

Abstract
For regulatory approval of a new medicine, the gold standard for demonstration of efficacy has traditionally been a minimum of two positive, adequate, and well‐controlled clinical trials. Nevertheless, drugs to treat cancer and rare diseases are usually approved based on a single and often uncontrolled pivotal trial. In contrast, little is known about single pivotal trial approvals for non‐orphan, non‐oncology drugs. Between 2012 and 2016, 23 novel therapeutic drugs were approved by the US Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA) for 27 non‐orphan, non‐oncology indications each based on a single pivotal trial. Although there was considerable variation in the nature and strength of the efficacy evidence supporting these drug approvals, the majority (85%) of the pivotal trials were randomized and controlled. For all superiority trials, the primary outcome was met with a statistical significance of P ≤ 0.005. Most approvals were supported by additional efficacy data from nonpivotal studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ To our knowledge, this is the first analysis of US Food and Drug Adminstration (FDA) and European Medicines Agency (EMA) single pivotal trial approvals that focuses specifically on drugs targeting conditions other than cancer and orphan diseases.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What is the nature and strength of efficacy evidence supporting single pivotal clinical trial approvals for non‐oncology, non‐orphan drugs?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study shows that, although the nature and strength of efficacy evidence supporting single pivotal trial approvals for the heterogeneous group of non‐oncology, non‐orphan drugs varies widely, superiority trials were statistically very convincing (P ≤ 0.005) and mostly supported by additional data from various other nonpivotal trials.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These learnings from regulatory precedence may supplement the available FDA and EMA guidance pertaining to applications with a single pivotal trial. The findings can help sponsors understand the FDA and EMA standards for single pivotal trial approvals of non‐orphan, non‐oncology drugs and, hence, inform design of the clinical development plan.
The regulatory approval of new drugs is based on an assessment of the available evidence of benefits and risks. Given the limited drug exposure at the time of drug approval, the assessment of benefits and particularly risks, especially in relation to rare adverse events, comprises some degree of uncertainty. Hence, a continuous benefit/risk evaluation is an accepted approach to confirm the safety—and, in some cases, also the efficacy—profile of a new drug in the postapproval setting depending on the medical need.1, 2, 3, 4
Traditionally, a minimum of two positive, adequate, and well‐controlled clinical trials have been considered the gold standard for demonstration of efficacy of a novel therapeutic drug.3, 5 However, the EU and US legislations allow for approvals based on a single pivotal clinical trial5, 6, 7 potentially permitting drugs for treatment of serious diseases with a high medical need to reach the patients faster. Both the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) approve more than a third of novel therapeutic drugs based on a single pivotal trial with orphan (i.e., drugs developed specifically to treat rare medical conditions) and oncology drugs accounting for the majority.8, 9
It has been widely debated whether regulators should accept a higher level of uncertainty concerning benefits and risks for life‐threatening or severely debilitating conditions with high medical needs, especially in the case of rare diseases.1, 10, 11, 12 Several analyses of marketing authorizations of oncology and orphan drugs suggest that the approvals are often based on limited evidence of efficacy.13, 14, 15, 16, 17 As such, it has been argued that there is a widening gap in the regulatory requirements for approval of such drugs compared with other disease areas.1
In the case of new drugs intended to treat more prevalent and not necessarily immediately life‐threatening diseases, the regulatory flexibility in terms of acceptability of a single pivotal trial to support approval in the United States and the European Union is less well known. Hence, the aim of this study was to analyze and better understand the strength of the clinical efficacy evidence supporting approvals based on single pivotal clinical trial for novel therapeutic drugs intended to treat diseases other than oncology and orphan conditions.
We identified non‐oncology, non‐orphan New Active Substances (EU) or New Molecular Entities (US; collectively referred to as novel therapeutic drugs) approved from 2012−2016 by the FDA and/or the EMA based on a single pivotal clinical trial. The clinical evidence of efficacy submitted in support of the applications for marketing authorization was analyzed in terms of design characteristics and results of the single pivotal trial along with requests for postapproval efficacy data. In addition, the availability of supportive evidence of efficacy from nonpivotal trials was explored.
Methods
This is a retrospective, descriptive, cross‐sectional study. Data sources included publicly available information from the FDA18 and EMA19 websites, including the FDA review documents and approval letters (collectively abbreviated FDA‐Rs), US Prescribing Information (USPI), and European Public Assessment Reports (EPARs). Originally approved Summary of Product Characteristics were retrieved via the Cortellis Database.20
All data were reviewed and verified by at least two independent authors, and discrepancies were resolved by discussion and consensus.
Selection criteria
For the EU, New Active Substances approved via the EMA Centralized Procedure between 2012 and 2016 for indication(s) supported by a single pivotal clinical trial were identified as described by Morant and Vestergaard.9
For the US, New Molecular Entities approved by the FDA Center for Drug Evaluation and Research between 2012 and 2016 were identified using the FDA Novel Drugs Summaries from 2012 through 2016.21 Initial indications approved based on a single pivotal clinical trial were identified using the original USPI and FDA‐Rs. Generics, biosimilars, diagnostic products, and drugs approved by informed consent or via the hybrid application pathways (also termed 505(b)(2) in the United States) were excluded. Only indications authorized as part of the initial approval were included. Drugs with an EMA or FDA Orphan Drug Status as well as oncology drugs (i.e., drugs belonging to the Anatomical Therapeutic Chemical classification group L—antineoplastic and immunomodulating agents—and indicated for treatment of cancer) were excluded.
