

Abstract
We summarize the progress in whole-genome sequencing and analyses of primate genomes. These emerging genome datasets have broadened our understanding of primate genome evolution revealing unexpected and complex patterns of evolutionary change. This includes the characterization of genome structural variation, episodic changes in the repeat landscape, differences in gene expression, new models regarding speciation, and the ephemeral nature of the recombination landscape. The functional characterization of genomic differences important in primate speciation and adaptation remains a signiflcant challenge. Limited access to biological materials, the lack of detailed phenotypic data and the endangered status of many critical primate species have significantly attenuated research into the genetic basis of primate evolution. Next-generation sequencing technologies promise to greatly expand the number of available primate genome sequences; however, such draft genome sequences will likely miss critical genetic differences within complex genomic regions unless dedicated efforts are put forward to understand the full spectrum of genetic variation.
Keywords: genome, sequencing, variation, gene comparison, speciation, diversity
INTRODUCTION
As new primate genomes become sequenced and are compared within the context of the primate phylogeny (Figure 1), scientists are provided with an unprecedented opportunity to reconstruct the evolutionary history of every basepair of the human genome [see (44, 50, 60) for reviews]. This review focuses on how multiple primate genomes have provided a framework to understand the mode and tempo of primate genome evolution, including an understanding of our gene repertoire and chromosomal organization. We discuss how this information has provided a new understanding of the extent of primate molecular adaptations and stimulated new hypotheses regarding selection in primate genomes. We focus on mounting evidence that gene regulation and expression as opposed to amino acid changes have driven primate adaptations to new environments. Comparisons among primate genomes have begun to reveal more basepairs affected by genome structural variation (IN/DELS, duplications, deletions, insertions, and bursts of retrotransposition events) than single nucleotide changes (16, 28, 165). Such changes occur preferentially within complex regions of the primate genome wherein, paradoxically, newly minted genes and gene families of unknown function have been discovered (77, 79, 124). Comparison of primate genome sequences has provided new insights into the molecular mechanisms that have contributed to chromosomal evolution within our species (51, 86), their influences on recombination (128, 162), and their relationship to natural selection within primates (18). Finally, we discuss the future of primate genomics and the need to strike the right balance between the number of genomes versus the quality of the sequence assemblies. We conclude with a discussion of the challenges of performing functional studies and obtaining material from other primate species that are endangered.
Figure 1.

Primate genome sequencing status. Primate whole-genome sequencing projects (10/25/08) with respect to the generally accepted primate phylogeny and an outgroup (mouse). Divergence times are estimated based on millions of years according to Goodman et al. 1998.
PRIMATE GENOME SEQUENCING: RATIONALE AND PROGRESS
With the initial draft sequence assembly of the human genome in 2001 followed by the mouse genome in 2002 (94, 160), other primate species became high priorities for whole-genome sequencing. A series of proposals and white papers (41) identified less than a dozen key primate species as sequencing targets (Figure 1). From the beginning, species choice was based on two different (sometimes antagonistic and sometimes complementary) rationales: biomedical relevance and the species position within the primate phylogeny with respect to human. The first considered primate species as models of disease and biomedical research. The baboon and macaque were justified because of their use in studying the genetic basis of numerous diseases (e.g., diabetes, cardiovascular disease, hypertension) and as models of transplantation, reproduction, infection, immunity, and pharmacology. Similarly, the squirrel monkey is used as a model for understanding primate neurobiology and infectious disease (in particular, malarial research). Since many of the genes and gene families underlying biological processes such as immunity, reproduction, and drug detoxification change rapidly over short periods of evolutionary time, complete genome sequences would reveal these idiosyncratic aspects and thereby enhance the utility of these models for understanding disease. More importantly, high-quality genomes provide an immediate framework upon which to map genetic traits associated with diseases and to develop transgenic models of disease (164).
The second major rationale was to improve annotation of the human genome. Based on our current understanding of the primate phylogeny (57, 142), there are seven key points of evolutionary transition (Figure 1) with respect to human. Sequencing index species from each of these phylogenetic nodes (i.e., chimpanzee, orangutan, gibbon, etc.) has two advantages. Comparative sequencing will determine the ancestral and derived states of every single basepair within the human genome sequence, thereby assigning mutational events to different time points during human evolution. Second, multiple primate genome comparisons will highlight differences of functional import (bursts of regulatory changes or amino-acid changes within specific lineages) (15). We should emphasize that these fundamental motivations contrast with that of other genome projects. Most mammalian genome projects, for example, aim to identify conserved regulatory elements, exonic sequence and other regions of potential functional significance (102, 152). This focus on the differences demands a unique quality of product to eliminate potential artifacts. High-quality comparative sequencing of primate genomes is necessary in order to provide a balanced view of genome variation (including regions of structural variation, segmental duplications, lineage-specific events and chromosomal variation) and to identify true genetic differences (as opposed to sequencing or assembly artifacts) between the species.
In addition to continued refinement and sequencing of additional human genomes, 12 primate genomes are currently in progress for whole-genome sequencing (Table 1). In most cases, the representative individuals are females to provide adequate coverage of the X chromosome, albeit with the concomitant loss of Y chromosome genetic information. Two working draft assemblies (chimpanzee and macaque) have been published (32, 51) and three additional genome assemblies (orangutan, gibbon, and common marmoset) have been generated and are being analyzed. Although none of the genomes will currently be sequenced to the same standard as the human genome, the need for higher-quality draft assemblies has been recognized owing in part to the frustration of distinguishing artifacts from true sequence differences in the low-coverage three-fold chimpanzee draft assembly (32).
Table 1.
Primate genome sequencing projects1
| Species | Scientiftc name | Individual | Sequencing center(s) | Target | Assembly status | WGS | Query |
|---|---|---|---|---|---|---|---|
| Human | Homo sapiens | RPCI-11 (M) | Consortium | Finishing genome | Build37 | 78,410,449 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “HOMO SAPIENS” |
| Chimpanzee | Pan troglodytes | Clint #C0471 (M) | WUGSC and BI/MIT | Draft2 | PanTro2 | 31,339,291 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “PAN TROGLODYTES” |
| Gorilla | Gorilla gorilla | Kamillah (F) | Sanger | Hybrid∗ | Not released∗ | 8,302,852 | TRACE_TYPE_CODE = “WGS” and (SPECIES_CODE = “GORILLA GORILLA” or SPECIES_CODE = “GORILLA GORILLA GORILLA”) |
| Orangutan | Pongo pygmaeus | Susie ISIS#71 (F) | BCM and WUGSC | Draft2 | Ponabe2 | 14,726,737 | TRACE TYPE_CODE = “WGS” and (SPECIES_CODE = “PONGO ABELII” or SPECIES_CODE = “PONGO PYGMAEUS” or SPECIES_CODE = “PONGO PYGMAEUS ABELII”) |
| Gibbon | Nomascus leucogenys | Asia #0098 (F) | BCM and WUGSC | Draft2 | In progress | 28,885,424 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “NOMASCUS LEUCOGENYS” or SPECIES_CODE = “NOMASCUS LEUCOGENYS LEUCOGENYS” |
| Rhesus macaque | Macaca mulatta | #25311 (F) | BCM, JCVI, WUGSC | Draft2 | Rhemac2 | 23,017,908 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “MACACA MULATTA” |
| Cynomolgus macaque | Macaca fascicularis | ND | WUGSC | Draft1 | Not started | ||
| Baboon | Papio hamadryas | ND | BCM | Draft1 | In progress | 12,717,107 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “PAPIO HAMADRYAS” |
| Vervet | Chlorocebus aethiops | ND | WUGSC | Draft1 | Not started | ||
| Marmoset | Callithrix jacchus | SFPRC#17066 | BCM and WUGSC | Draft2 | Caljach1 | 25,615,089 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “CALLITHRIX JACCHUS” |
| Squirrel monkey | Saimiri boliviensis∗∗ | ND | BI/MIT | Draft1 | Not started | 690,374 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “SAIMIRI BOLIVIENSIS” or SPECIES_CODE = “SAIMIRI BOLIVIENSIS BOLIVIENSIS” |
| Galago/bushbaby | Otolemur garnetti | ND | BI/MIT | Draft1 | Low coverage 2X | 13,953,011 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “OTOLEMUR GARNETTII” |
| Mouse lemur | Microcebus murinus | ND | BCM and BI/MIT | Draft1 | Low coverage 2X | 7,749,934 | TRACE_TYPE_CODE = “WGS” and SPECIES_CODE = “MICROCEBUS MURINUS” |
For each genome project, we identify the specific individual whose genome is being sequenced, the responsible genome sequencing center, and number of available capillary whole-genome shotgun sequences (WGS) in the trace repository. As of 10/1/2008, Draft1 = genomes with 6–8 coverage WGS only; Draft2 = genomes with 6–8X coverage with targeted sequencing of BACs for difficult regions.
Gorilla genome was approved by Wellcome Trust 12/2005, 2X capillary (3730) WGS data and 12 X WGS Solexa/Illumina WGS data were generated as of 12/2007; only 2X 3730 has been released as of 10/2008; Goal has shifted from WGSA of capillary reads to a hybrid assembly using next-generation technology, NIH approved targeted sequencing of complex regions within BACs.
Specific squirrel monkey species is not yet definite.
These initial analyses stressed the importance of independent assemblies of nonhuman primate genomes instead of simply mapping reads against the human reference sequence. As a result, typical working draft assemblies now target six- to eight-fold coverage in capillary whole-genome shotgun sequence reads. In five cases, these whole-genome shotgun sequence assemblies will be further supplemented by the sequencing of large-insert BAC clones mapping to structurally complex or biomedically relevant regions of the genome (a procedure known as genome refinement). This is especially crucial with respect to the phylogenetic index species such as chimpanzee, orangutan, gibbon, macaque, and squirrel monkey (indicated as Draft 2 in Table 1) where duplications and structural variation are now thought to account for more genetic variation than single basepair differences. Unfortunately, the gorilla genome assembly is not slated for the same standard. The Sanger Center has opted to generate an experimental hybrid assembly consisting of 12X Solexa sequencing data combined with 2X coverage of whole-genome assembly data. BAC refinement has been proposed but will be carried out as part of an NIH initiative.
FEATURES OF PRIMATE GENOME VARIATION
Although relatively few genome-wide comparisons have been completed to date (32, 51), analyses of the human, macaque, and chimpanzee genomes, as well as targeted resequencing of specific genomic segments, have revealed some important features and general principles of primate genome evolution. The alignment of the majority of genomic sequence from closely related primates is relatively trivial (38, 152) and shows a neutral pattern of single-nucleotide variation consistent with the primate phylogeny (Figure 2), although the rate of single-nucleotide variation has varied by a factor of three-fold within different lineages (42, 96, 143). Notably, the pattern of single-nucleotide variation also varies as a function of chromosome structure and organization (32, 51). Metacentric and acrocentric chromosomes show significant differences in variation, and regions within 10 Mb of telomeres evolve more rapidly perhaps as a consequence of biased gene conversion (Figure 2). On average, 10% of the genomic sequence has proven more elusive in terms of orthologous alignment. This includes segmental duplications, subtelomeric regions, pericentromeric regions, and lineage-specific repeats. Such regions typically cannot be resolved strictly by whole-genome sequence assembly (WGSA), thus larger insert clones (i.e., BAC clones) provide better substrates for resolving these areas.
Figure 2.

Human and primate sequence similarity. Whole-genome shotgun sequences from a human (blue), chimpanzee (red), and macaque (brown) were aligned against the human reference genome and the nucleotide divergence was computed in 100-kbp windows along (a) chromosome 2 and (b) chromosome 7. Note the increase in sequence divergence within 10 Mb of the telomere.
