

Abstract
The natural compound Alternol was shown to induce profound oxidative stress and apoptotic cell death preferentially in cancer cells. In this study, a comprehensive investigation was conducted to understand the mechanism for Alternol-induced ROS accumulation responsible for apoptotic cell death. Our data revealed that Alternol treatment moderately increased mitochondrial superoxide formation rate, but it was significantly lower than the total ROS positive cell population. Pre-treatment with mitochondria specific anti-oxidant MitoQ, NOX or NOS specific inhibitors had not protective effect on Alternol-induced ROS accumulation and cell death. However, XDH/XO inhibition by specific small chemical inhibitors or gene silencing reduced total ROS levels and protected cells from apoptosis induced by Alternol. Further analysis revealed that Alternol treatment significantly enhanced XDH oxidative activity and induced a strong protein oxidation-related damage in malignant but not benign cells. Interestingly, benign cells exerted a strong spike in anti-oxidant SOD and catalase activities compared to malignant cells after Alternol treatment. Cell-based protein-ligand engagement and in-silicon docking analysis showed that Alternol interacts with XDH protein on the catalytic domain with two amino acid residues away from its substrate binding sites. Taken together, our data demonstrate that Alternol treatment enhances XDH oxidative activity, leading to ROS-dependent apoptotic cell death.
Keywords: XDH, XOR, Alternol, ROS, apoptosis, prostate cancer
Graphical Abstract
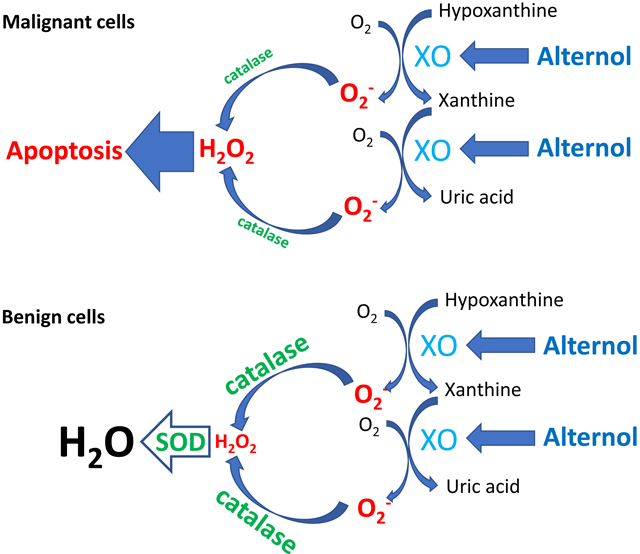
Introduction
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are chemically reactive molecules that are essential for living organisms and consist of multiple species such as superoxide (O2•−), hydrogen peroxide (H2 O2), nitric oxide (NO•), hydroxyl radical (•OH), peroxynitrite (ONOO−) and lipid peroxyl radical (LOO•) [1]. There are several ROS-generating mechanisms in mammalian cells and the major players are mitochondrial respiratory chain, membrane-bound NADPH oxidase (NOX), cytoplasmic xanthine dehydrogenase (XDH) and nitric oxide synthase (NOS). Meanwhile, there are several anti-oxidant mechanisms in cells operating through enzymatic reactions (e.g. superoxide dismutase, catalase and peroxidase), small redox proteins such as thioredoxin, and non-enzymatic small molecule antioxidants (vitamin E/C, glutathione, etc.) [2,3]. While a moderate increase of ROS levels promotes cell proliferation and differentiation, excessive accumulation of ROS species induces oxidative damage to lipids, proteins and DNA molecules [4,5]. It is conceivable that cancer cells are much more vulnerable to an acute increase of intracellular ROS levels in comparison to benign counterparts due to a significantly higher basal ROS level [6]. In fact, this property of cancer cells was recently utilized to develop new anti-cancer therapies [6,7].
XDH, also called xanthine oxidase (XO), is the rate-limiting enzyme for purine degradation, metabolizing hypoxanthine/xanthine to uric acid [8,9]. In the clinic, XDH is the therapeutic target of drugs like Allopurinol and febuxostat for gout or hyperuricemia [10,11]. During XDH-mediated purine metabolic reactions, ROS, including superoxide anion (O•−2) and hydrogen peroxide (H2O2), are produced [12]. The XDH gene is constitutively expressed in most tissues and it is regulated at the transcriptional level by multiple factors including hormones, growth factors and inflammatory cytokines, as well as irritative stimuli (reviewed in ref. [13]). Post-transcriptionally, XDH can be converted to xanthine oxidase (XO) via an irreversible proteolysis or a reversible cysteine oxidation to disulfide [14]. Although both XDH and XO can generate superoxide, XDH predominantly reduces NAD+ while XO predominantly reduces O2, generating superoxide and H2O2 [15].
In human cancers, studies on the significance of XDH expression were not widely reported. Limited literature showed that XDH protein expression and activity are much lower in tumor tissues compared to normal counterparts in gastrointestinal, breast, lung, kidney, bladder and ovary tissues, in which XDH protein levels are normally expressed at a higher level (reviewed in [16]). Meanwhile, lower XDH levels in patient tumors are associated with a worse prognosis of cancer-specific survival in several types of cancers [17–20]. These clinical observations suggest XDH as a tumor suppressor. Consistent with this notion, in chemically induced animal tumors, XDH protein expression and XO enzymatic activity were markedly reduced in tumor tissues and XDH/XO inhibition increased breast xenograft tumor growth in nude mice [16]. However, it is not clear if enhancing XDH/XO activity would reduce tumor growth or tumor cell survival.
