in a polystyrene microtiter plate, and the absorbance at 570 nm was determined with a BioTek Synergy II reader.
To test for biofilm formation in the presence of saliva, pooled human saliva was centrifuged at 4300 RPM (Beckman SX4750 rotor) for five minutes at 4°C, supernatants were aliquoted into 1.7 mL microfuge tubes, and centrifuged at 15000 RPM (Eppendorf 5415D) for 20 minutes at 4°C. Saliva aliquots were re-pooled, and 200 μl of clarified saliva was added to each microtiter plate well. Plates were incubated for one hour at room temperature on an orbital shaker, then the saliva was aspirated from each well and replaced with growth medium and bacteria. Negative control saliva-coated wells produced biofilms ≤ 5% of WT in assays.
Human-derived multispecies oral microbial community biofilm
Supragingival plaque from the maxillary molars of 4 healthy volunteers was collected approximately 24 hours after tooth brushing. The plaque was washed with pre-reduced sterile PBS and combined for overnight growth in SHI medium (59) at 37°C anaerobically (10% H2, 10% CO2, 80% N2). The complex microbial community was frozen in 10% glycerol and served as the stock for our biofilm integration experiments. For biofilm integration, the ex vivo plaque community was grown overnight in SHI medium and diluted to an absorbance of 0.1 (λ=600 nm) in fresh SHI medium. S. gordonii strains containing the JHMD1 cassette were inoculated at an absorbance of 0.0001 (λ=600 nm). Biofilms were grown overnight using saliva-coated 12-well polystyrene culture plates (Corning). The planktonic cells were aspirated, and the surface attached biofilm was washed once with 500 μL of pre-reduced sterile PBS. Biofilms that remained attached to the wells were collected for DNA isolation with DNeasy PowerLyzer PowerSoil Kit (Qiagen). S. gordonii presence in the ex vivo community was determined by qPCR amplification of the JHMD1 cassette with primer pairs Fwd and Rev (table S4) and normalized to total 16s rRNA gene DNA, which was amplified with primer pair (BAC1 and BAC2) (59). The ΔΔCt method was used to calculate relative abundance.
Identification and co-transcription analysis by RT-PCR of SGO_1180 and SGO_1181
To identify all of the S. gordonii sensor HKs, we queried the S. gordonii genome in the MIST2 database (http://mistdb.com) and analyzed the predicted HKs of signal transduction proteins (60). The query yielded 16 potential sensor HKs (53, 61, 62) (table S1). To identify the most probable intramembrane sensing HK, we filtered the S. gordonii HKs by removing from consideration HKs with a large extracellular region (greater than 60 amino acids), such as SGO_0484 or SGO_1761, and HKs with multiple exposed loops that could function as ligand binding sites (SGO_1732, SGO_1896, and SGO_2146.) We also removed HKs without transmembrane helices (SGO_0641 and SGO_1259), as these were considered to be cytosolic sensors. Of the 16 candidate HKs, six remained as potential intramembrane or membrane-bound cytosolic sensors (table S1). We then screened the six HKs for potential cytosolic sensing regions using the benchmark of approximately 60 amino acids for an independent folding domain in the absence of metal binding. Based on these inclusion and exclusion criteria, one HK was predicted to be only an intramembrane sensor, SGO_1180, and was selected for further study.
An overnight culture of S. gordonii DL1 in BHI broth was pelleted by centrifugation. RNA was extracted from the cells using the FastRNA Pro Blue Kit per manufacturer’s instructions, with one modification: cells were subjected to three rounds of FastPrep processing (2 × 45 sec and 1 × 30 sec, at speed 6). Purified total RNA was treated with TURBO DNase (Life Technologies) following the manufacturer’s protocol. DNA-free RNA was re-purified using the Qiagen RNeasy Minikit following manufacturer’s instructions and the concentration of RNA was determined using a NanoDrop 2000 spectrophotometer. RNA (500 ng) was reversed transcribed into cDNA using SuperScript III reverse transcriptase (RT) (Invitrogen) according to the manufacturer’s instructions. Separate reactions were set up for pavB-SGO_1181 or SGO_1181-SGO_1180 using PavBRTFor or SGO_1181RTFor oligonucleotide primers, respectively. Control reactions without RT enzyme were also included. Each reaction was diluted 1:100 and 1:1000 in nuclease-free water. The PCR reaction was performed using Crimson Taq DNA polymerase (New England Biolabs) according to the manufacturer’s instructions. The oligonucleotide primer pairs PavBRTFor/SGO_1181RTRev and SGO_1181RTFor/SGO_1180RTRev (table S4) were used with the pavB-SGO_1181 and SGO_1181-SGO_1180 cDNA reactions, respectively. PCR reactions were analyzed by electrophoresis in a 2% agarose gel and visualized by staining with ethidium bromide.