Comparative sequence data highlight the value of genomic sequence from nonhuman primates to determine the ancestral and derived status of human alleles (27, 81). There have been some surprises. Phylogenetic analysis of resequenced regions among humans and the great apes reveal that as many as 18% of genomic regions are inconsistent with the Homo-Pan clade, and, rather, support a Homo-Gorilla clade (27). This has been taken as evidence of lineage-sorting and/or an ancestral hominid population size greater than five times that of the effective human population size (n = 10,000). Another surprise has been the identification of ancestral allelic variants that now occur as disease alleles within the human population, i.e., pyrin and familial Mediterranean fever (51, 136). Such findings suggest that the functional and selective effects of mutations change over time, perhaps as a result of environmental changes or compensatory genetic mutations.
COMPARATIVE GENE ANALYSES
The unique attributes of the primate order, and more specifically that of human, may be explained either as a consequence of key amino-acid changes within the coding sequences of a subset of critical genes or as a result of dramatic changes in how genes are regulated both temporally and spatially. Genome-wide analyses have provided traction for both of these views, although not with equal levels of support. A critical component of these analyses has been the construction of rigorous multiple-sequence alignments among primate genes. Despite the ease at which genomic sequences can be aligned among primate genomes, the number of genes that can be assigned to 1:1:1 orthologous group has changed only slightly with the first two nonhuman primate genomes sequenced. A three-way comparison involving chimp-human-mouse identified 7645 orthologues [by the estimate of (31)] as compared to 10,376 by human-chimp-macaque (51) over the total estimated 20,000 genes in the human genome. It follows then that a large fraction of human genes have not been subjected to three-way orthologous comparisons, and the pattern of selection (and its directionality) operating on ∼50% of genes has not yet been adequately interrogated. With this caveat, we consider the lessons learned.
Evolution of Coding Sequences
The anthropocentric view of primate evolution, in which humans have acquired multiple idiosyncratic features needed for our success as a species, led to the tacit assumption that differences in a subset of key genes would explain the evolution of our species within the context of the primate order. Most of the first genome-wide studies of natural selection have thus been focused on coding sequence and estimates of omega (dN/dS)—the ratio of nonsynonymous versus synonymous substitutions as a metric of the signal of the intensity of such selection. The first three-way primate genome comparisons suggested that human and chimpanzee genes had similar average omega values but significantly larger values than macaque or rodents (human = 0.169, chimpanzee = 0.175, macaque = 0.124, nonprimate mammals ∼0.11) (51). This was explained by a relaxation of purifying selection during hominoid evolution as a consequence of smaller effective population sizes (see below, and see Figure 7).
Figure 7.

Demographic parameters and speciation times of humans and great apes. Current population sizes (red; IUCSN list of threatened species at http://www.iucnredlist.org/), effective population sizes, and estimated ancestral population sizes (blue) are shown in thousands of individuals for each hominid species and subspecies. Divergence times in either millions of years (Mya) or thousands of years (Kya) are indicated. Population genetic parameters are based on diversity data presented in the following papers: Wall (158), Won & Hey (163), Becquet & Przeworski (13), Hobolth et al. (66), Burgess & Yang (19), Steiper (141), and Caswell et al. (26). ∗ correspond to most of the studies but Burgess & Yang (19) suggested a lower bond of 14 Mya to 22 Mya.
Earlier studies that used mouse coding sequences as an outgroup (31) reported the first global estimates of genes under positive selection in human and chimpanzee, identifying more than 500 genes under positive selection (especially glycophorin C, protamines, and a gene family related to nociception). The posterior refinement with the macaque assembly, however, reduced that number to only ∼200 genes (32, 51). Looking at pervasive adaptive evolution in genes within the same category led groups to identify gene ontologies enriched for positive selection (51, 115). The common conclusion of most of these studies is an overrepresentation of rapidly evolving genes in biological processes associated with immunity, olfaction, host defense, and reproduction. These enrichments, however, are not particular to human but represent adaptive changes more generally important in evolution of the primate and broadly other mammalian lineages.
A recent report using macaque as an out-group has suggested that it is the chimpanzee lineage with an excess of positive selection compared to humans (7). Bakewell and colleagues found that even after multiple test correction, the chimpanzee has an excess of genes under positive selection (∼40% more than human), although these conclusions should be tempered by the incomplete nature of the chimpanzee genome assembly and the potential for sequence errors to confound such estimates asymmetrically. Nevertheless, this view was also supported by the most comprehensive genomic comparison to date including six genomes (human, chimpanzee, macaque, mouse, rat, and dog). Given the increased power of this six-way comparison, it was possible to identify genes involved in biological processes rapidly diverging within the primate lineage in contrast to rodents. Once again, genes related to sensory perception (e.g., pain receptor NPFF2 and color vision genes OPN1SW), immunity (CCR1), and defense were overrepresented among positively selected genes (93). The previous study also found mRNA transcription, stress response, and protein metabolism as the main biological processes with excess of amino-acid replacements in chimpanzee but not in human. Despite some discrepancies, these and other studies, in general, did not find an enrichment of positively selected genes related to brain development or size.
Gene Regulation and Expression
After considerable scrutiny of coding sequences searching for traces of adaptive evolution, it has become increasingly apparent that regulatory differences must be playing a key role in specifying primate adaptations (25). This is not a new concept. Thirty years ago Wilson & King suggested that the phenotypic changes between humans and great apes were too dramatic to be explained by the rather limited degree of protein variation (91). There are many layers of complexity ranging from changes in gene expression, chromatin regulation, and patterns of alternative splicing. For example, a whole-genome comparison of alternative splicing between human and chimpanzee found that as much as 6%–8% of the analyzed exons showed evidence of differential alternative splicing (23).
Numerous microarray studies have focused on gene-expression differences between human and chimpanzee (21, 43, 84, 154) or even between male and female individuals of different primates (129). There has been a strong emphasis on the investigation of genes acting in the brain especially in light of human cognitive specializations. In this regard, however, it should be noted that chimps may, in fact, have some mental skills that are enhanced, such as faster hand-eye signaling of character recognition (69) (see http://www.pri.kyoto-u.ac.jp/ai/video/video_library/project/project.html for a library of impressive videos on the project). Significant gene-expression differences between human and chimpanzee brains have been reported, but, surprisingly, other tissues (such as heart or liver) showed a larger number of differences (see (126) for a review on the use of microarrays on primate gene expression). Since the divergence of human and chimpanzee, ∼100 genes have altered their gene expression patterns. Some studies suggest that gene expression differences in the brain have been biased toward the human lineage as a result of upregulation in humans (21, 43) (Table 2), although other studies contradict these results (52). In contrast, sex differences in brain gene expression have been a conserved feature over the course of primate evolution (129).
Table 2.
List of genes upregulated in humans1
| #ID Gene symbol | RefSeq ID | Gene description | Chr (Build 35) | Start | End | Direction | Tissue | Source | dN/dS Chimp (ENSEMBL) |
|---|---|---|---|---|---|---|---|---|---|
| BAT8 | NM_025256 | LA-B associated transcript 8 BAT8 isoform b | chr6 | 31,955,518 | 31,973,443 | Up | Liver | Gilad et al. 2006 | 0.0862 |
| BTAF1 | NM_003972 | BTAF1 RNA polymerase II, B-TFIID transcription | chr10 | 93,673,715 | 93,780,062 | Up | Liver | Gilad et al. 2006 | 0.0000 |
| C21orf33 | NM_004649 | es1 protein isoform Ia precursor | chr21 | 44,382,191 | 44,384,261 | Up | Brain | Enard et al. and Caceres et al. | 0.1464 |
| C4ORF1 | #N/A | #N/A | Up | Liver | Gilad et al. 2006 | 0.1059 | |||
| CA2 | NM_000067 | Carbonic anhydrase II | chr8 | 86,578,696 | 86,580,973 | Up | Brain | Enard et al. and Caceres et al. | 2.1364 |
| COL6A1 | NM_001848 | Collagen, type VI, alpha 1 precursor | chr21 | 46,226,090 | 46,249,391 | Up | Brain | Enard et al. and Caceres et al. | 0.1162 |
| CROC4 | NM_006365 | Transcriptional activator of the c-fos promoter | chr1 | 153,187,128 | 153,212,257 | Up | Brain | Enard et al. and Caceres et al. | 0.9643 |
| CTCF | NM_006565 | CCCTC-binding factor | chr16 | 66,153,964 | 66,230,589 | Up | Liver | Gilad et al. 2006 | 0.0000 |
| DUSP6 | NM_022652 | Dual specificity phosphatase 6 isoform b | chr12 | 88,265,972 | 88,270,394 | Up | Liver | Gilad et al. 2006 | 0.0683 |
| ENTPD6 | NM_001247 | Ectonucleoside triphosphate diphosphohydrolase 6 | chr20 | 25,157,785 | 25,160,569 | Up | Brain | Enard et al. and Caceres et al. | 0.4350 |
| FCN3 | NM_173452 | Ficolin 3 isoform 2 precursor | chr1 | 27,568,218 | 27,573,883 | Up | Liver | Gilad et al. 2006 | #N/A |
| FLJ10326 | #N/A | #N/A | chr1 | 216,656,117 | 216,709,777 | Up | Liver | Gilad et al. 2006 | 0.0826 |
| FOXO1A | NM_002015 | Forkhead box O1 | chr13 | 40,027,817 | 40,138,734 | Up | Liver | Gilad et al. 2006 | 0.0000 |
| GM2A | NM_000405 | GM2 ganglioside activator precursor | chr5 | 150,628,953 | 150,631,513 | Up | Brain | Enard et al. and Caceres et al. | 0.4397 |
| GOSR1 | NM_001007024.1 | Golgi SNAP receptor complex member 1 isoform 3 | chr17 | 25,828,552 | 25,877,957 | Up | Liver | Gilad et al. 2006 | 0.2787 |
| GOSR1 | NM_001007024.1 | Golgi SNAP receptor complex member 1 isoform 3 | chr17 | 25,828,552 | 25,877,957 | Up | Brain | Enard et al. and Caceres et al. | 0.2787 |
| GTF2I | NM_032999 | General transcription factor II, i isoform 1 | chr7 | 73,516,681 | 73,619,673 | Up | Brain | Enard et al. and Caceres et al. | 0.0410 |
| HSPA2 | NM_021979 | Heat shock 70-kDa protein 2 | chr14 | 64,077,212 | 64,079,708 | Up | Brain | Enard et al. and Caceres et al. | 0.1539 |
| LGALS4 | NM_006149 | Galectin-4 | chr19 | 43,984,158 | 43,995,409 | Up | Liver | Gilad et al. 2006 | 0.2471 |
| NAGPA | NM_016256 | N-acetylglucosamine-1-phosphodiester | chr16 | 5,014,872 | 5,023,920 | Up | Brain | Enard et al. and Caceres et al. | 0.0677 |
| NPD009 | #N/A | #N/A | chr16 | 8,784,670 | 8,785,932 | Up | Liver | Gilad et al. 2006 | 0.4530 |
| OSBPL8 | NM_001003712.1 | Oxysterol-binding protein-like protein 8 isoform | chr12 | 75,269,707 | 75,477,720 | Up | Brain | Enard et al. and Caceres et al. | 0.0000 |
| PDE4DIP | NM_014644 | Phosphodiesterase 4D interacting protein isoform | chr1 | 143,562,782 | 143,706,379 | Up | Brain | Enard et al. and Caceres et al. | 0.4444 |
| PMS2L5 | NM_174930 | Postmeiotic segregation increased 2-like 5 | chr7 | 73,751,552 | 73,766,505 | Up | Brain | Enard et al. and Caceres et al. | #N/A |
| PRDX6 | NM_004905 | Peroxiredoxin 6 | chr1 | 171,713,108 | 171,724,569 | Up | Brain | Enard et al. and Caceres et al. | 0.0000 |
| RB1 | NM_000321 | Retinoblastoma 1 | chr13 | 47,775,911 | 47,954,123 | Up | Liver | Gilad et al. 2006 | 0.1163 |
| RGL1 | NM_015149 | Ral guanine nucleotide dissociation | chr1 | 181,871,830 | 182,164,289 | Up | Brain | Enard et al. and Caceres et al. | 0.1225 |
| SF3A3 | NM_006802 | Splicing factor 3a, subunit 3 | chr1 | 38,195,785 | 38,229,180 | Up | Brain | Enard et al. and Caceres et al. | 0.0000 |
| SMAD1 | NM_001003688 | Sma- and Mad-related protein 1 | chr4 | 146,760,556 | 146,837,928 | Up | Brain | Enard et al. and Caceres et al. | 0.1754 |
| SPTLC1 | NM_006415 | Serine palmitoyltransferase subunit 1 isoform a | chr9 | 93,852,933 | 93,917,459 | Up | Brain | Enard et al. and Caceres et al. | 0.1333 |
| STAT6 | NM_003153 | Signal transducer and activator of transcription | chr12 | 55,775,457 | 55,791,428 | Up | Liver | Gilad et al. 2006 | 0.4141 |
| THBS4 | NM_003248 | Thrombospondin 4 precursor | chr5 | 79,366,746 | 79,414,866 | Up | Brain | Enard et al. and Caceres et al. | 0.0719 |
| TIMP3 | NM_000362 | Tissue inhibitor of metalloproteinase 3 | chr22 | 31,521,362 | 31,583,581 | Up | Liver | Gilad et al. 2006 | 0.0000 |
| WIRE | NM_133264 | WIRE protein | chr17 | 35,629,100 | 35,691,965 | Up | Brain | Enard et al. and Caceres et al. | #N/A |
| ZFP36L2 | NM_006887 | Zinc finger protein 36, C3H type-like 2 | chr2 | 43,361,192 | 43,365,396 | Up | Brain | Enard et al. and Caceres et al. | #N/A |
Examples of genes showing increased gene expression in human compared to chimpanzee. Data were taken from References 52, 126 and references therein. Only one gene is found consistently upregulated in both studies (GOSR1). The Gene Symbol, RefSeq ID, chromosomal location (Human Build35), and the tissue studied in every study are also reported. dN/dS (a measure of the selection pressure acting upon the sequence) was retrieved from ENSEMBL. Only one gene could be a candidate to have been under positive selection (CA2, ω = 2.13).
