

Abstract
Chimeric antigen receptor T‐cell (CART) therapy is a new and promising cancer therapy. However, severe toxicity due to cytokine release syndrome (CRS) in CART‐treated patients highlighted the possible danger of this new therapy. Disease burden and CART doses are the potential factors associated with CRS but the detail relationships between these factors and the severity of the CRS remain largely unknown. In this study, the quantitative systems pharmacology (QSP) approach is used to quantify the complex relationships among CART doses, disease burden, and pro inflammatory cytokines in human subjects and to gain relevant insights into the determinant of clinical toxicity/efficacy in development of CART therapy. The expansion of CART and elimination of B cells are more highly correlated with disease burden than the administered CART doses. To our best knowledge, this is the first QSP model that can describe the observed clinical data from CART‐treated patients with cancer. This QSP model is a valuable tool for deepening our understanding of how the mechanism of action connects to the clinical outcomes and, therefore, may serve as an important model‐based platform to guide the development and personalized dosing of the CART therapy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑Chimeric antigen receptor T cell (CART) therapy is a new and promising therapy for cancer. However, unexpected toxicity, such as cytokine release syndrome (CRS), following CART therapy can cause significant organ dysfunction and death. The disease burden and CART doses have been proposed to be associated with the CRS, but the quantitative relationships between these factors and the CRS associated with CART therapy remain largely unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑We built the first quantitative systems pharmacology (QSP) model that can describe the complex relationships of CART and pro inflammatory cytokines associated with CRS for CART therapy. By using this model, we have quantified relationships among CART dose, disease burden, and CRS associated with CART therapy.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑This study shows that CART is a “living drug” that exhibits unique target‐mediated drug disposition characteristics (i.e., the CART cells “live and grow” in the presence of the target cells), which is in contrast to most drugs that accelerate the elimination from the body after binding to the targeted receptors. Furthermore, the magnitude of the inflammatory cytokine (e.g., interleukin (IL‐6), IL‐10, and interferonγ) elevation is more related with baseline disease burden than the administered doses.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCES?
☑This QSP model is a valuable tool for deepening our understanding of how the mechanism of action connects to the clinical outcomes and, therefore, may serve as an important model‐based platform to guide the development and personalized dosing of the CART therapy.
Chimeric antigen receptor T cells (CART) are engineered T cells with a single‐chain variable fragment on the cell membrane that recognizes the tumor antigen and intracellular signaling domain that can promote T cells’ antitumor function.1, 2 CART therapy demonstrates remarkable efficacy in some blood cancers and represents a paradigm shift in oncology treatment.2, 3, 4, 5, 6 Currently, two anti‐CD19 CART therapies have been approved by the US Food and Drug Administration: tisagenlecleucel‐T (Kymriah; Norvatis) for pediatric and young adults (up to 25 years) with relapsed or refractory B‐cell acute lymphoblastic leukemia (ALL) and axicabtagene ciloleucel (Yescarta; Kite Pharma/Gilead Sciences) for adults with relapsed or refractory B‐cells lymphoma, including diffuse large B‐cell lymphoma.2, 3, 4 The CART therapy pipeline is rapidly expanding and is expected to capture the largest share of drug sales in pediatric and young adult patients with ALL and diffuse large B‐cell lymphoma.2 However, unexpected organ damage and deaths following CART therapy have been reported and, thus, highlight the possible danger of this new treatment.1 Cytokine release syndrome (CRS), a systemic inflammatory response caused by pro inflammatory cytokines released by infused CART, is the major and most common source of toxicity that can cause widespread significant organ dysfunction and death.1, 3, 7 As a result, both tisagenlecleucel‐T and axicabtagene ciloleucel are available through a restricted program that includes a risk evaluation mitigation strategy to address the severe and life‐threatening toxicity of CRS.2 Although potential risk factors, such as disease burden (DB), CART doses, and T‐cell expansion levels after CART infusion, are expected to be associated with the severity of the CRS, the quantitative relationships between these factors and the severity of the CRS reported in CART‐treated patients remains largely unknown.1 Therefore, further investigation is needed to elucidate the quantitative relationship of CRS and the DB/CART dose in order to design an optimal dosing strategy for the prevention of the CRS. Recently, several mathematical models have been developed to provide theoretical speculation for the biologic of CART therapy, but none of these models has been used to describe the kinetic profiles of pro inflammatory cytokine and CART observed in patients with cancer treated with CART.8, 9, 10 Hence, in this study, we develop the first quantitative systems pharmacology (QSP) model that can describe the observed kinetic profiles of CART, DB, and pro inflammatory cytokines in a patient with advanced chronic lymphocytic leukemia (CLL) treated with anti‐CD19 CART therapy.
