

Abstract
Background
We asked whether, after excluding familial hypercholesterolemia, individuals with high low‐density lipoprotein cholesterol (LDL‐C) or triacylglyceride levels and a family history of the same hyperlipidemia have greater coronary artery disease risk or different lipidomic profiles compared with population‐based hyperlipidemias.
Methods and Results
We determined incident coronary artery disease risk for 755 members of 66 hyperlipidemic families (≥2 first‐degree relatives with similar hyperlipidemia) and 19 644 Finnish FINRISK population study participants. We quantified 151 circulating lipid species from 550 members of 73 hyperlipidemic families and 897 FINRISK participants using mass spectrometric shotgun lipidomics. Familial hypercholesterolemia was excluded using functional LDL receptor testing and genotyping. Hyperlipidemias (LDL‐C or triacylglycerides >90th population percentile) associated with increased coronary artery disease risk in meta‐analysis of the hyperlipidemic families and the population cohort (high LDL‐C: hazard ratio, 1.74 [95% CI, 1.48–2.04]; high triacylglycerides: hazard ratio, 1.38 [95% CI, 1.09–1.74]). Risk estimates were similar in the family and population cohorts also after adjusting for lipid‐lowering medication. In lipidomic profiling, high LDL‐C associated with 108 lipid species, and high triacylglycerides associated with 131 lipid species in either cohort (at 5% false discovery rate; P‐value range 0.038–2.3×10−56). Lipidomic profiles were highly similar for hyperlipidemic individuals in the families and the population (LDL‐C: r=0.80; triacylglycerides: r=0.96; no lipid species deviated between the cohorts).
Conclusions
Hyperlipidemias with family history conferred similar coronary artery disease risk as population‐based hyperlipidemias. We identified distinct lipidomic profiles associated with high LDL‐C and triacylglycerides. Lipidomic profiles were similar between hyperlipidemias with family history and population‐ascertained hyperlipidemias, providing evidence of similar and overlapping underlying mechanisms.
Keywords: coronary artery disease, family study, high‐risk populations, hypercholesterolemia, hypertriglyceridemia, lipids and lipoproteins
Subject Categories: Lipids and Cholesterol, Epidemiology, Cardiovascular Disease, Coronary Artery Disease
Clinical Perspective
What Is New?
Beyond familial hypercholesterolemia, the impact of hyperlipidemic family history on coronary artery disease risk is debated.
Coronary artery disease risk was comparable in our hyperlipidemic subjects (low‐density lipoprotein cholesterol or triacylglycerides >90th population percentile) with family history and subjects with population‐ascertained hyperlipidemias.
The lipidomic profiles of such hyperlipidemias were independent of family history, providing evidence for similar and/or overlapping metabolic pathways.
What Are the Clinical Implications?
Our results do not support different screening for those with a family history of hyperlipidemia and sporadically discovered hyperlipidemic cases.
Introduction
High levels of low‐density lipoprotein cholesterol (LDL‐C) and triacylglycerides have been identified as causal risk factors for atherosclerotic cardiovascular disease (ASCVD).1, 2 These hyperlipidemias may arise through lifestyle factors, but they are also highly heritable.3, 4, 5, 6 An estimated half of patients with premature coronary artery disease (CAD) have dyslipidemia with a family history of dyslipidemia, most of which are characterized by increases in LDL‐C and/or triacylglycerides.7
Whether dyslipidemias with family history should be diagnosed and managed differently from hyperlipidemias observed in randomly ascertained individuals in the general population is uncertain. Clinical guidelines emphasize their identification but, with the exception of familial hypercholesterolemia (FH), refrain from strong management recommendations.8, 9 The monogenic FH patients with rare high‐impact LDL‐C–elevating variants have a higher risk of developing CAD than noncarriers with similar lipid levels.10 This is potentially related to lifelong exposure to high LDL‐C levels and suggests that these individuals may benefit from earlier or more aggressive LDL‐C–lowering therapy. In contrast with FH, many other hyperlipidemias with family history appear genetically similar to population‐ascertained hyperlipidemias.11, 12, 13 Whether such hyperlipidemias with family history also confer additional elevation in ASCVD risk is not known.
Herein, we assess incident ASCVD risk associated with familial aggregation of high LDL‐C and triacylglycerides, excluding individuals with FH. We also ask whether their circulating lipid phenotypes are similar compared with population‐ascertained hyperlipidemias. Recent technological advancements have allowed replicable and simultaneous quantification of hundreds of lipid species, the main constituents of LDL and triacylglyceride‐rich lipoproteins, through lipidomic profiling.14, 15 We test whether detailed phenotypic differences in lipidomic profiles, which might reflect different pathophysiological features and ASCVD susceptibility, exist between hyperlipidemias with family history and population‐ascertained hyperlipidemias. Using a direct infusion platform that combines absolute quantification with high throughput, we were able to overcome problems that have hampered many previous studies.16
In this study, we first estimated the CAD risk associated with high LDL‐C or triacylglyceride levels with family history and population‐ascertained hyperlipidemias with similarly high LDL‐C or triacylglycerides. Second, we characterized the lipidomic profiles associated with elevated plasma levels of LDL‐C and triacylglycerides. Finally, we compared the lipidomic profiles of hyperlipidemias with family history and population‐ascertained hyperlipidemias to assess their potential differences.
Materials and Methods
Subjects and Clinical Ascertainment
The Finnish hyperlipidemia families included in this cohort study (74 families, n=1445 individuals with LDL‐C and triacylglyceride measures) were identified as part of the EUFAM (European Multicenter Study on Familial Dyslipidemias in Patients With Premature Coronary Heart Disease) project. Initial recruitment aimed to identify families with familial combined hyperlipidemia (at least 2 family members with total cholesterol and/or triacylglycerides ≥90th population percentile) or families with aggregation of low high‐density lipoprotein cholesterol. Classic FH was excluded on the basis of an in‐house functional LDL receptor test for the probands and later genotyping of selected Finnish FH mutations in other family members with high LDL‐C; further recruitment was not pursued in putative FH pedigrees.17 For the present study, designation of “high LDL‐C with family history” or “high triacylglycerides with family history” was made if at least 2 first‐degree relatives had LDL‐C or triacylglyceride levels, respectively, that were >90th age‐ and sex‐specific Finnish 1997 population percentiles (Table S1) without being affected by diabetes mellitus or other relevant comorbidities (Figure S1). More detailed information is given in Data S1.
Individuals from the Finnish National FINRISK study were used as a Finnish population‐based comparison group. A total of 19 644 individuals from the FINRISK study 1992 to 2002 cohorts and 755 individuals from EUFAM families were linked with the national hospital discharge and causes‐of‐death registries. Clinical incident CAD event end points were defined as either myocardial infarction or coronary revascularization (coronary angioplasty or coronary artery bypass grafting). CVD was defined as CAD or stroke, excluding subarachnoid hemorrhage. Mean (range) follow‐up time from baseline to CAD end point, death, or end of registry follow‐up was 16.1 (0.1–20.1) years in EUFAM and 12.6 (0.02–19.0) years in the FINRISK study. More detailed information is given in Data S1.