Purification of total RNA for next-generation RNA sequencing
Overnight cultures were diluted 1:100 in 10 mL of sterile BHI in 100 mm tissue culture-treated dishes (Sarstedt) for biofilm grown cells or 15 mL polypropylene conical vials (Sarstedt) for free-growing cells and incubated for 4 hours at 37°C in 5% CO2. Free-growing cultures were pelleted at 4,300 RPM at 4°C, medium removed, cells resuspended in 1 mL of TRIzol reagent (Ambion), and frozen at −80°C. For biofilm cultures, medium was removed, and each plate was gently washed twice with 10 mL of ice-cold PBS to remove loosely attached cells. After washing, 1 mL of TRIzol was placed in each dish and swirled to coat the cells. Each biofilm was carefully removed with a sterile cell-scraper, transferred to a microfuge tube, and frozen at −80°C.
Each sample was thawed as needed and lysed as described (10). Following lysis, total RNA was purified using the Direct-Zol RNA MiniPrep kit (Zymo Research) according to the manufacturer’s protocol. Purified total RNA was treated with TURBO DNase (Life Technologies) following the manufacturer’s protocol. Total RNA was re-purified using the Quick-RNA MiniPrep kit (Zymo Research) and quantified using a NanoDrop2000 spectrophotometer.
Next-generation transcriptomics analysis
To assay sample quality, each isolate of total RNA was quantified using a fluorimetric RiboGreen assay. Total RNA integrity was assessed using capillary electrophoresis (Agilent BioAnalyzer 2100), generating an RNA Integrity Number (RIN). To pass the initial QC step, samples needed an RIN of 8 or greater quantified from at least 100 ng. Samples were then converted to Illumina sequencing libraries.
Sequencing library creation
Total RNA samples were converted to Illumina sequencing libraries using Illumina’s Truseq Stranded mRNA Sample Preparation Kit. In summary, 70 ng of mRNA was fragmented and then reverse transcribed into cDNA. The cDNA was adenylated and then ligated to dual-indexed (barcoded) adaptors and amplified using 15 cycles of PCR. Final library size distribution was validated using capillary electrophoresis and quantified using fluorimetry (PicoGreen). Indexed libraries were then normalized and pooled.
Cluster generation and sequencing
Truseq libraries were hybridized to a single read flow cell and individual fragments were clonally amplified by bridge amplification on board the HiSeq2500 Rapid instrument. Once clustering was completed, the flow cell was sequenced using Illumina’s SBS chemistry. Upon completion of read 1, an 8 base-pair index read for Index 1 was performed. The Index 1 product was then removed and the template re-annealed to the flow cell surface. The run continues with 7 chemistry-only cycles, followed by an 8 base-pair index read to read Index 2.
Primary analysis and de-multiplexing
Base call (.bcl) files for each cycle of sequencing were generated by Illumina Real Time Analysis (RTA) software. The base call files and run folders were then exported to servers maintained at the Minnesota Supercomputing Institute. Primary analysis and de-multiplexing were performed using Illumina’s CASAVA software 1.8.2. The end result of the CASAVA workflow was de-multiplexed FASTQ files.
Quality control, read mapping, and differential gene expression analysis
Reads were trimmed using Trimmomatic (v 0.33) enabled with the optional “-q” option; 3bp sliding-window trimming from 3’ end requiring minimum Q30. Quality control checks on raw sequence data for each sample were performed with FastQC. Read mapping was performed via Bowtie (v2.2.4.0) using the genome NC_009785 as reference (63). Differential Gene Expression (DGE) analysis was performed via CLCGWB using an edgeR implementation of the algorithm. DGE lists were generated using Absolute 2-Fold Change (FC) and a FDR p-value ≤ 0.05 was considered significant.
Verification of next-generation RNA sequencing results using RT-qPCR reverse transcription. Total RNA was purified as described for RNA sequencing. RNA (500 ng) was reverse transcribed into cDNA using ProtoScript II First Strand cDNA Synthesis kit (New England Biolabs; Ipswich, MA) with random hexamer primers according to the manufacturer’s protocol.
Multiplex RT-qPCR
Real-time quantitative PCR amplification, detection, and analysis were performed by the Mx3000 Real-Time PCR system (Stratagene). Multiplex real-time PCR was performed in 20 μL reaction mixtures (1X PrimeTime qPCR low-ROX Master Mix [IDT DNA], 2 μL 1:10 cDNA, and 100 nM each gene-specific forward and reverse primer and probe). The PCR conditions were (i) 95°C for 3 minutes, and (ii) 40 cycles of 95°C for 15 sec and 60°C for 60 sec. The ΔΔCt method was used to calculate relative gene expression.