Evidence of efficacy supporting approval
Information on the clinical studies providing evidence of efficacy in support of the individual indications was retrieved from the EPARs and FDA‐Rs. For evaluation of whether a clinical trial was considered “pivotal” for the approved indication, the assessment of the FDA and the EMA as described in the FDA‐Rs and the EPARs was adopted as described by Morant and Vestergaard.9 In case of discrepancy between the two regulatory agencies, the novel therapeutic drug was included in the analysis only from the perspective of the agency that based the approval of the indication on a single pivotal clinical trial. In three cases, the pivotal clinical trial consisted of two distinct parts conducted under a single study protocol (patiromer, vedolizumab, and sucroferric oxyhydroxide); these were considered a single pivotal trial for this analysis.
The single pivotal trials were evaluated in terms of study population, trial design features (randomization, blinding, and comparator), number of patients randomized/enrolled, and outcome of the primary efficacy analysis. The primary efficacy end points were classified as clinical outcome, clinical scale, or surrogate marker based on the principles suggested by Pease et al.2: clinical outcomes measure mortality or reflects/characterizes a patient's symptoms, overall mental state, or the effects of a disease or condition on how the patient functions; clinical scales serve as quantitative gradations of an outcome that reflects/characterizes a patient's symptoms, overall mental state, or the effects of a disease or condition on how the patient functions; surrogate markers measure an intermediate biological characteristic/outcome that is expected to consistently and accurately predict a clinical outcome (clinical benefit or harm). In context of the approved indication, a surrogate marker was deemed “endorsed” if clearly supported by the regulatory agencies in context of the indication (e.g., in therapeutic guidelines or—in the case of the United States—the FDA “Table of Surrogate Endpoints That Were the Basis of Drug Approval”22 and/or in the regulatory review documents (FDA‐R or EPAR, as applicable)).
Nonpivotal trials providing additional support for the indication were classified based on clinical development phase and trial design features (i.e., population, randomization, and blinding). Trials were considered supportive if one of the following criteria was fulfilled: the FDA‐Rs/EPARs (i) explicitly mentioned the trials as “supportive for the indication,” or (ii) described the data as “supportive” or as “additional” efficacy evidence or as demonstrating a “clinically meaningful effect size,” and/or (iii) thoroughly described both design and efficacy results of the trial. Data were not considered supportive if merely mentioned as supportive for “dose‐selection” and/or “dose‐titration.”
Requests for postapproval efficacy data (excluding pediatrics) in the form of postauthorization measures (EU) were extracted from the EPARs, and postmarketing requirements/commitments (US) from the FDA Approval Letters and FDA‐Rs.
Topics addressed during the regulatory review were evaluated to indirectly assess uncertainties regarding the approvability of the novel therapeutic drugs in relation to efficacy/benefit; safety‐related uncertainties were beyond the scope of this analysis. We focused on efficacy issues that were subject to differences in opinions (i.e., one or more FDA reviewers explicitly raised concerns in relation to the approval of the drug or the EPAR described Committee for Medicinal Products for Human Use divergent positions).
Results
Excluding oncology and orphan drugs, 23 novel therapeutic drugs were approved in the European Union and/or the United States from 2012−2016 for one or more indications (27 indications in total) each supported by a single pivotal clinical trial (Figure 1; Table S1). Seven of these novel therapeutic drugs were approved in both the European Union and the United States (Figure 2); one drug was approved for different indications in the European Union and the United States (daclatasvir; Table S1) each supported by a single pivotal trial.
Figure 1.
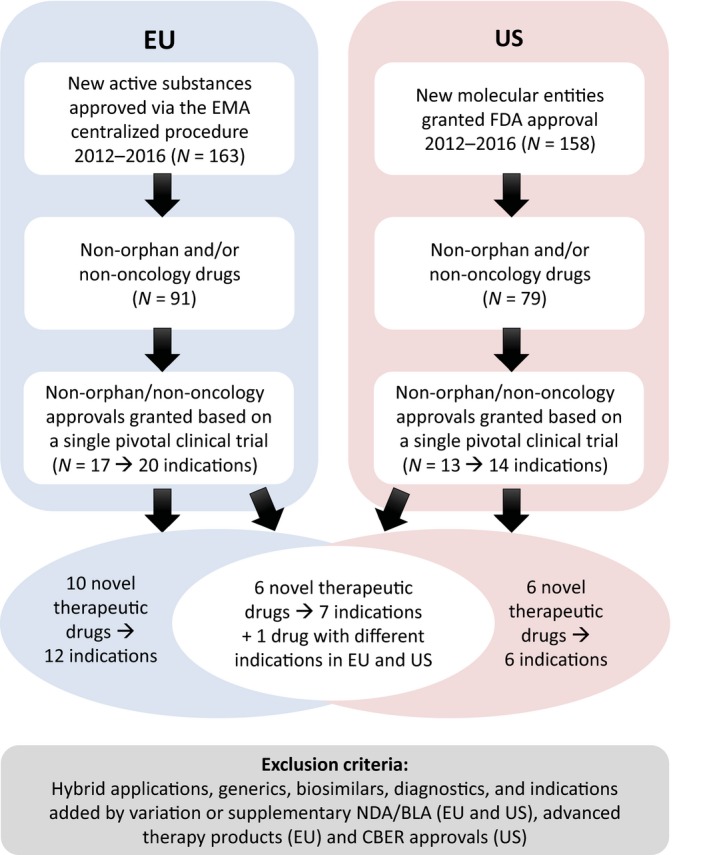
Selection criteria for the analysis. Overview of the selection criteria for identification of novel therapeutic drugs for which one or more indications were approved based on a single pivotal clinical trial in the United States (US) and European Union (EU) between 2012 and 2016. CBER, Center for Biologics Evaluation and Research; FDA, US Food and Drug Administration; EMA, European Medicines Agency; NDA/BLA, new drug application/biologics license application.