Written informed consent was obtained from all participants, except the 1992 FINRISK study survey, for which verbal informed consent was obtained, as required by legislation and ethics committees at the time. All samples were collected in accordance with the Declaration of Helsinki, and study protocols were approved by the ethics committees of the participating centers (The Hospital District of Helsinki and Uusimaa Coordinating Ethics committees, approval No. 184/13/03/00/12). Because of the consent given by the study participants, the data cannot be made publicly available. The data are available through the Institute for Molecular Medicine Finland Data Access Committee for authorized researchers who have an institutional review board/ethics approval and an institutionally approved study plan. For more details, please contact the Institute for Molecular Medicine Finland Data Access Committee (fimm-dac@helsinki.fi).
Lipidomics Measurements
Lipidomic profiling of circulating lipid species was performed for 550 EUFAM family members (all members with available plasma samples) and for 897 individuals from the FINRISK 2012 study cohort, after excluding individuals with predefined comorbidities (Data S1) and individuals known to use lipid‐lowering medication or sex hormones at the time of the measurements. Mass spectrometry–based lipid analysis was performed at Lipotype GmbH (Dresden, Germany), as described.14 Plasma and serum lipids were extracted with methyl tert‐butyl ether/methanol (7:2, v/v).18 Samples were analyzed by direct infusion in a QExactive mass spectrometer (Thermo Scientific) equipped with a TriVersa NanoMate ion source (Advion Biosciences). Samples were analyzed in both positive and negative ion modes in a single acquisition.
Data were analyzed with in‐house–developed lipid identification software based on LipidXplorer.19, 20 Reproducibility was assessed by the inclusion of reference plasma samples. The median coefficient of variation was <10% across all batches. A total of 151 species were detected in ≥80% of both EUFAM and FINRISK study samples and were included in subsequent analyses. Right‐skewed lipidomics measures were natural logarithm transformed before normalization. More detailed information is given in Data S1.
Statistical Analyses
To assess the risk of incident CAD associated with the hyperlipidemias, we used Cox proportional hazards models using age as the time scale, stratified by sex and clustered by family, to estimate hazard ratios (HRs) for incident CAD (or CVD) events, excluding individuals with prevalent CAD (or CVD). Additional models were also adjusted by lipid‐lowering medication and smoking. The statistical significance of intercohort differences in HRs was estimated on the basis of an interaction term between hyperlipidemia status and cohort designation.
We used linear mixed models to estimate the association between lipidomic measurements and predictors of interest (hyperlipidemia status or continuous lipid measurement), as implemented in MMM (version 1.01).21 Age, age2, and sex were used as additional fixed‐effect covariates.
To account for relatedness among individuals, an empirical genetic relationship matrix was included as the covariance structure of a random effect. Statistical significance was evaluated using the Benjamini‐Hochberg method at the 5% level to account for multiple comparisons similarly to recent lipidomics studies of CVDs.22, 23 R (version 3.4.3) was used for data transformations and other analyses.24 Detailed information is given in Data S1.
Results
Clinical Characteristics and CAD Risk of Individuals With High Levels of LDL‐C or Triacylglycerides
We first assessed the risk of developing CAD associated with high levels of LDL‐C or triacylglycerides in individuals from the Finnish FINRISK study population survey and in hyperlipidemic families ascertained as part of the EUFAM (Figure 1; Table S2; Figure S1A). Individuals with LDL‐C >90th percentile had an increased risk of incident CAD in the FINRISK study population surveys (n=19 644 individuals) compared with other individuals (HR, 1.74; 95% CI, 1.48–2.05) (Figure 1). The members of hyperlipidemic families with high LDL‐C had a similar HR for CAD compared with their relatives without high LDL‐C in 47 “high LDL‐C” families (n=625 individuals) (HR, 1.71; 95% CI, 0.94–3.10). The HRs did not differ between the cohorts (P=0.84). The mean age at incident CAD diagnosis was similar among individuals with high LDL‐C in the hyperlipidemic families (62.8 years) and in the population cohort (63.5 years). We also observed increased CAD risk in individuals with high triacylglycerides in the population (HR, 1.38; 95% CI, 1.09–1.75) and a similar HR in 35 “high triacylglyceride” families (n=371 individuals) (HR, 1.35; 95% CI, 0.52–3.51). The HRs did not differ between the cohorts (P=0.82). The results remained similar after adjusting for lipid‐lowering medication use and smoking (Figure S2 and Table S3) and body mass index (Table S3). Furthermore, we found no differences between the cohorts in the risk of incident CVD (P=0.42–0.98; Figure S3A and S3B; Table S3). Meta‐analyses of HRs closely approximated estimates derived from the population cohort.
Figure 1.
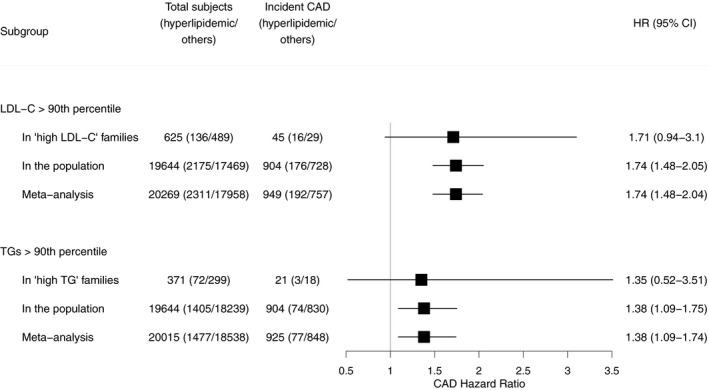
Risk of incident coronary artery disease (CAD) in hyperlipidemias with family history and population‐ascertained hyperlipidemias. To assess the risk of incident CAD associated with the hyperlipidemia types, we used Cox proportional hazards models using age as the time scale, stratified by sex and clustered by family, to estimate hazard ratios (HRs) for incident CAD events, excluding individuals with prevalent CAD. Further details on the participants are presented in Table S2. LDL‐C indicates low‐density lipoprotein cholesterol; TG, triacylglycerides.
We then characterized the detailed lipidomic profiles of 550 individuals from 73 hyperlipidemic families and 897 individuals from the FINRISK population study (Methods; Tables S4, S5 and S6). These included 105 individuals (23%) of 463 family members in 53 high LDL‐C families who had LDL‐C levels >90th percentile (mean±SD, 5.2±0.8 mmol/L) and 64 individuals (22%) of 287 family members in 39 high triacylglyceride families who had triacylglycerides >90th percentile (mean±interquartile range, 3.6±1.8 mmol/L). Using similar cutoffs in the population, 56 individuals (6%) and 65 individuals (7%) of 897 were affected by high LDL‐C levels (mean±SD, 5.3±1.1 mmol/L) and high triacylglycerides (mean±interquartile range, 3.5±1.9 mmol/L), respectively. Both high LDL‐C and triacylglyceride levels were observed in 31 individuals (6%) in the family cohort and 9 individuals (1%) in the population cohort.