Alternol is a small compound isolated from fermentation products of a mutant micro-organism [21]. We have demonstrated that Alternol induces a profound ROS response and apoptotic cell death in prostate cancer cells but not in benign prostate epithelial cells [22]. To understand the mechanism of Alternol-induced ROS response, we analyzed the ROS response with different fluorescent probes together with various pharmacological inhibitors of cellular ROS-generating enzymes. Our data revealed that XDH-specific inhibitors Febuxostat and Allopurinol abrogated Alternol-induced ROS accumulation and subsequent apoptotic cell death. In contrast, inhibitors for NOS and NOX had no significant effect on Alternol-induced ROS accumulation and cell death. Meanwhile, although Alternol moderately increased mitochondrial superoxide level, the mitochondria-specific ROS scavenger MitoQ had no protective effect on Alternol-induced cell death. In-silico analysis determined that Alternol interacts with the XDH protein catalytic domain without interfering its co-factor binding. Alternol treatment increased XDH protein level, promoted XDH proteolytic processing and enhanced its oxidative activity in prostate cancer cells but not in benign cells. Interestingly, Alternol treatment significantly enhanced cellular superoxide dismutase (SOD) and catalase activities in benign cells compared to malignant cells. Taken together, our data demonstrated that Alternol induced XDH/XO activation specifically in cancer cells that is a groundbreaking approach to treat malignances without harming normal tissues.
2. Materials and Methods
2.1. Cell culture, special chemical reagents, enzymatic assay kits and antibodies
The origin and culture condition for human prostate cancer PC-3, 22RV1, C4–2, LNCaP and DU145 cell lines and benign prostate epithelial cell line BPH1 were described in our recent publications [22,23]. Alternol (99.9% purity) was obtained from Sungen Biosciences (Shantou, China). Chemicals of ROS scavengers n-acetylcysteine (N-Ac) and dihydrolipoic acid (DHLA) NOX inhibitors Apocynin and Diphenyleneiodonium (DPI), NOS inhibitors L-NMMA, L-NAME and 1400W, MitoQ, thioredoxin reductase inhibitor Auranofin, XDH inhibitors allopurinol and febuxostat were obtained from Cayman Chemicals (Ann Arbor, MI). Pre-verified XDH siRNA, the negative control siRNA, sulforhodamine B (SRB), biotin hydrazide and sodium borohydride (NaBH4) were purchased from Santa Cruz Biotech (Santa Cruz, CA). ROS-ID® total ROS or NO detection kits and the Glutathione (GSH/GSSG) detection kit were purchased from Enzo Life Sciences (Farmingdale, NY). MitoSOX™ Red mitochondrial superoxide indicator and the RNAiMax reagent were purchased from Invitrogen (Carlsbad, CA). Xanthine oxidase activity, uric acid and H2O2 fluorescent assay kits were obtained from Cayman Chemicals. Antibodies for HRP-Biotin, caspase-3, PARP and Actin were from Cell Signal Tech (Danvers, MA). XDH antibody was purchased from Proteintech (catalog #55156, Rosemont, IL). SOD (K335) and catalase (K773) activity colorimetric assay kits were purchased from BioVision (Milpitas, CA).
2.2. Cytotoxicity, siRNA transfection, and intracellular total ROS, superoxide or NO levels
Cells were seeded at 1 × 104 cells/well in 24-well plates overnight, followed by treatment with the solvent or Alternol plus or minus other inhibitors as indicated in the figure legend. Cell death population was determined using the trypan blue exclusion assay as described [22]. Alternatively, cell survival was evaluated using the SRB-based assay as reported [24]. Transfection of XDH or the negative control siRNAs was conducted using the RNAiMax reagent as described in our publications [22,25].
Intracellular ROS, NO or mitochondrial superoxide levels were assessed with the corresponding fluorescent detection kits. Briefly, cells were seeded in a 24-well culture plate overnight and then were loaded with the fluorescent detection solution for 30 min. After removing the detection solution, cells were treated with the solvent or Alternol in the presence or absence of the inhibitors as indicated in the figure legends. ROS-positive cells were counted under a fluorescent microscope as described [22].
2.3. XO, SOD and catalase activity assays and glutathione, uric acid and H2O2 levels
Cells were seeded in 6-well plates and treated with the solvent or Alternol as indicated. Cells were harvested in the assay buffers from the kits for XO, SOD and catalase activities, or for the detection of cellular GSH/GSSG and uric acid levels. XO activity was assessed by quantifying a fluorescent end-product resorufin converted from ADHP (10-acetyl-3,7-dihydroxyphenoxazine) in the presence of horseradish peroxidase and H2O2, whereas the H2O2 is produced during xanthine oxidase-mediated oxidation of hypoxanthine (Cayman Chemical kit #10010895). SOD activity was assessed as an inhibitory effect on a water-soluble formazan dye production from WST-1 by H2O2 (BioVision kit #K335). Catalase activity was assessed by quantifying the residual levels of H2O2 that reacts with a chemical probe OxiRed™. The signal intensity is reversely proportional to catalase activity in the sample (BioVision kit #773). The glutathione assay kit utilized purified active glutathione reductase to reduce oxidized GSSG to form GSH that reacts on DTNB (5,5’-dithiobis-2-nitrobenzoic acid) to yield a yellow chemical TNB (5-thio-2-nitrobenzoic acid) as a readout (Enzo Life kit #ADI-900–160). Uric acid assay was based on the conversion of uric acid to allantoin plus H2O2, the later was captured by a fluorescent trap resorufin as described earlier (Cayman Chemical Kit #700320). Cell culture media were collected for the detection of H2O2 levels with the fluorescent trap resorufin (Cayman Chemical kit, #600050) by following the manufacturer’s manual. All results were normalized with total protein levels in each corresponding sample.
2.4. Western blotting, qPCR, protein biotin derivatization and nitration assays
Western blot assay of protein expression was conducted as descried [23]. Briefly, total cellular protein lysates were extracted using RIPA buffer supplemented with protease inhibitor cocktail. Equal amount of proteins was subjected to SDS-PAGE separation, followed by transferring onto PVDF membrane. After blocking in 5% nonfat milk, the membranes were incubated with primary antibodies overnight at 4C. Protein bands were visualized using HRP-linked secondary antibody and ECL solution from Santa Cruz Biotech.