One interesting example of a differential gene expression between human and chimpanzee is the two- to nearly sixfold increase in gene expression of THBS2 and THBS4 (thrombospondin 2 and 4) within specific parts of the human brain when compared to chimpanzee or macaque (22). In this study, the authors showed that the levels of THBS2 and THBS4 mRNA are highly differentiated in the forebrain (cortex and caudate) but not in the cerebellum or other analyzed tissues. As these genes are involved in synaptogenesis, there was speculation that these changes in gene expression may be underlying some of the human cognitive specializations.
Recent reports suggest that the hypothetical rapid relative rate of change in gene expression in the human brain has occurred in the context of a decreased rate of coding sequence evolution (compared to the genomic average) (159). Genes whose expression is restricted to the brain show greater conservation than genes expressed in the brain along with other tissues, perhaps as a consequence of stronger selective constraint operating within brain biochemical networks. Despite this constraint, there are some interesting examples of adaptive evolution for brain-expressed genes. The glutamate dehydrogenase (GDH) gene (20) shows both evidence of increased copy as a result of duplication and traces of positive selection within the human lineage. It encodes a protein that plays an important role in neurotransmitter recycling.
There has been renewed interest on the role of posttranscriptional regulation via microRNA (miRNA) and transcription factors. The study of miRNA, for example, has become increasingly relevant because of the central role miRNA plays in regulating the transcriptome of plants and animals (8). A whole-genome comparison of brain miRNA of human and chimpanzee found that more than 10% of the human brain miRNA are primate specific whereas only 1% is human specific. Although such differences might affect a large number of potential gene targets, no bias appears to exist within the human lineage (14). For example, the authors reported 14 human- and 15 chimpanzee-specific miRNA expansions. In contrast, the evolution of protein transcription factors may have not have evolved so uniformly (17). Gilad and colleagues (52) analyzed the expression profile of ∼1000 orthologous genes in human, chimpanzee, orangutan, and macaque by cDNA array hybridization and found transcription factors enriched among genes upregulated in humans. Similarly, as a class, transcription factors, in particular C2H2 zinc finger genes, show the greatest excess of amino-acid replacements within the human lineage (32), providing further support for the notion that gene expression changes have been pivotal during human evolution. These data suggest a concerted effect of positive selection and gene expression on transcription factors in altering gene expression.
Concomitant with positive selection and changes in expression of transcription factors, it follows that cis-regulatory regions (promoters, enhancers, etc.) might similarly show evidence of accelerated evolution. Using the background substitution rate of intronic regions, Haygood and colleagues developed a method to detect an excess of single-basepair changes within human promoters (compared to chimpanzee and macaque). Several genes were detected by this method, including an apparent excess of genes related to neural development and nutrition (63). Another approach has been to study highly conserved noncoding DNA that shows a burst of evolutionary changes within the human lineage (123). Among such regions, HAR1 (human accelerated region 1) has the distinction of being one of the most accelerated unique regions of the genome and is itself part of a novel RNA gene (HAR1F) that is highly expressed within the cortex of the human brain (see Figure 3). Similarly, Prabhakar and colleagues identified a 546-basepair element (HACNS1) conserved among terrestrial vertebrate genomes that had acquired 16 specific changes within the human lineage. In vivo transgenic experiments showed that a subset of these human-specific changes confers patterns of expression strongly associated with limb development relative to chimpanzees or macaques. Their results implicate changes within the conserved noncoding sequences in creating a de novo enhancer associated with anterior wrist and proximal thumb development. Although the role of this enhancer with respect to nearby genes CENTG2 and GBX2 is unknown, the authors speculate that this burst of nucleotide changes may have contributed to the dexterity of the human hand or opposability of the thumb (125) (Figure 4).
Figure 3.

A human-accelerated region of single-basepair substitution, HAR1F. (a) A highly conserved noncoding DNA segment, HAR1F, shows an excess of substitutions within the human lineage based on a multiple sequence alignment (123). (b) HAR1F is transcribed into a noncoding RNA. Many of the single-basepair substitutions correspond to compensatory changes within the context of RNA secondary structure ( yellow–blue denotes compensatory mutations, purple denotes substitutions in unpaired regions, and red denotes noncompensatory changes).
Figure 4.

HACNS1 human-accelerated mutations create a limb enhancer. (a) Multiple sequence alignment of the putative enhancer HACNS1 with other vertebrate genomes shows an excess of 13 human-specific substitutions (red ). (b) Transgenic mouse lacZ expression pattern using an enhancer in which the 13 human-specific substitutions were introduced against the orthologous chimpanzee sequence background. (c) Expression pattern using an enhancer reverting the substitutions in the human sequence to the nucleotide states in chimpanzee and rhesus. These results show that those substitutions drive strong gene expression within the limbs.
In total, several lines of evidence strongly suggest that gene-expression differences have been a catalyst of primate evolution and adaptive specializations. These findings raise the possibility that there has been an overemphasis on coding sequence differences and, perhaps, too much attention paid to the brain (as opposed to other morphological traits). Proving that these gene regulatory differences played a major role in primate adaptation remains a significant challenge, especially in the absence of “relevant” model organisms where these mutational effects can be directly tested. However, the discovery of previously unrecognized evolutionary targets potentially important in neurodevelopment and hand dexterity predicts that mutations in these may underlie neurocognitive disease in humans. The discovery of de novo mutations in association with human disease for these transcripts or cis-acting elements would provide strong evidence of their functional import.
PRIMATE GENOME EVOLUTION
Genomes are highly dynamic entities that evolve rapidly and nonuniformly through time; the genomes of primates are no exception. As genome-wide data and new technologies have become available, structural variation and copy-number differences have emerged as another important aspect of primate genetic variation (16, 28). With limited exceptions such as the gibbon (108), there are relatively few cytological differences among primate chromosomes. Most of the cytogenetic differences between humans and apes (167) have now been well characterized and most resolved at the molecular level (24, 55, 59, 87, 88, 89, 98, 133, 149–151). However, the majority of structural changes smaller than a few megabasepairs preclude detection by standard cytogenetic approaches. With the sequencing of the non-human primate genomes, the extent of this submicroscopic variation became more evident. A comparison of human and chimpanzee genomes estimated more than ∼90 Mb of DNA affected by insertion, deletion, duplication, and inversion (being ∼40–45 Mb in each lineage) (28, 107). Although single-basepair changes are far more numerous, structural variants have been estimated to affect 3–4 times the number of basepairs between human and great ape (i.e., 90 million basepairs of structural variation versus 30 million basepairs of single nucleotide difference between human and chimpanzee).
Due to the limitations of the early draft assemblies, these estimates likely represent a lower bound. Newman and colleagues (114), for example, mapped chimpanzee fosmid end-pairs against the human genome assembly and detected more than 500 insertion, deletion, and inversion events, ranging in length from 12 to 40 kbp. Most of these were previously unknown. In another study comparing the human and chimpanzee genome, a total of 1576 putative regions of inversion were detected (47). Using the gorilla genome as an outgroup, many of the validated sites were found to be relatively young events and some were polymorphic within the human lineage. Similarly, Szamalek and colleagues (148) performed gene-order comparisons for more than 10,000 orthologous genes between human and chimpanzees and identified 71 putative microre-arrangements. The importance of structural variation and copy-number polymorphism in human disease and disease susceptibility and its abundance suggest that such variation may have had profound repercussions in the evolution of human and great ape primates. The role of these events in altering the pattern of gene expression and chromatin organization has not yet been addressed.
Two features of primate genomes may account for the abundance of large-scale structural variation observed in primate genomes, namely segmental duplications and retrotransposons. Segmental duplications (SDs) are segments of DNA greater than 1 kbp in length with high sequence identity (>90%) that typically map to two or more locations in the genome.
Recent comparative work among humans and non-human great-apes has shown that the human and great-ape lineage are particularly enriched for interspersed duplications with a suggested burst occurring in the common ancestor of the human and African great-apes (105). Breakpoints of large-scale structural variation between and within species preferentially map to regions of segmental duplications (1, 4, 121). When compared to other sequenced mammalian genomes, primate (especially human and great ape) segmental duplications tend to be larger, more complex, and more interspersed [(5, 29, 77, 137, 138; for a detailed description of the organization and distribution of primate SDs see (6)]. These peculiar features promote further genomic instability leading to extensive copy-number variation both within and between species enhanced by long-distance nonallelic homologous recombination and other less well understood mechanisms of genetic exchange (78). As a result of this constant genomic turnover, evolutionarily shared duplications often show as much copy-number variation as lineage-specific duplication events (120, 121).