High LDL‐C and Lipidomic Profiles
To characterize the lipidomic profiles associated with elevated values of LDL‐C, we compared individuals with high LDL‐C levels with those without. In the hyperlipidemic families, individuals with a high LDL‐C had significantly elevated levels of 99 lipid species spread out across most of the studied lipid classes. Reduced levels among the high LDL‐C individuals were observed for 3 lysophospatidylcholine, 2 lysophosphatidylethanolamine, and 1 phosphatidylcholine‐ether (PCO) species (Figure 2A; Table S7). Similar trends were seen in the population cohort, in which the levels of 51 lipid species were elevated among high LDL‐C individuals (Figure 2B; Table S7). The effect estimates correlated strongly across all lipid species between the hyperlipidemic families and the population cohorts (Pearson's r=0.80; Figure 3). Furthermore, we observed no significant differences in the effect estimates between the cohorts at the 5% false discovery rate (FDR).
Figure 2.
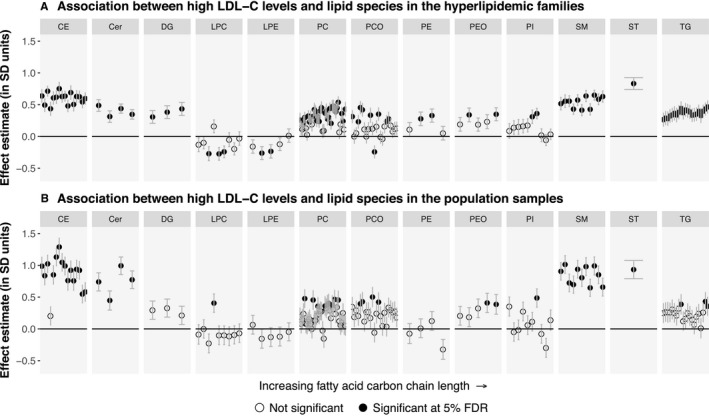
Associations between high low‐density lipoprotein cholesterol (LDL‐C) status and the levels of 151 lipid species. A, Individuals affected by high LDL‐C levels (n=105) were compared with their unaffected relatives (n=358) in the 53 “high LDL‐C” families. B, Individuals affected by high LDL‐C (n=56) were compared with other individuals (n=841) in the FINRISK study population cohort. The association of high LDL‐C status with the lipid species was estimated using linear mixed models with age, age2, and sex as the other fixed‐effect covariates. Statistical significance was evaluated using the Benjamini‐Hochberg method at a 5% false discovery rate (FDR). The ordering of the lipid species within each class is the same as in Table S7. Cer indicates ceramide; DG, diacylglyceride; FDR, false discovery rate; LDL‐C, low‐density lipoprotein cholesterol; LPA, lysophosphatic acid; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PC, phosphatidylcholine; PCO, phosphatidylcholine‐ether; PE, phosphatidylethanolamine; PEO, phosphatidylethanolamine‐ether; PI, phosphatidylinositol; CE, cholesteryl ester; SM, sphingomyelin; ST, sterol; TG, triacylglyceride.
Figure 3.
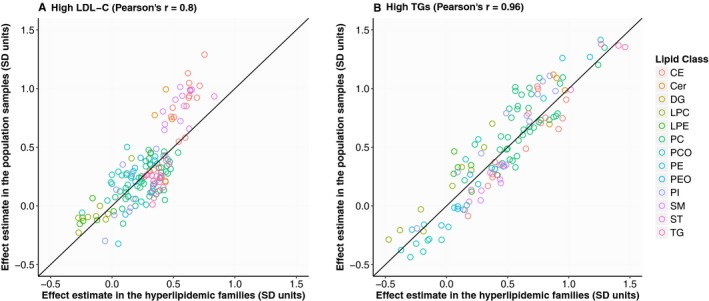
Correlation of effect estimates for hyperlipidemia status between the hyperlipidemic families and the population samples. The correlation between the effect estimates observed in the family and population cohorts is presented for high low‐density lipoprotein cholesterol (LDL‐C) (effect estimates presented in Figure 2; A) and for high triacylglycerides (effect estimates presented in Figure 4; B). Cer indicates ceramide; DG, diacylglyceride; LDL‐C, low‐density lipoprotein cholesterol; LPA, lysophosphatic acid; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PC, phosphatidylcholine; PCO, phosphatidylcholine‐ether; PE, phosphatidylethanolamine; PEO, phosphatidylethanolamine‐ether; PI, phosphatidylinositol; CE, cholesteryl ester; SM, sphingomyelin; ST, sterol; TG, triacylglyceride.
We also studied the association of high LDL‐C levels with the degree of saturation of fatty acids in each lipid class. In the hyperlipidemic families, high LDL‐C levels were associated with increased saturation of lysophospatidylcholines and ceramides, as well as reduced saturation of lysophosphatidylethanolamines, phosphatidylcholines, PCOs, and phosphatidylinositols (P‐value range=0.019–0.0014) (Figure S4). In the population cohort, the trends were similar, although there was an association for increased lysophospatidylcholine saturation only (P=7.2×10−4). The effect estimates did not differ significantly between the hyperlipidemic families and the population sample at the 5% FDR. Overall, the lipidomic profiles associated with high LDL‐C levels appeared similar in the hyperlipidemic families and the general population.
High Triacylglycerides and Lipidomic Profiles
In the hyperlipidemic families, individuals with high triacylglycerides had elevated levels of 107 lipid species covering all studied lipid classes with the exception of lysophosphatidylethanolamines. In addition, we observed reduced levels of 7 PCO, 2 lysophospatidylcholine, and 1 phosphatidylinositol species (Figure 4A; Table S7). Similar profiles were seen in the population when comparing individuals with high triacylglycerides with those without, including elevated levels of 108 species and reduced levels of 10 PCO and 1 lysophospatidylcholine species (Figure 4B; Table S7). The effect estimates correlated highly across all species between the families and the population cohort (Pearson's r=0.96; Figure 3). Furthermore, we observed no significant differences in the effect estimates between the cohorts at the 5% FDR.
Figure 4.
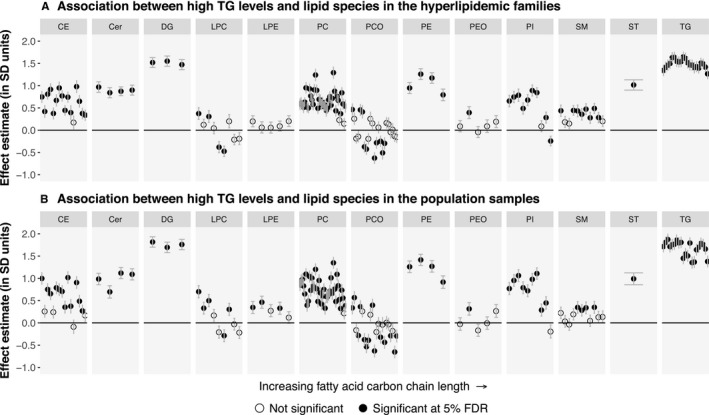
Associations between high triacylglyceride status and the levels of 151 lipid species. A, Individuals affected by high triacylglycerides (n=64) were compared with their unaffected relatives (n=223) in 39 “high TG” families. B, Individuals affected by high triacylglycerides (n=65) were compared with other individuals (n=832) in the FINRISK study population cohort. The association analyses were performed similarly to Figure 2. Cer indicates ceramide; DG, diacylglyceride; FDR, false discovery rate; LDL‐C, low‐density lipoprotein cholesterol; LPA, lysophosphatic acid; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PC, phosphatidylcholine; PCO, phosphatidylcholine‐ether; PE, phosphatidylethanolamine; PEO, phosphatidylethanolamine‐ether; PI, phosphatidylinositol; CE, cholesteryl ester; SM, sphingomyelin; ST, sterol; TG, triacylglyceride.
