

Abstract
Objective:
Exposure to organophosphates (OPs) and OP nerve agents (NAs) causes status epilepticus (SE) and irreversible brain damage. Rapid control of seizure activity is important to minimize neuronal injury and the resulting neurological and behavioral disorders; however, early treatment will not be possible after mass release of OPs or NAs.
Methods:
We utilized a delayed-treatment model of OP exposure in adult rats by administration of diisopropyl fluorophosphate (DFP) to study the relationship between the antiseizure and neuroprotective effects of the “standard-of-care” benzodiazepine, midazolam (MDZ), when given at 30, 60, and 120 min after SE onset. After electroencephalogram (EEG) recordings, neural damage in serial brain sections was studied with Fluoro-Jade B staining.
Results:
DZ-induced seizure suppression was equivalent in magnitude regardless of treatment delay (i.e., seizure duration). When assessed globally (i.e., normalized across 10 different brain regions) for each treatment delay, MDZ administration only resulted in non-significant reductions in neuronal death. However, when data for MDZ treatment were combined from all three delay times, a small but significant reduction in global neuronal death was detected when compared to vehicle treatment, which indicated that the substantive MDZ-induced seizure suppression only led to a small reduction in neuronal death.
Significance:
In conclusion, MDZ significantly reduced DFP-induced SE intensity when treatment was delayed 30, 60, and even up to 120 min; however, this reduction in seizure intensity had no detectable effect on neuronal death at each individual delay time. These data show that although MDZ suppressed seizures, additional neuroprotective therapies are needed to mitigate the effects of OP exposure.
Keywords: status epilepticus, diisopropyl fluorophosphate, seizures, neuropathology, Fluoro-Jade B
INTRODUCTION
Accidental or intentional exposure to organophosphate (OP) insecticides, or to the more lethal militarized OP nerve agents (NAs), causes significant morbidity and mortality worldwide. The accumulation of acetylcholine in peripheral synapses after cholinesterase inhibition results in muscle tremors, cardiovascular dysfunction, hypotension, and bronchial spasms 1,2. The mortality immediately after exposure is most commonly due to respiratory failure after OP/NA-induced bronchospasm, bronchorrhea, and paralysis of respiratory muscles 2,3. The accepted therapy to ameliorate these lethal peripheral responses is administration of the antidote compounds atropine, pyridostigmine, and an oxime, such as pralidoxime (2-pyridine aldoxime methyl chloride or 2-PAM) 4–9.
In addition to the severe peripheral effects, OPs act on the central nervous system (CNS) to cause intense, continuous seizures (i.e., status epilepticus, SE). Prolonged SE, regardless of etiology, induces widespread neuronal injury and neuronal death throughout the brain 10–14. This neuronal damage within the CNS leads to chronic neurological and behavioral deficits that have been recapitulated in animal models of SE 12,15–18. The severity of adverse neurological outcomes after SE generally depends on the duration and intensity of the seizures, which also correlates with the extent of CNS neuropathology 13. Consequently, rapid control of SE is critical in order to reduce neuronal death and prevent long-term neurological dysfunction 13.
First-line therapy for treatment of SE involves administration of benzodiazepines, such as diazepam, lorazepam or midazolam (MDZ) 19–26. However, the anticonvulsant efficacy of these agents is thought to progressively decrease as seizure duration increases 22,27–30. In a hospital setting, intravenous (IV) infusion is the preferred route of administration for lorazepam 31, however, in the event of an OP/NA mass release, IV administration is unrealistic. A recent randomized, double-blind clinical trial, the Rapid Anticonvulsant Medication Prior to Arrival Trial (RAMPART), determined that intramuscular (IM) MDZ in a pre-hospital setting was noninferior to IV lorazepam, as determined by absence of seizures upon arrival at an emergency department 25.
For animal models of OP/NA exposure, the precise relationships between the duration of SE and both the anticonvulsive and neuroprotective effects of MDZ have not been consistently defined, particularly for delayed-treatment paradigms. Therefore, to develop a delayed-treatment model of realistic exposure delays for studies aimed at evaluating the therapeutic efficacy of standard-of-care or novel compounds in the treatment of OP-induced SE, we studied the relationship between SE duration, anticonvulsant efficacy, and neuronal death following administration of MDZ after 30, 60 and 120 min of DFP-induced SE. In addition to being a standard-of-care, in a direct head-to-head comparison of benzodiazepines in the control of NA-induced SE, MDZ was determined to provide the most rapid seizure control 32. Importantly, MDZ has been shown to be safe and effective in control of SE when administered via intramuscular autoinjector in the prehospital setting 33. We found that MDZ was effective in reducing the intensity of SE regardless of the time point at which it was given (up to 120 min). When neuronal death was assessed globally with Fluoro-Jade B (FJB) across 10 brain sites after these MDZ-induced reductions in SE intensity, MDZ had no detectable effect at reducing neuronal death when analyzed for each individual delay time. Evidence for a small MDZ-induced reduction in neuronal death was only detected when the data from all three delay times were combined.