Figure 2.
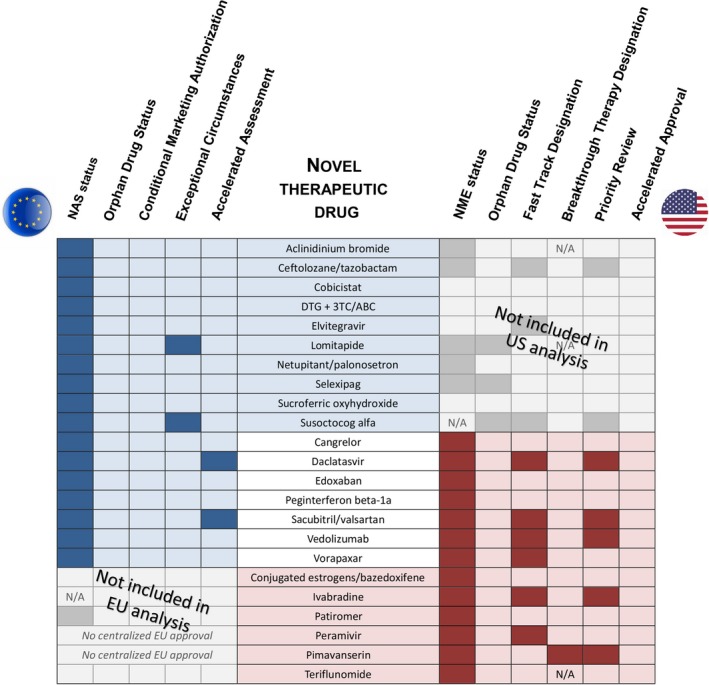
Regulatory overview of novel therapeutic drugs approved based on a single pivotal trial. Overview of regulatory pathways and designations for novel therapeutic drugs for which one or more indications were approved based on a single pivotal trial by the US Food and Drug Administration Center for Drug Evaluation and Research and/or via the European Medicines Agency Centralized Procedure between 2012 and 2016. Color shading signifies drugs fulfilling study selection criteria in the European Union (EU) only (blue shading), in both regions (no shading), and United States (US) only (red shading). 3TC, Lamivudine; ABC, Abacavir; DTG, dolutegravir; NAS, New Active Substance; NME, New Molecular Entity; N/A, not applicable.
For the novel therapeutic drugs included in this study, regulatory expedited pathway designations were less frequently used in the European Union compared with the United States (Figure 2) in line with previous reports.23, 24 None of the drugs received EU Conditional Marketing Authorization or US Accelerated Approval (Figure 2).
Each of the 27 indications was approved based on a single pivotal multicenter trial (Table 1). The majority (23; 85%) was randomized and included a control arm. Thirteen of these (48%) were placebo‐controlled, whereas 10 (37%) included an active comparator. The remaining four pivotal trials (15%) were open‐label and uncontrolled. The median number of patients was 828, ranging from 29 (lomitapide and susoctocog alfa) to 26,449 (vorapaxar).
Table 1.