When contrasting the profiles observed for the 2 types of hyperlipidemias, we saw that high triacylglyceride levels were more uniquely reflected in a range of circulating lipid classes, including triacylglycerides, diacylglycerides, phosphatidylethanolamines, phosphatidylcholines, PCOs, and phosphatidylinositols. However, associations with sphingomyelin species appeared more unique to high LDL‐C levels.
Next, we studied the association of high triacylglyceride levels with the degree of saturation of fatty acids in each lipid class. In both the hyperlipidemic families and the population, having high triacylglycerides was associated with increased saturation of triacylglycerides, diacylglycerides, lysophospatidylcholines, and cholesteryl esters (CEs) (P‐value range=0.0012–5.9×10−11) (Figure S5). The effect estimates did not differ significantly between the hyperlipidemic families and the population sample at the 5% FDR. Overall, we observed great similarity in the lipidomic profiles associated with high triacylglycerides in the hyperlipidemic families and in the general population.
Independent Associations of LDL‐C and Triacylglyceride Values With the Lipid Species
We then tested if the variation in the lipid species was driven by both LDL‐C and triacylglyceride levels or if either was dominating the profiles. For this, we estimated the independent associations of LDL‐C and triacylglyceride levels with each lipid species in coadjusted models (Figure 5; Table S7). In these analyses, many of the observed associations with LDL‐C were greatly diluted in magnitude, most notably for triacylglyceride, diacylglyceride, and phosphatidylcholine species. LDL‐C levels remained most strongly associated with CE, sphingomyelin, ceramide, phosphatidylcholine, and PCO species in both cohorts. A total of 83 species in the hyperlipidemic families and 91 species in the population were independently associated with LDL‐C at the 5% FDR. In contrast, triacylglycerides remained strongly associated with a wide range of lipid species, including all individual triacylglyceride species, diacylglycerides, phosphatidylcholines, phosphatidylethanolamines, phosphatidylinositols, ceramides, and a subset of CEs in both cohorts. A total of 125 species in the hyperlipidemic families and 124 species in the population were independently associated with triacylglycerides at the 5% FDR. Overall, only 13 species were uniquely associated with LDL‐C in either cohort, whereas 42 species were uniquely associated with triacylglycerides (Figure 6).
Figure 5.
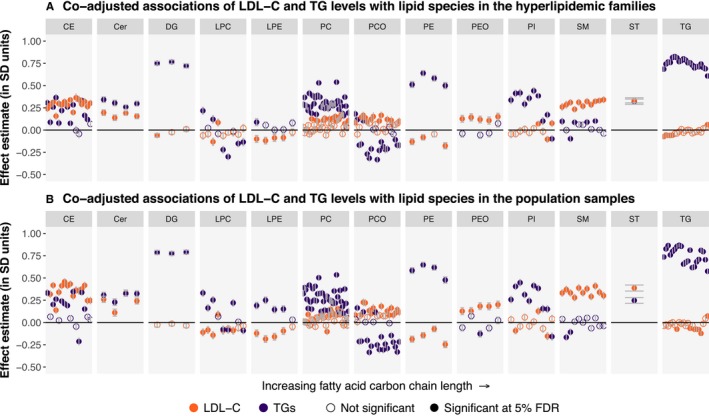
Independent (coadjusted) associations of low‐density lipoprotein cholesterol (LDL‐C) and triacylglycerides with 151 lipid species. Effect estimates for LDL‐C and triacylglycerides were derived from linear mixed models with the lipid species as outcomes and LDL‐C, log(triacylglycerides), age, age2, and sex as fixed‐effect covariates. The effect estimates were derived separately in the hyperlipidemic families (n=550 individuals; A) and the FINRISK study population cohort (n=897 individuals; B). Effect estimates are presented for LDL‐C in orange and triacylglycerides in purple. Statistical significance was evaluated using the Benjamini‐Hochberg method at a 5% false discovery rate (FDR). The ordering of the lipid species within each class is the same as in Table S7. Cer indicates ceramide; DG, diacylglyceride; FDR, false discovery rate; LDL‐C, low‐density lipoprotein cholesterol; LPA, lysophosphatic acid; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PC, phosphatidylcholine; PCO, phosphatidylcholine‐ether; PE, phosphatidylethanolamine; PEO, phosphatidylethanolamine‐ether; PI, phosphatidylinositol; CE, cholesteryl ester; SM, sphingomyelin; ST, sterol; TG, triacylglyceride.
Figure 6.
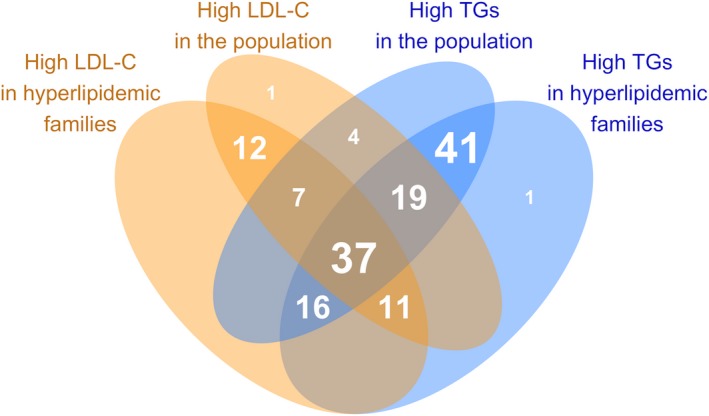
Overlap of the statistically significant independent (coadjusted) associations of low‐density lipoprotein cholesterol (LDL‐C) and triacylglycerides with 151 lipid species. Each shaded area shows the number of lipid species associated with the corresponding types of hyperlipidemias. More detailed methods are presented in Figure 5 legend. TG indicates triacylglyceride; LDL, low‐density lipoprotein cholesterol.
Discussion
Recent lipidomic approaches have identified several hundreds of different lipid species in the human circulation, some of which could be better prognostic biomarkers for ASCVD than the traditional clinical chemistry measurements. In this study, we used a mass spectrometric lipidomics platform to assess the lipidomic profiles in individuals with high LDL‐C and/or triacylglyceride levels. We found that individuals affected by high levels of LDL‐C or triacylglycerides had CAD HRs between 1.35 and 1.74 in the family and population cohorts and exhibited distinct lipidomic profiles with clear variation between lipid classes. In total, of 151 lipidomic species, 108 were significantly associated with high LDL‐C and 131 with high triacylglyceride levels in at least one cohort. Of these species, 96 were associated with both high LDL‐C and triacylglycerides. In addition, we observed highly similar lipidomic profiles between the hyperlipidemias with family history and population‐ascertained hyperlipidemias. The present study is the most comprehensive lipidomic profiling of common hyperlipidemias to date.