New insights regarding the evolution of human SDs have recently come to light (77, 109). Using a computational graph-theory and phylogenetic approach, Jiang et al. revealed that human SDs are frequently organized around “core” elements that show greater EST and exon density when compared to flanking segmental duplications (77). Detailed comparative sequencing of one of these core elements on chromosome 16 in human and ape showed that the complexity of these regions has emerged as a result of the serial accretion of duplicated segments centered around the core duplication segment (Figure 5). In different primate lineages, the core segments have moved or been copied to new locations or entirely different chromosomes leading to a completely different suite of segmental duplications all centered around the same core duplication (78). This feature helps to explain why unique genes mapping in close proximity to these duplication blocks have a ten-fold increased probability of being duplicated when compared to a random model of genome duplication (28, 40, 140)—a phenomenon known as duplication shadowing (28).
Figure 5.
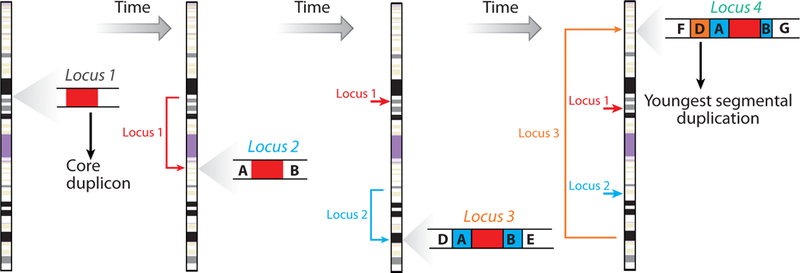
Core duplication hypothesis of human segmental duplications. Duplicative transposition or biased gene conversion duplicates copies to new locations with different unique sequences flanking the duplicated copy of the core; a secondary duplication moves into a third location but also carries flanking duplicons from the second locus to the third; the process repeats itself during the course of evolution, leading to the formation of large complex blocks of intrachromosomal segmental duplication along the chromosome that can now promote nonallelic homologous recombination. These events occur before and after speciation, leading to both lineage-specific and shared duplication blocks between closely related species. Segmental duplications located at the periphery are more likely to be evolutionary young or lineage specific (e.g., duplication D), whereas the core is common to all the duplication blocks. Paralogous sequence exchanges can occur between loci changing the sequence and erasing evolutionary history. Data based on primate comparative sequencing (78) and evolutionary reconstruction of human segmental duplications (77).
In addition to being hotspots of genomic structural variation, segmental duplications are substrates for the emergence of new genes and gene families (39, 79). Primate segmental duplications are enriched for exons when compared to mouse segmental duplications (137). Most of the primate gene expansions correspond to regions of segmental duplication (28, 36). This includes the olfactory receptor gene family (153), which has been subjected to sudden decline in catarrhine primates (53, 54), although it may still play a critical role in kin recognition (71).
In general, gene expansions seem to have been common in primate evolution (36, 61), and many notable examples have been found [such as PRAME (expressed antigen of melanoma) within the human lineage and PFKP (sugar metabolism) and DIP2C (segmentation patterning) within the macaque lineage]. Examples of reported human-specific expansions include NEK2 or ANAPC1 encoding centrosome-related proteins and aquaporin 7 (AQP7), which has been suggested as a candidate for a human endurance running (36). Perhaps some of the most conspicuous gene family expansions map to the core duplicons described above (Figure 5) and include gene families such as NPIP, DUF1220, RANBP2, and TRE2 (Table 3) (30, 79, 119, 124, 156). These hominoid-specific gene families frequently show evidence of remarkable signatures of positive selection, are associated with bursts of segmental duplication, and demonstrate dramatic changes in their expression profile. The function(s) of these novel genes are largely unknown.
Table 3.
Great ape and human gene family expansions1
| Gene family name | Representative gene | Gene info | Evidence of positive selection | Gene expression | Estimated human copy number | Chromosome | Comment | Reference |
|---|---|---|---|---|---|---|---|---|
| Morpheus | NPIP | Nuclear pore complex interacting protein | ++ | Widely expressed | 18 | 16 | 50X dN/dS | (79) |
| DUF1220 (NBPF) | NBPF11 | Neuroblastoma breakpoint family, member 11 | + (but see 155) | Neurons (among other tissues) | 11 | 1 | Expressed in neurons | (124, 155) |
| RGP | RANBP2 | RAN binding protein 2 | + | Testis/cortex | 8 | 2 | Positive selection | (30) |
| LRRC37 | LRRC37 | Leucine-rich repeat containing 37 | Unknown | Ubiquitous | 3 | 17 | (77) | |
| TBC1D3 | TRE2/USP6 | Biquitin-specific protease 6 | Unknown | Testis | 4 | 17 | Result of a recent evolutionary fusion of two other genes—TBC1D3 and USP32 | (119) |
Examples of gene families embedded in “core” duplicon (77) that have been greatly duplicated specifically in great apes (NPIP, RANBP2, TRE2, NBPF11) or in primates (LRRC37). Evidence of positive selection as well as information on the gene function and expression or the estimated copy number in human (according to the number of WGS reads mapping to the region) is also provided.
In addition to segmental duplications, mobile elements, particularly retrotransposons, have played a significant role in altering the landscape of primate genomes. Primates are distinguished from all other mammalians by the presence of Alu retroposons (169). Similar to segmental duplications, there is strong evidence of a burst of Alu activity. For example, over one third of existing human elements are thought to have retrotransposed over a 10-million-year window of evolution (30–40 Mya) (11, 35, 97).
Notably, Alu repeats are preferentially found at the boundaries of segmental duplications (4, 83) and map to gene-rich and GC-rich chromosomal regions. It has been postulated that the abundance of interspersed segmental duplications in primates may have been precipitated by the potential for 100,000s of Alu repeats to incur double-strand breaks and to provide microhomology. In this model of cascading repeat instability, the burst of Alu activity would have promoted segmental duplications and, in turn, segmental duplications promoted copy-number variation—a domino effect of increasing structural variation over time.
Although most retrotranspon activity has waned in recent human evolution, sequencing of other primate genomes has uncovered lineage-specific expansions of other elements. For example, a retroviral expansion of 100–200 copies of the retroviral PTERV1 element was discovered after sequencing of the chimpanzee genome (Figure 6) (82, 122, 165).
Figure 6.

A burst of PTERV1 retroviral insertions in African great apes and Old World monkey species but not orangutan or human. (a) Whole-genome landscape of PTERV1 integration sites is shown using the human chromosomes as a reference (build35). (b) A detailed view on one chromosome (chromosome 2) is shown. While some species has been massively plagued by the retrovirus that integrated into the germline (chimpanzee, gorilla, macaque, and baboon), other species (human and orangutan) are unusually devoid. Furthermore, 95% of these sites were found to be in nonorthologous locations when compared between species, suggesting episodic events early in the history of each of these species potentially from an exogenous source.
PTERV1 is found in both the gorilla and chimpanzee genome but the map locations are largely nonorthologous. Moreover, no copies of the sequence have been identified in orangutan or human, although both macaque and baboon genomes carry multiple copies—albeit at locations different from each other and the gorilla and chimpanzee. These data have been used to argue that PTERV1 arose from an external viral source that integrated into the germline. Mutations in the TRIM5α (a protein active in restriction of retroviral insertion) have been posited to explain differences in the distribution of this retrovirus. For example, the human variant of TRIM5α has been shown to be capable of restricting the ancestrally reconstructed version of the PTERV1 retrovirus (82).
TESTS OF MODELS OF SPECIATION ON GENOMIC DATA
The complex process, tempo, and mode of primate speciation has been the subject of considerable debate. However, if a limited understanding has been produced regarding divergence from a common ancestor of humans and chimpanzees, even less is known concerning the speciation within other great apes. Taking advantage of genome-wide datasets, at least two different approaches have been put forward to answer this question. One (a test of a chromosomal speciation model) focuses on the hypothetical effect of chromosomal rearrangements and suppressed recombination within primates [see review of (3)], whereas another approach focuses on identifying traces of speciation by using scans of divergence across different regions of the genome (see for instance Reference 118). Both are highly controversial but have engendered considerable discussion regarding the events underlying separation of our species from nonhuman primates.
Chromosomal Rearrangements and Speciation
Polymorphic genome structural variants (such as inversions, fusions, and fissions) may promote speciation events between contiguous (parapatric) or partly overlapping (sympatric) populations. Among the several models of chromosomal speciation (see 161), the so-called suppressed-recombination model of speciation suggests that chromosomal rearrangements serve as a genetic barrier between populations and, hence, substitutions linked with the rearranged chromosomes cannot be freely exchanged among karyotypically distinct subpopulations, eventually leading to incompatibilities and thus to speciation (112). Studies within a variety of different lineages such as Drosophila, Anopheles, murids, shrews, or sunflowers (3, 10, 116, 131, 132) have provided some support for this model of speciation.
In the first study to test predictions of suppressed-recombination chromosomal speciation models in primates, Navarro & Barton reported an association between chromosomal rearrangements and higher evolutionary rates based on an analysis of a limited set of 115 autosomal orthologous genes from humans and chimpanzees (113). This and other observations (such as a lower level of polymorphism in rearranged chromosomes) were consistent with the predictions of the model. Several interpretations for the results were given, the most controversial being the suggestion that under the hypothesis tested, the results would hold only if the chromosomal rearrangements had been barriers in parapatry for no less than half of the time of divergence between humans and chimpanzees (130). This conclusion was striking because it ran counter to both anthropological data and the molecular dating of rearrangement events (151).
Subsequent analyses have questioned the results and inferences from the initial paper. For example, Lu and colleagues found that rapidly evolving genes may not have a homogeneous distribution among chromosomes and that rearranged chromosomes may have been linked to rapidly evolving genes due to factors unrelated to speciation. An alternative explanation was that the GenBank sequences used by the initial study and by Lu et al. (99) were biased and not representative of the rest of the genome. This latter explanation seemed to be the main reason for the initial result since subsequent genome-wide studies (32, 51, 64) found evolutionary rates threefold smaller than the original dataset. Similarly, studies (mainly from noncoding DNA) found that the average nucleotide divergence was in fact lower in rearranged chromosomes when compared to colinear—a result opposite to the predictions of Navarro & Barton (168). Other studies (32, 155) found no genome-wide differences.
Within the past year, additional studies have revisited the topic with more complete datasets. All these studies found slightly less divergence within the pericentromeric inversions that distinguish human and chimpanzee (106, 147) even when considering the effect of duplicated regions (104). Although genome-wide analyses of positive selection have provided little support for this model, analogous comparisons of gene expression differences between humans and chimpanzees for colinear and rearranged chromosomes have, however, yielded contradictory results. Based on earlier gene-expression studies of the cerebral cortex (21, 43, 90), the average gene expression difference between human and chimpanzee was statistically higher in rearranged chromosomes when compared to colinear chromosomes (103). This observation was replicated by Khaitovich and colleagues (90) but not by Zhang and colleagues (168), although the latter considered only a subset of genes (genes with statistical different expression pattern in human and chimpanzee).
Overall, it appears that there is little evidence in support of human-chimpanzee speciation via suppressed-recombination. Chromosomal speciation in primates, however, cannot be definitively ruled because (a) chromosomal speciation does not have to involve all rearranged chromosomes, and (b) speciation might have involved other noncoding functional elements, such as genes that do not encode proteins (microRNAs, for example) or other regulatory elements (such as transcription factor binding sites). The discrepancy between gene expression and positive selection data may provide some support for this view.