Characteristics of single pivotal trials supporting the approval of novel therapeutic drugs
| Novel therapeutic drug | Study population (disease or condition) | Study design | Treatment arms | Number of randomized patients | Primary end point | Results of the primary efficacy analysis for the approved dosage(s) |
|---|---|---|---|---|---|---|
| Aclidinium bromide (AB) | Chronic obstructive pulmonary disease | Randomized, double‐blind, placebo‐controlled |
Grp 1: AB 200 μg Grp 2: AB 400 μg Grp 3: placebo |
828 | Change in forced expiratory volume |
Mean difference from placebo: Grp 1: 0.099 L (95% CI: 0.057, 0.141); Grp 2: 0.128 L (95% CI: 0.085, 0.170); P < 0.0001 (both groups) |
| Cangrelor | Coronary artery disease | Randomized, double‐blind, active‐controlled |
Grp 1: cangrelor Grp 2: clopidogrel |
11,145 | Death or CV event |
Grp 1: 4.7%; Grp 2: 5.9%; OR = 0.78 (95% CI: 0.66−0.93); P = 0.005 |
| Ceftolozane/tazobactam (C/T) | Complicated intra‐abdominal infections | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: C/T + metronidazole Grp 2: meropenem |
993 | Clinical cure rate |
Grp 1: 94.1%; Grp 2: 94.0%; 0.0% (99% CI: −4.16%, 4.30%) |
| Complicated urinary tract infections including pyelonephritis | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: C/T Grp 2: levofloxacin |
1,083 | Microbiological response rate |
Grp 1: 84.7%; Grp 2: 75.4%; 9.4% (99% CI: 1.54%, 17.12%) |
|
| Cobicistat (COBI) | HIV | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: COBI/ATV/ FTC/TDF/placebo Grp 2: RTV/ATV/FTC/TDF/placebo |
698 | Proportion of subjects with HIV‐1 RNA below predefined cutoff |
Grp 1: 85.2%; Grp 2: 87.4%; −2.2% (95.2% CI: −7.4%, 3.0%) |
| Conjugated estrogens/bazedoxifene (CE/B) | Vasomotor symptoms associated with menopause | Randomized, double‐blind, placebo‐controlled |
Grp 1: CE/B Grp 2: placebo |
332 | Change from baseline in frequency and severity of hot flushes (coprimary end points) |
Mean difference from placebo: Frequency: W4: −3.1 (95% CI: −4.4, −1.7); W12: −2.7 (95% CI: −3.8, −1.6); Severity: W4: −0.5 (95% CI: −0.7, −0.3); W12: −0.6 (95% CI: −0.9, −0.4); P < 0.001 (all primary end points) |
| Daclatasvir | EU: Hepatitis C virus infection genotypes 1, 2, and 3 | Randomized, open label | 10 groups stratified by prior treatment, viral genotype, and treatment regimen | 211 | Rate of sustained virologic response | > 90% in all treatment arms |
| US: Hepatitis C virus genotype 3 infection | Single arm, open label | Daclatasvir/sofosbuvir | 152 (treated) | Proportion of treated subjects with sustained virologic response | 89% (95% CI: 83%−93%) | |
| DTG + 3TC/ABC | HIV | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: DTG + 3TC/ABC Grp 2: EFV/FTC/TDF |
844 | Proportion of subjects with HIV‐1 RNA below predefined cutoff |
Grp 1: 88%; Grp 2: 81%; 7.4% (95% CI: 2.5%, 12.3%); P = 0.003 (superiority) |
| Edoxaban (EDOX) | Stroke and systemic embolism in pts with nonvalvular atrial fibrillation | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: 30 mg EDOX Grp 2: 60 mg EDOX Grp 3: warfarin |
21,105 | All strokes and systemic embolic event |
Grp 2: 1.18%/year; Grp 3: 1.50%/year; HR = 0.79 (97.5% CI: 0.63, 0.99) (Grp 2) |
| Venous thromboembolism and/or pulmonary embolism | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: 60 mg EDOX Grp 2: warfarin |
8,292 | Symptomatic recurrent venous thromboembolism |
Grp 1: 3.2%; Grp 2: 3.5%; HR = 0.89 (95% CI: 0.703, 1.128) |
|
| Elvitegravir | HIV | Randomized, double‐blind, active‐controlled (noninferiority) |
Grp 1: elvitegravir Grp 2: raltegravir |
724 | Proportion of subjects with HIV‐1 RNA below predefined cutoff |
Grp 1: 59%; Grp 2: 57.8%; 1.1% (95% CI: −6.0%, 8.2%) |
| Ivabradine | Chronic heart failure | Randomized, double‐blind, placebo‐controlled |
Grp 1: ivabradine Grp 2: placebo |
6,558 | Time to first adjudicated CV death or hospitalization |
Incidence rates: Grp 1: 24.5%; Grp 2: 28.7%; HR = 0.82 (95% CI: 0.75, 0.90); P < 0.0001 |
| Lomitapide | Homozygous familial hyper‐cholesterolemia | Non‐randomized, single‐arm, open label | Lomitapide | 29 (treated) | Change from baseline in low‐density lipoprotein cholesterol | −3.8 mmol/L mean change from baseline, i.e., −40% (P < 0.001 on the mean percent change from baseline) |
| Netupitant (NETU)/palonosetron (PALO) | Emesis associated with highly emetogenic chemotherapy | Randomized, double‐blind, placebo‐controlled |
Grp 1: PALO alone Grp 2: PALO + NETU 100 mg Grp 3: PALO + NETU 200 mg Grp 4: PALO + NETU 300 mg Grp 5 (exploratory): combination of licensed agents |
694 | Complete response rate |