METHODS
Animals
Male Sprague-Dawley rats were obtained from Charles River Laboratories, and housed in our temperature-controlled vivarium on a 12-hr light, 12-hr dark cycle with ad libitum access to food and water. All surgical and experimental procedures used in these studies were reviewed and approved by the University of Utah Institutional Animal Care and Use Committee.
Electroencephalographic Recording Electrodes
To implant electrodes for recording of the electroencephalogram (EEG), rats were anesthetized with 2-4% isoflurane and placed in a stereotaxic instrument. A midline incision was made in the scalp to expose the skull, and the scalp was then retracted laterally. Next, six 500-μm holes were drilled through the skull; three on each side of the midline (approximately 2 mm from center), equally spaced between bregma and lambda. Small screws were placed in the top two and bottom left holes for anchoring of the electrode headset. Then, two 2-3 mm-long recording electrodes (MS333-3-B, Plastics One, Roanoake, VA) were placed in two of the remaining holes on the right side of the midline, and a ground electrode was positioned through the remaining hole on the left side of the skull. These electrodes were positioned to touch the dura for differential recording of EEG activity. The headset was then secured in place with dental cement surrounding the support screws and electrodes. Finally, the wound was closed with sutures, and rats returned to their home cages for 7 days prior to testing.
EEG Recordings
After a 1-week recovery from these surgical procedures, between 8:00 AM and 11:00 AM on “treatment day”, the conscious, unrestrained rats were placed into individual Plexiglas recording chambers, and the implanted electrodes were connected to spring-covered EEG cables (Plastics One, Roanoke, VA). EEG100 amplifiers (high-pass filter, 1 Hz; low-pass, 100 Hz; notch filter at 60 Hz; 5000× gain) amplified the signals, which were then digitized at 500 Hz with an MP150 digital-to-analog converter. The digitized EEG signals were recorded using AcqKnowledge software (BioPac Systems, Inc. Santa Barbara, CA) and stored for subsequent analysis.
Experimental Procedures
A delayed-treatment rodent model of OP exposure inducing SE with DFP was used in these experiments (Schematic in Figure 1). In order to decrease mortality due to the peripheral, potentially lethal effects of the OP, all rats (220-280 g) were given pyridostigmine bromide (0.026 mg/kg, IM) 30 min prior to DFP (6.5-7.7 mg/kg, SC), and atropine methyl nitrate (2 mg/kg, IM) plus 2-PAM (25 mg/kg, IM) 1 min after DFP. After DFP administration, rats were directly observed until the first tremor was noted. Thirty, 60, or 120 min after the first tremors, rats were treated with equal volume vehicle (saline) or MDZ (2.0 mg/kg) IM. For these studies, the vehicle was not pH-matched to the injectable MDZ solution (pH 3.3-3.5). EEG was recorded continuously for 24 hr after DFP administration. DFP, pyridostigmine bromide and 2-PAM were purchased from Sigma-Aldrich (St. Louis, MO); MDZ was purchased from Akorn Pharmaceuticals (Vernon Hills, IL); atropine methyl nitrate was purchased from Spectrum Chemicals (New Brunswick, NJ).
Figure 1. Schematic timeline representation of the protocol.
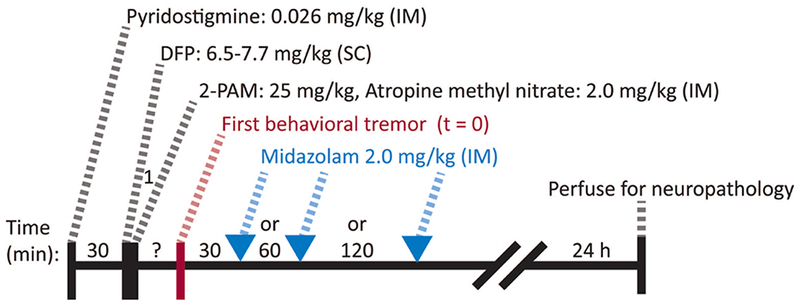
After a baseline recording period, the experiment began with administration of pyridostigmine (0.026 mg/kg, IM). Thirty minutes later, rats were given diisopropyl fluorophosphate (DFP, 6.5-7.7 mg/kg, SC) followed by 2-PAM (25 mg/kg, IM) and atropine methyl nitrate (2 mg/kg, IM) after 1 min. Rats were then observed until they began to show behavioral signs of DFP-induced intoxication (i.e. tremors), and this was used to indicate “time zero” for the countdown until treatment. Treatment in the form of intramuscular MDZ (2.0 mg/kg) was then given at 30 min, 60 min or 120 min. Twenty-four hours after DFP, rats were transcardially perfused with formalin and brains were harvested for neuropathology.
EEG Analysis
Seizure activity was quantified by analyzing EEG data obtained during the baseline and first 20 hr after administration of DFP using two previously described quantitative algorithms for calculation of power spectral density in the 20-60 Hz (gamma) bandwidth 34 and spike frequency 35. The changes in total power in the gamma band and in spike frequency throughout each recording were determined by subtracting the calculated value in each sequential 15-min bin from a baseline period recorded prior to DFP administration. The mean changes in gamma power and spike rate, and the 95% confidence intervals were plotted for comparison.