Yellow fluorescent protein labeling of the SGO_0707 C-pep
To construct the SGO_0707 C-pep Venus YFP strain, the gBlock 0707Up-Venus-YFP-JHMD1 was synthesized (IDT DNA). This gBlock contains the coding sequence for the penultimate codon and preceding 165 codons of sgo_0707, full Venus YFP I152L coding sequence without a stop codon, and an overlapping sequence complementary to the 5’ terminus of the JHMD1 cassette. This gBlock, along with a PCR amplified region from DL1 genomic DNA containing the sgo_0707 stop codon and approximately 500 bp downstream, was attached to the either end of the JHMD1 cassette using SOE-PCR, and transformed into S. gordonii DL1 as described above. To resolve the YFP insertion, the sgo_0707Up-Venus segment was amplified by PCR using the gBlock as template and attached using SOE-PCR to a downstream PCR product containing the sgo_0707 stop codon and downstream region of homology. Resolved mutants were verified by diagnostic PCR, the production of a fluorescent signal, and DNA sequencing.
Structured-illumination microscopy (SIM)
SIM was performed using a custom-built system (64) as modified for live organisms (65). In brief, SGO_0707-YFP cells were cultured for 4 h at 37°C with the lipid dye, FM® 4–64FX (F34653, ThermoFisher Scientific) at a final concentration of 15 ng/μl, then fixed in 1% paraformaldehyde supplemented with 20 μg/ml Hoechst 33342 for 10 min at 4°C. Cells were then plated on a 25 mm glass coverslip #1.5 with TetraSpeck™ Microspheres 0.1 μm (T7279, ThermoFisher Scientific), which were used as fiducials for post-processing registration. The coverslip was then sealed with another coverslip using ProLong® Diamond (P36965, ThermoFisher Scientific). YFP was excited using the 488 nm laser line and detected using a GFP filter (BP 525/45). FM 4–64FX was excited using the 561 nm laser line and filtered with a far-red filter (LP 635). Hoechst 33342 was excited with the 405 nm laser line and detected with a DAPI filter (BP 480/40). Data acquisition and post-processing was performed as described (65).
In vivo bi-fluorescent complementation
Using a series of PCRs and SOE-PCR, the non-fluorescent N-terminal or C-terminal halves of Venus YFP (66), amplified from the 0707Up-Venus-YFP-JHMD1 gBlock, was placed between codons two and three of SGO_1180 (cytoplasmic side of TM1, creating strains 1180-TM1-V-N and 1180-TM1-V-C) or codons 61 and 62 (cytoplasmic side of TM2, creating strains 1180-TM2-V-N and 1180-TM2-V-C). Subsequently, the mated N- or C-fragments were placed in-frame between the penultimate codon and stop codon of sgo_0707 separately and in combination with the SGO_1180 N- or C- fragment strains.
SGO_1180 and SspA C-pep purification
To obtain purified SGO_1180 HK and SspA C-pep, the corresponding ORFs were cloned into an E. coli expression system. Sgo_1180 was cloned into pET28 vector, creating pET1180, and the sequence was verified by DNA sequencing. pET1180 was subsequently transformed into E. coli BL21(DE3) cells, creating strain BL1180. An overnight culture of BL1180 was diluted into fresh LB medium. Upon reaching an OD600nm = 1, IPTG was added to a final concentration of 1 mM to induce production of SGO_1180. The culture was incubated at 25°C for 16 hours. Cells were collected by centrifugation and lysed by sonication (3 cycles x 2 minutes). Cellular debris and unlysed cells were pelleted by centrifugation at 8,000 × g for 20 min. The membranes were then recovered by ultracentrifugation at 138,000 × g (Type 45Ti rotor) for 60 min. The proteins were solubilized from membranes using 50 mL of solubilization buffer (50 mM Tris, pH 7.4; 300 mM NaCl; 1 mM PMSF; 10% glycerol [v/v]) by slowly adding dodecyl-β-maltoside (DDM) (Anatrace) to a final detergent concentration of 1.0% [v/v]. The solubilization mixture was incubated at 4°C for 60 min with stirring. Insoluble material was pelleted by ultracentrifugation at 138,000 × g for 60 min. The soluble fraction was mixed with 5 mL of Ni NTA agarose (Qiagen) (pre-equilibrated with 50 mM Tris, pH 7.4; 300 mM NaCl; 0.1%DDM; 1 mM PMSF; 10% glycerol [v/v]) and incubated with gentle agitation at 4°C for 60 min. The resin was then packed in a chromatography column and washed with 30 mL of wash buffer (0.1% DDM [v/v]; 50 mM Tris, pH 7.4; 10% glycerol [v/v]; 300 mM NaCl; 30 mM imidazole) to remove nonspecifically bound material. Bound material was eluted with elution buffer (0.1 DDM [v/v]; 50 mM Tris, pH 7.4; 10% glycerol [v/v]; 300 mM NaCl; 300 mM imidazole). The eluted sample was concentrated and purified by size-exclusion chromatography (Superdex 75; 0.05% DDM; 50 mM HEPES, pH 7.4; 300 mM NaCl). The sample was digested with thrombin overnight at 4°C to remove the 6xHis tag and re-purified by size-exclusion chromatography.