Ancient Hybrid Models
Coalescent models have recently been applied to the question of human and chimpanzee speciation. Based on the hypothesis that allopatric models predict similar divergence times among different regions of the genome, Osada & Wu found that coding regions shared deeper genealogies than intergenic regions as suggested by parapatric models, because coding regions are more likely to have been involved in hybrid incompatibilities or adaptive evolution (117). The analysis supported a complex parapatric view of speciation between human and chimps. Patterson and colleagues (118) addressed the question by analyzing the heterogeneity of human chimpanzee divergencies in DNA sequence based on analysis of genomic sequence from human, chimpanzee, gorilla, and other related species (orangutan or macaque). Human-chimpanzee genetic divergences varied from ∼84% of the average to up to 147%, a range of more than 4 million years. Notably, they observed a lower divergence between X-chromosomal sequences than for autosomal sequences, a circumstance they suggest could be explained if human and chimpanzee initially diverged, and then later exchanged genes before separating permanently.
They proposed that humans and chimpanzees would be more closely related through the X chromosome, by the following process: If human and chimpanzee ancestors initially speciated and then interbred, male hybrids might be partly sterile, as Haldane’s rule on heterogametic sex suggested (62). A viable population could then only have arisen if the fertile females mated back to one of the ancestral populations, producing fertile male hybrids that then transmitted X chromosomes derived almost entirely from the initial population. Several concerns, however, have risen with respect to the interpretation of these results. Barton (9) noted that the scenario proposed by Patterson and colleagues is not the only compatible explanation. In fact, the heterogeneity in divergence observed between human and chimpanzee genomes is consistent with quick speciation (allopatric) from a large ancestral population (Ne ∼ 45,000), a view supported by others (66). Moreover, a similar level of X-chromosome divergence could also be explained by the amount of time that copies of the X chromosomes spent in the male/female lineages, which of course depends on the male/female ratio (α) (100, 157). Additionally, Burgess & Yang (19) analyzed 7.4 Mb aligned for human, chimp, gorilla, orangutan, and macaque showing that the peculiar reduction of X-chromosome diversity may have arisen as a result of specific selective sweeps on the X chromosome prior to the human/chimp speciation (19).
In summary, applications of genome-wide datasets to develop models of primate speciation have provided no clear consensus on the underlying mechanisms. The controversy and confusion are largely a reflection of the fact that sequence divergence data are much less uniform than anticipated. As population genetic models evolve to more adequately incorporate these data from additional primate genomes, greater insight into speciation should emerge.
PRIMATE DIVERSITY AND RECOMBINATION
Primate Diversity
Analysis of genetic variation within and between primate populations is a powerful tool to understand the evolutionary history, demography, and effective ancestral population size of primate populations. High-throughput analyses of SNPs, haplotypes, and CNVs, for example, have significantly resolved the population ancestry and geographic origin of humans (72, 95, 127). Although studies of nonhuman primates are less developed, the availability of reference genomes and the sampling of genetic diversity (largely SNPs) from multiple individuals of different species have begun to provide other contrasting models of primate demography. From the first restriction fragment length polymorphism (RFLP) studies of ape mitochondrial DNA (46) suggesting that chimpanzees and gorillas possessed two to three times more genetic diversity than humans, it has been evident that humans have less genetic diversity than that of our closest great-ape relatives. Later analysis of nuclear loci confirmed the existence of fundamentally different levels of genetic diversity, although at a lower level—approximately 50% more than humans (76, 80).
Wild primate populations are notoriously difficult to census, suggesting that genetic approaches to estimating effective population size can help understand the evolution of current populations and their demographic history. Previous studies of chimpanzee genetic diversity suggested that chimpanzees can be differentiated in three main subpopulations: Eastern (Pan troglodytes schweinfurthii), Central (Pan troglodytes troglodytes), and Western (Pan troglodytes verus). From all three subpopulations, Central chimpanzees show the greatest genetic diversity and the largest effective population size (12, 13, 26, 48, 49, 80, 144, 163, 166). The effective population size of Central chimpanzees has been estimated to be much higher than human and Western chimpanzees perhaps as a result of three- to four-fold population expansion after separation from the Western chimpanzees (Figure 7). Other studies suggest an even larger effective population size (∼100,000) (26). Given that the estimated effective population size for that common ancestor of bonobo and chimp is approximately 25,000 (13,000 to 30,000), which in turn is smaller than that estimated from human and chimpanzee (∼100,000) (13, 19, 26, 66, 158, 163), the data suggest that human and chimpanzees have experienced a drastic reduction in their effective population sizes.
Similarly, the genetic diversity of Western gorillas and Eastern gorillas and orangutan subspecies also suggests lower effective population sizes than their ancestral population (∼40,000 and ∼87,000, respectively) (13, 19, 66) and lower than the common ancestral human, chimp, gorilla, and orangutan population sizes (70,000 and up to 127,000) (19). On a much smaller scale, and using an approach that allows the ratio of past and present population effective size to be estimated (145), Goossens and colleagues (58) analyzed microsatellite allelic variation in endangered Kinabantangan (Bornean) orangutans and inferred a drastic reduction in population size over the past several decades. In this instance, the reduction in population size has been attributed to direct effects of recent human activities, especially habitat reduction through logging and agricultural activities. Taken together, a consensus emerges that the ancestral effective population size in hominoid lineage was five- to tenfold higher than almost all primate effective population sizes, suggesting a continuous reduction of effective individuals during great-ape evolution.
The recent sequence of the macaque genome has been accompanied by a detailed diversity analysis of a few Kb (∼150 Kb) in 47 macaque individuals from two populations (9 from Chinese and 38 from Indian populations) (51, 65). This survey showed that Chinese macaques have an excess of rare variants when compared to the Indian population where inter-mediate frequency variants predominate. These findings contrasted with earlier studies based on mtDNA and a much smaller set of SNPs that showed much more modest levels of differentiation (45, 139). Demographic inferences of these findings indicate that the Chinese population was first expanded and that later there was a drastic reduction of population size in the Indian population (population size in the Chinese population is now estimated to be one order of magnitude higher than the Indian population). These findings have immediate practical application in mapping of genetic traits. The patterns of LD (linkage disequilibrium) decay suggest that Indian macaques are especially useful for disease association studies since their LD patterns extend even farther than humans, hence fewer markers would be required to discover significant associations. On the other hand, the Chinese rhesus would be more useful in winnowing the interval associated with any phenotypic trait (65) as a consequence of greater decay in the patterns of LD disequilibrium.
Recombination
Among the forces shaping primate genomes are adaptations of cellular mechanisms that facilitate reproduction, including the structural components of recombination pathways and impacts of genome organization. Recombination hotspots are defined as narrow regions of the genome (1–2 Kb) in which recombination occurs to a higher degree than the genome average. Recombination has been assessed from direct genotyping of recombination events in sperm (2, 74) or inferred by population genetic analysis of genome sequence data (33). The former method, although time-consuming, identified the existence of recombination hotspots in human males (2, 74). An alternative approach is to make use of the patterns of LD generated to estimate the recombination rate required to produce the population distribution of observed haplotypes. Importantly, the majority of hotspots detected by sperm typing have been also detected by LD-based approaches (75; but see 85). Myers et al. (110) proposed that the human genome contains ∼25,000 hotspots (approximately one hotspot every 50 Kb) and that there are sequence motifs associated with those hotspots that could be experimentally demonstrated to modulate the intensity of recombination (110). These results suggested that recombination rates could be regulated (at least in part) by cis-acting sequences, and motifs have been identified associated with both allelic and non-allelic recombination (111). Genetic linkage maps in baboon, green monkey, and macaque have shown that the recombination maps are significantly shorter than the human (34, 73, 134). The available results suggest that humans and, perhaps, hominoids more generally have evolved higher rates of recombination with implications of greater genetic diversity in a background of a reduced rates of single nucleotide substitution (42, 56, 143).
One of the most interesting findings that has emerged from comparative maps of fine-scale recombination has been that recombination hotspots are not necessarily conserved between human and chimpanzee (128, 162) (Figure 8). These findings establish that positional recombination rates may change rapidly over time, a concept reinforced by the finding that recombination rates are highly variable among different humans (92). However, when analyzed on a broader scale (∼50 Kb), a weak correlation in rates of recombination exists between humans and chimpanzees (128, 162) and among other mammals (37). This has opened the possibility that the regional background rate of recombination remains relatively constant but that the actual hotspots are transient, perhaps as a result of competition. Yet this difference in recombination must occur in a background where the sequence (and thus the sequence motifs) are nearly identical, suggesting that other structural changes in trans-regulation, epigenetic factors, or selection against alternative alleles in the motifs affect the specificity of recombination and ultimately rates of recombination in different primate lineages.
Figure 8.
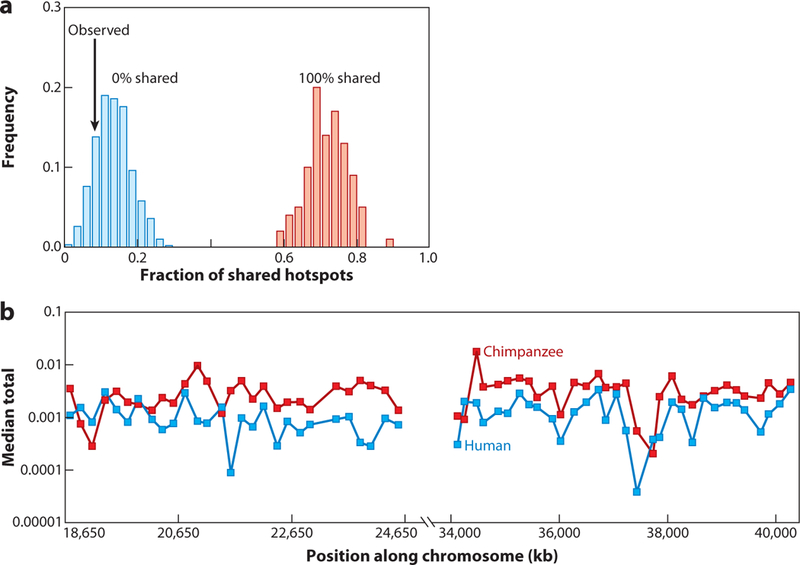
Recombination hotspots vary between chimpanzee and human. (a) Distribution of shared hotspots under two statistical models. In the first model (blue) all the hotspots are independent and in the second (red ) all hotspots are shared. The observed proportion of shared hotspots (8%) suggests that allelic hotspots of recombination evolve rapidly and change over short periods of evolutionary time. (b) On a broader scale, the recombination rates (measured in 50-Kb windows) correlate, suggesting that the landscape of recombination remains relatively constant.
THE FUTURE
A complete framework for understanding of the human genome requires knowledge of its evolution, necessitating comparative studies with other species, including closely related primates. With the emergence of next-generation sequencing technology (101) and the concomitant reduction in sequencing costs, it is no longer outside the realm of possibilities that “complete” genome sequencing of all species of the primate order will be achieved within the next ten years. The prospects of a “500 Primate Genomes Sequencing” project should be tempered with the need to sequence the genomes to a standard of high quality. In light of the complex organization of the human genome and the finding that so much of the genetic difference maps to these complex regions, simply aligning sequencing reads to a reference genome and cataloguing the single-nucleotide differences between and within species should not be the end goal. As evidenced by the sequencing of the human and chimpanzee genomes, sequencing primate genomes well is nontrivial. Even whole-genome sequence assembly of primate genomes using “Sanger” capillary sequences leads to the significant loss of information, including the absence of entire genes and gene families (Figure 9). The excitement of sequencing more primate genomes thus should be balanced by insisting that the most dynamic regions are not excluded as part of the process. This requires a greater investment of resources and careful attention to details, which in this era of “slash-and-burn” genomics is becoming a dying art.