These findings allow us to draw several conclusions. First, the CAD risks are highly similar regardless of whether hyperlipidemic individuals were identified from families with a high prevalence of similar hyperlipidemia or from the general population. Earlier studies have found higher CAD risk in relatives of familial combined hyperlipidemia probands compared with spouses.25, 26, 27 Our study, however, compares the estimates between family members and individuals with similar lipid levels from the population to quantify the effect of familiality. We also studied the risk associated with elevated LDL‐C and triacylglycerides separately. Our estimates for CAD risk caused by high LDL‐C with family history are lower than typically reported for monogenic FH, despite comparable differences in LDL‐C levels.10, 28, 29 In the present study, we excluded probands with monogenic FH based on a functional LDL receptor test and genetic testing in the families. Excepting monogenic FH, hyperlipidemias with family history of high LDL‐C and/or triacylglyceride levels have been reported to be highly polygenic.11, 12, 13, 30 The pleiotropic effects of diverse genes and pathways, in contrast with the single affected pathway in monogenic FH, may partly explain why we did not observe increased CAD risk caused by familiality in our study.
Second, to more deeply characterize potential differences between hyperlipidemias with family history and population‐ascertained hyperlipidemias, we performed precise phenotyping of circulating lipid species known to be associated with ASCVD risk.22, 23, 31 Individual lipid species, including sphingolipids, glycerophospholipids, glycerolipids, and CEs, have previously been associated with ASCVD incidence or event risk over traditional risk factors.22, 23, 31, 32 Major differences in the metabolic pathways underlying different types of hyperlipidemias would thus be expected to be reflected in different lipidomic profiles. As an example, individuals with low high‐density lipoprotein cholesterol levels have previously been shown to have low phosphatidylethanolamine‐plasmalogen levels in high‐density lipoprotein particles, a putative marker of high‐density lipoprotein antioxidative capacity.33 Herein, in contrast, we observed similar profiles in hyperlipidemias with family history and population‐ascertained hyperlipidemias, highlighting the biochemical similarity of the conditions.
We started by characterizing the lipid profiles associated with high LDL‐C and triacylglyceride levels. Many of the associations were not specific to LDL‐C but were rather caused by combined dyslipidemia. LDL particles are generated in circulation as downstream metabolic products from the triacylglyceride‐rich lipoproteins and their postlipolytic remnants by the action of 2 lipases, lipoprotein lipase and hepatic lipase.34, 35 A proportion of the core lipids, especially cholesterol esters, and the particle surface phospholipids thus remains in the generated LDL particles. The actions of the CE transfer protein and phospholipid transfer protein, however, further modulate the constituents of triacylglyceride‐rich and LDL particles.36 Percentual lipid compositions have been reported for different lipoprotein classes, but they do not directly reflect variation in plasma LDL‐C or triacylglyceride concentrations. For example, phosphatidylcholines have been estimated to constitute 12% of all lipids in LDL particles versus 3% to 9% in triacylglyceride‐rich lipoproteins.37 However, in our study, phosphatidylcholines were overall more strongly associated with triacylglyceride levels than with LDL‐C levels. Nevertheless, LDL‐C remained positively associated with a range of species, including CEs, ceramides, sphingomyelins, phosphatidylcholines, and PCOs. Among the strongly increased species, CE(14:0), CE(16:0), CE(16:1), CE(18:0), sphingomyelin(34:1;2), sphingomyelin(34:2;2), sphingomyelin(42:2;2), ceramide(42:1;2), and ceramide(42:2;2) have previously been associated with the risk of ASCVD.22, 23
Elevated triacylglyceride levels were associated with differences in the levels of lipid species across most of the studied classes. More important, most of these associations appeared to be independent of LDL‐C levels. Among the lipid species that were strongly correlated with high triacylglycerides after correction for LDL‐C levels were several species that have previously been associated with risk of ASCVD.22, 23, 31 These include the species CE(14:0), CE(16:0), CE(16:1), CE(18:0), triacylglyceride(50:1), triacylglyceride(50:2), triacylglyceride(50:3), triacylglyceride(52:2), triacylglyceride(52:3), triacylglyceride(52:5), triacylglyceride(56:5), triacylglyceride(56:6), ceramide(42:1;2), and ceramide(42:2;2). Furthermore, high triacylglycerides were associated with increased saturation of fatty acids in the triacylglyceride, diacylglyceride, CE, and lysophospatidylcholine classes. Such differences in the relative fatty acid concentrations can be partly related to dietary intake and reflected in liver‐derived very low‐density lipoprotein particles, but they are also influenced by endogenous metabolism.38 Overall, a larger proportion of the lipid species previously linked with increased ASCVD risk was more strongly associated with elevated triacylglycerides rather than with elevated LDL‐C. This suggests that the levels of these lipid biomarkers are more closely linked with circulating triacylglyceride‐rich lipoprotein metabolism than with LDLs.
Third, several lipid species, such as specific CEs, ceramides, and PCOs, remained independently associated with both elevated LDL‐C and triacylglycerides. Among these species, the ceramides ceramide(42:1;2) (presumably ceramide[d18:1/24:0]) and ceramide(42:2;2) (presumably ceramide[d18:1/24:1]), the sterol esters CE(16:1) and CE(18:0), and the sphingomyelin(34:1;2) may have added value in ASCVD prediction over traditional lipid measurements.22, 23, 31 Plasma ceramides have been reported to be independent predictors of cardiovascular events in addition to LDL‐C in the population and in patients with CAD.31, 39, 40 Both LDL‐C and triacylglycerides remained independently associated with all 4 ceramides quantified in our study, and LDL‐C was additionally associated with increased saturation of ceramides. Unlike most CE species, CE(16:1) was more strongly associated with the concentration of triacylglycerides than with LDL‐C in our study. Sphingomyelin(34:1;2) was the only sphingomyelin species that was negatively associated with triacylglycerides; and this association became evident only after adjusting for LDL‐C levels. In addition, some species, such as ceramide(42:1;2) and triacylglyceride(56:6), which were positively associated with hyperlipidemias in our sample, have previously been reported to be associated with decreased risk of ASCVD events.23, 31 These coassociations and discordances between reported associations might explain why some lipid species can improve risk prediction. Consequently, there is an urgent need for a better understanding of the potential underlying signaling and metabolic pathways.
Finally, the lipidomic profiles associated with high LDL‐C or triacylglyceride levels were comparable between hyperlipidemias with family history and population‐ascertained hyperlipidemias. We observed no differences in either the levels of individual lipid species or the saturation of fatty acids within lipid classes. Our findings are in line with a pediatric study of hypercholesterolemia, in which similar nuclear magnetic resonance metabolite profiles (including lipoprotein parameters and circulating fatty acids) were seen for FH and for continuous LDL‐C measures in healthy children.41 These results support the hypothesis that hyperlipidemias with family history and population‐ascertained hyperlipidemias have similar, overlapping, and heterogeneous pathophysiological features. Our results are also reassuring for studies that combine familial and population‐based hyperlipidemic samples to increase statistical power.
