

Abstract
Testing novel drugs on fellow human beings is fraught with potential ethical concerns; however, developing drugs to treat the wide spectrum of human diseases and disorders is a moral imperative. How do we best navigate the balance between protecting the individual vs. the greater good? Global government regulatory bodies are accountable for ensuring that medical experiments on human subjects are appropriately justified and subject to close oversight. In this article, we focus on two major global health authorities, the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), and the path to legally treating humans with new investigational products.
The US Food and Drug Administration and the European Medicines Agency
The US Food and Drug Administration (FDA) and European Medicines Agency (EMA) are health authority bodies that regulate the use of investigational drugs within the United States and the European Union, respectively. In addition, investigational review boards (IRBs) in the United States and ethics committees (ECs) in the European Union must approve the use of drugs in humans; these entities are separate from health authorities, and their primary responsibility is ensuring the protection of human subjects who participate in studies testing new drugs.
The FDA is a part of the executive branch of the US government under the Department of Health and Human Services. As per their mission statement, “The Food and Drug Administration is responsible for protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drugs, biological products, and medical devices; and by ensuring the safety of our nation's food supply, cosmetics, and products that emit radiation.”
The EMA was inaugurated in 1993 in an effort to ensure harmonization of the assessments and approval of drugs in Europe, and ensure fair access to innovative drugs to all European patients with no distinction of borders. Over the years, the European pharmaceuticals system has been evolving toward centralization under the EMA's umbrella, always with patient safety and access as highest priority.
Basis of ethical human medical experimentation
Global health authorities (HAs), including the FDA and the EMA, adhere to a set of principles called Good Clinical Practices (GCPs), which are the basis of modern human medical experiments and were developed to ensure the protection of human subjects participating in clinical trials. GCPs are rooted in, among other precedents, the Nuremberg Code (see Box 1: Nuremberg Code). The Nuremberg Code, which describes the tenets of ethical human research, was developed as a result of the Nuremberg trials of 1947.1 During these trials, Nazi physicians claimed that horrific experiments conducted on prisoners were not illegal, as they adhered to common international research practice. In response, the Code outlines the conditions under which medical experimentation on human subjects is acceptable.
Box 1. The Nuremberg Code.
| (1) The voluntary consent of the human subject is absolutely essential. |
| (2) The experiment should be such as to yield fruitful results for the good of society, unprocurable by other methods or means of study, and not random and unnecessary in nature. |
| (3) The experiment should be so designed and based on the results of animal experimentation and a knowledge of the natural history of the disease or other problem under study that the anticipated results will justify the performance of the experiment. |
| (4) The experiment should be so conducted as to avoid all unnecessary physical and mental suffering and injury. |
| (5) No experiment should be conducted where there is an a priori reason to believe that death or disabling injury will occur; except, perhaps, in those experiments where the experimental physicians also serve as subjects. |
| (6) The degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment. |
| (7) Proper preparations should be made and adequate facilities provided to protect the experimental subject against even remote possibilities of injury, disability, or death. |
| (8) The experiment should be conducted only by scientifically qualified persons. The highest degree of skill and care should be required through all stages of the experiment of those who conduct or engage in the experiment. |
| (9) During the course of the experiment, the human subject should be at liberty to bring the experiment to an end if he has reached the physical or mental state where continuation of the experiment seems to him to be impossible. |
| (10) During the course of the experiment, the scientist in charge must be prepared to terminate the experiment at any stage, if he has probable cause to believe in the exercise of the good faith, superior skill, and careful judgment required of him, that a continuation of the experiment is likely to result in injury, disability, or death to the experimental subject. |
Today, GCPs are detailed in a guidance document, International Conference on Harmonization (ICH) E6(R2),2 generated by a consortia of global HAs. In addition to guidance documents, such as ICH, detailed requirements governing all aspects of drug development, including manufacturing, nonclinical studies, clinical trials, safety monitoring, efficacy assessments, marketing, and postmarketing surveillance, are codified in the laws and regulations of global regulatory bodies.3
In this article, we discuss the information required by the FDA4 and the EMA5 to initiate and conduct medical experiments in human subjects. To enable the study of investigational drugs in human subjects, documentation must be submitted and reviewed by these HAs prior to administering an investigational drug to a human subject. In the United States, the initial submission to permit use of an investigational drug in a clinical setting is called an investigational new drug (IND) application. In the European Union, this documentation is submitted within a clinical trial application (CTA).
Data Required to Support Initial Clinical Trials
Administering a new drug to humans has inherent risk. Scientists, clinicians, and regulators strive to continuously improve how drug candidates are evaluated prior to the first‐in‐human (FIH) administration to minimize this risk as much as possible.
Years of research in multiple specialties are required to produce the totality of evidence necessary to support advancing a new drug into human trials. The company sponsoring the development of a new drug (Sponsor) is required to provide a robust data set describing how the drug is made and determined to be pure and potent, the results of testing the effects of the drug in animals, and the plans for exploring exposure in humans as safely as possible. Here, we review the data required to support filing an IND in the United States6, 7 or a CTA in the European Union.8, 9
Chemistry, manufacturing, and control information
Before a new drug can be administered to humans, the Sponsor must assure regulators that the drug is made under controlled conditions, with ongoing tests to ensure that the drug meets prospective criteria (e.g., identity, purity, potency, and stability). These data are required to show that a drug is what it is supposed to be, without any contaminants, and that it will maintain its purity and potency for at least the duration of the studies. Information on both the drug substance (the active pharmaceutical ingredient) and the drug product (the formulated drug ready for administration) must be submitted to the regulators. These data evolve over time as the Sponsor optimizes production and formulation of the drug.