Figure 9.
Limitations of whole-genome shotgun sequencing of primate genomes. A comparison of two genome assembly methods for human chromosome 16 is shown (138). Top line represents the chromosome 16 assembly based on hierarchical sequencing of large insert clones (IHGSC, 2005) (94) while bottom line represents the genome based strictly on whole-genome shotgun sequence assembly of sequence reads from capillary sequencers (70). The WGSA assembly is ~ 20 Mbp shorter than the clone-based assembly and is missing primarily duplicated sequences (middle line). The missing material is highly duplicated (28 distinct regions), carries a rapidly evolving human-great ape gene family (78), and is a breakpoint for microdeletions associated with mental retardation and autism. The WGSA assembly is missing most of this material and fails to map most of the loci. This effect is expected to be exacerbated with next-generation sequence technology where the average read length is currently significantly shorter than capillary sequencing methods.
The opportunity, however, to sequence the true extent of primate diversity is time-limited. It is a commentary on the success of human beings that the rapid population expansion that has taken place in our species over the past approximately 10,000 years has led to dramatic reductions in other primate species. Fully 48% of all primate species are threatened; 70% of Asian primates face extinction and two dozen are critically endangered (http://www.primate-sg.org/redlist08.htm; 68). All of the great apes, to which humans are most closely related (Figure 1)—comprising chimpanzees, bonobos, gorillas, and orangutans—are endangered or critically endangered. Declining populations of primates are the object of conservation efforts to secure habitat and reduce threats to population viability. Knowledge of the genetic structure of primate populations, their demo-graphic history, and significant life-history attributes that can be inferred from assessments of genetic diversity can contribute to efforts to conserve self-sustaining wild primate populations. Thus, there is clear opportunity of comparative genomics studies to contribute to primate conservation actions (135).
Additional whole-genome sequencing projects for primates serve societal interests in identifying the functional elements of the human genome and provide a basis for evaluating genetic diversity and nucleotide sequence evolution not only for species whose genomes have been sequenced, but for closely related species as well. The practical consideration of obtaining appropriate samples for preliminary studies, partial or complete genome sequencing, and postassembly resequencing has received relatively little attention, yet represents a significant community need. The U.S. National Science Foundation recently established the Integrated Non-human Primate Biomaterials and Information Resource (www.ipbir.org) to assemble, characterize and distribute high-quality DNA samples of known provenance with accompanying demographic, geographic, and behavioral information in order to stimulate and facilitate research in primate genetic diversity and evolution, comparative genomics, and population genetics. Many samples in the IPBIR were derived from primates in zoos or U.S. National Primate Centers, and a large proportion of the cell cultures serving as sources of DNA for distribution came from the Frozen Zoo® of the Zoological Society of San Diego.
In addition to access to DNA from index individuals, nucleic acid preparations from additional unrelated individuals are essential for evaluating genetic diversity, SNP discovery, copy-number variation, recombination parameters, effective population size, and parameters of selection, including selective sweeps. SNP identification and analysis is of direct interest as SNP assays are a basic currency of genetic variation, relevant to population genetics, disease association, and phylogeographic studies alike. The potential utility of high-throughput SNP assays is manifested in the aforementioned studies of geographic population of rhesus macaques and the effort to identify factors associated with variability in responses to SIV infection that distinguish rhesus macaques of Indian and Chinese origin (45, 65). Recently, comparison of SNPs in cynomolgus (Macaca fascicularis) and rhesus macaque (M. mullata) identified shared polymorphisms, raising the possibility that these two well-recognized species may be more closely related than suggested by mitochondrial DNA or morphological comparisons (146). This unexpected finding serves as an additional example of the significance of genomic studies for the understanding of evolutionary processes in primate speciation and phenotypic diversification, relevant to medical primatology and conservation of wild primate populations alike.
Among the primate order of mammals, comparative genomic studies have advanced more rapidly for taxa closely related to humans, chimpanzees, macaques, and baboons. As complete genome-sequencing projects advance for other primate families, including the New World monkeys (Cebidae) and strepsirhine primates (lemurs, lorises, aye-aye, pottos, and galagos), new insights are anticipated as, particularly for a lemur genome project, new information about primate adaptations and evolution can be anticipated (67).
The availability of complete genome sequences from additional primate species and the elucidation of intraspecific and population diversity at the nucleotide sequence and cytogenetic levels can be applied to conservation efforts for wild populations of threatened primates. Constraints in sample collection may require exploration of new technological approaches as, for example, current studies of intraspecific variation in wild populations of great apes depend on noninvasive samples such as feces and hair. Genome sequencing projects for primates should typically include components evaluating genetic diversity of geographically distinct populations. Ideally, new partnerships between field researchers, governments, conservation organizations, and genome biologists can advance both the understanding of genomic mechanisms of primate adaptations and evolution, while enriching the understanding of primatology and contributing to primate conservation.
ACKNOWLEDGMENTS
We are indebted to J. Pedersen, K. Pollard, S. Salama, M. Caceres, T. Preuss, J. Noonan, M. Oldham, D. Geschwind, Z. Cheng, and S. Ptak for providing access to their data and figures. We would like to thank T. Brown for comments during the preparation of the manuscript. T. M-B is funded by a Marie Curie Fellowship. O.R. acknowledges support from NSF BCS-0094993, the Caesar Kleberg Wildlife Foundation. and the John and Beverly Stauffer Foundation. E.E.E. is an investigator of Howard Hughes Medical Institute. E.E.E. acknowledges support from NIH grant GM058815.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Armengol L, Pujana MA, Cheung J, Scherer SW, Estivill X. 2003. Enrichment of segmental duplications in regions of breaks of synteny between the human and mouse genomes suggest their involvement in evolutionary rearrangements. Hum. Mol. Genet 12:2201–8 [DOI] [PubMed] [Google Scholar]
- 2.Arnheim N, Calabrese P, Nordborg M. 2003. Hot and cold spots of recombination in the human genome: the reason we should find them and how this can be achieved. Am. J. Hum. Genet 73:5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayala FJ, Coluzzi M. 2005. Chromosome speciation: humans, Drosophila, and mosquitoes. Proc. Natl. Acad. Sci. USA 102(Suppl. 1):6535–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey JA, Baertsch R, Kent WJ, Haussler D, Eichler EE. 2004. Hotspots of mammalian chromosomal evolution. Genome Biol 5:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey JA, Church DM, Ventura M, Rocchi M, Eichler EE. 2004. Analysis of segmental duplications and genome assembly in the mouse. Genome Res 14:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey JA, Eichler EE. 2006. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet 7:552–64 [DOI] [PubMed] [Google Scholar]
- 7.Bakewell MA, Shi P, Zhang JZ. 2007. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc. Natl. Acad. Sci. USA 104:7489–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–97 [DOI] [PubMed] [Google Scholar]
- 9.Barton NH. 2006. Evolutionary biology: How did the human species form? Curr. Biol 16:R647–50 [DOI] [PubMed] [Google Scholar]
- 10.Basset P, Yannic G, Brunner H, Hausser J. 2006. Restricted gene flow at specific parts of the shrew genome in chromosomal hybrid zones. Evolution 60:1718–30 [PubMed] [Google Scholar]
- 11.Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat. Rev. Genet 3:370–79 [DOI] [PubMed] [Google Scholar]
- 12.Becquet C, Patterson N, Stone AC, Przeworski M, Reich D. 2007. Genetic structure of chimpanzee populations. PLoS Genet 3:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becquet C, Przeworski M. 2007. A new approach to estimate parameters of speciation models with application to apes. Genome Res 17:1505–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, et al. 2006. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet 38:1375–77 [DOI] [PubMed] [Google Scholar]
- 15.Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, et al. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299:1391–94 [DOI] [PubMed] [Google Scholar]
- 16.Britten RJ. 2002. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc. Natl. Acad. Sci. USA 99:13633–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brivanlou AH, Darnell JE. 2002. Transcription—signal transduction and the control of gene expression. Science 295:813–18 [DOI] [PubMed] [Google Scholar]
- 18.Bullaughey K, Przeworski M, Coop G. 2008. No effect of recombination on the efficacy of natural selection in primates. Genome Res 18:544–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgess R, Yang Z. 2008. Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol 25:1979–94 [DOI] [PubMed] [Google Scholar]
- 20.Burki F, Kaessmann H. 2004. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet 36:1061–63 [DOI] [PubMed] [Google Scholar]
- 21.Caceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, et al. 2003. Elevated gene expression levels distinguish human from nonhuman primate brains. Proc. Natl. Acad. Sci. USA 100:13030–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caceres M, Suwyn C, Maddox M, Thomas JW, Preuss TM. 2007. Increased cortical expression of two synaptogenic thrombospondins in human brain evolution. Cereb. Cortex 17:2312–21 [DOI] [PubMed] [Google Scholar]
- 23.Calarco JA, Xing Y, Caceres M, Calarco JP, Xiao X, et al. 2007. Global analysis of alternative splicing differences between humans and chimpanzees. Genes Dev 21:2963–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbone L, Vessere GM, ten Hallers BFH, Zhu BL, Osoegawa K, et al. 2006. A high-resolution map of synteny disruptions in gibbon and human genomes. PLoS Genet 2:2162–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll SB. 2005. Evolution at two levels: on genes and form. PLoS Biol 3:1159–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caswell JL, Mallick S, Richter DJ, Neubauer J, Gnerre CSS, et al. 2008. Analysis of chimpanzee history based on genome sequence alignments. PLoS Genet 4:e1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen FC, Li WH. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet 68:444–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Z, Ventura M, She X, Khaitovich P, Graves T, et al. 2005. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature 437:88–93 [DOI] [PubMed] [Google Scholar]
- 29.Cheung J, Wilson MD, Zhang J, Khaja R, MacDonald JR, et al. 2003. Recent segmental and gene duplications in the mouse genome. Genome Biol 4:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P. 2005. Complex genomic rearrangements lead to novel primate gene function. Genome Res 15:343–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, et al. 2003. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302:1960–63 [DOI] [PubMed] [Google Scholar]
- 32.Consortium CSaA. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87 [DOI] [PubMed] [Google Scholar]