Methods
Distinct kinetic profiles of the CART and inflammatory cytokines (interleukin‐6 (IL‐6), IL‐10, and interferon gamma (IFNγ) in peripheral blood (PB) from two patients with CLL were used to develop the QSP model (Figure 1 a). Kinetic data of pro inflammatory cytokines were obtained from the supplemental tables in Kalos et al.,7 and the kinetic data of CART were obtained from the same study using software WebPlotDigitizer (version 3.8; https://automeris.io/WebPlotDigitizer/). The clinical study was conducted according to the guidelines described in the Declaration of Helsinki, and detailed information about the study design has been reported previously.7 In brief, three patients with advanced, chemotherapy‐resistant CLL were enrolled in the study and the treatments were scheduled as an i.v. dose of CART cells with 3‐day split dose regimen (10%, 30%, and 60%). Corticosteroid therapy can inhibit the expression and action of most cytokine.11 UPN2 was excluded from the analysis because this patient had a different and subdued CART and cytokine kinetic profile compared with the other two subjects (UPN1 and UPN3),7 which was possibly due to the cytokine‐masking effects of concurrent corticosteroid treatment. As a result, only two subjects (UPN1 and UPN3) were included in the QSP model development in this study.
Figure 1.
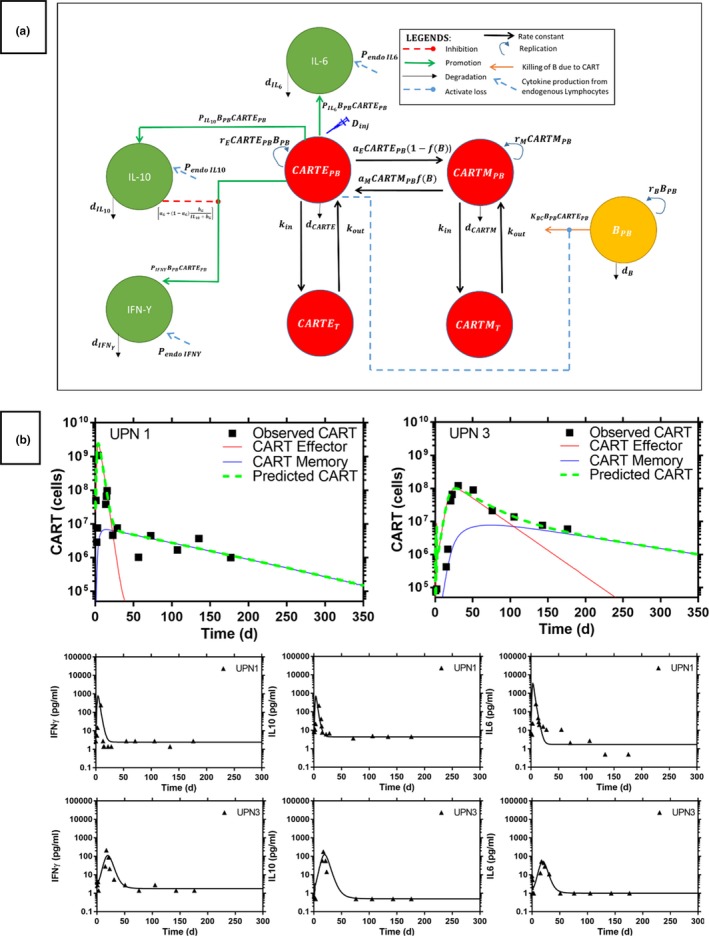
(a) The quantitative systems pharmacology model of anti‐CD19 chimeric antigen receptor T cells (CART) in a patient with chronic lymphatic leukemia (CLL). B PB represent the B‐cell CLL in peripheral blood (PB). CART effector (CARTE) and CART memory (CARTM) cells are distributed between PB (CARTEPB/CARTMPB) and tissue (CARTET/CARTMT) compartments via rate constant k in and k out after i.v. infusion. d CARTE and d CARTM are the death rate constants of CART from the PB. CARTE and CARTM are activated via activation rate a E and a M. P endo IL6, P endo IL10, and P endo IFNγ are endogenous synthesis rate and d IL6, d IL10, and d IFNγ are the natural death rate of interleukin (IL)‐6, IL‐10, and gamma interferon (IFN γ), respectively. P IL6, P IL10, and P INFγ represent the production rate of IL‐6, IL‐10, and IFN γ by the activated CART, respectively. KBC is the CARTE‐mediated B‐cell CLL degradation rate constant in peripheral blood. a G and b G are the inhibitory parameters of IL10 on IFN γ production. (b) Observed CART and cytokines kinetic profiles and model prediction in an anti‐CD19 CART‐treated CLL patient. Symbol—observed CART and cytokines. Solid line—model prediction.