Information on the drug substance should include the proper identification, quality, purity, and strength of the active ingredient, with an emphasis on the identification and control of raw materials and the new drug substance.
Information on the drug product is also provided, and similar to the drug substance section, this should include data supporting the assays and acceptable results for assessing its identity, strength, quality, and purity. It is also necessary to demonstrate stability (evidence on how the quality varies with time under the influence of a variety of environmental factors, such as temperature, humidity, light, etc.) for at least the duration of the clinical trial to inform the drug product shelf life and recommended storage conditions. Early in development, the drug product may be in a variety of forms; for example, it may be simply the drug substance handfilled into capsules, or it may be a biologic purified from a small‐scale production.
Information on the placebo, if included in the clinical study, is also required.
Nonclinical pharmacokinetics, pharmacology, and toxicology information
A multitude of issues must be explored prior to dosing a human with a new drug. What happens biologically when a person takes a new drug? Does the drug stay in the body long enough to have any effect? Is it effectively delivered to the site of action? Is the drug metabolized, and what is the potential impact of the metabolites? Does the drug have an impact on how the body functions? If so, what body systems are impacted, and at what doses? Does the drug have any long‐term positive or negative effects? Does the drug have potential to treat a disease or condition? If a drug has potential therapeutic benefit, do these benefits outweigh the potential risks?
Given the risks of administering a new drug to humans, a range of studies are conducted beforehand in test tubes, human and animal‐derived cell lines, and animal models to explore the effects of the drug.
Distribution, metabolism, and pharmacokinetics
Pharmacokinetics refers to how a drug is processed through the body. These studies explore how the drug is absorbed once administered, where it distributes to in the body, how it is metabolized, and how it is excreted. These studies range from the identification of small molecule drug metabolites in vitro to the measurement of the drug in the blood, urine, and feces obtained from animals that are administered the drug.10, 11
Pharmacology
Pharmacology12, 13 is how a body responds to a drug and can be broken down into three categories: primary pharmacodynamics, secondary pharmacodynamics, and safety pharmacology.
Primary pharmacodynamics explores whether the drug has the intended effect in vitro: if it is a kinase inhibitor, does it inhibit the kinase in a test tube or cell line? If the drug is supposed to bind a cell‐surface receptor, does it bind that receptor expressed on a human cell line? In other words, does the drug have the primary effect one would anticipate and at what concentration?
Secondary pharmacodynamics explores additional effects of the drug: does it bind any other proteins or antigens or receptors, and with what affinity? Does it inhibit any enzymes other than the intended target, and is the inhibition competitive or not? Secondary pharmacodynamics are key to assessing whether the drug has additional impact other than the intended effect, which may contribute to a better understanding of the overall safety profile of the drug. For example, this may include a dose‐ranging study to assess drug binding with other human substrates in vitro. In the case of a kinase inhibitor, these studies would reveal what other enzymes to which the drug may bind and at what concentration. This information is critical to understanding the drug's possible “off‐target” effects or the unintended side effects of drug treatment or overexposure (see Box 2: Bial‐Portela Study).
Box 2. Bial‐Portela study26, 27 .
| In 2016, Bial‐Portela was conducting a single‐ascending dose (SAD) and multiple‐ascending dose (MAD) study with BIA‐102474‐101, the 10th fatty acid amide hydroxylase to enter the clinic, as a potential treatment for pain. A total of 90 subjects completed treatment without incident in the SAD and first four MAD cohorts. In the fifth MAD cohort, one subject became ill after the fifth dose and was admitted to the hospital. Despite this, remaining subjects in the cohort continued to be dosed. Four of the five subjects who were dosed were eventually hospitalized, and the first subject incurred brain injury that resulted in death. BIA‐102474‐101 binds the enzyme fatty acid amide hydrolase (FAAH); however, it is a relatively unselective drug and, at higher exposures, it may bind other targets as well (known as “off‐target binding”). Doses administered in the affected cohort of the BIA‐102474‐101 study were several‐fold higher than required to fully inhibit FAAH. This tragedy led to a revision to the EMA's first‐in‐human (FIH) guidance, with revised recommendations for Sponsors, such as considering all FIH drugs as high risk; incorporating detailed stopping rules at the subject, cohort, and study levels; and better integration of the pharmacokinetic/pharmacodynamic data and modeling to determine the appropriate doses and schedule.28 |
Safety pharmacology experiments are designed to assess the impact of the drug on vital functions and are generally conducted in animal models, such as rodents and nonhuman primates. The core battery of these assessments includes the central nervous system, cardiovascular system, and respiratory system in animal species that are considered pharmacologically relevant. Selection of the pharmacologically relevant species is based on a number of factors, including whether the drug acts on the animal's system in a way that is similar to how it would act in humans (see Box 3: TeGenero Study for case study). The Sponsor may also conduct targeted safety pharmacology studies in other systems if there may be a known or suspected impact.