Gene expression was assessed using SYBR Green-based quantitative RT-PCR assay. Total RNA samples were isolated using Trizol solution from prostate cancer cells and frozen specimens obtained from radical prostatectomy with matched benign counterparts as described in our previous report [25]. PCR primer pair for human XDH gene is: forward 5’-AGCACTAAC ACTGTGCCCAA-3’; reverse 5’-TGGTCTGACAAGCCGCATAG-3’. Human 18S rRNA primer pair is: forward 5’-CTACCACATCCAAGGAAGCA-3’ and reverse 5’-TTTTTCGTCA CTACCTCCCCG-3’ as internal control.
Protein carbonylation was evaluated using biotin derivatization assay as described [26]. Briefly, after treatment in P100 dishes, cells were harvested in cold PBS and cellular proteins were extracted under native condition in G-lysis buffer (Guanidine HCl 6.0 M, Tis 50 mM, pH 8.3, EDTA 3.0 mM, Triton-X100 0.5% (v/v), sodium iodoacetate 50 mM) as described [27]. Protein lysate was then incubated in the dark with 5.0 mM biotin hydrazide for 2 h at room temperature. Biotin-conjugated proteins were then reduced with 10.0 mM NaBH4 for 1 h. After removing the excessive salty chemicals with the Ultracel®−3K centrifugal filter (Merk Millipore), eluted proteins were loaded onto SDS-PAGE gel and carbonylated proteins on PVDF membrane were detected using the HRP-linked anti-Biotin antibodies.
Protein nitration was assessed using the fluorogenic tagging approach as described in our publication [28]. Briefly, after treatment, cells were harvested in cold PBS and total proteins were extracted in 1% SDS added PBS followed by sonication. After removing cell debris by centrifugation, equal amounts of soluble protein from each treatment were subjected to 4-aminomethyl-benzenesulfonate (ABS) derivatization and sodium dithionite (SDT) reduction. Fluorescence spectra were then recorded using a Shimadzu RF-5000U fluorescence spectrophotometer.
2.5. Cellular thermal stability assay (CETSA) and in-silicon docking analysis
Cell-based CETSA assay was conducted as described in our recent publication [23,29]. Briefly, after treatment, PC-3 cells were harvested in cold PBS. Aliquoted cell pallets were heated at various temperatures (37–69C with a 4C interval) for 3 min and cooled down at room temperature for 3 min. Cell pallets were then subjected to three freeze-thaw cycles. After centrifugation, soluble proteins were used in western blot assays. XDH protein band density was acquired using ImageJ software and data were plotted using GraphPad Prism software (La Jolla, CA).
The in-silicon docking analysis was conducted as described in our recent report [23]. Briefly, XDH crystal structure (PBD 2CKJ) obtained from Research Collaboratory for Structural Bioinformatics Protein Data Bank was used for docking analysis with Alternol after removing the solvent and ligand FAD. Re-docking the crystal ligands were able to recover docked conformations very close to what was reported in the crystal structure. The endogenous ligand FAD showed a better affinity score of −10.39457 than Alternol at −7.2096448, indicating Alternol docked to XDH homodimers with a moderate affinity.
2.6. Statistical analysis
Quantitative data were present as the mean ± SEM from at least three experiments. Representative images of non-quantitative data were shown from multiple experiments. Statistical analysis was conducted using ANOVA analysis followed by student t-test to compare two groups with SPSS software (Chicago, IL). P value of 0.05 or less was considered as a significant difference.
3. Results
3.1. Mitochondrial superoxide is not the main player in Alternol-induced cancer death
We recently demonstrated that the natural compound Alternol induces ROS-dependent apoptosis preferentially in cancer cells over benign cells [22]. To elucidate the major sources for Alternol-induced ROS response, we first examined mitochondrial superoxide levels because it is considered as the major ROS generating machinery [3]. Mitochondrial superoxide levels were monitored using the MitoSOX fluorescent probe as described [30]. Total cellular ROS levels were monitored at the same time as described [22]. As shown in Fig 1A, although MitoSOX-positive cells increased moderately (about 25%) after Alternol treatment compared to the solvent control, this increase was significantly lower than total ROS-positive population. A similar effect was also observed in benign BPH1 cells (Supplemental Figure). To determine the significance of mitochondrial superoxide accumulation in cell death, we tested the effect of a mitochondrial-specific anti-oxidant MitoQ [31] on Alternol-induced cell death. As shown in Fig 1B, MitoQ had no obvious protective effect on Alternol-induced cell death, while ROS scavenger N-Ac and DHLA abolished Alternol-induced ROS accumulation and cell death as reported [22]. These data suggest that mitochondrial superoxide formation is not the main player in Alternol-induced ROS-dependent cell death.
Fig 1.


Mitochondrial superoxide, NOX and NOS are not involved in Alternol-induced ROS-dependent cell death in PC-3 cells. A & C; PC-3 cells were seeded in 24-well pallets overnight and then loaded with MitoSOX (5 μM) or total ROS fluorescent probe for 30 min. After media replacement, cells were treated with Alternol plus or minus ROS scavengers (N-Ac 5 mM, DHLA 0.25 mM) or NOX inhibitors (DPI 10 μM and Apocynin 1 μM) for 4 h. Fluorescent positive cells with MitoSOX or total ROS probe were counted under fluorescent microscope as described [22].
B & D; PC-3 cells seeded in 24-well plates were treated with Alternol plus or minus N-Ac (5 mM), DHLA (0.25 mM) or MitoQ (5 μM) for 8 h. Cell death rate was determined using trypan blue exclusion assay as described [22].
E; PC-3 cells seeded in 24-well plates were loaded with total ROS fluorescent probe for 30 min and then treated with Alternol (10 μM) or Auranofin (5 μM) as indicated for 8 h. Cell populations for total ROS-positive or dying cells were determined as described above [22].
F; PC-3 cells seeded in 24-well plates were treated with Alternol (10 μM) plus or minus NOS inhibitors (L-NAME 10 μM, L-NMMA 10 μM, 1400W 1 μM) for 8 h. Cell survival rate was determined using SRB assay as described [24].