- 33.Coop G, Przeworski M. 2007. An evolutionary view of human recombination. Nat. Rev. Genet 8:23–34 [DOI] [PubMed] [Google Scholar]
- 34.Cox LA, Mahaney MC, Vandeberg JL, Rogers J. 2006. A second-generation genetic linkage map of the baboon (Papio hamadryas) genome. Genomics 88:274–81 [DOI] [PubMed] [Google Scholar]
- 35.Deininger PL, Batzer MA. 1999. Alu repeats and human disease. Mol. Genet. Metab 67:183–93 [DOI] [PubMed] [Google Scholar]
- 36.Dumas L, Kim YH, Karimpour-Fard A, Cox M, Hopkins J, et al. 2007. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res 17:1266–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dumont BL, Payseur BA. 2008. Evolution of the genomic rate of recombination in mammals. Evolution 62:276–94 [DOI] [PubMed] [Google Scholar]
- 38.Ebersberger I, Metzler D, Schwarz C, Paabo S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet 70:1490–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eichler EE. 2001. Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet 17:661–69 [DOI] [PubMed] [Google Scholar]
- 40.Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, et al. 1997. Interchromosomal duplications of the adrenoleukodystrophy locus: a phenomenon of pericentromeric plasticity. Hum. Mol. Genet 6:991–1002 [DOI] [PubMed] [Google Scholar]
- 41.Eichler EE, DeJong PJ. 2002. Biomedical applications and studies of molecular evolution: a proposal for a primate genomic library resource. Genome Res 12:673–78 [DOI] [PubMed] [Google Scholar]
- 42.Elango N, Thomas JW, Yi SV, Progra NCS. 2006. Variable molecular clocks in hominoids. Proc. Natl. Acad. Sci. USA 103:1370–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enard W, Khaitovich P, Klose J, Zollner S, Heissig F, et al. 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296:340–43 [DOI] [PubMed] [Google Scholar]
- 44.Enard W, Paabo S. 2004. Comparative primate genomics. Annu. Rev. Genomics Hum. Genet 5:351–78 [DOI] [PubMed] [Google Scholar]
- 45.Ferguson B, Street SL, Wright H, Pearson C, Jia Y, et al. 2007. Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). BMC Genomics 8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferris SD, Brown WM, Davidson WS, Wilson AC. 1981. Extensive polymorphism in the mitochondrial-DNA of apes. Proc. Natl. Acad. Sci. USA Biol. Sci 78:6319–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feuk L, MacDonald JR, Tang T, Carson AR, Li M, et al. 2005. Discovery of human inversion poly-morphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet 1:489–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer A, Pollack J, Thalmann O, Nickel B, Paabo S. 2006. Demographic history and genetic differentiation in apes. Curr. Biol 16:1133–38 [DOI] [PubMed] [Google Scholar]
- 49.Fischer A, Wiebe V, Paabo S, Przeworski M. 2004. Evidence for a complex demographic history of chimpanzees. Mol. Biol. Evol 21:799–808 [DOI] [PubMed] [Google Scholar]
- 50.Gagneux P, Varki A. 2001. Genetic differences between humans and great apes. Mol. Phylogenet. Evol 18:2–13 [DOI] [PubMed] [Google Scholar]
- 51.Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316:222–34 [DOI] [PubMed] [Google Scholar]
- 52.Gilad Y, Oshlack A, Smyth GK, Speed TP, White KP. 2006. Expression profiling in primates reveals a rapid evolution of human transcription factors. Nature 440:242–45 [DOI] [PubMed] [Google Scholar]
- 53.Gilad Y, Wiebe V, Przeworski M, Lancet D, Paabo S. 2007. Correction: Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates (vol. 2, p. 120, 2004). PLoS Biol 5:1383–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilad Y, Wiebel V, Przeworski M, Lancet D, Paabo S. 2004. Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol 2:120–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goidts V, Szamalek JM, Hameister H, Kehrer-Sawatzki H. 2004. Segmental duplication associated with the human-specific inversion of chromosome 18: a further example of the impact of segmental duplications on karyotype and genome evolution in primates. Hum. Genet 115:116–22 [DOI] [PubMed] [Google Scholar]
- 56.Goodman M 1985. Rates of molecular evolution—the hominoid slowdown. BioEssays 3:9–14 [DOI] [PubMed] [Google Scholar]
- 57.Goodman M, Porter CA, Czelusniak J, Page SL, Schneider H, et al. 1998. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol. Phylogenet. Evol 9:585–98 [DOI] [PubMed] [Google Scholar]
- 58.Goossens B, Chikhi L, Ancrenaz M, Lackman-Ancrenaz I, Andau P, Bruford MW. 2006. Genetic signature of anthropogenic population collapse in orang-utans. PLoS Biol 4:285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gross M, Starke H, Trifonov V, Claussen U, Liehr T, Weise A. 2006. A molecular cytogenetic study of chromosome evolution in chimpanzee. Cytogenet. Genome Res 112:67–75 [DOI] [PubMed] [Google Scholar]
- 60.Hacia JG. 2001. Genome of the apes. Trends Genet 17:637–45 [DOI] [PubMed] [Google Scholar]
- 61.Hahn MW, Demuth JP, Han SG. 2007. Accelerated rate of gene gain and loss in primates. Genetics 177:1941–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haldane JBS. 1922. Sex ratio and unidirectional sterility in hybrid animals. J. Genet 58:237–42 [Google Scholar]
- 63.Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Awray G. 2007. Promoter regions of many neural-and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet 39:1140–44 [DOI] [PubMed] [Google Scholar]
- 64.Hellmann I, Zollner S, Enard W, Ebersberger I, Nickel B, Paabo S. 2003. Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res 13:831–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hernandez RD, Hubisz MJ, Wheeler DA, Smith DG, Ferguson B, et al. 2007. Demographic histories and patterns of linkage disequilibrium in Chinese and Indian rhesus macaques. Science 316:240–43 [DOI] [PubMed] [Google Scholar]
- 66.Hobolth A, Christensen OF, Mailund T, Schierup MH. 2007. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet 3:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horvath JE, Willard HF. 2007. Primate comparative genomics: lemur biology and evolution. Trends Genet 23:173–82 [DOI] [PubMed] [Google Scholar]
- 68.IUCN. 2008. 2008 IUCN Red List of Threatened Species Gland, Switz. [Google Scholar]
- 69.Inoue S, Matsuzawa T. 2007. Working memory of numerals in chimpanzees. Curr. Biol 17:R1004–5 [DOI] [PubMed] [Google Scholar]
- 70.Istrail S, Sutton GG, Florea L, Halpern AL, Mobarry CM, et al. 2004. Whole-genome shotgun assembly and comparison of human genome assemblies. Proc. Natl. Acad. Sci. USA 18;101:1916–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacob S, McClintock MK, Zelano B, Ober C. 2002. Paternally inherited HLA alleles are associated with women’s choice of male odor. Nat. Genet 30:175–79 [DOI] [PubMed] [Google Scholar]
- 72.Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, et al. 2008. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451:998–1003 [DOI] [PubMed] [Google Scholar]
- 73.Jasinska AJ, Service S, Levinson M, Slaten E, Lee O, et al. 2007. A genetic linkage map of the vervet monkey (Chlorocebus aethiops sabaeus). Mamm. Genome 18:347–60 [DOI] [PubMed] [Google Scholar]
- 74.Jeffreys AJ, Holloway JK, Kauppi L, May CA, Neumann R, et al. 2004. Meiotic recombination hot spots and human DNA diversity. Philos. Trans. R. Soc. London Ser. B 359:141–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jeffreys AJ, Neumann R, Panayi M, Myers S, Donnelly P. 2005. Human recombination hot spots hidden in regions of strong marker association. Nat. Genet 37:601–6 [DOI] [PubMed] [Google Scholar]
- 76.Jensen-Seaman MI, Deinard AS, Kidd KK. 2001. Modern African ape populations as genetic and demographic models of the last common ancestor of humans, chimpanzees, and gorillas. J. Hered 92:475–80 [DOI] [PubMed] [Google Scholar]
- 77.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, et al. 2007. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat. Genet 39:1361–68 [DOI] [PubMed] [Google Scholar]
- 78.Johnson ME, Cheng Z, Morrison VA, Scherer S, Ventura M, et al. 2006. Recurrent duplication-driven transposition of DNA during hominoid evolution. Proc. Natl. Acad. Sci. USA 103:17626–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, et al. 2001. Positive selection of a gene family during the emergence of humans and African apes. Nature 413:514–19 [DOI] [PubMed] [Google Scholar]
- 80.Kaessmann H, Wiebe V, Paabo S. 1999. Extensive nuclear DNA sequence diversity among chimpanzees. Science 286:1159–62 [DOI] [PubMed] [Google Scholar]
- 81.Kaessmann H, Wiebe V, Weiss G, Paabo S. 2001. Great ape DNA sequences reveal a reduced diversity and an expansion in humans. Nat. Genet 27:155–56 [DOI] [PubMed] [Google Scholar]
- 82.Kaiser SM. 2007. Restriction of an extinct retrovirus by the human TRIM5 alpha antiviral protein. Science 317:1036–36 [DOI] [PubMed] [Google Scholar]
- 83.Kapitonov VV, Jurka J. 2005. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol 3:e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Karaman MW, Houck ML, Chemnick LG, Nagpal S, Chawannakul D, et al. 2003. Comparative analysis of gene-expression patterns in human and African great ape cultured fibroblasts. Genome Res 13:1619–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kauppi L, Stumpf MP, Jeffreys AJ. 2005. Localized breakdown in linkage disequilibrium does not always predict sperm crossover hot spots in the human MHC class II region. Genomics 86:13–24 [DOI] [PubMed] [Google Scholar]
- 86.Kehrer-Sawatzki H, Cooper DN. 2008. Molecular mechanisms of chromosomal rearrangement during primate evolution. Chromosome Res 16:41–56 [DOI] [PubMed] [Google Scholar]
- 87.Kehrer-Sawatzki H, Schreiner B, Tanzer S, Platzer M, Muller S, Hameister H. 2002. Molecular characterization of the pericentric inversion that causes differences between chimpanzee chromosome 19 and human chromosome 17. Am. J. Hum. Genet 71:37588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kehrer-Sawatzki H, Sandig C, Chuzhanova N, Goidts V, Szamalek JM, et al. 2005. Breakpoint analysis of the pericentric inversion distinguishing human chromosome 4 from the homologous chromosome in the chimpanzee (Pan troglodytes). Hum. Mutat 25(1):45–55 [DOI] [PubMed] [Google Scholar]
- 89.Kehrer-Sawatzki H, Szamalek JM, Tanzer S, Platzer M, Hameister H. 2005. Molecular characterization of the pericentric inversion of chimpanzee chromosome 11 homologous to human chromosome 9. Genomics 85:542–50 [DOI] [PubMed] [Google Scholar]
- 90.Khaitovich P, Muetzel B, She X, Lachmann M, Hellmann I, et al. 2004. Regional patterns of gene expression in human and chimpanzee brains. Genome Res 14:1462–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188:107–16 [DOI] [PubMed] [Google Scholar]
- 92.Kong A, Barnard J, Gudbjartsson DF, Thorleifsson G, Jonsdottir G, et al. 2004. Recombination rate and reproductive success in humans. Nat. Genet 36:1203–6 [DOI] [PubMed] [Google Scholar]
- 93.Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, et al. 2008. Patterns of positive selection in six mammalian genomes. PLoS Genet 4:e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 95.Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319:1100–4 [DOI] [PubMed] [Google Scholar]
- 96.Li WH, Tanimura M. 1987. The molecular clock runs more slowly in man than in apes and monkeys. Nature 326:93–96 [DOI] [PubMed] [Google Scholar]
- 97.Liu G, Zhao SY, Bailey JA, Sahinalp SC, Alkan C, et al. 2003. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res 13:358–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Locke DP, Archidiacono N, Misceo D, Cardone MF, Deschamps S, et al. 2003. Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biol 4:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu J, Li WH, Wu CI. 2003. Comment on “Chromosomal speciation and molecular divergence—accelerated evolution in rearranged chromosomes.” Science 302:988. [DOI] [PubMed] [Google Scholar]
- 100.Makova KD, Li WH. 2002. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416:624–26 [DOI] [PubMed] [Google Scholar]
- 101.Mardis ER. 2008. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet 9:387–402 [DOI] [PubMed] [Google Scholar]