Box 3. TeGenero study29, 30 .
| In 2006, TeGenero AG initiated a phase I study in healthy human volunteers with a single intravenous dose of their anti‐CD28 antibody, TGN1412. TGN1412 was a candidate for the treatment of B‐cell lymphomas and autoimmune disorders based on its potential to expand the T‐cell population in the absence of T‐cell receptor activation. TGN1412 had been assessed extensively in nonclinical studies; importantly, these studies did not appropriately predict the human immune response. The starting dose in humans was 500× below the no‐observed‐adverse‐effect level (NOAEL). Six healthy volunteers received TGN1412 doses within minutes of each other. Within 90 minutes, the subjects began to feel ill and, within 24 hours, all 6 were hospitalized with cytokine‐release syndrome that resulted in organ failure. All subjects survived, although they may have long‐term disability, including one subject who required amputation of fingers and toes. The analysis of this tragedy informed the European Medicines Agency's subsequent guidance on first‐in‐human trials.28, 31 The guidance included recommendations for Sponsors to consider using the minimal anticipated biological effect level rather than NOAEL in determining the human starting dose; staggering drug administration in subjects; ensuring that dosed subjects are observed for adverse events prior to initiating dosing in subsequent subjects; including additional risk mitigations for drugs that are considered to be “high risk”; and ensuring that phase I studies are conducted by qualified investigators with access to emergency medical services. |
Toxicology
Toxicology14, 15, 16 studies are conducted primarily in animal models and are designed to predict, as much as possible, any potential toxicities (i.e., adverse or negative outcomes) in humans who may be exposed to the drug. The extent of these studies varies by indication: drugs designed to treat life‐threatening diseases may not require the same level of evidence to support FIH studies as drugs designed to treat a more chronic disorder. For example, prior to initiating clinical trials, most drugs are evaluated for the risk of genotoxicity (when the drug changes the recipient's DNA such that a mutation could be passed to their offspring) using a bacteria‐based assay, whereas oncology drugs are not required to evaluate genotoxicity until prior to marketing.
Generally, toxicology studies are required for all drugs to explore the adverse effects of the drug when administered to one or more animal models as a single dose and after multiple doses. These studies attempt to predict the highest dose that could potentially be administered to humans before encountering toxicities. Once toxicities are identified in animals, these studies assess whether or not the toxicities are reversible. For oncology drugs, data from studies of 28 days in two different animal species support continuous dosing in patients, as long as the patient is deriving benefit. Toxicology studies with a duration similar to that in the planned clinical trial are required for nononcology drugs. Results from these studies are critical in evaluating whether the potential benefit outweighs the potential risk of administering the drug to humans.
Additional toxicology studies, including the drug's risk of causing cancer or reproductive harm, may be evaluated later in the lifecycle of the drug development.
Clinical protocol
The clinical protocol is the step‐by‐step detailed plan of exactly how the new drug is to be evaluated in humans. The critical chemistry, manufacturing, and control (CMC) and nonclinical safety assessment data collected to date are briefly summarized and then integrated into the study plan. For example, the chemistry of a small molecule drug will determine whether the drug can be formulated into an orally available pill or if it will be administered intravenously. Predictions from pharmacokinetic studies will drive how often the drug is administered, and the nonclinical safety findings in animal models from the toxicology studies will inform the drug dose and how human subjects are monitored for potential adverse side effects.
Key aspects of the protocol include the following:
Study population: In designing the clinical trial in which a new drug is administered to humans for the first time (an FIH study), Sponsors decide whether it is safe to test the drug in healthy volunteers or whether it would be more appropriate to test the drug in the intended patient population, where the acceptable risk tolerance may be higher. For chronic diseases, it is critical that the clinical safety profile supports long‐term administration. It may be preferable to initially evaluate these drugs in healthy volunteers, where the safety assessment is not complicated by the underlying disease. In contrast, many oncology drugs have significant adverse effects, and may only be initially tested in patients with late‐stage cancer, in whom the risk/benefit profile of the drug is acceptable. Another aspect to assess is whether the drug may have negative long‐term consequences in the treated population. For example, in the 1990s, a study was conducted in patients with renal failure who were administered recombinant human erythropoietin to treat anemia. Some of these patients generated antibodies against their endogenous erythropoietin in response to treatment with the recombinant erythropoietin, leading to an autoimmune disorder called pure red cell aplasia.17
Dose selection: Based on the nonclinical safety assessments, a starting dose that is unlikely to result in adverse side effects is selected (see Table 1). A common FIH study design is to test the drug at a low dose in a small number of subjects and assess for safety concerns for a duration determined by the drug's pharmacokinetic and pharmacodynamic profile. If the safety is acceptable, a higher dose level is similarly assessed in another small group of subjects. This dose‐escalation paradigm is continued until an appropriate pharmacodynamic dose level is safety attained, unacceptable safety findings are noted, or the dose level approaches an exposure at which unacceptable safety findings were observed in nonclinical studies. In some studies, additional subjects are assessed at this dose level to obtain further safety data. In some patient populations, such as rare diseases, in which there are only a small number of patients in which to assess the drug's safety and efficacy, it may be important to select a dose that may have the potential for direct benefit for patients enrolled in an FIH study.18
Safety monitoring plan: A clinical safety monitoring plan is designed to assess subjects for any toxicity that may have been identified by the nonclinical studies, may be of particular importance to the patient population, or may be a class effect of drugs that hit the same target or pathway. The Sponsor will list which potential adverse events may arise, at what exposures, and at what duration after the drug is administered. Based on this, subjects will undergo safety assessments during the conduct of the clinical study.