All experiments were reaped in triplicates and the data were presented as MEAN. The error bar represents the SEM of the MEAN. A statistical significance (Student t-test) was indicated with the asterisk (p < 0.01) or the hashtag (p < 0.05) compared to the solvent/DMSO control.
3.2. NOX and NOS activities are not involved in Alternol-induced ROS-dependent cancer death
To determine the possible involvement of NOX activity in Alternol-induced ROS accumulation and cell death, we tested two NOX inhibitors DPI [32] and apocynin [33]. Similar to the MitoQ, NOX inhibitors had no protective effect on Alternol-induced ROS and cell death (Fig 1C &1D). Notably, NOX inhibitor DPI induced a moderate increase in ROS levels and cell death (Fig 1C and 1D), possibly related to its off-target effect on thioredoxin reductase (TRxR) [34]. Indeed, TRxR inhibition with Auranofin induced a significant increase of ROS levels and enhanced Alternol-induced cell death (Fig 1E). However, benign BPH1 cells were also resistant to Auranofin-induced cell death (Supplemental Figure), same as the response to Alternol [22]. In addition, Alternol treatment did not induce any detectable NO signal (data not shown) and NOS inhibition with multiple specific inhibitors had no protective effect on cell survival after Alternol treatment (Fig 1F).
3.3. XO activity is involved in Alternol-induced ROS-dependent cancer death
Then, we examined if xanthine oxidase (XO) is involved in Alternol-induced oxidative stress and cell death. The levels of XO end-products, uric acid and H2O2, were utilized as the readouts. As shown in Fig 2A & 2B, Alternol treatment markedly increased the levels of uric acid and H2O2 in a time-dependent manner in PC-3 cells but not in BPH1 cells. In contrast, only a slight increase of H2O2 levels was induced by Doxorubicin that was reported to induce XO-dependent oxidative stress (Fig 2C) [35]. Consistent with the results of uric acid and H2O2 levels, cellular XO activities were significantly elevated after Alternol treatment in malignant (PC-3 and C4–2) but not benign BPH1 cells (Fig 2D). These data clearly indicated that XO is activated specifically in malignant cells after Alternol treatment.
Fig 2.

Alternol activates XDH/XO in PC-3 but not in BPH1 cells. A-D; Cells were seeded in 6-well plates and treated with Alternol (10 μM) or Doxorubicin (2.5 μM) as indicated. Cells were harvested for uric acid or XDH/XO activity assays and cell culture media were collected for H2O2 assay as described in the text.
E-F; Cells were seeded in 6-well plates and treated with Alternol (10 μM) plus or minus Febuxostat (20 μM) or Allopurinol (200 μM) for 4 h. Cells were harvested for uric acid assay and cell culture media were collected for H2O2 assay.
All data were from triplicated experiments and were presented as MEAN ± SEM. The asterisk indicates a statistical significance compared to the 0 h or the solvent/DMSO control (Student t-test, p < 0.01). The hashtag indicates a significant difference between indicated groups (ANOVA analysis, p < 0.05)
To verify the significance of XO activation in Alternol-induced ROS accumulation, XO inhibitors Allopurinol [10] and Febuxostat [11] were used together with or without Alternol. As shown in Fig 2E & 2F, Allopurinol and Febuxostat completely blocked Alternol-induced increases of uric acid and H2O2 levels, although allopurinol itself induced a moderate increase of H2O2 (Fig 2F), consistent with the notion that Allopurinol induces ROS when it initially binds to the enzyme XO as a suicide inhibitor [36]. In addition, Febuxostat also dramatically reduced Alternol-induced total ROS levels (Fig 3A & 3B) and subsequent cell death (Fig 3C). Western blot analysis revealed that XO inhibition by Febuxostat or siRNA-mediated XDH gene silencing abolished Alternol-induced apoptosis, as evidenced by reduced caspase-3 processing and PARP cleavage (Fig 3D & 3E). These data demonstrated that XO activity is enhanced in Alternol-treated malignant cells, which plays a critical role in Alternol-induced ROS accumulation and apoptotic cell death.
Fig 3.

XDH/XO inhibition blocks Alternol-induced ROS accumulation and apoptosis. A-C; PC-3 cells were seeded in 24-well plates overnight and were then loaded with total ROS fluorescent probe for 30 min. Cells were treated with Alternol (10 μM) or Febuxostat (20 μM) as indicated for 4 h and cell nuclear were stained with Hoechst 33342 (1.0 μg/ml) for 15 min before assessment for ROS-positive cell population under a fluorescent microscope. Microscopic images were taken from each treatment and the representative images were shown in panel A. Quantitative data were summarized in panel B as described [22]. Cell death rate was determined after 8 h following drug treatment by trypan blue exclusion assay as shown in panel C. A statistical significance was indicated by the asterisk compared to the solvent control or by the hashtag compared to the Alternol alone (Student t-test, p < 0.05).
D-E; PC-3 cells with or without XDH siRNA (100 nM) transfection for 2 days were treated with Alternol (10 μM) or Febuxostat (20 μM) for 12h as indicated. A scramble siRNA was used as negative control in panel E. Cells were harvested in cold PBS and protein lysates were used for Western blots with the primary antibodies as indicated on the left side of the panel. Actin blots served as protein loading control. Data represent two separate experiments.
3.4. Alternol interacts with XDH protein and increases its protein levels
To examine if Alternol interacts with XDH, CETSA assay [37] was conducted to evaluate ligand-protein engagement. As shown in Fig 4A & 4B, Alternol treatment dramatically shifted XDH melting curve, the same as Allopurinol’s effect. As expected, Allopurinol did not engage with DLAT, an Alternol target protein as described in our recent publication [23]. Detailed parameters for ligand-protein interaction were summarized in Tab 1.
Fig 4.


Alternol interacts with XDH protein. A-D; PC-3 cells treated with DMSO, Alternol (10 μM) or Allopurinol (200 μM) for 2 h were harvested in cold PBS and then subjected to CETSA ass as described [23].