Although we present the most comprehensive characterization of CAD risk and circulating lipid species in common hyperlipidemias with family history to date, our study has limitations. Although we were unable to observe significant differences in CAD risk caused by hyperlipidemic family history, the large CIs in the family samples do not preclude their possibility. Careful exclusion of individuals with comorbidities or using lipid‐lowering medication reduced our sample size but enabled more robust analyses. We could not perform detailed analyses on individuals with FH as the original study protocol led to their exclusion from further ascertainment. Clinical ascertainment was based on 90th population lipid percentiles; different cutoffs have also been used in other family studies. Some of the individuals surveyed in population cohorts might, in fact, have a family history of hyperlipidemia, including FH, as we could not fully rule out such cases. It is also unclear how well our results can be generalized to other populations than Finns. The field of lipidomics is still relatively young, and concerns have been raised about the replicability of individual lipidomics platforms. The platform used herein overcomes these problems by using direct infusion mass spectrometry for high‐throughput screening studies. The similarity of lipidomic profiles between the 2 independent cohorts also supports the replicability of the platform. Furthermore, the lipid species included in our analyses are heritable and associated with both known and novel genetic lipid loci with similar effect sizes in the 2 cohorts.42 We excluded poorly captured lipid species from the analyses; future advances in lipidomics technology might enable their detection. The blood samples from the hyperlipidemic families were obtained after overnight fasting, whereas participants in the FINRISK population study were advised to fast for 4 hours before the examination and avoid heavy meals earlier during the day. In this light, the similarity of lipidomic profiles between the cohorts becomes even more striking. Moreover, recent recommendations support routine use of nonfasting blood samples for the assessment of plasma lipid profiles.43
In conclusion, our results highlight the similarity between hyperlipidemias with family history and population‐based hyperlipidemias in terms of both CAD risk and detailed lipidomic profiles. Except for FH, our results do not support different screening for sporadically discovered cases and those with a family history of hyperlipidemia. Additional work is needed to confirm the validity of this hypothesis in clinical settings.
Sources of Funding
This work was supported by National Institutes of Health (grant HL113315 to Drs Ripatti, Taskinen, Freimer, and Palotie); Finnish Foundation for Cardiovascular Research (to Drs Ripatti, Salomaa, Taskinen, Jauhiainen, and Palotie); Academy of Finland Center of Excellence in Complex Disease Genetics (grants 213506 and 129680 to Drs Ripatti, Pirinen and Palotie); Academy of Finland (grants 251217 and 285380 to Dr Ripatti and grant 286500 to Dr Palotie); Jane and Aatos Erkko Foundation (to Dr Jauhiainen); Sigrid Jusélius Foundation (to Drs Ripatti, Palotie, and Taskinen); Biocentrum Helsinki (to Dr Ripatti); Horizon 2020 Research and Innovation Programme (grant 692145 to Dr Ripatti); EU (European Union)‐project RESOLVE (EU 7th Framework Program) (grant 305707 to Dr Taskinen); HiLIFE Fellowship (to Dr Ripatti); Helsinki University Central Hospital Research Funds (to Dr Taskinen); Magnus Ehrnrooth Foundation (to Dr Jauhiainen); Leducq Foundation (to Dr Taskinen); Ida Montin Foundation (to Dr Ripatti); MD‐PhD Programme of the Faculty of Medicine, University of Helsinki (to Dr Rämö); Doctoral Programme in Population Health, University of Helsinki (to Drs Rämö and Ripatti); Finnish Medical Foundation (to Dr Rämö); Emil Aaltonen Foundation (to Drs Rämö and Ripatti); Biomedicum Helsinki Foundation (to Dr Rämö); Paulo Foundation (to Dr Rämö); Idman Foundation (to Dr Rämö); and Veritas Foundation (to Dr Rämö). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures
Dr Gerl is an employee of Lipotype GmbH. Dr Klose is a shareholder and employee of Lipotype GmbH. Dr Simons is a shareholder and chief executive officer of Lipotype GmbH. Dr Surma is a shareholder of Lipotype GmbH and an employee of Polish Center for Technology Development (PORT). This does not alter the authors’ adherence to all policies on sharing data and materials. The remaining authors have no disclosures to report.
Supporting information
Data S1. Supplemental methods.
Table S1. Sex‐ and Age‐Specific 90th Population Percentiles for LDL‐C and TGs Based on the FINRISK 1997 Cohort
Table S2. Clinical and Metabolic Characteristics of the Study Individuals Included in the Analyses of Incident CAD Risk
Table S3. Risk of Incident CAD or CVD in Hyperlipidemias With Family History and Population‐Ascertained Hyperlipidemias
Table S4. Clinical and Metabolic Characteristics of the Study Individuals Included in the Analyses of Circulating Lipidomics Profiles
Table S5. Median Concentrations of the 151 Lipid Species in the Family and Population Cohorts
Table S6. Effect Estimates in SD Units (±SE) and P‐Values From Linear Mixed Models for Each Lipid Species
Figure S1. Overlap of families with family histories of high LDL‐C and high TGs.
Figure S2. Risk of incident CAD in hyperlipidemias with family history and population ascertained hyperlipidemias, adjusted by lipid lowering medication usage and smoking.
Figure S3. A, Risk of incident CVD in hyperlipidemias with family history and population‐ascertained hyperlipidemias. B, Risk of incident CVD in hyperlipidemias with family history and population‐ascertained hyperlipidemias, adjusted for lipid lowering medication usage and smoking.
Figure S4. Association of high LDL‐C status and weighted saturation averages within each class.
Figure S5. Association of high TG status and weighted saturation averages within each class.
Acknowledgments
We would like to thank Sari Kivikko, Huei‐Yi Shen, and Ulla Tuomainen for management assistance. The FINRISK study data used for the research were obtained from THL Biobank. We thank all study participants for their generous participation in the FINRISK study and the EUFAM (European Multicenter Study on Familial Dyslipidemias in Patients With Premature Coronary Heart Disease). Drs Ripatti and Rämö acknowledge support from the Doctoral Programme in Population Health, University of Helsinki.
(J Am Heart Assoc. 2019;8:e012415 DOI: 10.1161/JAHA.119.012415.)