Table 1.
Determining a safe starting dose (FDA and EMA Guidance)
| Step 1: Determine the NOAEL |
| NOAEL is the highest dose tested in a nonclinical study that does not produce a significant increase in adverse effects compared to the control group. |
| Alternatively, Sponsors may use MABEL, which is typically more conservative than NOAEL, as it is based on any pharmacological activity and not toxicity and may be used for drugs with higher risk. |
| Step 2: Calculate the HED |
| HED is calculated from the exposure defining the NOAEL in each nonclinical toxicology study most commonly by normalizing to body surface area or body weight. |
| Alternatives include scaling based on drug levels if dose is limited by local toxicity (e.g., topical drugs) or based on volume if administration is limited by compartment (e.g., intrathecal administration). |
| Step 3: What is the most appropriate species? |
| Sponsors will have several HEDs based on various nonclinical studies. The most appropriate HED is selected based on the most appropriate pharmacologically relevant species. |
| Interestingly, the most sensitive species is not always the most relevant (e.g., low doses of NSAIDs causes gastrointestinal lesions in beagles but are well tolerated in humans).32 |
| Step 4: The safety factor |
| Nonclinical toxicology studies may not perfectly predict the human adverse event profile. To account for differences between nonclinical models and humans, Sponsors apply a safety factor, typically 10‐fold, to the HED to get the recommended starting dose. |
| This safety factor may be increased for higher risk drugs (e.g., if there is a nonmonitorable toxicity or nonlinear pharmacokinetics). Alternatively, this factor may be reduced for a drug that belongs to a well‐characterized class or if toxicities are monitorable, predictable, and reversible. |
| Step 5: Consider the estimated PAD |
| Sponsors should consider the estimated PAD to determine a starting dose. The FDA suggests that the PAD may be a more sensitive estimate of possible toxicities and may warrant lowering the starting dose to below the PAD. The EMA recommends that the starting dose is below the PAD in healthy volunteer studies. |
Investigator's Brochure
The Investigator's Brochure (IB) is similar to prescribing information (or drug label), but for an investigational drug. It summarizes all of the known nonclinical and clinical safety and efficacy information of the drug; the intent is to inform clinicians of the potential toxicity of the drug. A key section of the IB defines the adverse events that are considered “expected” for the purposes of expedited safety reporting for the drug based on observations to date. The IB is updated at least annually for as long as the drug is undergoing clinical trials to ensure the information is current.
INDs AND CTAs: Similarities and Differences
Although the purpose of both INDs and CTAs is to enable studies of investigational drugs in people, the two types of submissions fulfill different requirements and are thus composed of overlapping yet nonidentical components.
In the United States, the initial IND includes multiple forms specific to the FDA, all nonclinical study reports (including validation reports of bioanalytical methods), nonclinical summaries (key information from the reports summarized concisely), detailed CMC information, as well as the protocol and IB (see Data to Support Initial Clinical Trials section above and 21 Code of Federal Regulations (CFR) 312.23).19 Once an IND has been cleared by the FDA (see details below), multiple studies can be conducted under the same IND, as per the FDA's legal requirements, the CFRs (21 CFR 312.22).19 These studies must use the same investigational drug and be used in patients with the same disease (i.e., the same indication), but after the initial clearance, subsequent protocols can be initiated immediately after submission to the IND without a waiting period (subject to approval by an IRB) and assuming appropriate supporting documents are available, such as nonclinical reports (21 CFR 312.30).19 The majority of the IND, including the nonclinical and clinical summaries, is only submitted once as part of the initial IND. Subsequent new protocols submitted to the IND are considered IND amendments. Amendments are made to the IND throughout its lifecycle and also encompass submissions to update the drug product information and new nonclinical and clinical reports (see IND Maintenance section below).
Some research studies may not require the filing of an IND (see Figure 1).20 For studies that require an IND, there are two IND categories: commercial and research INDs, differentiated by the entity that submits the IND and the purpose of the clinical research (see Figure 1). Commercial INDs are usually submitted by biopharmaceutical drug companies with the intent of eventually submitting a marketing application to sell the drug commercially. Research INDs are submitted by investigators to test a new dose or indication of an existing drug, but the data to be generated from the study are not intended to be used for a subsequent application for market approval.
Figure 1.
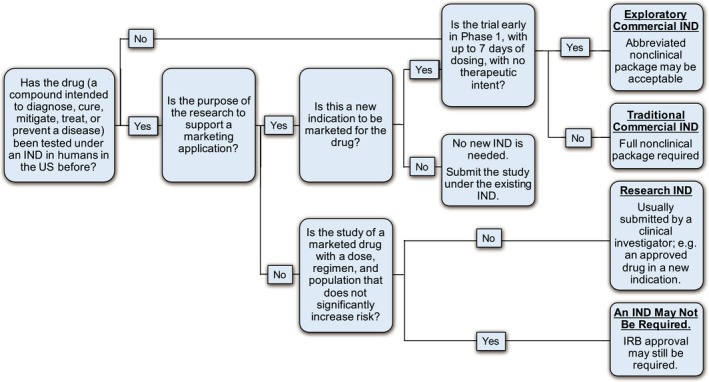
To (IND) or not to IND… IRB, instutional review board. Note: Exceptions are possible. Information summarized from refs. 20, 21, 22.