The IL‐6, IL‐10, and IFN‐γ were selected because these cytokines played an important role in the inflammatory responses. Elevation of these pro inflammatory cytokines was associated with the severity of CRS and other toxicities after CART infusion.1, 12 Figure 1 a shows the developed QSP model based on the current understanding of the complex interactions among CART, B cells, IL‐6, IL‐10, and IFNγ in human subjects. The detailed description of the QSP model implementation is included in the Supplementary File . In brief, CART was divided into two phenotypes of T cells (i.e., activated/effector (CARTE) and memory T cells (CARTM). Injected CART were assumed to be activated after interacting with CLL B‐cells and were expanded via growth rate r E. CARTE was eliminated from the body with death rate d CARTE. Activated CART were converted into long‐life CARTM via activation rate a E, which related to the CLL B‐cells saturation function f(B). On the other hand, during the fast expansion of CARTE, CARTM were converted into short‐life CARTE to kill the CLL cancer via activation rate a M. CARTM were expanded via natural growth rate r M and were eliminated from the body with death rate d CARTM. Distribution of both CARTE and CARTM between PB and tissue was described by the rate constants k in and k out.
The B cells in patients with CLL were assumed to have exponential growth with production rate r B and natural death rate d B in PB. Activated CARTE cells killed the B cells via killing rate K BC and secreted IL‐6, IL‐10, and IFNγ into the blood to induce the inflammatory responses and CRS. The baseline cytokine levels in patients with CLL before anti‐CD19 CART therapy were maintained with the endogenous synthesis rate of P endo IL6, P endo IL10, and P endo IFNγ, and natural death rate of d IL6, d IL10, and d IFNγ for IL‐6, IL‐10, and IFNγ, respectively. Known inhibitory effects of IL‐10 on the secretion of IFNγ by activated T cells was included in the model. Most model parameters were obtained from the published literature ( Table S1 ) except for several CART‐related parameters that had never been reported in the literature and, therefore, had to be estimated by fitting the QSP model to all observed data simultaneously. These CART‐related parameters included CART‐induced IL‐6, IL‐10, and IFNγ secretion (P IL6, P IL10, and P INFγ), growth rate of CARTE cells (r E), the killing rate of CARTE cells to B cell CLL in PB (K BC), activation rate a M, and CARTE death rate d CARTE.
The ability of the final QSP model in describing the relationships between disease burden/CART doses and CART/cytokine kinetic profiles in PB was examined with two external validation data sets that consisted of (i) maximal CART peak plasma concentration (Cmax) and area under the curve of the time‐concentration curve from t = 0 to t = 28 days (AUC0–28) in patients with CLL treated with CART,13 and (ii) Cmax of IFNγ and CART observed in CART‐treated adult subjects with ALL.13, 14 The details of the external validation process is described in the Supplementary File . The final QSP model was then used to simulate the kinetic profiles of CART cells, B cells, and pro inflammatory cytokines in patients with CLL with different DB and CART doses. Due to the limitation of the data used for QSP model development, the DB and CART doses up to twofold differences from the actual values in UPN1 and UPN3 were used in the simulation studies.