Histopathology Analysis
Twenty-four hours after DFP injections, rats were anesthetized with isoflurane and cardiac perfused with ice-cold saline followed by 10% buffered-formalin. The fixed brains were removed and cryoprotected with 30% sucrose, subsequently frozen and 30-μm coronal sections were cryostat-sectioned between bregma coordinates −2.3 and −6.3 mm. The sections were mounted on glass slides and stained with Fluoro-Jade B (FJB, Histo-chem Inc, Jefferson, AR) 36. For staining, sections were first incubated in 0.06% KMnO4, and then in 0.001% FJB. A Hamamatsu Nanozoomer 2.0 HT (Olympus Corporation, Japan) was used to obtain digital images.
FJB staining was evaluated by an observer blinded to the experimental treatment of each animal. An unbiased random-sampling technique to quantify the number of FJB positive neurons in selected brain sections was used to estimate the neuropathological effects of DFP-induced SE. Specifically, four sections/slides from each brain area, separated by a minimum of 60 μm, were randomly selected for evaluation. For areas with uniform distribution of neurons (thalamus, amygdala, piriform cortex, parietal cortex, and entorhinal cortex), a randomly placed grid of 175-μm × 175-μm squares was positioned over the entire image. The image was rotated when necessary to align the laminar architecture of the cortex parallel with one direction of the grid. The counting region of interest (ROI) was then demarcated by the counter and required to include a pre-set number of squares defined by the area of each brain ROI. Then, within each of these selected sections, three of the 175-μm × 175-μm square counting boxes were chosen by a random number generator, and an observer (blinded to the experimental treatment of the animal) counted the number of FJB positive neurons in each of the counting areas. This was repeated for 4 sections for each brain region so that 12 squares were counted per region in total.
Statistical Analyses
Statistically significant changes in gamma power and spike-rate frequency were evaluated at hourly intervals. For these variables, comparisons between two means were done with a t-test, and when comparing more than two groups a Levene test was first used, followed by either an ANOVA or a Welch ANOVA. Differences between individual group means were determined with a Games-Howell post-hoc test. Region-specific neuron counts were compared by ANOVA and normalized histopathology data were evaluated using a Wilcoxon signed rank test. A probability of P < 0.05 was considered significant.
RESULTS