SspA C-pep was purified using essentially the same protocol, with minor modifications. A DNA sequence consisting of GFP-TEV-SspA C-pep (IDT DNA) was cloned into the pE-SUMOpro-Amp vector. After purification by Ni-NTA chromatography, the fusion protein was sequentially treated with ULP1 protease to cleave the SUMO tag and TEV protease to cleave the GFP from SspA C-pep. The SspA C-pep was purified by size-exclusion chromatography (Superdex 75; 50 mM Tris, pH 7.4; 200 mM NaCl; 0.1% DDM).
Solid-state Nuclear Magnetic Resonance
For Magic Angle Spinning (MAS) solid-state NMR experiments, SGO_1180 dimer in the absence and presence of C-pep was reconstituted in 2H 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipids. Specifically, recombinant 15N, 13C uniformly labeled SGO_1180 was solubilized in 0.1% n-dodecyl-β-D-maltopyranoside (DDM) and added to a suspension of DMPC. The detergent was removed by addition of 50:1 (w:w) of biobeads®. After centrifugation and removal of biobeads®, the proteolipids were pelleted using two ultracentrifugation steps: the first one for 30 min at 30,000 rpm and a second at 80,000 rpm for 12–20 hours. Subsequently, the wet proteolipidic preparation was inserted in a 3.2 mm MAS rotor. For the preparation of the SGO_1180:C-pep complex, both proteins were solubilized in 0.1% DDM and added to the DMPC lipid vesicle preparation reaching a final stoichiometry of 1:1 SGO_1180:C-Pep (2 C-pep per per SGO_1180 dimer). A 50:1 (w:w) suspension of biobeads® was used to remove DDM and the complex was pelleted by ultracentrifugation prior to insertion in the MAS rotor. Solid-state NMR experiments were performed on an Agilent ssNMR spectrometer operating at a 1H Larmor frequency of 600 MHz equipped with 3.2 mm scroll MAS probe. For all the experiments, MAS rate (ωr) was set to 12 kHz, with acquisition time and recycle delay respectively set to 20 ms and 2 s. During acquisition, Spinal-64 decoupling was applied (67). For CP and INEPT experiments, the temperature was respectively set to and 2 and 25oC (68). The 90o pulse lengths for 1H and 15N were 2.5 and 6 μs, respectively. The CP experiments were acquired with 32 scans, with the Hartman-Hahn contact time set to 500 μs during which the 15N RF amplitude set to 35 kHz, whereas 1H RF amplitude was linearly ramped from 80% to 100 % with the center of the slope set at 59 kHz (69). The 1D and 2D rINEPT spectra of SGO_1180 in the absence of SspA C-pep were acquired with 592 scans, whereas for the 1D rINEPT spectrum in the presence of SspA C-pep was acquired with 784 scans.
SGO_1180 ortholog cloning and biofilm complementation analysis
To determine whether the SGO_1180 orthologs identified via bioinformatics and S. gordonii SGO_1180 functioned similarly, the orthologous genes of S. pneumoniae, S. agalactiae, and Staphylococcus aureus were cloned into the E. coli-Streptococcus shuttle vector, pDL278. Each ortholog was synthesized (IDT, Coralville, IA) with the S. gordonii ldh promoter and optimized Gram-positive RBS preceding the open reading frame. The DNA sequence encoding a 3×TY1 epitope tag was placed into the sequences corresponding to the C-terminal cytoplasmic side of the second transmembrane domain for each ortholog as described for the Venus YFP fragments in the BiFC section. We had determined that SGO_1180 was rendered non-functional after placement of an N-terminal 3×TY1 epitope tag. Each construct was synthesized with 5’ phosphorylation for blunt-end cloning. The vector pDL278 was digested with SmaI, treated with rSAP (New England Biolabs Inc., Ipswich, MA), and linear pDL278 was purified from a 1.2% agarose gel after electrophoresis using QIAquick Gel Extraction kit (Qiagen). Each synthetic construct (30 ng) and 1 ng of purified linear rSAP-treated pDL278 were mixed in a T4 ligase (New England Biolabs) reaction and incubated at room temperature overnight. Each reaction mix was transformed into NEB5alpha competent E. coli cells and recombinants were screened on LBspec agar, 1 mM IPTG, and 200 μg/mL X-gal. White colonies were subsequently screened by colony PCR for insert orientation in pDL278 using primers ldhFor/pDL278For and OneTaq 2X PCR Green Master Mix (New England Biolabs) according to the manufacturer’s instructions. The pDL278 control and correctly oriented ortholog complementation constructs were purified from E. coli using a QIAprep Spin Miniprep kit (Qiagen). Each construct was separately transformed into DL1ΔSGO_1180ΔSspAB and selected on THAspec agar.