Grp 1: 76.5%; Grp 4: 89.6%; P = 0.004 |
| Emesis associated with moderately emetogenic chemotherapy | Randomized, double‐blind, active‐controlled |
Grp 1: PALO + NETU 300 mg Grp 2: PALO alone |
1,455 | Complete response rate |
Grp 1: 76.9%; Grp 2: 69.5%; OR = 1.48 (95% CI: 1.16, 1.87); P = 0.001 |
|
| Patiromer | Chronic kidney disease with hyperkalemia |
Part A: single arm, single‐blind, open‐label Part B: randomized, single‐blind, placebo‐controlled |
Part A: patiromer Part B: Grp 1: patiromer Grp 2: placebo |
Part A: 243 (treated) Part B: 107 |
Change in serum potassium from baseline |
Part A: −1.01 mEq/L (95% CI: −1.07, −0.95) Part B: Difference from placebo: −0.72 mEq/L (95% CI: −0.46, −0.99); P < 0.001 |
| Peginterferon beta‐1a (PEG) | Relapsing remitting multiple sclerosis | Randomized, double‐blind, placebo‐controlled |
Grp 1: placebo Grp 2: PEG Q4W Grp 3: PEG Q2W |
1,516 | Annualized relapse rate at 1 year |
Grp 1: 0.397; Grp 3: 0.256; RR = 0.644 (95% CI: 0.500, 0.831); P = 0.0007 |
| Peramivir (PRV) | Acute uncomplicated influenza | Randomized, double‐blind, placebo‐controlled |
Grp 1: PRV 300 mg Grp 2: PRV 600 mg Grp 3: placebo |
300 | Time to alleviation of symptoms |
Difference from placebo: Grp 1: −22.7 hours; Grp 2: −21.9 hours; P = 0.001 (pooled doses) |
| Pimavanserin (PIMV) | PD with psychosis | Randomized, double‐blind, placebo‐controlled |
Grp 1: PIMV Grp 2: placebo |
199 | PD‐adapted scale for the assessment of positive symptoms |
Difference from placebo: −3.06 (95% CI: −4.91, −1.20); P = 0.001 |
| Sacubitril/valsartan (SBT/VAL) | Heart failure with reduced ejection fraction | Randomized, double‐blind, active‐controlled |
Grp 1: SBT/VAL Grp 2: enalapril |
8,442 | Time to first occurrence of either CV death or heart failure hospitalization |
Incidence rates: Grp 1: 21.8%; Grp 2: 26.5%; HR = 0.80 (95% CI: 0.73, 0.87); p = 0.0000002 |
| Selexipag | Pulmonary arterial hypertension | Randomized, double‐blind, placebo‐controlled |
Grp 1: selexipag Grp 2: placebo |
1,156 | Time to first Critical Event Committee ‐confirmed morbidity and/or mortality event |
Incidence rates: Grp 1: 24.4%; Grp 2: 36.4%; HR = 0.60 (99% CI: 0.46, 0.78); P < 0.0001 |
| Sucroferric oxyhydroxide (SUC) | Chronic kidney disease on maintenance dialysis | Randomized, open label (stage 1: active‐controlled; stage 2: “surrogate placebo”‐controlled) |
Stage 1 (non‐inferiority): Grp 1: SUC Grp 2: sevelamer Stage 2 (superiority): Grp 1: SUC maintenance dose Grp 2: SUC low dose |
Stage 1: 1,059 Stage 2: 99 |
Change in serum phosphorus levels from baseline |
Stage 1: Grp 1: −0.7 mmol/L; Grp 2: −0.8 mmol/L; 0.08 mmol/L (97.5% CI: ‐infinity, 0.15); P = 0.011 (superiority) Stage 2: Grp 1: 0.08 mmol/L; Grp 2: 0.62 mmol/L; difference between doses: 0.54 mmol/L (95% CI: 0.37, 0.71); P < 0.001 |
| Susoctocog alfa | Hemophilia A with serious bleeding episode | Open label, single arm | Susoctocog alfa | 29 (treated) | Proportion of serious bleeding episodes responsive to therapy | 100% |
| Teriflunomide (TER) | Relapsing multiple sclerosis | Randomized, double‐blind, placebo‐controlled |
Grp 1: TER 7 mg Grp 2: TER 14 mg Grp 3: placebo |
1,086 | Annualized relapse rate |
Grp 1: 0.370 (95% CI: 0.318, 0.432); P = 0.0002; Grp 2: 0.369 (95% CI: 0.308, 0.441); P = 0.0005; Grp 3: 0.539 (95% CI: 0.466, 0.623) |
| Vedolizumab (VDZ) | Ulcerative colitis | Randomized, double‐blind, placebo‐controlled (induction and maintenance) |
Induction: Grp 1: VDZ Grp 2: placebo Maintenance: Grp 1: VDZ Q4W Grp 2: VDZ Q8W Grp 3: placebo |
Induction: 374 Maintenance: 373 |
Induction: % patients with clinical response Maintenance: % patients in remission |
Induction: Difference from placebo: 21.7% (95% CI: 11.6, 31.7); P < 0.0001
Maintenance: Difference from placebo: Grp 1 (EU only): 29.1% (95% CI: 17.9−40.4); Grp 2: 26.1% (95% CI: 14.9, 37.2); P < 0.0001 (both dose regimens) |
| Vorapaxar | Atherosclerosis | Randomized, double‐blind, placebo‐controlled |
Grp 1: vorapaxar Grp 2: placebo |
26,449 | Time to the first CV event |
Grp 1: K‐M = 11.2%; Grp 2: K‐M = 12.4%; HR = 0.88 (95% CI: 0.82, 0.95); P = 0.001 |
3TC, lamivudine; ABC, Abacavir; ATV, atazanavir; CI, confidence interval; CV, cardiovascular; DTG, dolutegravir; EFV/FTC/TDF, efavirenz/emtricitabine/tenofovir; EU, European Union; FTC/TDF, emtricitabine/tenofovir; Grp, group; HR, hazard ratio; K‐M, Kaplan‐Meier event rate; OR, odds ratio; PD, Parkinson's disease; RR, rate ratio; RTV, ritonavir; US, United States; Q2W, Once every two weeks; Q4W, Once every four weeks; Q8W, Once every eight weeks; W12, week 12; W4, week 4.