DFP administration induced behavioral tremors in all rats within minutes, which were followed shortly by persistent electrographic seizure activity in the EEG recordings that lasted for several hours 37. Figure 2 shows representative EEG recordings from a rat given vehicle or MDZ at 30 min after the initial behavioral indication of seizure activity (Figure 2A). As can be seen easily by comparison of the compressed raw traces, MDZ rapidly terminated the SE induced by DFP (N = 16 MDZ-treated compared to N = 16 vehicle-treated). Expanded traces demonstrate the intensity of the electrographic activity immediately preceding treatment (Figure 2B, left) and the reduction in activity at 2 hr post-treatment with MDZ (Figure 2B, right). These data are quantified for 20 hr by measurement of the change in power of the activity in the gamma band (20-60 Hz, Figure 2C, left) and the change in mean spike frequency (Figure 2C, right) by automated analysis, as previously described 34. MDZ treatment at 30 min after the start of SE significantly reduced both the change in power as well as the change in spike frequency for up to 10 hr (Figure 2C, dashed red lines, P < 0.05, Student’s t-test), at which time both MDZ- and vehicle-treated rats continue to have significantly diminished electrographic SE.
Figure 2. Acute treatment of DFP-induced electrographic SE with MDZ at 30 min.
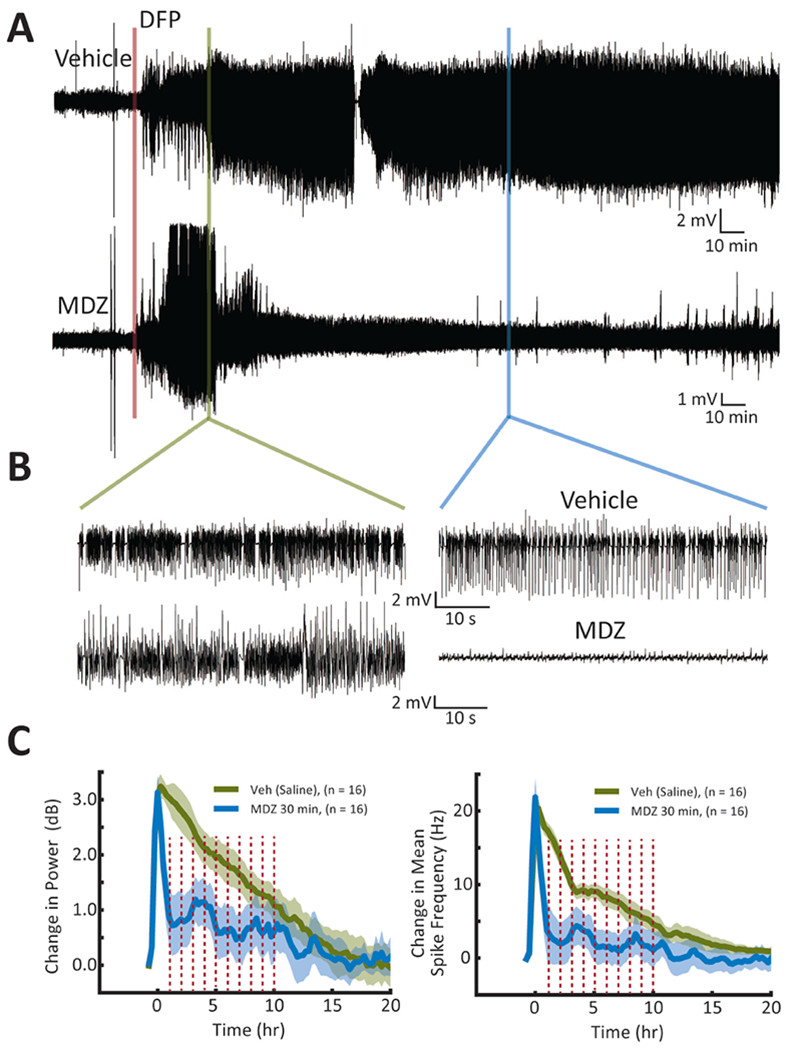
(A) Representative EEG recordings showing the effects of treatment with vehicle at 30 min (top) or MDZ at 30 min (bottom) after onset of SE. For this Figure, and Figures 3 and 4, DFP administration is indicated by the red bar, and the time of treatment is shown immediately after the green bar. (B) Expanded traces immediately prior to treatment and 2 hr after treatment demonstrate that the initial electrographic activity was similar for each rat and that MDZ treatment dramatically reduced that activity. (C) The average change in power (left) and change in mean spike frequency (right) are plotted for saline (solid green line) and MDZ (solid blue line) treated rats to demonstrate the reduction in electrographic activity observed after treatment with MDZ. Shading indicates 95% confidence intervals. Red dashed bars indicate statistically significant differences at those time points (P < 0.05, Student’s t-test).
Similarly, administration of MDZ at 60 min (Figure 3) produced a robust reduction of seizure intensity as demonstrated by the conspicuous reduction in the amplitude of activity in the raw traces (Figure 3A and B). Quantification of these data demonstrated that the reduction in the change in power produced by administration of MDZ at 60 min (Figure 3C, left, N = 19 MDZ-treated compared to N = 19 vehicle-treated) was similar in duration to MDZ administered at 30 min, but the duration of the reduction in mean spike frequency was shortened to only a few hours (Figure 3C, right).
Figure 3. Delayed treatment of DFP-induced electrographic SE with MDZ at 60 min.
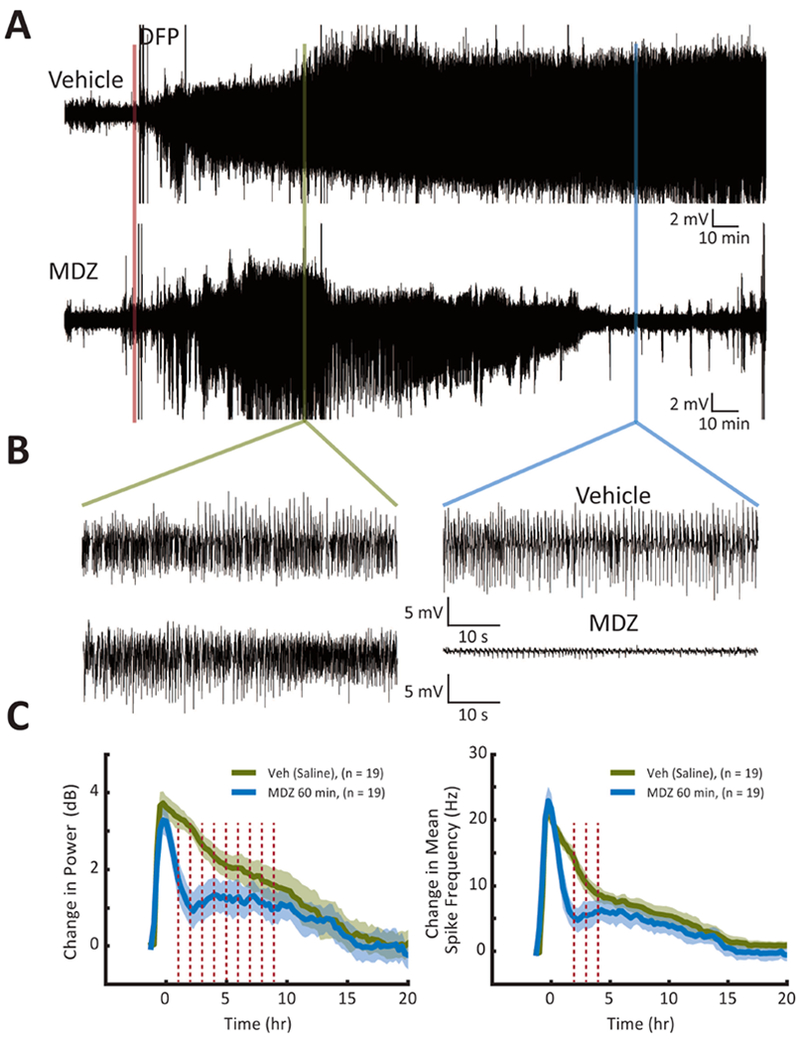
(A) Representative EEG recordings showing the effects of treatment with vehicle at 60 min (top) or MDZ at 60 min (bottom) after the beginning of SE. (B & C) See legend for Figure 2.