To verify complementation construct production, S. gordonii transformants were cultured overnight, centrifuged and lysed as described (53). Lysates were mixed 3:1 with SDS-PAGE loading buffer, boiled, and cooled to room temperature. After cooling and allowing lysing matrix beads to settle, a 20-μl aliquot of the supernatant was loaded into a pre-cast 4–20% SDS-polyacrylamide gel (Bio-Rad) and electrophoresed until the loading buffer dye front reached the bottom of the gel. The gel was electro-blotted onto 0.45 μm nitrocellulose membrane using a Bio-Rad Trans-Blot Semi-Dry Transfer Cell. The membrane was blocked with 5% dry milk in PBS-Tween 20 (0.05% v/v, PBS-T) for one h at RT. The membrane was incubated with 1:1000 dilution of rabbit anti-TY1 antibody (GenScript) for one h at RT and then washed three times with PBS-T. The membrane was incubated with a 1:15,000 dilution of anti-rabbit 800CW antibody (LICOR), washed three times with PBS-T and imaged in using an Odyssey Imaging System (LICOR). S. gordonii WT strains, carrying the empty vector and a chromosomally tagged 3xTY1-sgo_1180 served as negative and positive immune-blotting controls, respectively. Verified ortholog-producing S. gordonii transformants were cultured overnight, and assayed for biofilm formation in the saliva-modified, biofilm assay as described above.
Data Analysis
Numerical data were analyzed in GraphPad Prism version 8. Data was analyzed for normality using a D’Agostino and Pearson test. Parametric data was analyzed using Student’s t-test or ANOVA with Tukey’s multiple comparisons test as reported in the corresponding figure legend. Non-parametric data was analyzed using a Kruskal-Wallis test with Dunn’s multiple comparisons test. An p-value ≤ 0.05 was considered significant with * = ≤ 0.05, ** = ≤ 0.01, *** = ≤ 0.001, **** = ≤ 0.0001. The data and materials that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgements:
We thank Michael Shyne of the Biostatistical Design & Analysis Center (BDAC) at the University of Minnesota for expert statistical consultation and suggestions and Alita Caldwell for studies on the co-transcription of pavB-1180–1181. Appreciation is extended to Howard Jenkinson, University of Bristol, for providing wild-type Streptococcus gordonii DL1 and Justin Merritt, Oregon Health Sciences University, for helpful discussions about construction of the pJHMD1 mutagenesis cassette. We thank Brian D. Guenther for identification of potential S. gordonii histidine kinases and Juan Abrahante of the University of Minnesota Genomics Center for expert assistance with next-gen transcriptomics analyses. We extend appreciation to Gary Dunny of the Department of Microbiology and Immunology at the University of Minnesota for advice about the conduct of this study and the writing of this manuscript. We also thank the former and current members of the Herzberg and Svenstater labs (Malmö University) for their critical reading and feedback on this manuscript.
Funding: This work was supported by the National Institute of Dental & Craniofacial Research grants 1R01DE02561801A1 (to MCH), T90DE0227232 and F32DE02578 (to JWH), 1K08DE027705-01A1 (to BL), GM 64742 (to GV), the University of Minnesota Summer Dental Student Research Fellowship Program (to APC), and the NIDCR-NIH intramural program (to GGH). The NMR experiments were carried out at the Minnesota NMR Center. The transcriptomics analysis was completed at the University of Minnesota Genomics Center. Support to MCH is also acknowledged from the University of Minnesota Office of the Vice President for Research and the School of Dentistry.
Footnotes
Competing Interests: The authors declare that they have no competing interests.