- 102.Margulies EH, Blanchette M, Haussler D, Green ED, Progra NCS. 2003. Identification and characterization of multi-species conserved sequences. Genome Res 13:2507–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marques-Bonet T, Caceres M, Bertranpetit J, Preuss TM, Thomas JW, Navarro A. 2004. Chromosomal rearrangements and the genomic distribution of gene-expression divergence in humans and chimpanzees. Trends Genet 20:524–29 [DOI] [PubMed] [Google Scholar]
- 104.Marques-Bonet T, Cheng Z, She X, Eichler EE, Navarro A. 2008. The genomic distribution of intraspecific and interspecific sequence divergence of human segmental duplications relative to human/chimpanzee chromosomal rearrangements. BMC Genomics 9:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, et al. 2009. A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457:877–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marques-Bonet T, Sanchez-Ruiz J, Armengol L, Khaja R, Bertranpetit J, et al. 2007. On the association between chromosomal rearrangements and genic evolution in humans and chimpanzees. Genome Biol 8:R230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mikkelsen TS, Hillier LW, Eichler EE, Zody MC, Jaffe DB, et al. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87 [DOI] [PubMed] [Google Scholar]
- 108.Muller S, Hollatz M, Wienberg J. 2003. Chromosomal phylogeny and evolution of gibbons (Hylobatidae). Hum. Genet 113:493–501 [DOI] [PubMed] [Google Scholar]
- 109.Murphy WJ, Larkin DM, Everts-van de Wind A, Bourque G, Tesler G, et al. 2005. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 309:613–17 [DOI] [PubMed] [Google Scholar]
- 110.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310:321–24 [DOI] [PubMed] [Google Scholar]
- 111.Myers S, Freeman C, Auton A, Donnelly P, McVean G. 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet 40:1124–29 [DOI] [PubMed] [Google Scholar]
- 112.Navarro A, Barton NH. 2003. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57:447–59 [DOI] [PubMed] [Google Scholar]
- 113.Navarro A, Barton NH. 2003. Chromosomal speciation and molecular divergence—accelerated evolution in rearranged chromosomes. Science 300:321–24 [DOI] [PubMed] [Google Scholar]
- 114.Newman TL, Tuzun E, Morrison VA, Hayden KE, Ventura M, et al. 2005. A genome-wide survey of structural variation between human and chimpanzee. Genome Res 15:1344–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, et al. 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3:976–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Noor MA, Grams KL, Bertucci LA, Reiland J. 2001. Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. USA 98:12084–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Osada N, Wu CI. 2005. Inferring the mode of speciation from genomic data: a study of the great apes. Genetics 169:259–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. 2006. Genetic evidence for complex speciation of humans and chimpanzees. Nature 441:1103–8 [DOI] [PubMed] [Google Scholar]
- 119.Paulding CA, Ruvolo M, Haber DA. 2003. The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc. Natl. Acad. Sci. USA 100:2507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, et al. 2006. Hotspots for copy number variation in chimpanzees and humans. Proc. Natl. Acad. Sci. USA 103:8006–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, et al. 2008. Copy number variation and evolution in humans and chimpanzees. Genome Res 18:698–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Polavarapu N, Bowen NJ, McDonald JF. 2006. Identification, characterization and comparative genomics of chimpanzee endogenous retroviruses. Genome Biol 7:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, et al. 2006. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443:167–72 [DOI] [PubMed] [Google Scholar]
- 124.Popesco MC, Maclaren EJ, Hopkins J, Dumas L, Cox M, et al. 2006. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science 313:1304–7 [DOI] [PubMed] [Google Scholar]
- 125.Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, et al. 2008. Human-specific gain of function in a developmental enhancer. Science 321:1346–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Preuss TM, Caceres M, Oldham MC, Geschwind DH. 2004. Human brain evolution: insights from microarrays. Nat. Rev. Genet 5:850–60 [DOI] [PubMed] [Google Scholar]
- 127.Price AL, Weale ME, Patterson N, Myers SR, Need AC, et al. 2008. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet 83:132–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, et al. 2005. Fine-scale recombination patterns differ between chimpanzees and humans. Nat. Genet 37:429–34 [DOI] [PubMed] [Google Scholar]
- 129.Reinius B, Saetre P, Leonard JA, Blekhman R, Merino-Martinez R, et al. 2008. An evolutionarily conserved sexual signature in the primate brain. PLoS Genet 4:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rieseberg LH, Livingstone K. 2003. Evolution. Chromosomal speciation in primates. Science 300:267–68 [DOI] [PubMed] [Google Scholar]
- 131.Rieseberg LH, Vanfossen C, Desrochers AM. 1995. Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 375:313–16 [Google Scholar]
- 132.Rieseberg LH, Whitton J, Gardner K. 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152:713–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roberto R, Capozzi O, Wilson RK, Mardis ER, Lomiento M, et al. 2007. Molecular refinement of gibbon genome rearrangements. Genome Res 17:249–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rogers J, Garcia R, Shelledy W, Kaplan J, Arya A, et al. 2006. An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics 87:30–38 [DOI] [PubMed] [Google Scholar]
- 135.Ryder OA. 2005. Conservation genomics: applying whole genome studies to species conservation efforts. Cytogenet. Genome Res 108:6–15 [DOI] [PubMed] [Google Scholar]
- 136.Schaner P, Richards N, Wadhwa A, Aksentijevich I, Kastner D, et al. 2001. Episodic evolution of pyrin in primates: Human mutations recapitulate ancestral amino acid states. Nat. Genet 27:318–21 [DOI] [PubMed] [Google Scholar]
- 137.She X, Cheng Z, Zollner S, Church DM, Eichler EE. 2008. Mouse segmental duplication and copy number variation. Nat. Genet 40:909–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.She X, Liu G, Ventura M, Zhao S, Misceo D, et al. 2006. A preliminary comparative analysis of primate segmental duplications shows elevated substitution rates and a great-ape expansion of intrachromosomal duplications. Genome Res 16:576–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith DG, McDonough J. 2005. Mitochondrial DNA variation in Chinese and Indian rhesus macaques (Macaca mulatta). Am. J. Primatol 65:1–25 [DOI] [PubMed] [Google Scholar]
- 140.Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR. 2004. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res 14:2209–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Steiper ME. 2006. Population history, biogeography, and taxonomy of orangutans (Genus: Pongo) based on a population genetic meta-analysis of multiple loci. J. Hum. Evol 50(5):509–22 [DOI] [PubMed] [Google Scholar]
- 142.Steiper ME, Young NM. 2006. Primate molecular divergence dates. Mol. Phylogenet. Evol 41:384–94 [DOI] [PubMed] [Google Scholar]
- 143.Steiper ME, Young NM, Sukarna TY. 2004. Genomic data support the hominoid slowdown and an Early Oligocene estimate for the hominoid-cercopithecoid divergence. Proc. Natl. Acad. Sci. USA 101:17021–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stone AC, Griffiths RC, Zegura SL, Hammer MF. 2002. High levels of Y-chromosome nucleotide diversity in the genus Pan. Proc. Natl. Acad. Sci. USA 99:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Storz JF, Beaumont MA. 2002. Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution 56:154–66 [DOI] [PubMed] [Google Scholar]
- 146.Street SL, Kyes RC, Grant R, Ferguson B. 2007. Single nucleotide polymorphisms (SNPs) are highly conserved in rhesus (Macaca mulatta) and cynomolgus (Macaca fascicularis) macaques. BMC Genomics 8:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Szamalek JM, Cooper DN, Hoegel J, Hameister H, Kehrer-Sawatzki H. 2007. Chromosomal speciation of humans and chimpanzees revisited: studies of DNA divergence within inverted regions. Cytogenet. Genome Res 116:53–60 [DOI] [PubMed] [Google Scholar]
- 148.Szamalek JM, Cooper DN, Schempp W, Minich P, Kohn M, et al. 2006. Polymorphic microinversions contribute to the genomic variability of humans and chimpanzees. Hum. Genet 119:103–12 [DOI] [PubMed] [Google Scholar]
- 149.Szamalek JM, Goidts V, Chuzhanova N, Hameister H, Cooper DN, Kehrer-Sawatzki H. 2005. Molecular characterization of the pericentric inversion that distinguishes human chromosome 5 from the homologous chimpanzee chromosome. Hum. Genet 117:168–76 [DOI] [PubMed] [Google Scholar]
- 150.Szamalek JM, Goidts V, Cooper DN, Hameister H, Kehrer-Sawatzki H. 2006. Characterization of the human lineage-specific pericentric inversion that distinguishes human chromosome 1 from the homologous chromosomes of the great apes. Hum. Genet 120:126–38 [DOI] [PubMed] [Google Scholar]
- 151.Szamalek JM, Goidts V, Searle JB, Cooper DN, Hameister H, Kehrer-Sawatzki H. 2006. The chimpanzee-specific pericentric inversions that distinguish humans and chimpanzees have identical breakpoints in Pan troglodytes and Pan paniscus. Genomics 87:39–45 [DOI] [PubMed] [Google Scholar]
- 152.Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424:788–93 [DOI] [PubMed] [Google Scholar]
- 153.Trask BJ, Friedman C, Martin-Gallardo A, Rowen L, Akinbami C, et al. 1998. Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum. Mol. Genet 7:13–26 [DOI] [PubMed] [Google Scholar]
- 154.Uddin M, Wildman DE, Liu GZ, Xu WB, Johnson RM, et al. 2004. Sister grouping of chimpanzees and humans as revealed by genome-wide phylogenetic analysis of brain gene expression profiles. Proc. Natl. Acad. Sci. USA 101:2957–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vallender EJ, Lahn BT. 2004. Effects of chromosomal rearrangements on human-chimpanzee molecular evolution. Genomics 84:757–61 [DOI] [PubMed] [Google Scholar]
- 156.Vandepoele K, Van Roy N, Staes K, Speleman F, van Roy F. 2005. A novel gene family NBPF: intricate structure generated by gene duplications during primate evolution. Mol. Biol. Evol 22:2265–74 [DOI] [PubMed] [Google Scholar]
- 157.Wakeley J 2008. Complex speciation of humans and chimpanzees. Nature 452:E3–4; discussion E4 [DOI] [PubMed] [Google Scholar]
- 158.Wall JD. 2003. Estimating ancestral population sizes and divergence times. Genetics 163:395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang HY, Chien HC, Osada N, Hashimoto K, Sugano S, et al. 2007. Rate of evolution in brain-expressed genes in humans and other primates. Plos Biol 5:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–62 [DOI] [PubMed] [Google Scholar]
- 161.White MJD. 1978. Modes of Speciation San Francisco: Freeman [Google Scholar]
- 162.Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, et al. 2005. Comparison of fine-scale recombination rates in humans and chimpanzees. Science 308:107–11 [DOI] [PubMed] [Google Scholar]
- 163.Won YJ, Hey J. 2005. Divergence population genetics of chimpanzees. Mol. Biol. Evol 22:297–307 [DOI] [PubMed] [Google Scholar]
- 164.Yang SH, Cheng PH, Banta H, Piotrowska-Nitsche K, Yang JJ, et al. 2008. Towards a transgenic model of Huntington’s disease in a nonhuman primate. Nature 453:921–U56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yohn CT, Jiang ZS, McGrath SD, Hayden KE, Khaitovich P, et al. 2005. Lineage-specific expansions of retroviral insertions within the genomes of African great apes but not humans and orangutans. Plos Biol 3:577–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yu N, Jensen-Seaman MI, Chemnick L, Kidd JR, Deinard AS, et al. 2003. Low nucleotide diversity in chimpanzees and bonobos. Genetics 164:1511–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yunis JJ, Prakash O. 1982. The origin of man: a chromosomal pictorial legacy. Science 215:1525–30 [DOI] [PubMed] [Google Scholar]
- 168.Zhang J, Wang X, Podlaha O. 2004. Testing the chromosomal speciation hypothesis for humans and chimpanzees. Genome Res 14:845–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zietkiewicz E, Richer C, Sinnett D, Labuda D. 1998. Monophyletic origin of Alu elements in primates. J. Mol. Evol 47:172–82 [DOI] [PubMed] [Google Scholar]