For drugs that are in very early development, an abbreviated IND, called an exploratory IND, may be submitted to allow drug sponsors to evaluate up to five chemical entities or formulations simultaneously, supported by limited nonclinical data (see Figure 1).21 Exploratory INDs provide the opportunity to study pharmacokinetic and target interaction early in drug development. When a lead candidate drug has been selected, the exploratory IND is closed, and a traditional IND is opened.
In the European Union, each interventional clinical study requires a new CTA.22 Because of the different purposes of these submissions, the documentation required for a CTA is not identical to that for an IND. For a CTA, the four main documents are the protocol, informed consent form, IB, and Investigational Medicinal Product Dossier (IMPD), which contains CMC data. Additional information such as European Union–specific forms, questionnaires, or patient diaries to be used in the trial, and insurance certificates, must also be included.
Lifecycle of an IND and CTA
IND initial clearance
INDs are not “approved”; they are “cleared.” The FDA reviews initial INDs in 30 days (21 CFR 312.20).19 An IND can be opened with a study of any phase (i.e., phase I, II, or III; 21 CFR 312.21).19 Questions from the FDA that arise during the review of the IND are communicated to the Sponsor, usually during the last 2 weeks of the 30‐day review. A teleconference may be needed to clarify these issues. The Sponsor then addresses the FDA's concerns by providing additional information and/or revising the IND documents as needed. If the FDA's concerns are adequately addressed such that they consider the study safe to proceed, the IND is cleared after 30 days. If, however, the FDA's concerns remain, they may place the IND on full or partial clinical hold (21 CFR 312.42).19 A full hold means that no clinical study can be initiated under the IND until the FDA's issues are satisfactorily addressed. A partial hold means the clinical study and any other studies submitted under the IND may proceed with certain constraints (e.g., the investigational drug may not be administered above a certain dose). The Sponsor must then provide a complete response to the clinical hold, which also has a 30‐day review period. The FDA may lift the hold if the response addresses the identified issues and clears the IND, thereby allowing studies under the IND to proceed, or the FDA may maintain the clinical hold. Because future protocols submitted under an IND are allowed to proceed without the review, the FDA may be cautious when clearing the initial IND. However, the FDA may place an IND on clinical hold if any concerns arise at any time during the lifecycle of the IND; this prerogative is not restricted to the first 30 days of the IND submission.
CTA initial approval
Review, approval, and maintenance of CTAs in the European Union are currently ruled by Directive 2001/20/EC, a nonbinding set of rules for the European Union Member States (MS; countries that are part of the European Union) to interpret and implement with a relative degree of freedom. In replacing this Directive with Clinical Trial Regulation (CTR) 536/2014,8 which will go into effect in 2020, the European Commission aims to enhance the harmonization of the CTA process across MS for a more efficient and consistent supervision of clinical trials in the European Union.
The shift from Directive to Regulation is significant and will enable simultaneous submissions of HA and EC applications. Beyond the pure procedural changes, the new CTR will address other key issues, such as increased transparency and access and more efficient linkage to the EudraVigilance database for a more consolidated generation and monitoring of safety data. (EudraVigilance is the EMA's system for monitoring the safety of medicines by facilitating electronic reporting of suspected adverse reactions and enabling the early detection of potential safety issues.)
Current directive 2001/20/EC
The Directive is applicable to all MS, where national laws apply as well. This leads to variance in interpretation and local practice (e.g., between individual MS HAs and ECs), nuances of format and content, review timelines, and more.
The introduction of the Voluntary Harmonisation Procedure (VHP) in 2009 was the first attempt of better alignment between MS. The VHP enables a Sponsor to submit a CTA to multiple MS in parallel and perform a single combined scientific review. Although single‐country national CTAs are still an available option, here, we will focus on VHP as a precursor to the upcoming CTR 536/2014.
The VHP procedure extends through three phases:
Request for VHP application and validation
Assessment by selected MS
Formal submission of the VHP‐approved CTA to the local HAs
The average timelines for a national CTA is 60 days plus any additional time the Sponsor requires to respond to any questions from the HA, whereas the average time for CTAs via VHP from start to finish was 52.5 days, including time for the Sponsor to respond to questions (see Figure 2).23, 24
Figure 2.
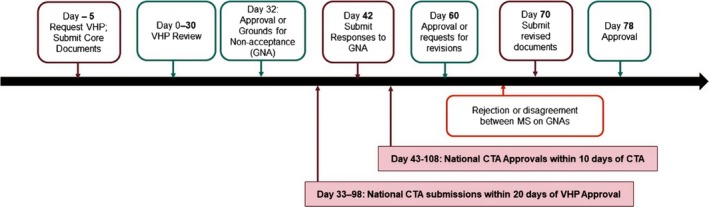
Standard Voluntary Harmonisation Procedure timelines. Average timelines for national clinical trial application (CTA) evaluation: 60 days + clock stop for questions. Average timeline for Voluntary Harmonisation Procedure (VHP): 52.5 days, including time for questions. Information summarized from refs. 23, 24.
New CTR 536/2014
There are multiple innovations and efficiencies with the new CTR. Besides enabling submissions through one gateway and using a single set of documents, the CTR permits concurrent and harmonized involvement of the ECs, which will still be governed by local rules but will need to adhere to the new procedure timelines. This will lead to more reliable and predictable review and approval timelines for the European Union CTAs.