Data and Materials Availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Brandtzaeg P 2009. Mucosal immunity: induction, dissemination, and effector functions. Scand. J. Immunol. 70: 505–515. [DOI] [PubMed] [Google Scholar]
- 2.Doss M, White MR, Tecle T, and Hartshorn KL. 2010. Human defensins and LL-37 in mucosal immunity. J. Leukoc. Biol. 87: 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belyakov IM, and Ahlers JD. 2009. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J. Immunol. Baltim. Md 1950. 183: 6883–6892. [DOI] [PubMed] [Google Scholar]
- 4.Otto M 2008. Staphylococcal Biofilms In Bacterial Biofilms. Current Topics in Microbiology and Immunology Romeo T, ed. Springer Berlin Heidelberg; 207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boles BR, and Horswill AR. 2011. Staphylococcal biofilm disassembly. Trends Microbiol. 19: 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nobbs AH, Lamont RJ, and Jenkinson HF. 2009. Streptococcus Adherence and Colonization. Microbiol. Mol. Biol. Rev. 73: 407–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jakubovics NS, Kerrigan SW, Nobbs AH, Strömberg N, van Dolleweerd CJ, Cox DM, Kelly CG, and Jenkinson HF. 2005. Functions of Cell Surface-Anchored Antigen I/II Family and Hsa Polypeptides in Interactions of Streptococcus gordonii with Host Receptors. Infect. Immun. 73: 6629–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreth J, Merritt J, and Qi F. 2009. Bacterial and Host Interactions of Oral Streptococci. DNA Cell Biol. 28: 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen RN, Ganeshkumar N, and Kolenbrander PE. 1993. Cloning of the Streptococcus gordonii PK488 gene, encoding an adhesin which mediates coaggregation with Actinomyces naeslundii PK606. Infect. Immun. 61: 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Lei Y, Nobbs A, Khammanivong A, and Herzberg MC. 2005. Inactivation of Streptococcus gordonii SspAB Alters Expression of Multiple Adhesin Genes. Infect. Immun. 73: 3351–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei Y, Zhang Y, Guenther BD, Kreth J, and Herzberg MC. 2011. Mechanism of adhesion maintenance by methionine sulphoxide reductase in Streptococcus gordonii. Mol. Microbiol. 80: 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneewind O, and Missiakas D. 2014. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1843: 1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneewind O, Model P, and Fischetti VA. 1992. Sorting of protein a to the staphylococcal cell wall. Cell 70: 267–281. [DOI] [PubMed] [Google Scholar]
- 14.Schneewind O, Mihaylova-Petkov D, and Model P. 1993. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J. 12: 4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies JR, Svensäter G, and Herzberg MC. 2009. Identification of novel LPXTG-linked surface proteins from Streptococcus gordonii. Microbiology 155: 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nobbs AH, Vajna RM, Johnson JR, Zhang Y, Erlandsen SL, Oli MW, Kreth J, Brady LJ, and Herzberg MC. 2007. Consequences of a sortase A mutation in Streptococcus gordonii. Microbiology 153: 4088–4097. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi Y, Konishi K, Cisar JO, and Yoshikawa M. 2002. Identification and Characterization of hsa, the Gene Encoding the Sialic Acid-Binding Adhesin of Streptococcus gordonii DL1. Infect. Immun. 70: 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nobbs AH, Zhang Y, Khammanivong A, and Herzberg MC. 2007. Streptococcus gordonii Hsa Environmentally Constrains Competitive Binding by Streptococcus sanguinis to Saliva-Coated Hydroxyapatite. J. Bacteriol. 189: 3106–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Z, Okinaga T, Qi F, Zhang Z, and Merritt J. 2011. Cloning-Independent and Counterselectable Markerless Mutagenesis System in Streptococcus mutans. Appl. Environ. Microbiol. 77: 8025–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller BK, Subramaniam S, and Senes A. 2014. A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα–H hydrogen bonds. Proc. Natl. Acad. Sci. U. S. A. 111: E888–E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim S, Jeon T-J, Oberai A, Yang D, Schmidt JJ, and Bowie JU. 2005. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 102: 14278–14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li E, Wimley WC, and Hristova K. 2012. Transmembrane helix dimerization: Beyond the search for sequence motifs. Biochim. Biophys. Acta BBA - Biomembr. 1818: 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, and Miyawaki A. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20: 87–90. [DOI] [PubMed] [Google Scholar]
- 24.Zschiedrich CP, Keidel V, and Szurmant H. 2016. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 428: 3752–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mascher T 2006. Intramembrane-sensing histidine kinases: a new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol. Lett. 264: 133–144. [DOI] [PubMed] [Google Scholar]