E-G; in silicon-docking analysis for Alternol interaction with XDH protein. The crystal structure for XDH 2CKJ was extracted from RCSB PDB database and the docking analysis were conducted as described in the text. The image on the right side shows the interaction diagram of Alternol with the amino acid residues (Lys755 and Arg787) on XDH protein. The green arrows indicate sidechain acceptor; The blue arrows indicate backbone donor. The images on the left side show the cartoon view of the interaction site.
Tab 1.
CETSA data Summary
| Treatment | Tm50 | ΔTm50 | R2 value | |
|---|---|---|---|---|
| PC-3 XDH | DMSO | 52.2 | 0.9908 | |
| Alternol | 46.15 | −6.05 | 0.9913 | |
| Allopurinol | 44.39 | −7.81 | 0.9917 | |
| PC-3 DLAT | DMSO | 56.21 | 0.9908 | |
| Allopurinol | 53.08 | −1.47 | 0.9919 | |
| C4–2 XDH | DMSO | 52.13 | 0.988 | |
| Alternol | 47.43 | −4.7 | 0.9673 | |
| Allopurinol | 48.41 | −3.72 | 0.9901 | |
| C4–2 DLAT | DMSO | 54.24 | 0.9604 | |
| Allopurinol | 53.91 | −0.33 | 0.9921 |
In addition, an in-silicon docking analysis was performed to understand how Alternol interacts with XDH. The results showed that Alternol binds to XDH protein with two amino acid residues K755 and R787 within its catalytic molybdenum binding domain (Fig 4E–4G). This interaction did not interfere XDH binding with its co-factor FAD (Fig 4E). The docking affinity is −7.2096448 for Alternol and −10.39457 for FAD, indicating that Alternol-XDH interaction is less affinitive compared to its natural co-factor FAD.
3.5. Alternol enhances XDH protein expression and protein oxidative damage
To further investigate the nature of Alternol-induced XDH/XO activation, we first assessed XDH gene expression in patient specimens and several commonly used prostate cancer cell lines. Consistent with previous report [16], XDH gene expression in patient specimens was significantly lower than that in benign counterparts (Fig 5, left panel), although XDH gene expression varied among different cell lines.
Fig 5.

XDH gene expression is down-regulated in prostate cancer specimens. Quantitative RT-PCR was conducted to assess XDH gene expression in prostate cancer specimens (n = 30) and their matched benign compartments (left panel) or prostate cell lines (right panel) using the cDNA samples generated from total RNAs used in our previous publication [25]. PCR counts from XDH gene were normalized with 18S rRNA levels in each sample. Cell line data were shown as MEAN from triplicated reactions and the error bar represents the SEM of the MEAN. The asterisk indicates a statistical significance compared to the benign group (Student t-test, p < 0.05).
We then examined the alteration of XDH protein expression after Alternol treatment. Human XDH/XO exists as a homodimer of ~300 KD and appears as four fragments with variable molecular weights between of 54–157 KD on reducing SDS-PAGE gel [38]. As shown in Fig 6A, XDH/XO protein mainly appeared at a molecular weight around 125 kD as detected with a c-terminal specific antibody in prostate-derived cell lines. Interestingly, this band exerted as a doublet only in malignant cells, indicating a post-translational modification in cancers. Alternol treatment dramatically increased XDH/XO protein levels in malignant PC-3 and C4–2 cells in time- and dose-dependent manners (Fig 6B–6D).
Fig 6.


Alternol increases XDH protein expression and induces cellular protein damages. A; Exponentially grown cells from each line as indicated were harvested for whole cell lysis. Equal amount of proteins was used in anti-XDH western blot assay. Actin blot served as protein loading control.
B-D; After treatment with Alternol or the solvent as indicated or at 10 μM for 4h, cells were harvested in cold PBS and cellular proteins were subjected to anti-XDH western blot assay. Actin blot served as protein loading control.
E-G; After treatment with Alternol (10 μM), N-Ac (5 μM) or Febuxostat (20 μM) as indicated, cells were harvested in cold PBS and cellular proteins were extracted in G-lysis buffer as described in the text. Biotin derivatized proteins were separated on 15% SDS-PAGE gel and visualized using HRP-linked Biotin antibodies.
H. After Alternol treatment as indicated, cells were harvested in cold PBS and subjected to 3-NT labeling and measurement as described [28]. Quantitative data were summarized from three independent measurements. The asterisk indicates a statistical significance compared to the DMSO control (Student t-test, p <0.01).
Since Alternol-induced oxidative stress is a key player in cancer cell death, we analyzed protein oxidative damages via assessing protein carbonylation and nitration of amino acid residues [39–41]. The results revealed that Alternol treatment induced a dramatic increase of protein carbonylation in PC-3 (Fig 6E) and C4–2 cells (Fig 6F). On the other hand, only a slight increase was observed in BPH1 cells (Fig 6G). Consistent with the data shown before, N-Ac and Febuxostat significantly eliminated Alternol-induced protein carbonylation.
Protein 3-nitrotyrosine (3-NT) contents was assessed with the fluorogenic tagging approach using 4-aminomethyl-benzenesulfonate (ABS) derivatization following reduction with sodium dithionite (SDT) as described in our report [28]. The results showed a significant increase of 3-NT contents in PC-3 cells but not in BPH1 cells after Alternol treatment (Fig 6H). These data indicate that Alternol-induced oxidative induced a significant protein damage that is associated with cancer cell death.