Results and discussion
The parameters in the final QSP model are estimated with good precision (percentage of coefficient of variation < 50) and the final QSP model is able to describe the observed CART kinetic and pro inflammatory cytokine profiles in patients with CLL (Figure 1 b, and Table S2 and Figure S1). In general, CARTs differentiate and expand rapidly in the presence of the targeted B cell after infusion into patients with CLL. Then these activated CARTE target and destroy the B cell CLL, resulting in rapid decline of the DB. As the DB decline, the CARTE decay rapidly. Depending on the baseline tumor burden and administered CART doses, this model is able to describe two distinct CART and pro inflammatory cytokine profiles observed in patients with CLL (Figure 1 b). In Subject UPN1 who had 4.5‐fold higher baseline disease burden and received 79‐fold higher CART doses compared with subject UPN3, more CARTE are activated and are expanded rapidly in the presence of higher DB and administered CART doses. Then the CARTE undergo a rapid decline, as many of these cells are used to destroy the large DB. This rapid formation and elimination of activated CARTE coupled with a relatively slow formation of the CARTM cells15 contribute to the observed biexponential CART kinetic profiles of subject UPN1 (Figure 1 b). In contrast, relatively low DB and administered CART doses are associated with a slow formation and elimination of the activated CARTE that contribute to the lower and less distinct bi‐exponential CART kinetic profiles observed in subject UPN3 (Figure 1 b). CART are a “living drug” that exhibit unique target‐mediated drug disposition characteristics.16, 17, 18 In contrast with most drugs that are eliminated from the body after administration, the CART “live and grow” in the presence of the target cells. As the disease burden remains low, the CARTE populations lose its feeding sources and continue to decline and, thus, predisposing the patient to potential relapse. The inflammatory response characterized by elevation of the cytokine IL‐6, IL‐10, and IFNγ reaches its peak between 15 and 23 days postinfusion7 and the magnitude of the cytokine elevation is highly correlated with the baseline DB. As the DB declines, the inflammation subsides1, 3 (Figure 1 b).
The ability of the final QSP model in describing the relationships between the DB/CART doses and CART kinetic profiles in peripheral blood of the patients with CLL is examined with the external validation data set. As clearly shown in Figure 2 a, the log‐transformed Cmax and AUC0–28 (mean ± SD) from the simulation using our QSP model is similar to the observed values in patients with CLL treated with CART.13 The quantitative effect of DB on cytokine/CART kinetic profiles in the peripheral blood of patients with CLL treated with CART has never been reported in published literature. Therefore, we decided to compare the trends of the simulated relationships between blood DB and Cmax of the CART/pro inflammatory cytokine IFNγ in peripheral blood of patients with CLL to the reported relationships between bone marrow DB and Cmax of the CART/pro inflammatory cytokine IFNγ in peripheral blood of patients with ALL.13, 14 The simulated relationships between disease burden and Cmax of CART and IFNγ using our QSP model in peripheral blood of patients with CLL share a similar trend with those observed in patients with ALL13, 14 (i.e., higher DB is associated with higher Cmax of the CART and IFNγ; Figure S2 and Figure S3). Taken together, these results suggested that the ability of our QSP model in describing the relationships between the DB/CART doses and CART/cytokine kinetic profiles are relatively consistent with the observed clinical findings in cancer patients treated with CART.
Figure 2.
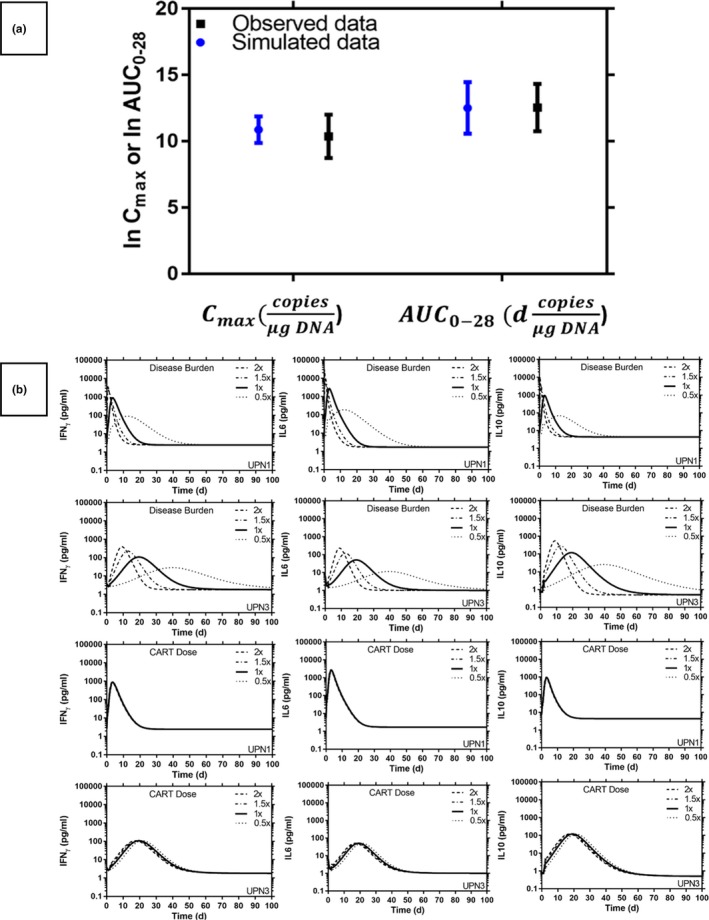
(a) External validation for the final quantitative systems pharmacology (QSP) model using the mean maximal concentration of chimeric antigen receptor T‐cell (CART) peak plasma concentration (Cmax) and the mean area under the curve of the time‐concentration curve from t = 0 to t = 28 days (AUC 0–28) of the typical kinetics of CART in chronic lymphatic leukemia (CLL). Symbols and bars represent the predicted and observed log‐transformed mean and SD values. (b) Simulated CART and cytokine kinetic profiles with different B‐cell disease burden and anti‐CD19 CART doses in a patient with CLL, CART baseline in peripheral blood (PB): 1.1 × 109 cells (UPN1) and 1.4 × 107 cells (UPN3), disease burden baseline in PB: 4.14 × 109 cells (UPN1) and 9.3 × 108 cells (UPN3). IFNγ, gamma interferon; IL, interleukin.