- 26.Mascher T, Helmann JD, and Unden G. 2006. Stimulus Perception in Bacterial Signal-Transducing Histidine Kinases. Microbiol. Mol. Biol. Rev. 70: 910–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensch I, Gámez G, Rothe M, Ebert S, Fulde M, Somplatzki D, Bergmann S, Petruschka L, Rohde M, Nau R, and Hammerschmidt S. 2010. PavB is a surface-exposed adhesin of Streptococcus pneumoniae contributing to nasopharyngeal colonization and airways infections. Mol. Microbiol. 77: 22–43. [DOI] [PubMed] [Google Scholar]
- 28.Song X-M, Connor W, Hokamp K, Babiuk LA, and Potter AA. 2009. The growth phase-dependent regulation of the pilus locus genes by two-component system TCS08 in Streptococcus pneumoniae. Microb. Pathog. 46: 28–35. [DOI] [PubMed] [Google Scholar]
- 29.Throup JP, Koretke KK, Bryant AP, Ingraham KA, Chalker AF, Ge Y, Marra A, Wallis NG, Brown JR, Holmes DJ, Rosenberg M, and Burnham MK. 2000. A genomic analysis of two-component signal transduction in Streptococcus pneumoniae. Mol. Microbiol. 35: 566–576. [DOI] [PubMed] [Google Scholar]
- 30.Liang X, Yu C, Sun J, Liu H, Landwehr C, Holmes D, and Ji Y. 2006. Inactivation of a Two-Component Signal Transduction System, SaeRS, Eliminates Adherence and Attenuates Virulence of Staphylococcus aureus. Infect. Immun. 74: 4655–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mrak LN, Zielinska AK, Beenken KE, Mrak IN, Atwood DN, Griffin LM, Lee CY, and Smeltzer MS. 2012. saeRS and sarA Act Synergistically to Repress Protease Production and Promote Biofilm Formation in Staphylococcus aureus. PLoS ONE 7: e38453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stock AM, Robinson VL, and Goudreau PN. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69: 183–215. [DOI] [PubMed] [Google Scholar]
- 33.Giomarelli B, Visai L, Hijazi K, Rindi S, Ponzio M, Iannelli F, Speziale P, and Pozzi G. 2006. Binding of Streptococcus gordonii to extracellular matrix proteins. FEMS Microbiol. Lett. 265: 172–177. [DOI] [PubMed] [Google Scholar]
- 34.Kolenbrander PE, Andersen RN, and Ganeshkumar N. 1994. Nucleotide sequence of the Streptococcus gordonii PK488 coaggregation adhesin gene, scaA, and ATP-binding cassette. Infect. Immun. 62: 4469–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanna I, Gomathi G, Sambandam C, Jayalakshmi M, Premavathy R, and Shantha S. 2014. Determination of three dimensional structure of scaA protein, a virulence factor of Streptococcus gordonii by homology modelling and design of inhibitors for scaA protein. Am. J. PharmTech Res. 4. [Google Scholar]
- 36.Vickerman MM, Clewell DB, and Jones GW. 1991. Sucrose-promoted accumulation of growing glucosyltransferase variants of Streptococcus gordonii on hydroxyapatite surfaces. Infect. Immun. 59: 3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricker A, Vickerman M, and Dongari-Bagtzoglou A. 2014. Streptococcus gordonii glucosyltransferase promotes biofilm interactions with Candida albicans. J. Oral Microbiol. 6: 23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolenbrander PE, Andersen RN, Baker RA, and Jenkinson HF. 1998. The Adhesion-Associated sca Operon in Streptococcus gordonii Encodes an Inducible High-Affinity ABC Transporter for Mn2+ Uptake. J. Bacteriol. 180: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mhatre E, Troszok A, Gallegos-Monterrosa R, Lindstädt S, Hölscher T, Kuipers OP, and Kovács ÁT. 2016. The impact of manganese on biofilm development of Bacillus subtilis. Microbiology 162: 1468–1478. [DOI] [PubMed] [Google Scholar]
- 40.Holmes AR, McNab R, and Jenkinson HF. 1996. Candida albicans binding to the oral bacterium Streptococcus gordonii involves multiple adhesin-receptor interactions. Infect. Immun. 64: 4680–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakubovics NS, Smith AW, and Jenkinson HF. 2000. Expression of the virulence-related Sca (Mn2+) permease in Streptococcus gordonii is regulated by a diphtheria toxin metallorepressor-like protein ScaR. Mol. Microbiol. 38: 140–153. [DOI] [PubMed] [Google Scholar]
- 42.Kerppola TK 2006. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1: 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu C-D, Chinenov Y, and Kerppola TK. 2002. Visualization of Interactions among bZIP and Rel Family Proteins in Living Cells Using Bimolecular Fluorescence Complementation. Mol. Cell 9: 789–798. [DOI] [PubMed] [Google Scholar]
- 44.Rekas A, Alattia J-R, Nagai T, Miyawaki A, and Ikura M. 2002. Crystal Structure of Venus, a Yellow Fluorescent Protein with Improved Maturation and Reduced Environmental Sensitivity. J. Biol. Chem. 277: 50573–50578. [DOI] [PubMed] [Google Scholar]
- 45.Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, and Veglia G. 2013. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. 110: 17338–17343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baldus M, Petkova AT, Herzfeld J, and Griffin RG. 1998. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 95: 1197–1207. [Google Scholar]