3.6. Benign cells possess a strong anti-oxidant capacity over malignant cells
Lastly, we examined if Alternol alters the activities of cellular anti-oxidant mechanisms, including SOD and catalase activities, as well as the GSH/GSSH ratio [4]. Compared to PC-3 cells, BPH1 cells had a significant increase in cellular SOD activity after Alternol treatment, although its basal SOD activity is lower than PC-3 cells (Fig 7A). Interestingly, BPH1 cells showed a higher basal level of catalase activity than PC-3 cells, and both cell lines had a significant increase after Alternol treatment (Fig 7B). In addition, the basal level of GSH/GSSH ratio was significantly higher in BPH1 cells than that in PC-3 cells (Fig 7C), indicating a greater reducing capacity in benign cells. A rapid decrease in GSH/GSSH ratio was observed at 2 h after Alternol treatment in PC-3 cells but not in BPH1 cells. These data indicate that benign cells possess a stronger reducing equivalent or capacity compared to malignant cells either in the basal condition or after Alternol treatment, possibly responsible for Alternol-induced cancer cell-specific oxidative stress and apoptotic cell death.
Fig 7.

Alternol enhances anti-oxidant enzymatic activities in BPH1 cells. A-B; Cells were seeded in 6-well plates and treated with DMSO or Alternol (10 μM) for 4 h. Cellular proteins were extracted for SDO or catalase activity with the colorimetric assay kits from BioVision.
C; Cells were left untreated or treated with Alternol (10 μM) for 2–4 h and cellular contents of GSH and GSSG were assessed with the assay kit from Enzo Life as described in the text.
Data are presented as the MEAN from three separated experiments and the error bar indicates the SEM of the MEAN. The statistical significance (Student t-test, p <0.05) was indicated with the asterisk compared to the DMSO control or the hashtag compared to BPH1 cells.
4. Discussion
We recently demonstrated Alternol-induced strong oxidative stress and apoptosis preferentially in cancer cells over benign cells [22], which is supported by other report [42]. In present study, we determined that XO-derived ROS accumulation is the major mechanism of oxidative stress induced by Alternol. We also defined that Alternol interacts with XDH/XO protein on its catalytic domain via two amino acid residues K755 and R787. Alternol-induced XO-dependent oxidative stress resulted in a profound protein damages in carbonylation and nitration. Benign cells exerted a stronger anti-oxidant capacity in SOD and catalase activities, which might be responsible for Alternol-mediated killing preferentially in cancer cells but not in benign cells.
XDH is a metabolic enzyme for purine catalysis and xenobiotic substrate oxidation while generating ROS species [12,15,16]. Its activation has been implicated in ROS-dependent tissue damage under ischemia-reperfusion or hypoxia conditions [43,44]. So far, multiple mechanisms have been reported in regulating XDH activity at the transcriptional and post-transcriptional levels [45]. Besides proteolytic and cystine disulfide modifications [9], XDH/XO protein phosphorylation was also reported previously [46]. Interestingly, our XDH protein analysis by Western blot found a doublet band appearance after Alternol treatment, indicating a potential phosphorylation event on XDH protein, although a deep analysis is needed to confirm this hypothesis. In addition, cellular iron is also playing a role in regulation of XDH gene expression and enzymatic activity [47]. However, addition of an ion chelator Desferoxamine had not protective but enhancing effect on Alternol-induced ROS accumulation and cell death (data not shown).
Previous reports showed that structurally different substrates modulate XDH/XO enzymatic activity towards to other substrates via cooperative interaction between two subunits in the homodimer complex [48,49]. In our study, we found that Alternol interacts with XDH protein within the catalytic domain at K755/R787 residues, which had not interference with its FAD binding. Since XDH/XO executes its substrate binding and catalysis on residues of E803/R881/E1262 [50], we postulate that by binding to XDH/XO catalytic domain at K755/R787 Alternol might enhance its efficiency of catalytic activity on purine substrates but further analysis is needed to understand this mechanism.
In mammalian cells, redox homeostasis is maintained at multiple levels between pro-oxidant and anti-oxidant factors. Although excessive ROS accumulation has been detected in human cancer cells, high levels of anti-oxidants also exist [1]. Consistent with this notion, we found a higher level of SOD activity in malignant cells than that in benign cells. However, benign cells showed a strong spike on SOD and catalase activities compared to malignant cells, indicating a better compensation of anti-oxidant capacity in benign cells over malignant cells. This unique property supports the conceptual basics for developing ROS-inducing anti-cancer agents like Alternol [7] and provides a mechanistic insight for Alternol-induced cancer cell-specific killing.
Similar as reported from other cancers [16], we also found a significantly reduced expression of XDH gene in prostate cancers. In addition, by re-analyzing the cDNA microarray data on the OncoMine™ database, a significant reduction (mostly > 2.0-fold) of XDH mRNA expression was found in multiple types of human cancers such as colon, liver, breast, leukemia, urinary bladder and prostate compared to normal tissues (Supplemental data table).
For the enzymatic activity, we found a comparable level among benign and malignant cells, which is supported by a previous report showing no significant difference in XDH/XO activity between malignant and its benign counterpart tissues [51]. However, Alternol treatment significantly enhanced XDH/XO activity in malignant cells but not in benign cells. The mechanism behind this difference is under further investigation by our group. Interestingly, in non-small cell lung cancers, a better survival rate was achieved in patients with higher XDH expression after chemotherapy [52], indicating a potential role of XDH activation in chemo-drug induced anti-cancer effect. It is postulated that cancer cell-specific activation of XDH/XO enzyme might serve as a ground-breaking approach in cancer therapy. As the authors are aware, Alternol is the first compound that activates XDH/XO enzyme specifically in cancer cells, despite a retracted report showing berberine-induced XDH/XO activation in prostate cancer cells (PMID18275980).
5. Conclusion
In this study, we demonstrated XDH/XO activation in cancer cells after Alternol treatment, leading to ROS accumulation and apoptotic cell death. Alternol interacts with XDH protein on its catalytic domain and enhances its purine catalytic activity only in prostate cancer cells. It is concluded that Alternol acts as a novel and unique XDH/XO activator in malignant cells to induce oxidative stress and apoptosis.
Supplementary Material
Highlights.