Characteristics and outcomes of the pivotal trials supporting the approval of novel therapeutic drugs based on a single pivotal trial. Trial characteristics comprised overall trial design including study population, randomization, blinding, treatment arms, and number of randomized (or enrolled in case of no randomization) patients as well as nature and result of the primary outcome measure. Color shading signifies drugs fulfilling study selection criteria in the EU only (blue shading), in both regions (no shading); and US only (red shading).
The end points for primary analysis were clinical outcomes in 12 trials (44%), clinical scales in 5 trials (19%), and surrogate markers in 10 trials (37%). All surrogate markers were endorsed from a regulatory perspective in the context of the approved indications. For the 16 superiority trials, the primary end point was met with a statistical significance of P value equal to or below 0.005 (Table 1). For noninferiority trials, confidence intervals of 95% (or higher) were within the prespecified noninferiority margin. In the uncontrolled trials, the primary outcomes were established as very convincing virologic response rates of 89% or above (daclatasvir), a 100% response rate for serious bleeding episodes (P < 0.001; susoctocog alfa), or a 40% change from baseline in low‐density lipoprotein cholesterol (P < 0.001; lomitapide).
In almost all cases, evidence from one or more nonpivotal trials submitted as part of the application were classified as “supportive” for the assessment of efficacy in the FDA‐Rs/EPARs (Figure S1); the only exception was sacubitril/valsartan where the additional efficacy data described in the FDA‐Rs were not considered supportive as the described data were only mentioned as supportive for dosetitration. The nature of the supportive studies varied widely across indications (please refer to Figure S1 for examples). Many of the supportive trials were not designed nor powered to demonstrate efficacy but planned with primary objective of investigating pharmacokinetics and/or safety and tolerability. The supportive trials were not consistently described in the product label (Figure S1).
In only a few instances, postapproval studies were requested by the regulatory agencies to confirm efficacy (Figure S1). The EMA required postapproval studies only for the two drugs that were approved under Exceptional Circumstances (lomitapide and susoctocog alfa). The FDA requested postapproval studies to collect maintenance data for pimavanserin and to establish efficacy in specific patient populations for two antivirals (daclatasvir and peramivir).
Internal agency discrepancies pertaining to benefit/risk conclusions were observed for 2 of 17 drugs in the European Union (divergent opinions included in the EPARs) and 5 of 13 drugs in the United States (opposing views from one or more reviewers expressed in the FDA‐Rs).
Discussion
Our focus on non‐oncology, non‐orphan novel therapeutic drugs was based on two considerations. First, the regulatory requirements for approval of drugs to treat cancer and orphan indications seem to be subject to a higher degree of regulatory flexibility,1, 13, 14, 16, 17 and the evidence supporting approval of these drugs has already been widely debated.10, 11, 12, 14 Second, we wished to gain insight into the regulatory precedence for approvals based on a single pivotal trial for drugs intended to treat more prevalent and not immediately life‐threatening diseases knowing that the outcome would potentially represent a very heterogeneous group of drugs and therapeutic areas.
Our predefined selection criteria were based on the legal framework in the two regions rather than the medical perception. Three of the 23 drugs identified in our analysis were actually approved for treatment of rare diseases (lomitapide, susoctocog alfa, and selexipag); only they did not fulfill the regulatory criteria for Orphan Drug Status in the European Union as different regulatory eligibility criteria apply in the two regions. These three drugs were, therefore, included in the analysis only from a European Union perspective.
The fact that the majority of the drugs included in the analysis were approved within conditions that may arguably be categorized as life‐threatening or seriously debilitating (e.g., cardiovascular events, HIV infection, serious bacterial infections, and chronic hepatitis C) points to the seriousness of the disease as an important eligibility factor for approval based on a single pivotal trial. This is further supported by the fact that 7 of the 13 novel therapeutic drugs included in the US analysis had also been granted Fast Track or Breakthrough Therapy Designation by the FDA.
Most of the primary outcome measures in the single pivotal trials were overall objective efficacy measures (i.e., clinical outcomes or surrogate markers in 22 of 27 pivotal trials). Superiority trials were statistically very convincing with P values equal to or below 0.005 in line with the range previously proposed for approvals based on a single pivotal trial.25 For noninferiority trials, confidence intervals of minimum 95% were generally well away from the prespecified lower or upper noninferiority margin as recommended for single pivotal noninferiority trials7; for some of these, superiority to the active control group was demonstrated as well.
As our analysis mainly focused on the statistical persuasiveness of the primary outcome measure, it did not assess other important factors for interpretation of the clinical meaningfulness and robustness of the trial results, such as the therapeutic effect size, results of secondary outcome measures, and sensitivity analyses. Due to the lack of independent substantiation from a second pivotal trial, clinical meaningfulness, and robustness of the trial results become even more critical in the context of single pivotal trial approvals. However, it is difficult to put forward an overall conclusion on clinical meaningfulness of the therapeutic effect size in this descriptive study due to the heterogeneity in the outcome measures and therapeutic areas. As our analysis is entirely based on approved drugs, it is inherent that the benefit/risk assessment was deemed sufficiently positive from a regulatory perspective to grant approval based on a single pivotal trial. Hence, it was not our intent to evaluate and form an opinion on the regulatory or medical assessment of the clinical meaningfulness of the effect sizes observed in these trials.