References
- 1. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Boren J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen MR, Tokgozoglu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL. Low‐density lipoproteins cause atherosclerotic cardiovascular disease, 1: evidence from genetic, epidemiologic, and clinical studies: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordestgaard BG. Triglyceride‐rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118:547–563. [DOI] [PubMed] [Google Scholar]
- 3. Yuan G, Al‐Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. 2007;176:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, Stalenhoef AF; Endocrine Society . Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kathiresan S, Manning AK, Demissie S, D'Agostino RB, Surti A, Guiducci C, Gianniny L, Burtt NP, Melander O, Orho‐Melander M, Arnett DK, Peloso GM, Ordovas JM, Cupples LA. A genome‐wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8(suppl 1):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weiss LA, Pan L, Abney M, Ober C. The sex‐specific genetic architecture of quantitative traits in humans. Nat Genet. 2006;38:218–222. [DOI] [PubMed] [Google Scholar]
- 7. Genest JJ Jr, Martin‐Munley SS, McNamara JR, Ordovas JM, Jenner J, Myers RH, Silberman SR, Wilson PW, Salem DN, Schaefer EJ. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation. 1992;85:2025–2033. [DOI] [PubMed] [Google Scholar]
- 8. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, Ferranti Sd, Faiella‐Tommasino J, Forman DE, Goldberg R, Heidenreich PA, Hlatky MA, Jones DW, Lloyd‐Jones D, Lopez‐Pajares N, Ndumele CE, Orringer CE, Peralta CA, Saseen JJ, Smith SC, Sperling L, Virani SS, Yeboah J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol. Circulation. 2019;139:e1082–e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR, Reiner Ž, Riccardi G, Taskinen M‐R, Tokgozoglu L, Verschuren WMM, Vlachopoulos C, Wood DA, Zamorano JL, Cooney M‐T; ESC Scientific Document Group . 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;37:2999–3058. [DOI] [PubMed] [Google Scholar]
- 10. Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ripatti P, Ramo JT, Soderlund S, Surakka I, Matikainen N, Pirinen M, Pajukanta P, Sarin AP, Service SK, Laurila PP, Ehnholm C, Salomaa V, Wilson RK, Palotie A, Freimer NB, Taskinen MR, Ripatti S. The contribution of GWAS loci in familial dyslipidemias. PLoS Genet. 2016;12:e1006078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stitziel NO, Peloso GM, Abifadel M, Cefalu AB, Fouchier S, Motazacker MM, Tada H, Larach DB, Awan Z, Haller JF, Pullinger CR, Varret M, Rabes JP, Noto D, Tarugi P, Kawashiri MA, Nohara A, Yamagishi M, Risman M, Deo R, Ruel I, Shendure J, Nickerson DA, Wilson JG, Rich SS, Gupta N, Farlow DN, Neale BM, Daly MJ, Kane JP, Freeman MW, Genest J, Rader DJ, Mabuchi H, Kastelein JJ, Hovingh GK, Averna MR, Gabriel S, Boileau C, Kathiresan S. Exome sequencing in suspected monogenic dyslipidemias. Circ Cardiovasc Genet. 2015;8:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, Neil HA, Descamps OS, Langenberg C, Lench N, Kivimaki M, Whittaker J, Hingorani AD, Kumari M, Humphries SE. Use of low‐density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case‐control study. Lancet. 2013;381:1293–1301. [DOI] [PubMed] [Google Scholar]
- 14. Surma MA, Herzog R, Vasilj A, Klose C, Christinat N, Morin‐Rivron D, Simons K, Masoodi M, Sampaio JL. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. Eur J Lipid Sci Technol. 2015;117:1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han X. Lipidomics for studying metabolism. Nat Rev Endocrinol. 2016;12:668–679. [DOI] [PubMed] [Google Scholar]
- 16. Simons K. How can omic science be improved? Proteomics. 2018;18:e1800039. [DOI] [PubMed] [Google Scholar]
- 17. Cuthbert JA, East CA, Bilheimer DW, Lipsky PE. Detection of familial hypercholesterolemia by assaying functional low‐density‐lipoprotein receptors on lymphocytes. N Engl J Med. 1986;314:879–883. [DOI] [PubMed] [Google Scholar]
- 18. Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl‐tert‐butyl ether for high‐throughput lipidomics. J Lipid Res. 2008;49:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herzog R, Schuhmann K, Schwudke D, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. LipidXplorer: a software for consensual cross‐platform lipidomics. PLoS One. 2012;7:e29851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herzog R, Schwudke D, Schuhmann K, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. A novel informatics concept for high‐throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011;12:R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pirinen M, Donnelly P, Spencer CCA. Efficient computation with a linear mixed model on large‐scale data sets with applications to genetic studies. Ann Appl Stat. 2013;7:369–390. [Google Scholar]
- 22. Stegemann C, Pechlaner R, Willeit P, Langley SR, Mangino M, Mayr U, Menni C, Moayyeri A, Santer P, Rungger G, Spector TD, Willeit J, Kiechl S, Mayr M. Lipidomics profiling and risk of cardiovascular disease in the prospective population‐based Bruneck study. Circulation. 2014;129:1821–1831. [DOI] [PubMed] [Google Scholar]
- 23. Alshehry ZH, Mundra PA, Barlow CK, Mellett NA, Wong G, McConville MJ, Simes J, Tonkin AM, Sullivan DR, Barnes EH, Nestel PJ, Kingwell BA, Marre M, Neal B, Poulter NR, Rodgers A, Williams B, Zoungas S, Hillis GS, Chalmers J, Woodward M, Meikle PJ. Plasma lipidomic profiles improve on traditional risk factors for the prediction of cardiovascular events in type 2 diabetes mellitus. Circulation. 2016;134:1637–1650. [DOI] [PubMed] [Google Scholar]
- 24. R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. Available at: https://www.R-project.org/. [Google Scholar]
- 25. Voors‐Pette C, de Bruin TW. Excess coronary heart disease in familial combined hyperlipidemia, in relation to genetic factors and central obesity. Atherosclerosis. 2001;157:481–489. [DOI] [PubMed] [Google Scholar]
- 26. Luijten J, van Greevenbroek MMJ, Schaper NC, Meex SJR, van der Steen C, Meijer LJ, de Boer D, de Graaf J, Stehouwer CDA, Brouwers MCGJ. Incidence of cardiovascular disease in familial combined hyperlipidemia: a 15‐year follow‐up study. Atherosclerosis. 2019;280:1–6. [DOI] [PubMed] [Google Scholar]
- 27. Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, Brunzell JD, Motulsky AG. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: a 20‐year prospective study. Circulation. 2000;101:2777–2782. [DOI] [PubMed] [Google Scholar]
- 28. Lahtinen AM, Havulinna AS, Jula A, Salomaa V, Kontula K. Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis. 2015;238:64–69. [DOI] [PubMed] [Google Scholar]