CTR CTAs will be submitted based on a single dossier split in two modules, as shown in Table 2.
Table 2.
Modules for the New Clinical Trial Regulation CTAs
| Part 1: Study‐specific Assessed by all participating member states | Part 2: – Country and site‐specific Assessed by each member state separately |
|---|---|
| Application form | ICF |
| Protocol | Recruitment arrangements |
| IB | Rules of liability |
| IMPD | Suitability of investigators and trial sites |
| IMP manufacturing, labeling, and import | Financial compensation |
| Any HA advice received | Data protection requirements |
| PIP |
CTA, clinical trial application; HA, health authority; IB, Investigator's Brochure; ICF, informed consent form; IMP, investigational medicinal product; IMPD, Investigational Medicinal Product Dossier; PIP, pediatric investigational plan.
The two‐part assessment leads to a single approval per member state, replacing separate approvals by the local HA and EC.
The timelines for review and approval are supported by tacit approval, holding the participating MS accountable for their timeline commitment. The timelines for CTA approval under the CTR are expected to be comparable to current VHP timelines.
IND maintenance
Once an IND is in effect, there are three primary maintenance activities and responsibilities for Sponsors: amendments, safety reporting, and annual reports.
Amendments
The IND is often amended throughout its lifecycle. There are two types of IND amendments: Protocol Amendments and Information Amendments.
Protocol amendments are to ensure that the clinical investigations are conducted according to the protocols included in the application (21 CFR 312.30).19 Examples of protocol amendments include:
New protocol: As discussed above, an IND may contain multiple studies of the same investigational drug in the same patient population or indication. A protocol for a new clinical trial may be submitted to an IND that has cleared (i.e., an open IND). New studies may begin soon after the protocol has been submitted to the FDA and has been approved by the IRB. The IND submission should include a copy of the new protocol and a brief description of the most clinically significant differences from previous protocols.
Change in existing protocol: Sponsors must submit a protocol amendment to describe any changes in protocols that significantly affect the safety of subjects, the scope of the investigation, or the scientific quality of the study. A protocol change intended to eliminate an apparent safety hazard to subjects may be implemented immediately, provided that the FDA and the IRB are subsequently notified. The IND submission should include a brief description of the changes and a reference to the submission that contained the original protocol.
New investigator: The FDA should be notified within 30 days of the addition of a new investigator to conduct a study previously submitted to the IND (21 CFR 312.23).19 The submission should include the investigator's name, qualifications, reference to the previously submitted protocol, and other additional information.
Information amendments are any amendments to information essential to the investigational drug and can be categorized as relating to chemistry/microbiology, pharmacology/toxicology, clinical, statistics, or clinical pharmacology (21 CFR 312.31).19 These are submitted to the FDA as necessary but generally no more frequently than every 30 days. The submission should include a statement of the nature and purpose of the amendment.
Safety reporting
Sponsors must notify HAs and all participating investigators of potential serious risks associated with the use of the investigational drug based on prompt review of all relevant safety information (21 CFR 312.32).19 These include serious and unexpected suspected adverse reactions, findings from other studies, findings from animal or in vitro testing, or increased rate of occurrence of serious suspected adverse reactions. Each safety report, in narrative format, should be submitted as soon as possible but no later than 15 calendar days following the Sponsor's initial receipt of the information. Any unexpected fatal or life‐threatening suspected adverse reaction reports should be reported as soon as possible but no later than 7 calendar days following the Sponsor's initial receipt of the information. If applicable, relevant follow‐up information to an initial safety report must be submitted as a Follow‐up Safety Report as well.
Annual reports
Sponsors are expected to submit a brief report of the progress of the studies conducted under their IND application annually within 60 days of the anniversary date that the IND went into effect (21 CFR 312.33).19 This annual update and summary is intended to inform HAs of the progress of a drug's development program during the past year and identifies any potential issues or safety concerns in the program (including a summary of any issues beyond routine safety reporting), see Box 4: Annual Report Requirements. The Sponsor may report this information as outlined in the inset or use the Development Safety Update Report format as outlined in ICH E2F25 with prior approval from the FDA.
Box 4. Annual report requirements.
| Individual study information: A brief summary of each study under the investigational new drug (IND), the status (ongoing or completed) of each study, summary of subject enrollment to date, and overview of available results. |
| Summary information: Summary of clinical and nonclinical investigations, including the most frequent and serious adverse events, IND safety reports submitted in the past year, any deaths, subject discontinuations due to safety, information pertinent to an understanding of the drug's actions (e.g., dose response, bioavailability, etc.), list of nonclinical studies and findings, and any significant manufacturing or microbiological changes. |
| General investigational plan: Brief description of the overall plan for investigating the drug product for the following year, including rationale for study(ies), indication(s), general approach, kinds of clinical trials to be conducted, estimated number of subjects, and any risks of particular severity or seriousness anticipated on the basis of toxicological data. |
| Investigator's Brochure updates. |
| Significant phase I protocol modifications not reported to the IND in a protocol amendment. |
| Brief summary of significant foreign marketing developments. |
| If desired, a log of any outstanding business with response to the IND for which the Sponsor requests or expects a reply, comment, or meeting. |
IND withdrawal and inactivation
A Sponsor can withdraw an effective IND at any time without prejudice (21 CFR 312.38).19 The appropriate HAs should be notified, all investigations ended, all investigators notified, and all investigational drugs returned to the Sponsor or disposed of appropriately. If an IND is withdrawn for safety reasons, the Sponsor should inform the appropriate HAs, all investigators, and IRBs of the reason(s) for the withdrawal.