In addition to a statistically persuasive result of the single pivotal trial, independent substantiation from other clinical studies is important to reduce the probability of erroneously concluding that a drug is effective.6 The efficacy demonstrated in all single pivotal trials analyzed in this study was supported by data from one or more additional clinical trials. Although the quantity and quality of the supportive efficacy data varied across the indications, the supportive data allowed, at least to some extent, for an independent substantiation of the evidence demonstrated in the single pivotal clinical trial. Although sponsors may submit several trials designed and powered to demonstrate efficacy, it is the regulatory assessment that decides whether these are classified as pivotal. In fact, in several cases, the efficacy data packages did include additional studies that were not evaluated as pivotal or even supportive by the FDA reviewers and/or EMA assessors. Furthermore, discrepancies between the FDA and EMA were observed in a few instances in terms of classification of clinical trials as pivotal or not for the approved indication.
There are several limitations to this analysis in addition to those already discussed. First, we focused specifically on efficacy, and as such, the risk aspect was not analyzed as part of the benefit/risk equation.
Second, the study does not allow for an overall comparison between the FDA and EMA perspectives due to the limited sample, heterogeneity of the therapeutic areas, and differences in regulatory procedures and remits of the agencies. As information on rejected and withdrawn applications for marketing authorization is not systematically publicly available,26 learnings from nonapprovals also remain a deficiency. Hence, it is hard to draw firm conclusions on the policies of the FDA and EMA on this topic. Nevertheless, these learnings from regulatory precedence may supplement the available FDA and EMA guidance pertaining to applications with a single pivotal trial.
Third, in determining the strength of the evidence of efficacy, we focused only on the results of the primary analysis. According to both the FDA's and EMA's guidance,6, 7 similar findings of beneficial effect across a range of different end points (primary and secondary) would support the adequacy of a single pivotal trial for approval of a new drug.
Last, we did not analyze whether the company–regulator negotiations resulted in a restriction in the company‐proposed indication. Although this is an important element of the benefit/risk assessment, it is challenging to link directly to the assessment of the single pivotal trial because of confounding factors, including safety, study design, patient population, and nature of the company's originally proposed indication.
The main objective of this retrospective, descriptive, cross‐sectional study was to analyze the benefit aspects of the benefit/risk assessment for single pivotal trial approvals of novel therapeutic drugs. The eligibility for approval based on a single pivotal trial depends not only on known factors such as the seriousness of the disease and the availability of effective treatments but also on the ability to demonstrate a robust and convincing effect in one pivotal trial. Factors influencing this ability include the nature of the primary end point (e.g., objective, hard end points vs. subjective, soft end points; validated vs. nonvalidated outcome measures); understanding of the disease biology (well‐characterized underlying pathology vs. complex multifactorial pathologies) and strength of the pharmacological rationale and the ability to easily identify and diagnose the patients. Although other technical and non–data‐driven social factors, such as reviewer and company experience, company–regulator interactions, as well as interactions with opinion leaders and patients may also influence the regulatory decision‐making process, the main drivers for drug approval remain robust evidence of efficacy and the seriousness of the disease.27
CONCLUSION
Although it has been widely discussed whether approvals of oncology and orphan drugs are too often based on limited evidence of efficacy, our analysis shows that single pivotal trial approvals for novel therapeutic drugs targeting other disease areas are generally based on data from a randomized and controlled trial with a statistically very compelling result of the primary analysis. Our analysis supports that the seriousness of the disease and the availability of supportive evidence of efficacy from other clinical trials are also important eligibility factors for approvals based on a single pivotal trial.
Funding
No funding was received for this work.
Conflict of Interest
All three authors are employees of H. Lundbeck A/S.
Author Contributions
A.V.M. and H.T.V. wrote the manuscript. A.V.M. and H.T.V. designed the research. A.V.M., V.J., and H.T.V. performed the research. A.V.M., V.J., and H.T.V. analyzed the data.
Supporting information
Figure S1. Overview of supportive “confirmatory” evidence of efficacy.
Table S1. Indications approved for novel therapeutic drugs based on a single pivotal trial.
Acknowledgments
The authors are very grateful to Steffen Thirstrup, MD, PhD, NDA Advisory Services, Ltd, for a critical review, Anders Blædel Lassen, Director, Regulatory Science & Advocacy, H. Lundbeck A/S for valuable discussions, and to Brian Odlaug, Medical Advisor, H. Lundbeck A/S, for creative input to figure layouts.
References
- 1. Eichler, H. , Pignatti, F. , Flamion, B. , Leufkens, H. & Breckenridge, A. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat. Rev. Drug Disc. 7, 818–826 (2008). [DOI] [PubMed] [Google Scholar]
- 2. Pease, A.M. , Krumholz, H.M. , Downing, N.S. , Aminawung, J.A. , Shah, N.D. & Ross, J.S. Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ 357, j1680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Woodcock, J. Expediting drug development for serious illness: trade‐offs between patient access and certainty. Clin. Trials 15, 230–234 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Califf, R.M. Expedited and facilitated drug evaluations and evidence of benefit and risk: the cup is half‐full. Clin. Trials 15, 235–239 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Katz, R. FDA: evidentiary standards for drug development and approval. NeuroRx 1, 307–316 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. US Food and Drug Administration (FDA) . Guidance for industry: providing clinical evidence of effectiveness for human drugs and biological products. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072008.pdf> (1998).