Simulations are performed to investigate the effect of DB and CART dose to the cytokines level (Figure 2 b). Patients with a higher baseline of DB are associated with higher cytokine and CART Cmax after infusion and, therefore, are likely to experience severe CRS toxicity (Figure 2 b and Figure S3 ). Hence, our results demonstrate that the developed QSP model using a well‐known complex mechanistic interaction between CART and the inflammatory cytokine is sufficient to support the DB‐correlated toxicity observed in clinical studies.1 Our results also support the clinical findings that the CART dose alone has a minor effect on the kinetic profile of cytokines and correlates poorly with the degree of CRS toxicity.1, 8 The observation that toxicity is more related with baseline DB than the administered CART doses is contrary to what is observed with most cancer chemotherapy in which the administered dose is often the major factor in drug‐limiting toxicity, possibly due to the unique kinetic and dynamic characteristics of the CART in patients with CLL.
An activated macrophage is a major source of the IL‐6 and can play an important role in the CRS of CART therapy.19 However, due to lack of the kinetic profiles of the activated macrophage in our data, attempts to build the activated macrophages into our model are unsuccessful. Furthermore, other cytokines, such as monocyte chemo‐attractant protein‐1 and macrophage inflammatory protein‐1β, can play an important role in the CRS and other toxicities associated with CART therapy.20 It has been shown that DB in the bone marrow compartment is correlated with the CART level in blood and, hence, can play a role in the CART kinetics.13, 14 However, the attempt to add the bone‐marrow tumor compartment in our QSP model was unsuccessful due to lack of sufficient tumor burden kinetic data in the bone marrow for this study. Therefore, further study with more experimental data and validation is needed to refine this QSP model in order to increase the confidence and precision in model prediction.
Several mathematical models have been proposed to describe the kinetics of CRS in human subjects. Yiu et al.21 developed a complex mathematical model to describe the kinetics of nine cytokines in six human volunteers experiencing CRS during the phase I clinical trial of a monoclonal antibody that was designed to simulate a regulatory T‐cell response. However, 90 system parameters from a set of complex second‐order ordinary differential equations are needed to be optimized in order to describe the observed kinetics of the 9 cytokines. Therefore, it is unlikely that this complex model can be supported by the limited cytokine kinetic data included in our study. Mathematical models have been developed to describe the complex interaction between the activated neutrophils/monocyte and pro inflammatory cytokines in bacteria‐induced sepsis, but none of these models can be used to investigate the CRS and CART cells in human subjects without major modifications.22 Recently, several mathematical models have been developed to describe the biologics of the CART.8, 9, 10 However, no clinical data are used to develop and validate these models,8, 9, 10 and, therefore, the ability of these models in describing the observed kinetics of cytokines and CART is questionable.