An IND can be placed on inactive status by the FDA or upon request by the Sponsor if no subjects are entered into clinical studies for 2 years or more, or if all investigations under an IND remain on clinical hold for 1 year or more (21 CFR 312.45).19 As with an IND withdrawal, all investigators should be notified and all drugs should be returned to the Sponsor or disposed of appropriately. An IND Annual Report is not required to be submitted to an IND on inactive status. The IND may be reactivated by submitting an amendment containing the proposed general investigational plan for the coming year and appropriate protocol(s). If an IND is on inactive status for 5 years or more, it can be terminated by the FDA.
CTA maintenance
Many of the same aspects of IND maintenance are applicable to CTAs as well.
Protocol amendments: Substantial changes to the protocol must be approved by the relevant HA and EC prior to implementation, unless changes are required to immediately protect the safety of study subjects.
Safety reporting: Safety reporting requirements are similar to those in the United States. Safety data in the European Union are reported via the EudraVigilance system.
Annual reports: Annual reporting to the EMA is typically in Development Safety Update Report format.
In addition, changes to the CMC information are submitted to the relevant HAs as an amendment to the IMPD.
CTA end‐of‐study notification
After the official end of a clinical study, as defined in the protocol, typically the last visit by the last subject enrolled in the study, Sponsors are required to notify the relevant HAs and ECs that a clinical trial has concluded.
INDs vs. CTAs: Summary
Above, we have outlined the process and requirements for initiating clinical trials with an investigational drug in the United States and the European Union. Although both INDs and CTAs require the same basic data set to support initiation of clinical trials in humans, the documentation required for HA review and the process for application review and approval differs considerably between the two. CTAs contain fewer documents than INDs, requiring less preparation time. INDs have well‐defined timelines to clearance (30 days); in contrast, there can be considerable variability in the approval process between each MS's HA and EC (e.g., parallel vs. sequential review, set or limited submission times, variable review lengths, etc.). With INDs, there is no cost or time delay to amend or add new protocols (assuming sufficient nonclinical and CMC information are already present in the IND), whereas substantial protocol amendments require CTA approval, and new protocols require new/separate CTAs. CTAs do not carry potential risk for clinical hold like INDs do; the CTA is either approved (perhaps with mandatory changes) or rejected.
Conclusion
Sponsors are required to consolidate data from CMC, pharmacology, pharmacokinetics, nonclinical toxicology, and clinical development into a cohesive plan to evaluate the potential safety and efficacy of a potential new drug in humans, while taking every necessary precaution to protect clinical study subject safety. Whether provided in an IND or CTA, the data required to support the initiation of a clinical trial of a new drug in humans are justifiably substantial, and significant time and resources are required to enable a successful submission and approval.
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
Acknowledgment
The authors would like to thank Katherine McKiernan for her expert contributions to this article.
All authors contributed equally to this article.
References
- 1. Shuster, E. Fifty years later: the significance of the Nuremberg Code. N. Engl. J. Med. 337, 1436–1440 (1997). [DOI] [PubMed] [Google Scholar]
- 2. International Council on Harmonisation (ICH) . Harmonised Guideline. Integrated addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2). Current Step 4 version. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf> (2016). Accessed January 25, 2019.
- 3. International Council on Harmonisation (ICH) . Guidelines website. <https://www.ich.org/products/guidelines.html> Accessed February 18, 2019.
- 4. US Food and Drug Administration (FDA) . Guidance search website. <https://www.fda.gov/regulatoryinformation/guidances/> Accessed February 18, 2019.
- 5. European Medicines Agency (EMA) . Scientific guidelines. <https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines> Accessed February 18, 2019.
- 6. US Food and Drug Administration (FDA) . Guidance for Industry: Content and format of Investigational New Drug applications (INDs) for Phase 1 studies of drugs, including well characterized, therapeutic, biotechnology‐derived products. <https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071597.pdf> (1995). Accessed January 25, 2019.
- 7. US Food and Drug Administration (FDA) . Guidance for Industry: IND meetings for human drugs and biologics, chemistry, manufacturing, and controls information. <https://www.gmp-compliance.org/guidelines/gmp-guideline/fda-guidance-for-industry-ind-meetings-for-human-drugs-and-biologics-chemistry-manufacturing-and-controls-information> (2001). Accessed January 25, 2019.
- 8. Clinical Trials Regulation EU No. 536/2014 . <https://ec.europa.eu/health/human-use/clinical-trials/regulation_en> (2014). Accessed January 25, 2019.
- 9. European Medicines Agency (EMA) . Draft Guideline on the requirements to the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials. EMA/CHMP/QWP/545525/2017. <https://www.ema.europa.eu/en/requirements-chemical-pharmaceutical-quality-documentation-concerning-investigational-medicinal> (2017). Accessed January 25, 2019.
- 10. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S3A: Note for guidance on toxicokinetics: the assessment of systemic exposure in toxicity studies. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S3A/Step4/S3A_Guideline.pdf> (1994). Accessed January 25, 2019.