Alternol induces ROS accumulation and apoptosis preferentially in malignant cells;
Alternol-induced ROS stress is not due to mitochondrial superoxide accumulation;
NOS or NOX inhibitors did not block Alternol-induced ROS stress;
XDH/XO inhibition suppresses Alternol-induced ROS accumulation and apoptosis;
Alternol interacts with XDH on its catalytic domain
Alternol activates XDH oxidase activity in malignant cells;
Alternol treatment promotes XDH protein expression and modification.
XDH gene expression is reduced in malignant tissues.
Acknowledgements
We are very grateful for the generous gift of Alternol reagent from Dr Jiepeng Chen at Sungen Biosciences (Shantou, China). The in-silicon docking work was conducted by Dr Aaron Smalter, the former Director for the Molecular Graphics & Modeling Lab in the University of Kansas Molecular Structures Group.
Abbreviation:
- ADHP
10-acetyl-3,7-dihydroxyphenoxazine
- ABS
aminomethyl-benzenesulfonate derivatization
- CETSA
cellular thermal stability assay
- DHLA
dihydrolipoic acid
- DPI
Diphenyleneiodonium
- LOO
lipid peroxyl radical
- NaBH4
sodium borohydride
- N-Ac
n-acetylcysteine
- NAD
nicotinamide adenine
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- OH
hydroxyl radical
- ONOO
peroxynitrite
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SDT
sodium dithionite
- SEM
standard error of mean
- SOD
superoxide dismutase
- SRB
sulforhodamine B
- XDH
xanthine dehydrogenase
- XO
xanthine oxidase
- XOR
xanthine oxidoreductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None
Appendix A. Supplemental material
References
- [1].Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol 2018;80:50–64. [DOI] [PubMed] [Google Scholar]
- [2].Mahmood DF, Abderrazak A, El Hadri K, Simmet T, Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal 2013;19(11):1266–303. [DOI] [PubMed] [Google Scholar]
- [3].Mailloux RJ. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol 2015;4:381–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. Trends Mol Med 2017;23(5):411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Assi M The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol 2017;313(6):R646–R653. [DOI] [PubMed] [Google Scholar]
- [6].Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013;12(12):931–47. [DOI] [PubMed] [Google Scholar]
- [7].Raza MH, Siraj S, Arshad A, Waheed U, Aldakheel F, Alduraywish S, Arshad M. ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol 2017;143(9):1789–1809. [DOI] [PubMed] [Google Scholar]
- [8].Xu P, Huecksteadt TP, Hoidal JR. Molecular cloning and characterization of the human xanthine dehydrogenase gene (XDH). Genomics 1996;34(2):173–80. [DOI] [PubMed] [Google Scholar]
- [9].Okamoto K, Kusano T, Nishino T. Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Curr Pharm Des 2013;19(14):2606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Delgado JN, Cosgrove FP, Isaacson EI. Allopurinol: xanthine oxidase inhibitor. Tex Med 1966;62(1):100–1. [PubMed] [Google Scholar]
- [11].Osada Y, Tsuchimoto M, Fukushima H, Takahashi K, Kondo S, Hasegawa M, Komoriya K. Hypouricemic effect of the novel xanthine oxidase inhibitor, TEI-6720, in rodents. Eur J Pharmacol 1993;241(2–3):183–8. [DOI] [PubMed] [Google Scholar]
- [12].Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid Med Cell Longev 2016;2016:3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Battelli MG, Bortolotti M, Polito L, Bolognesi A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol 2019;21:101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nishino T, Okamoto K, Eger BT, Pai EF, Nishino T. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 2008;275(13):3278–89. [DOI] [PubMed] [Google Scholar]
- [15].Cantu-Medellin N, Kelley EE. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol 2013;1:353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med 2016;5(3):546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Linder N, Butzow R, Lassus H, Lundin M, Lundin J. Decreased xanthine oxidoreductase (XOR) is associated with a worse prognosis in patients with serous ovarian carcinoma. Gynecol Oncol 2012;124(2):311–8. [DOI] [PubMed] [Google Scholar]
- [18].Linder N, Martelin E, Lundin M, Louhimo J, Nordling S, Haglund C, Lundin J. Xanthine oxidoreductase - clinical significance in colorectal cancer and in vitro expression of the protein in human colon cancer cells. Eur J Cancer 2009;45(4):648–55. [DOI] [PubMed] [Google Scholar]
- [19].Linder N, Haglund C, Lundin M, Nordling S, Ristimaki A, Kokkola A, Mrena J, Wiksten JP, Lundin J. Decreased xanthine oxidoreductase is a predictor of poor prognosis in early-stage gastric cancer. J Clin Pathol 2006;59(9):965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Linder N, Lundin J, Isola J, Lundin M, Raivio KO, Joensuu H. Down-regulated xanthine oxidoreductase is a feature of aggressive breast cancer. Clin Cancer Res 2005;11(12):4372–81. [DOI] [PubMed] [Google Scholar]
- [21].Liu ZZ, Chen JP, Zhao SL, Li CL. [Apoptosis-inducing effect of alternol on mouse lymphocyte leukemia cells and its mechanism]. Yao Xue Xue Bao 2007;42(12):1259–65. [PubMed] [Google Scholar]
- [22].Tang Y, Chen R, Huang Y, Li G, Huang Y, Chen J, Duan L, Zhu BT, Thrasher JB, Zhang X and others. Natural compound Alternol induces oxidative stress-dependent apoptotic cell death preferentially in prostate cancer cells. Mol Cancer Ther 2014;13(6):1526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li C, He C, Xu Y, Xu H, Tang Y, Chavan H, Duan S, Artigues A, Forrest ML, Krishnamurthy P and others. Alternol eliminates excessive ATP production by disturbing Krebs cycle in prostate cancer. Prostate 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1(3):1112–6. [DOI] [PubMed] [Google Scholar]