- 7. European Medicines Agency . Points to consider on application with 1. Meta‐analyses; 2. One pivotal study. CPMP/EWP/2330/99. <https://www.ema.europa.eu/documents/scientific-guideline/points-consider-application-1meta-analyses-2one-pivotal-study_en.pdf> (2001).
- 8. Downing, N.S. , Aminawung, J.A. , Shah, N.D. , Krumholz, H.M. & Ross, J.S. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005‐2012. JAMA 311, 368–377 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morant, A.V. & Vestergaard, H.T. European marketing authorizations granted based on a single pivotal clinical trial: the rule or the exception? Clin. Pharmacol. Ther. 104, 169–177 (2018). [DOI] [PubMed] [Google Scholar]
- 10. Booth, C.M. & Del Paggio, J.C. Approvals in 2016: questioning the clinical benefit of anticancer therapies. Nat. Rev. Clin. Oncol. 14, 135–136 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Pignatti, F. , Johnsson, B. , Blumenthal, G. & Justice, R. Assessment of benefits and risks in development of targeted therapies for cancer – the view of regulatory authorities. Mol. Oncol. 9, 1034–1041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martinalbo, J. et al Early market access of cancer drugs in the EU. Ann. Oncol. 27, 96–105 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Winstone, J. , Chadda, S. , Ralston, S. & Sajosi, P. Review and comparison of clinical evidence submitted to support European Medicines Agency market authorization of orphan‐designated oncological treatments. Orphanet J. Rare Dis. 10, 139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Picavet, E. , Cassiman, D. , Hollak, C.E. , Maertens, J.A. & Simoens, S. Clinical evidence for orphan medicinal products – a cause for concern? Orphanet J. Rare Dis. 8, 164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hatswell, A.J. , Baio, G. , Berlin, J.A. , Irs, A. & Freemantle, N. Regulatory approval of pharmaceuticals without a randomised controlled study: analysis of EMA and FDA approvals 1999‐2014. BMJ Open 6, e011666 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis, C. , Naci, H. , Gurpinar, E. , Poplavska, E. , Pinto, A. & Aggarwal, A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009‐13. BMJ 359, j4530 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasinowski, F.J. , Panico, E.B. & Valentine, J.E. Quantum of effectiveness evidence in FDA's approval of orphan drugs: update, July 2010 to June 2014. Ther. Innov. Regul. Sci. 49, 680–697 (2015). [DOI] [PubMed] [Google Scholar]
- 18. US Food and Drug Administration (FDA) . Drugs@FDA website. <https://www.accessdata.fda.gov/scripts/cder/daf/>. Accessed August 2017 through June 2018.
- 19. European Public Assessment Reports. EMA website. <https://www.ema.europa.eu/en/medicines>. Accessed August 2017 through June 2018.
- 20. Cortellis database, clarivate analytics. Cortellis website. <https://www.cortellis.com/intelligence/login.do>. Accessed February through May 2018.
- 21. US Food and Drug Administration (FDA) . Novel drug summaries 2012 through 2016. FDA website. <https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm>. Accessed February through May 2018.
- 22. US Food and Drug Administration (FDA) . Table of surrogate endpoints that were the basis of drug approval or licensure. Last updated 23 July 2018. FDA website. <https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm613636.htm>.
- 23. Center for Innovation in Regulatory Science (CIRS) . New drug approvals in ICH countries 2005‐2014. R&D Briefing 57. 2015. CIRS website. <http://www.cirsci.org/sites/default/files/CIRS_R&D_57_ICH_%20approval_%20times_2005-2014_%2006072015.pdf>. Accessed June 19, 2018.
- 24. Center for Innovation in Regulatory Science (CIRS) . The impact of the evolving regulatory environment on the approval of new medicines across six major authorities 2006‐2015. R&D Briefing 59. 2016. CIRS website. <http://www.cirsci.org/wp-content/uploads/2016/05/CIRS_RD_-Briefing_59_23052016.pdf>. Accessed June 11, 2018.
- 25. Coutant, D. , Riggs, D. & Van Sant Hoffman, E. Substantial evidence: when is a single trial sufficient for approval and promotion? Drug Inf. J. 45, 253–263 (2011). [Google Scholar]
- 26. Lurie, P. , Chahal, H.S. , Sigelman, D.W. , Stacy, S. , Sclar, J. & Ddamulira, B. Comparison of content of FDA letters not approving applications for new drugs and associated public announcements from sponsors: cross sectional study. BMJ 350, h2758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liberti, L. , Breckenridge, A. , Hoekman, J. , McAuslane, N. , Stolk, P. & Leufkens, H. Factors related to drug approvals: predictors of outcome? Drug Discov. Today 22, 937–946 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overview of supportive “confirmatory” evidence of efficacy.
Table S1. Indications approved for novel therapeutic drugs based on a single pivotal trial.