When treatment was given at 120 min after the start of electrographic SE, MDZ was effective in reducing seizure intensity for only a few hours. Figure 4 demonstrates the remaining high-amplitude activity in the raw data and the expanded traces (Figure 4A and B). The quantified data for the cohort of rats treated at this time demonstrated that while the initial reduction in seizure intensity was significant (P < 0.05, Student’s t-test), the effect was short in duration (compared to 30 min treatment), lasting only a few hours (Figure 4C, N = 13 MDZ-treated compared to N = 9 vehicle-treated).
Figure 4. Long-delayed treatment of DFP-induced electrographic SE with MDZ at 120 min.
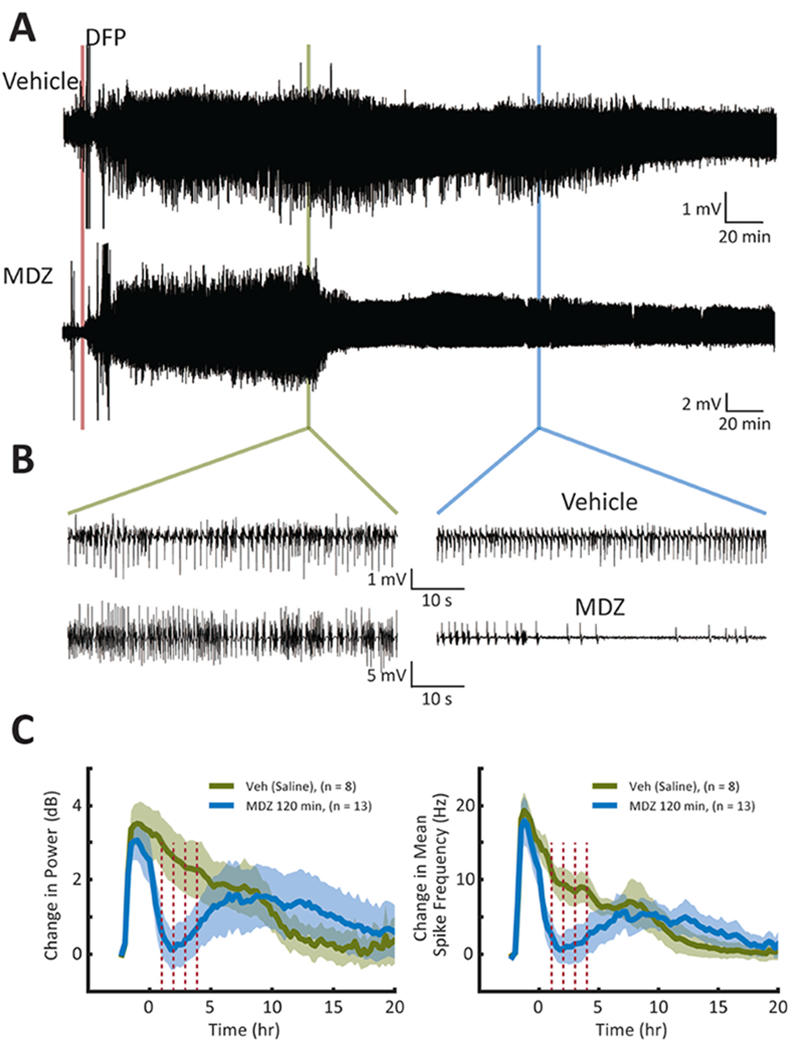
(A) Representative EEG recordings showing the effects of treatment with vehicle at 120 min (top) or MDZ at 120 min (bottom) after onset of SE. (B & C) See legend for Figure 2.
In each of the three sets of experiments presented above, the observed duration of the anti-seizure effect of MDZ appeared to decrease as the delay to treatment increased; however, due to the differences in the duration of SE, it is difficult to directly compare the effect of the treatment from one experiment to the other. In each data set the natural decrease in gamma power over time can be seen in the controls that were not given anti-seizure treatment at any time (vehicle treated at 30, 60 or 120 min). In order to compare the magnitude of the effect of the treatment with MDZ between the different treatment times, we subtracted the mean power in the MDZ group from the mean power in the untreated group for each set of data, and the remaining gamma power was plotted on the same time axis in Figure 5. The resulting plot of the remaining gamma power after treatment demonstrates that regardless of the time of treatment, MDZ reduced the EEG power to a similar amount (Figure 5). If MDZ had lost effectiveness with time, these plots would not be similar to each other.
Figure 5. Comparison of the remaining EEG power at each treatment time after administration of MDZ.