- [25].Chen R, Zeng X, Zhang R, Huang J, Kuang X, Yang J, Liu J, Tawfik O, Thrasher JB, Li B. Cav1.3 channel alpha1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol Oncol 2014;32(5):524–36. [DOI] [PubMed] [Google Scholar]
- [26].Colombo G, Clerici M, Garavaglia ME, Giustarini D, Rossi R, Milzani A, Dalle-Donne I. A step-by-step protocol for assaying protein carbonylation in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci 2016;1019:178–90. [DOI] [PubMed] [Google Scholar]
- [27].Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem 2003;278(35):33408–15. [DOI] [PubMed] [Google Scholar]
- [28].Sharov VS, Dremina ES, Galeva NA, Gerstenecker GS, Li X, Dobrowsky RT, Stobaugh JF, Schoneich C. Fluorogenic Tagging of Peptide and Protein 3-Nitrotyrosine with 4-(Aminomethyl)-benzenesulfonic Acid for Quantitative Analysis of Protein Tyrosine Nitration. Chromatographia 2010;71(1–2):37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].He C, Duan S, Dong L, Wang Y, Hu Q, Liu C, Forrest ML, Holzbeierlein JM, Han S, Li B. Characterization of a novel p110beta-specific inhibitor BL140 that overcomes MDV3100-resistance in castration-resistant prostate cancer cells. Prostate 2017;77(11):1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2007;2(9):2295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 2001;276(7):4588–96. [DOI] [PubMed] [Google Scholar]
- [32].Ellis JA, Mayer SJ, Jones OT. The effect of the NADPH oxidase inhibitor diphenyleneiodonium on aerobic and anaerobic microbicidal activities of human neutrophils. Biochem J 1988;251(3):887–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 1994;11(1):95–102. [DOI] [PubMed] [Google Scholar]
- [34].Leitsch D, Kolarich D, Duchene M. The flavin inhibitor diphenyleneiodonium renders Trichomonas vaginalis resistant to metronidazole, inhibits thioredoxin reductase and flavin reductase, and shuts off hydrogenosomal enzymatic pathways. Mol Biochem Parasitol 2010;171(1):17–24. [DOI] [PubMed] [Google Scholar]
- [35].Yee SB, Pritsos CA. Comparison of oxygen radical generation from the reductive activation of doxorubicin, streptonigrin, and menadione by xanthine oxidase and xanthine dehydrogenase. Arch Biochem Biophys 1997;347(2):235–41. [DOI] [PubMed] [Google Scholar]
- [36].Massey V, Komai H, Palmer G, Elion GB. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J Biol Chem 1970;245(11):2837–44. [PubMed] [Google Scholar]
- [37].Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013;341(6141):84–7. [DOI] [PubMed] [Google Scholar]
- [38].Sarnesto A, Linder N, Raivio KO. Organ distribution and molecular forms of human xanthine dehydrogenase/xanthine oxidase protein. Lab Invest 1996;74(1):48–56. [PubMed] [Google Scholar]
- [39].Moldogazieva NT, Lutsenko SV, Terentiev AA. Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res 2018;78(21):6040–6047. [DOI] [PubMed] [Google Scholar]
- [40].Galligan JJ, Smathers RL, Fritz KS, Epperson LE, Hunter LE, Petersen DR. Protein carbonylation in a murine model for early alcoholic liver disease. Chem Res Toxicol 2012;25(5):1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Franco MC, Estevez AG. Tyrosine nitration as mediator of cell death. Cell Mol Life Sci 2014;71(20):3939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zuo D, Zhou Z, Wang H, Zhang T, Zang J, Yin F, Sun W, Chen J, Duan L, Xu J and others. Alternol, a natural compound, exerts an anti-tumour effect on osteosarcoma by modulating of STAT3 and ROS/MAPK signalling pathways. J Cell Mol Med 2017;21(2):208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Anderson RF, Patel KB, Reghebi K, Hill SA. Conversion of xanthine dehydrogenase to xanthine oxidase as a possible marker for hypoxia in tumours and normal tissues. Br J Cancer 1989;60(2):193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Portugal-Cohen M, Kohen R. Exposure of human keratinocytes to ischemia, hyperglycemia and their combination induces oxidative stress via the enzymes inducible nitric oxide synthase and xanthine oxidase. J Dermatol Sci 2009;55(2):82–90. [DOI] [PubMed] [Google Scholar]
- [45].Battelli MG, Bolognesi A, Polito L. Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta 2014;1842(9):1502–17. [DOI] [PubMed] [Google Scholar]
- [46].Kayyali US, Donaldson C, Huang H, Abdelnour R, Hassoun PM. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J Biol Chem 2001;276(17):14359–65. [DOI] [PubMed] [Google Scholar]
- [47].Martelin E, Lapatto R, Raivio KO. Regulation of xanthine oxidoreductase by intracellular iron. Am J Physiol Cell Physiol 2002;283(6):C1722–8. [DOI] [PubMed] [Google Scholar]
- [48].Tai LA, Hwang KC. Regulation of xanthine oxidase activity by substrates at active sites via cooperative interactions between catalytic subunits: implication to drug pharmacokinetics. Curr Med Chem 2011;18(1):69–78. [DOI] [PubMed] [Google Scholar]
- [49].Tai LA, Hwang KC. Cooperative catalysis in the homodimer subunits of xanthine oxidase. Biochemistry 2004;43(16):4869–76. [DOI] [PubMed] [Google Scholar]
- [50].Yamaguchi Y, Matsumura T, Ichida K, Okamoto K, Nishino T. Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate. J Biochem 2007;141(4):513–24. [DOI] [PubMed] [Google Scholar]
- [51].Biri H, Ozturk HS, Kacmaz M, Karaca K, Tokucoglu H, Durak I. Activities of DNA turnover and free radical metabolizing enzymes in cancerous human prostate tissue. Cancer Invest 1999;17(5):314–9. [DOI] [PubMed] [Google Scholar]
- [52].Kim AW, Batus M, Myint R, Fidler MJ, Basu S, Bonomi P, Faber LP, Wightman SC, Warren WH, McIntire M and others. Prognostic value of xanthine oxidoreductase expression in patients with non-small cell lung cancer. Lung Cancer 2011;71(2):186–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.