- 29. Abul‐Husn NS, Manickam K, Jones LK, Wright EA, Hartzel DN, Gonzaga‐Jauregui C, O'Dushlaine C, Leader JB, Lester Kirchner H, Lindbuchler DAM, Barr ML, Giovanni MA, Ritchie MD, Overton JD, Reid JG, Metpally RPR, Wardeh AH, Borecki IB, Yancopoulos GD, Baras A, Shuldiner AR, Gottesman O, Ledbetter DH, Carey DJ, Dewey FE, Murray MF. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016;354:aaf7000. [DOI] [PubMed] [Google Scholar]
- 30. Hegele RA, Ginsberg HN, Chapman MJ, Nordestgaard BG, Kuivenhoven JA, Averna M, Boren J, Bruckert E, Catapano AL, Descamps OS, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Raal FJ, Ray KK, Santos RD, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg‐Hansen A, Watts GF, Wiklund O; European Atherosclerosis Society Consensus Panel . The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laaksonen R, Ekroos K, Sysi‐Aho M, Hilvo M, Vihervaara T, Kauhanen D, Suoniemi M, Hurme R, Marz W, Scharnagl H, Stojakovic T, Vlachopoulou E, Lokki ML, Nieminen MS, Klingenberg R, Matter CM, Hornemann T, Juni P, Rodondi N, Raber L, Windecker S, Gencer B, Pedersen ER, Tell GS, Nygard O, Mach F, Sinisalo J, Luscher TF. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL‐cholesterol. Eur Heart J. 2016;37:1967–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Demirkan A, van Duijn CM, Ugocsai P, Isaacs A, Pramstaller PP, Liebisch G, Wilson JF, Johansson A, Rudan I, Aulchenko YS, Kirichenko AV, Janssens AC, Jansen RC, Gnewuch C, Domingues FS, Pattaro C, Wild SH, Jonasson I, Polasek O, Zorkoltseva IV, Hofman A, Karssen LC, Struchalin M, Floyd J, Igl W, Biloglav Z, Broer L, Pfeufer A, Pichler I, Campbell S, Zaboli G, Kolcic I, Rivadeneira F, Huffman J, Hastie ND, Uitterlinden A, Franke L, Franklin CS, Vitart V; DIAGRAM Consortium , Nelson CP, Preuss M; CARDIoGRAM Consortium , Bis JC, O'Donnell CJ, Franceschini N; CHARGE Consortium , Witteman JC, Axenovich T, Oostra BA, Meitinger T, Hicks AA, Hayward C, Wright AF, Gyllensten U, Campbell H, Schmitz G; EUROSPAN Consortium . Genome‐wide association study identifies novel loci associated with circulating phospho‐ and sphingolipid concentrations. PLoS Genet. 2012;8:e1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laurila PP, Surakka I, Sarin AP, Yetukuri L, Hyotylainen T, Soderlund S, Naukkarinen J, Tang J, Kettunen J, Mirel DB, Soronen J, Lehtimaki T, Ruokonen A, Ehnholm C, Eriksson JG, Salomaa V, Jula A, Raitakari OT, Jarvelin MR, Palotie A, Peltonen L, Oresic M, Jauhiainen M, Taskinen MR, Ripatti S. Genomic, transcriptomic, and lipidomic profiling highlights the role of inflammation in individuals with low high‐density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2013;33:847–857. [DOI] [PubMed] [Google Scholar]
- 34. Olivecrona G. Role of lipoprotein lipase in lipid metabolism. Curr Opin Lipidol. 2016;27:233–241. [DOI] [PubMed] [Google Scholar]
- 35. Kobayashi J, Miyashita K, Nakajima K, Mabuchi H. Hepatic lipase: a comprehensive view of its role on plasma lipid and lipoprotein metabolism. J Atheroscler Thromb. 2015;22:1001–1011. [DOI] [PubMed] [Google Scholar]
- 36. Masson D, Jiang X‐C, Lagrost L, Tall AR. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J Lipid Res. 2009;50:S201–S206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Christinat N, Masoodi M. Comprehensive lipoprotein characterization using lipidomics analysis of human plasma. J Proteome Res. 2017;16:2947–2953. [DOI] [PubMed] [Google Scholar]
- 38. Raatz SK, Bibus D, Thomas W, Kris‐Etherton P. Total fat intake modifies plasma fatty acid composition in humans. J Nutr. 2001;131:231–234. [DOI] [PubMed] [Google Scholar]
- 39. Tarasov K, Ekroos K, Suoniemi M, Kauhanen D, Sylvanne T, Hurme R, Gouni‐Berthold I, Berthold HK, Kleber ME, Laaksonen R, Marz W. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J Clin Endocrinol Metab. 2014;99:E45–E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Havulinna AS, Sysi‐Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, Salomaa V, Laaksonen R. Circulating ceramides predict cardiovascular outcomes in the population‐based FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol. 2016;36:2424–2430. [DOI] [PubMed] [Google Scholar]
- 41. Christensen JJ, Ulven SM, Retterstøl K, Narverud I, Bogsrud MP, Henriksen T, Bollerslev J, Halvorsen B, Aukrust P, Holven KB. Comprehensive lipid and metabolite profiling of children with and without familial hypercholesterolemia: a cross‐sectional study. Atherosclerosis. 2017;266:48–57. [DOI] [PubMed] [Google Scholar]
- 42. Tabassum R, Rämö JT, Ripatti P, Koskela JT, Kurki M, Karjalainen J, Hassan S, Nunez‐Fontarnau J, Kiiskinen TTJ, Söderlund S, Matikainen N, Gerl MJ, Surma MA, Klose C, Stitziel NO, Laivuori H, Havulinna AS, Service SK, Salomaa V, Pirinen M, Jauhiainen M, Daly MJ, Freimer NB, Palotie A, Taskinen M‐R, Simons K, Ripatti S. Genetics of human plasma lipidome: understanding lipid metabolism and its link to diseases beyond traditional lipids. bioRxiv. 2018;457960. [Google Scholar]
- 43. Nordestgaard BG, Langsted A, Mora S, Kolovou G, Baum H, Bruckert E, Watts GF, Sypniewska G, Wiklund O, Boren J, Chapman MJ, Cobbaert C, Descamps OS, von Eckardstein A, Kamstrup PR, Pulkki K, Kronenberg F, Remaley AT, Rifai N, Ros E, Langlois M; European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Joint Consensus Initiative . Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cutpoints—a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Clin Chem. 2016;62:930–946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Sex‐ and Age‐Specific 90th Population Percentiles for LDL‐C and TGs Based on the FINRISK 1997 Cohort
Table S2. Clinical and Metabolic Characteristics of the Study Individuals Included in the Analyses of Incident CAD Risk
Table S3. Risk of Incident CAD or CVD in Hyperlipidemias With Family History and Population‐Ascertained Hyperlipidemias
Table S4. Clinical and Metabolic Characteristics of the Study Individuals Included in the Analyses of Circulating Lipidomics Profiles
Table S5. Median Concentrations of the 151 Lipid Species in the Family and Population Cohorts
Table S6. Effect Estimates in SD Units (±SE) and P‐Values From Linear Mixed Models for Each Lipid Species
Figure S1. Overlap of families with family histories of high LDL‐C and high TGs.
Figure S2. Risk of incident CAD in hyperlipidemias with family history and population ascertained hyperlipidemias, adjusted by lipid lowering medication usage and smoking.
Figure S3. A, Risk of incident CVD in hyperlipidemias with family history and population‐ascertained hyperlipidemias. B, Risk of incident CVD in hyperlipidemias with family history and population‐ascertained hyperlipidemias, adjusted for lipid lowering medication usage and smoking.
Figure S4. Association of high LDL‐C status and weighted saturation averages within each class.
Figure S5. Association of high TG status and weighted saturation averages within each class.