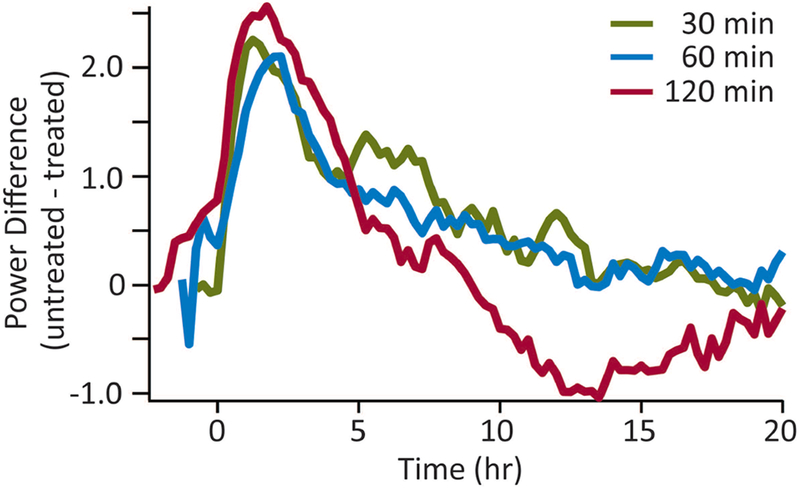
For each treatment time, the power in the treated rats was subtracted from the power in the untreated rats. Those values are plotted for 20 hr with time zero being the time of treatment. As can be seen from this comparison, treatment with MDZ at 30 min (green line), 60 min (blue line) or 120 min (red line) left a similar amount of EEG power indicating that the remaining electrographic activity was similar regardless of treatment time.
In contrast to the antiseizure effect of MDZ, no detectable effect on neuronal loss was found at any individual treatment-delay time compared to the control vehicle-treated rats. Representative images of FJB labeling in the counting panels of three brain regions for MDZ-treated and vehicle-treated rat can be seen in Figure 6A. Figure 6B shows data summarizing the neuronal death observed in each of the 10 brain regions, selected for their involvement in seizure circuits, at each delay time point of MDZ treatment (N = 15 vehicle 30 min; N = 13 MDZ 30 min; N = 10 vehicle 60 min; N = 10 MDZ 60 min; N = 7 vehicle 120 min; N = 13 MDZ 120 min). With the exception of the hilus, ventral CA3, thalamus, and parietal cortex at a few time points, no significant difference in the number of FJB+ neurons was found in rats treated with MDZ. Moreover, no consistent DFP-induced increase in neuronal loss was found at longer delay times of MDZ treatment (i.e., after a 120-min delay), as was predicted prior to conducting these experiments.
Figure 6. Neuronal loss across multiple brain regions.
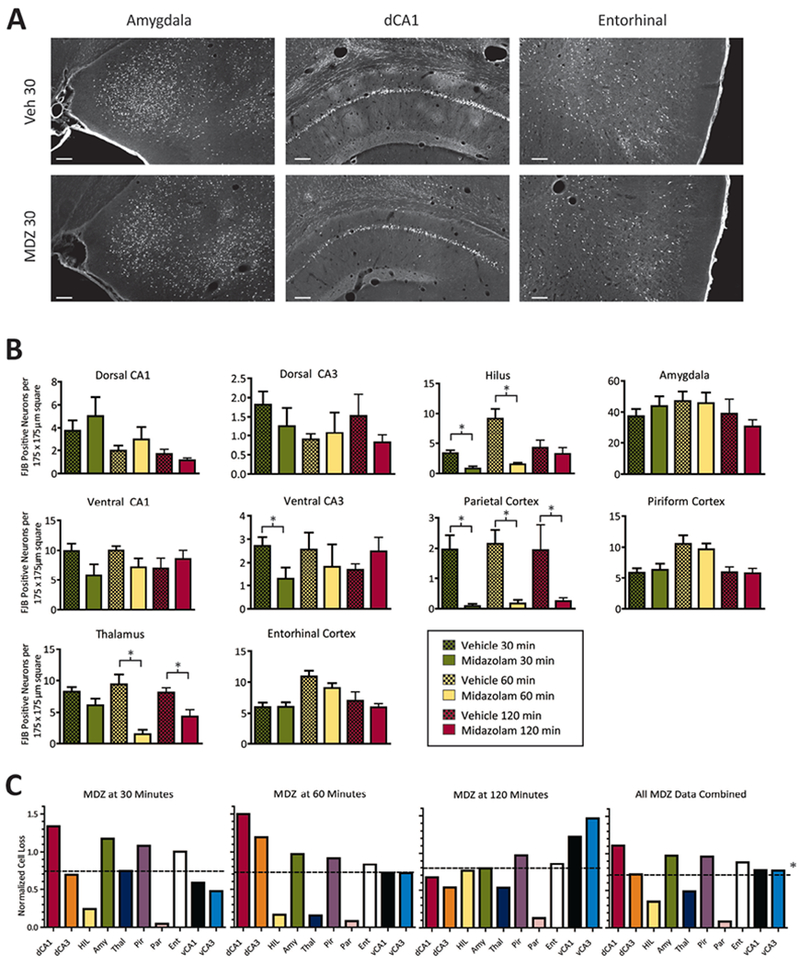
(A) Representative images of FJB labeled (bright green) neurons in three brain regions (Amygdala, dorsal CA1 and Entorhinal Cortex) for rats treated with vehicle (top row) or MDZ at 30 minutes (bottom row) from tissue taken at 24 hours after DFP. Scale bar = 250 μm. (B) Each bar represents the average number of FJB positive cells counted in 12 randomly placed 175 × 175 μm squares over 4 non-adjacent sections per brain region in each rat. For treatment groups: N = 15, vehicle 30 min; N = 13, MDZ 30 min; N = 10, vehicle 60 min; N = 10, MDZ 60 min; N = 7, vehicle 120 min; N = 13, MDZ 120 min. Asterisks indicate significant differences at P < 0.05, ANOVA, Tukey’s Multiple Comparison Test. Values plotted are mean + SEM. (C) To determine the global neuronal death at each timepoint of treatment, the average neuron counts from MDZ-treated rats (B) were normalized to the control rat counts for each brain region. This generated 10 normalized counts (one for each region) for each treatment time (C). These counts were then averaged to generate a single neuroprotection score for that treatment (dotted lines). Values below 1 represent less neuronal death than control (e.g., 1 is 100% of the neuronal death observed in the control). Dotted line values are: MDZ at 30 min = 0.74 ± 0.42, MDZ at 60 min = 0.72 ± 0.47, MDZ at 120 min = 0.79 ± 0.37, and MDZ combined = 0.71 ± 0.32. Combined data comparing treatment independent of time (right) resulted in a statistically significant effect of treatment with MDZ (asterisk, P < 0.05, Wilcoxon signed rank test). Abbreviations: dorsal CA1 (dCA1), dorsal CA3 (dCA3), hilus (HIL), amygdala (Amy), thalamus (Thal), piriform cortex (Pir), parietal cortex (Par), entorhinal cortex (Ent), ventral CA1 (vCA1), ventral CA3 (vCA3).