- 11. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S3B: Pharmacokinetics: guidance for repeated dose tissue distribution studies. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S3B/Step4/S3B_Guideline.pdf> (1994). Accessed January 25, 2019.
- 12. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S7A: Safety Pharmacology studies for human pharmaceuticals. <https://www.ich.org/products/guidelines/safety/safety-single/article/safety-pharmacology-studies-for-human-pharmaceuticals.html> (2000). Accessed January 25, 2019.
- 13. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S7B: The nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf> (2005). Accessed January 25, 2019. [PubMed]
- 14. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline M3 (R2): Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf> (2009). Accessed January 25, 2019.
- 15. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S6(R1): Preclinical Safety evaluation of biotechnology‐derived pharmaceuticals. <https://www.ema.europa.eu/documents/scientific-guideline/ich-s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals-step-5_en.pdf> (2011). Accessed January 25, 2019.
- 16. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline S9: Nonclinical evaluation for anticancer pharmaceuticals. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S9/Step4/S9_Step4_Guideline.pdf> (2009). Accessed January 25, 2019.
- 17. Means, R.T. Pure red cell aplasia. Blood 128, 2504–2509 (2016). [DOI] [PubMed] [Google Scholar]
- 18. DeGeorge, J.J. et al Regulatory considerations for preclinical development of anticancer drugs. Cancer Chemother. Pharmacol. 41, 173–185 (1997). [DOI] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration (FDA) . Code of Federal Regulations Title 21 (21 CFR). Subpart B–Investigational New Drug Application (IND) and Subpart C–Administrative Actions. <https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRsearch.cfm?CFRPart=312> (2018). Accessed January 25, 2019.
- 20. US Food and Drug Administration (FDA) . Guidance for Clinical Investigators, Sponsors, and IRBs Investigational New Drug Applications (INDs) — Determining Whether Human Research Studies Can Be Conducted Without an IND. <https://www.fda.gov/downloads/drugs/guidances/ucm229175.pdf> (2013). Accessed January 25, 2019.
- 21. US Food and Drug Administration (FDA) . Overview of the exploratory IND: Differences from the traditional IND: Improving the quality of cancer clinical trials. <http://www.nationalacademies.org/hmd/~/media/92B1B2A5B6A14D8496B3FF315DF50763.ashx> (2007). Accessed January 25, 2019.
- 22. Medicines and Healthcare Regulatory Agency (MHRA) . Algorithm: Is it a Clinical Trial of a Medicinal Product? From MHRA Guidance, Clinical trials for medicines: apply for authorisation in the UK. <https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/317952/Algothrim.pdf> (2018). Accessed January 25, 2019.
- 23. Clinical Trials Facilitation Group . Results of the Voluntary Harmonisation Procedure 2009–2014. <http://www.infarmed.pt/documents/281/1432055/Results+of+the+Voluntary+Harmonisation+Procedure+Jan2015.pdf/374e969c-9c86-492b-996f-a5cb0f3a8582> (2015). Accessed January 25, 2019.
- 24. Heads of Medicines Agencies . Clinical Trials Facilitation Groups Guidance document for sponsors for a Voluntary Harmonisation Procedure (VHP) for the assessment of multinational Clinical Trial Applications. <http://www.hma.eu/fileadmin/dateien/Human_Medicines/01-About_HMA/Working_Groups/CTFG/2013_06_CTFG_VHP.pdf?sc_site=website> (2013). Accessed January 25, 2019.
- 25. International Council on Harmonisation (ICH) . Harmonised Tripartite Guideline E2F: Development Safety Update Report. <https://www.ema.europa.eu/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-26.pdf> (2011). Accessed January 25, 2019.
- 26. Eddleston, M. , Cohen, A.F. & Webb, D.J. Implications of the BIA‐102474‐101 study for review of first‐into‐human clinical trials. Br. J. Clin. Pharmacol. 81, 582–586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Temporary Specialist Scientific Committee (TSSC) Report . “FAAH (Fatty Acid Amide Hydrolase)”, on the causes of the accident during a Phase 1 clinical trial in Rennes in January 2016. <https://ansm.sante.fr/var/ansm_site/storage/original/application/744c7c6daf96b141bc9509e2f85c227e.pdf>. Accessed January 25, 2019.
- 28. European Medicines Agency (EMA) . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/07 Rev. 1. <https://www.ema.europa.eu/documents/scientific-guideline/guideline-strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational_en.pdf> (2017). Accessed January 25, 2019.
- 29. Attarwala, H. TGN1412: from discovery to disaster. J. Young Pharm. 2, 332–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clair, E.W. The calm after the cytokine storm: lessons from the TGN1412 trial. J. Clin. Invest. 118, 1344–1347 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Expert Scientific Group (ESG) on Phase One Clinical Trials . Final report. <https://webarchive.nationalarchives.gov.uk/20130105143109 / http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_073165.pdf> (2006). Accessed January 25, 2019.
- 32. Bluemel J., Korte S., Schenck E. & Weinbauer G., eds. The Nonhuman Primate in Nonclinical Drug Development and Safety Assessment, 1st Ed Elsevier/Academic Press, Amsterdam: (2015). [Google Scholar]
- 33. US Food and Drug Administration (FDA) . Guidance for Industry: Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. <https://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf%23search=%27guidekines+for+industry+sfe+starting%27> (2005). Accessed January 25, 2019.