- 47.Morris GA, and Freeman R. 1979. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 101: 760–762. [Google Scholar]
- 48.Adhikari RP, and Novick RP. 2008. Regulatory organization of the staphylococcal sae locus. Microbiology 154: 949–959. [DOI] [PubMed] [Google Scholar]
- 49.Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, and Voyich JM. 2010. SaeR Binds a Consensus Sequence within Virulence Gene Promoters to Advance USA300 Pathogenesis. J. Infect. Dis. 201: 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X, Zhang X, Zhu Y, Niu L, Teng M, Sun B, and Li X. 2015. Structure of the DNA-binding domain of the response regulator SaeR from Staphylococcus aureus. Acta Crystallogr. Sect. D 71: 1768–1776. [DOI] [PubMed] [Google Scholar]
- 51.Huang KJ, Lan CY, and Igo MM. 1997. Phosphorylation stimulates the cooperative DNA-binding properties of the transcription factor OmpR. Proc. Natl. Acad. Sci. U. S. A. 94: 2828–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jenkinson HF 1992. Adherence, coaggregation, and hydrophobicity of Streptococcus gordonii associated with expression of cell surface lipoproteins. Infect. Immun. 60: 1225–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Lei Y, Khammanivong A, and Herzberg MC. 2004. Identification of a Novel Two-Component System in Streptococcus gordonii V288 Involved in Biofilm Formation. Infect. Immun. 72: 3489–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Whiteley M, Kreth J, Lei Y, Khammanivong A, Evavold JN, Fan J, and Herzberg MC. 2009. The two-component system BfrAB regulates expression of ABC transporters in Streptococcus gordonii and Streptococcus sanguinis. Microbiology 155: 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stewart RC 2010. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr. Opin. Microbiol. 13: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gushchin I, Melnikov I, Polovinkin V, Ishchenko A, Yuzhakova A, Buslaev P, Bourenkov G, Grudinin S, Round E, Balandin T, Borshchevskiy V, Willbold D, Leonard G, Büldt G, Popov A, and Gordeliy V. 2017. Mechanism of transmembrane signaling by sensor histidine kinases. Science eaah6345. [DOI] [PubMed] [Google Scholar]
- 57.Kilmury SLN, and Burrows LL. 2016. Type IV pilins regulate their own expression via direct intramembrane interactions with the sensor kinase PilS. Proc. Natl. Acad. Sci. 113: 6017–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sambrook J, Maniatis T, and Fritsch E. 1989. Molecular cloning: a laboratory manual,. Cold Spring Harbor Laboratory Press, New York. [Google Scholar]
- 59.Tian Y, He X, Torralba M, Yooseph S, Nelson KE, Lux R, McLean JS, Yu G, and Shi W. 2010. Using DGGE profiling to develop a novel culture medium suitable for oral microbial communities. Mol. Oral Microbiol. 25: 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ulrich LE, and Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38: D401–D407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Håvarstein LS, Gaustad P, Nes IF, and Morrison DA. 1996. Identification of the streptococcal competence-pheromone receptor. Mol. Microbiol. 21: 863–869. [DOI] [PubMed] [Google Scholar]
- 62.Guenzi E, Gasc AM, Sicard MA, and Hakenbeck R. 1994. A two-component signal-transducing system is involved in competence and penicillin susceptibility in laboratory mutants of Streptococcus pneumoniae. Mol. Microbiol. 12: 505–515. [DOI] [PubMed] [Google Scholar]
- 63.Vickerman MM, Iobst S, Jesionowski AM, and Gill SR. 2007. Genome-Wide Transcriptional Changes in Streptococcus gordonii in Response to Competence Signaling Peptide. J. Bacteriol. 189: 7799–7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhuvanendran S, Salka K, Rainey K, Sreetama SC, Williams E, Leeker M, Prasad V, Boyd J, Patterson GH, Jaiswal JK, and Colberg-Poley AM. 2014. Superresolution Imaging of Human Cytomegalovirus vMIA Localization in Sub-Mitochondrial Compartments. Viruses 6: 1612–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.York AG, Parekh SH, Nogare DD, Fischer RS, Temprine K, Mione M, Chitnis AB, Combs CA, and Shroff H. 2012. Resolution Doubling in Live, Multicellular Organisms via Multifocal Structured Illumination Microscopy. Nat. Methods 9: 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kodama Y, and Hu C-D. 2010. An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. BioTechniques 49: 793–805. [DOI] [PubMed] [Google Scholar]
- 67.Fung BM, Khitrin AK, and Ermolaev K. 2000. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. San Diego Calif 1997. 142: 97–101. [DOI] [PubMed] [Google Scholar]
- 68.Gopinath T, and Veglia G. 2018. Probing membrane protein ground and conformationally excited states using dipolar- and J-coupling mediated MAS solid state NMR experiments. Methods San Diego Calif 148: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hartmann SR, and Hahn EL. 1962. Nuclear Double Resonance in the Rotating Frame. Phys. Rev. 128: 2042–2053. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.