To our best knowledge, this is the first proposed QSP model that can describe the observed clinical data from patients with cancer treated with CART therapy. This QSP model is a valuable tool for deepening our understanding of how the mechanism of action connects to the clinical outcomes and, therefore, may serve as an important model‐based platform to guide the development and personalized dosing of the CART therapy. As shown by the well‐known “learn‐confirm” approach proposed by Sheiner,23 the QSP modeling and simulation can be used to learn from experience and confirm what has been learned in order to improve the efficiency of the clinical development of CART therapy. First, the clinical data collected in the early phase I data can be used to develop the basic QSP framework (phase I QSP model) to increase our understanding of how the mechanism of action of CART therapy connects to the toxicity (safety) and possible early efficacy signals in patients. Then the modeling and simulation results with the phase I QSP model are used to design a subsequent phase II study in order to acquire new and more diverse data to refine the QSP model for predicting future clinical outcomes of safety and efficacy. This refined phase I/II population QSP model should be able to quantify the effects of specific patient factors, such as disease and performance status, prior chemotherapy, and genetic predisposition on the QSP model parameters that govern the clinical safety and efficacy in the studied population. Hence, this population QSP model will define the exposure–response (safety and efficacy) relationships that offer insights into how the phase III study protocol may be modified based on individual patient characteristics in order to decrease toxicity, increase the persistence of CARTE population, and potentially decrease relapse rates of cancer.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
D.H. and C.M.N. wrote the manuscript. D.H. and C.M.N. designed the research. D.H. performed the research. D.H. and C.M.N. analyzed the data. C.M.N. contributed new reagents/analytical tools.
Supporting information
Supplementary Material, Tables and Figures.
References
- 1. Brudno, J.N. & Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yip, A. & Webster, R.M. The market for chimeric antigen receptor T cell therapies. Nat. Rev. Drug. Discov. 17, 161–162 (2018). [DOI] [PubMed] [Google Scholar]
- 3. Schuster, S.J. et al Chimeric antigen receptor T cells in refractory B‐cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neelapu, S.S. et al Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maude, S.L. et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tran, E. , Longo, D.L. & Urba, W.J. A milestone for CAR T cells. N. Engl. J. Med. 377, 2593–2596 (2017). [DOI] [PubMed] [Google Scholar]
- 7. Kalos, M. et al T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanson, S. et al Toxicity management in CAR T cell therapy for B‐ALL: mathematical modelling as a new avenue for improvement. bioRxiv, 1–7 (2016). 10.1101/049908. [DOI] [Google Scholar]
- 9. Hopkins, B. , Tucker, M. , Pan, Y. , Fang, N. & Huang, Z. A model‐based investigation of cytokine storm for T‐cell therapy. IFAC‐PapersOnLine 51, 76–79 (2018). [Google Scholar]
- 10. Mostolizadeh, R. , Afsharnezhad, Z. & Marciniak‐Czochra, A. Mathematical model of chimeric anti‐gene receptor (CAR) T cell therapy with presence of cytokine. Numer. Algebr. Control Optim. 8, 63–80 (2018). [Google Scholar]
- 11. Brattsand, R. & Linden, M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment. Pharmacol. Ther. 10 (suppl. 2), 81–90 (1996); discussion 91−92. [DOI] [PubMed] [Google Scholar]
- 12. Teachey, D.T. et al Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T‐cell therapy for acute lymphoblastic leukemia. Cancer Discov. 6, 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mueller, K.T. et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brentjens, R.J. et al CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Boer, R.J. & Perelson, A.S. Quantifying T lymphocyte turnover. J. Theor. Biol. 327, 45–87 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ng, C.M. , Bai, S. , Takimoto, C.H. , Tang, M.T. & Tolcher, A.W. Mechanism‐based receptor‐binding model to describe the pharmacokinetic and pharmacodynamic of an anti‐alpha5beta1 integrin monoclonal antibody (volociximab) in cancer patients. Cancer Chemother. Pharmacol. 65, 207–217 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Ng, C.M. , Stefanich, E. , Anand, B.S. , Fielder, P.J. & Vaickus, L. Pharmacokinetics/pharmacodynamics of nondepleting anti‐CD4 monoclonal antibody (TRX1) in healthy human volunteers. Pharm. Res. 23, 95–103 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Mager, D.E. Target‐mediated drug disposition and dynamics. Biochem. Pharmacol. 72, 1–10 (2006). [DOI] [PubMed] [Google Scholar]
- 19. Norelli, M. et al Monocyte‐derived IL‐1 and IL‐6 are differentially required for cytokine‐release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 24, 739–748 (2018). [DOI] [PubMed] [Google Scholar]
- 20. Hay, K.A. et al Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor‐modified T‐cell therapy. Blood 130, 2295–2306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yiu, H.H. , Graham, A.L. & Stengel, R.F. Dynamics of a cytokine storm. PLoS One. 7, e45027 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi, Z. , Wu, C.H. , Ben‐Arieh, D. & Simpson, S.Q. Mathematical model of innate and adaptive immunity of sepsis: a modeling and simulation study of infectious disease. Biomed. Res. Int. 2015, 504259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 61, 275–291 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material, Tables and Figures.