Given the variability in neuronal death from region to region at each treatment time point, we next asked if neuronal death could be assessed on a global scale. To determine the global change in neuronal death throughout the brain, we normalized the average number of dead neurons per region by dividing the MDZ-treated counts by the vehicle-treated counts for that particular area at that time. This generated a number that represents the neuronal death in the MDZ-treated group as a fraction of neuron death in the vehicle-treated group. Numbers less than 1.0 indicate less death in the MDZ-treated group and numbers more than 1.0 indicate more death in the MDZ-treated group. Those data were plotted in Figure 6C (these numbers were generated using the average neuronal death values plotted in Figure 6B and therefore do not have error values associated with them; i.e. they are the normalized average for the group, not the average of normalized values for each rat). These normalized neuronal death values were then converted to a single score for each treatment time, by calculating the average of the 10-region data set (dashed horizontal lines). At each time point, MDZ-treated rats had a similar reduction in neuronal death to 72-79% of control levels, but no individual treatment time was determined to be significantly different from control (Wilcoxon’s signed rank test). Because each treatment time produced a similar trend (i.e., non-significant) toward a reduction in neuronal loss, we next asked the question: Did treatment with MDZ have an effect on global neuronal death when data from all three of the delay times were combined (i.e., when the sample size was tripled by combining the three data sets)? For this analysis, all control and all MDZ-treated rats were grouped together (Figure 6C, right). This analysis demonstrated that on average, MDZ-treated rats had 71% of the neuronal death observed in the control rats (Figure 6C, right, dashed line), and this difference was statistically significant (P < 0.05, Wilcoxon’s signed rank test), indicating that MDZ intervention did cause a reduction in neuronal death compared to no treatment at all. However, in each data set this change in the mean of neuronal death was less than the standard deviation making the relative effect difficult to detect.
Finally, we observed no effect of MDZ treatment on mortality. For the total cohort of rats in this study, 4 vehicle-treated and 5 MDZ-treated rats died at some time after treatment prior to the end of our 24 hr duration of EEG recordings. These deaths were dispersed throughout each group so that there were 1-2 deaths in each treatment time and each treatment condition. Rats that did not survive the full duration of the recording were not included in our analysis.
DISCUSSION
These studies used a novel delayed-treatment protocol in the rat to evaluate the anticonvulsant and neuroprotective efficacy of MDZ administered after three different delays from the onset of OP-induced SE. MDZ reduced the intensity of the seizure activity when administered after 30, 60 and 120 min of DFP-induced SE. The duration of the antiseizure effect was diminished at the longest treatment delay (i.e., 120 min), but the magnitude of the effect was similar for treatment delays of 30, 60 and 120 min. When assessed on a global scale (i.e., across all 10 of the brain regions studied here), treatment with MDZ did not significantly decrease SE-induced neuronal death for any delay time when data were analyzed for each delay time individually; however, a neuroprotective effect was detected when data from the three different treatment delays were grouped together. These data showed that MDZ was effective at providing transient seizure suppression up to 2 hr after SE onset, but this only resulted in a barely detectable (possibly due to high variance) reduction in neuronal death when assessed 24 hr after treatment of DFP-induced SE.
Seizure suppression after delayed administration of MDZ
Substantial evidence supports the hypothesis that benzodiazepines progressively lose efficacy for the suppression of seizures with increasing durations of SE 22,27–30,38. We therefore expected to observe a distinct loss of efficacy from 30 to 60 min, with the potential for no effect when MDZ was administered after a delay of 120 min. Our data with DFP and MDZ do not support this hypothesis. Although our data against this hypothesis may relate to differences in experimental approaches with some of the previously published research, other studies have obtained results that also do not support this hypothesis. For instance, in a direct comparison of several benzodiazepines tested head-to-head for their efficacy in the treatment of soman-induced SE in guinea pigs, McDonough and coworkers found that each of the six benzodiazepines tested, including MDZ, were effective at stopping seizures when administered with a 40-min treatment delay 32. This study also found similar ED50s and seizure offset delays when treatment was given at a 5-min or 40-min delay (i.e. no loss of efficacy with the longer duration of SE). In another study, Chapman et al. found that high-dose MDZ (5 mg/kg) was effective at stopping sarin-induced seizures in rats when administered with a 60-min treatment delay 39. Taken together, our data support and extend these earlier studies that have previously provided evidence that MDZ shows considerable efficacy in the treatment of OP-induced SE, even after a long delay before treatment (i.e., after 2 hr, in the present study).
In this regard, our earliest administration of MDZ was already at a 30-min delay time, when benzodiazepine treatment has reportedly lost at least some of its efficacy. Studies reporting a time-dependent decrease in the efficacy of benzodiazepines have typically compared much shorter delay times for benzodiazepine administration, ranging from a few minutes to 30-40 min as the longest intervals (i.e., our shortest delay time was 30 min). For instance, in a study with a treatment paradigm that was very similar to ours, Wu et al observed a reduction in efficacy of MDZ in treatment of DFP-induced SE when treatment delay was increased from 10 min to 40 min 40,41, but they also found that MDZ treatment still partially suppressed DFP-induced SE when given after delays of 60 min and 120 min. Our dose of MDZ (2.0 mg/kg, IM) was similar to the above-mentioned studies 32,39,40, although these authors used a slightly different analysis of the EEG (spikes/10 min) and a behavioral measure (seizure stage). Therefore, treatment with MDZ at 10 min was far more effective than at the delay times of 40 and 60 min, but by both measures these authors found that MDZ treatment still had some effect on seizures at 40 min and 60 min. Our data clearly demonstrate a robust effect on electrographic SE at 30 min with apparently similar efficacy at 60 and 120 min.
Most previous studies that have found little or no effect of benzodiazepines on SE at long treatment delays have used diazepam treatment in non-OP chemoconvulsant models or used limited or no quantification of the EEG for comparison (i.e., relied on seizure termination as the outcome measure) 10,22,29,30. This is also consistent with our own experience with diazepam treatment of pilocarpine-induced SE, where diazepam lost effectiveness at 30 min compared to 15 min 38. Perhaps the most conservative explanation for the differences observed in these studies comes from the use of different chemoconvulsants. Several of the studies that have reported substantive effects with a delayed-treatment protocol used OP-induced SE, while the other studies used pilocarpine or kainate to induce SE. To our knowledge, no study has ever reported side-by-side testing of one treatment against different SE-inducing agents.
In this study, we used two objective, independent and quantifiable outcome measures to assess the time-dependent severity and temporal profile (e.g. duration) of the SE: power in the gamma band (i.e., 20-60 Hz) and spike rate. We have previously demonstrated that changes in gamma band power are similar to changes in total power 34; however, we acknowledge this analysis may have missed some changes in other bands in this study. These analyses were conducted with semi-automated methods and blind procedures, so the potential for experimenter bias is low. We considered blinded analyses of the EEG with human observers, since such an approach has the potential advantage of detecting more subtle differences, but we chose not to take this approach because of the added time required and the difficulty standardizing across human observers. We did not include any behavioral analyses as measures of seizure activity primarily because they are prone to bias and can lead to both false positives and false negatives (i.e., behaviors that may be considered representative of seizures may not actually correspond to seizure activity [false positives], and electrographic seizures can occur without any behavioral signs, particularly in the presence of antiseizure drugs [false negatives]). Furthermore, behavioral analyses of seizure activity are extremely difficult to automate, and they tend to be highly subjective and variable across observers, particularly when applied to seizure-like behaviors during SE. Despite not using behavior as an outcome measure, we routinely observed heavy sedation after treatment with MDZ that was consistent with the reduction in the EEG activity, but could have been mistaken for cessation of seizures despite continuous electrographic SE.
MDZ is standard-of-care for treatment of refractory status epilepticus
Despite the previous studies demonstrating a loss of efficacy of benzodiazepines with increasing duration of SE, it should be re-iterated that MDZ is a standard-of-care for treatment of refractory SE 42–44. Refractory SE is often defined as SE that has failed treatment with benzodiazepines (typically lorazepam, clonazepam or midazolam) and at least one antiepileptic drug (typically phenytoin, valproate or levetiracetam). At this point in treatment, at about 30 min after the start of SE, intravenous treatment with anesthetic doses of MDZ (0.2-0.6 mg/kg/h, human dosage) is recommended with propofol as an alternative and/or second option, followed by pentobarbital if seizures persist. In this study we used a human equivalent dose 45 of 0.324 mg/kg, which falls within the treatment range for MDZ use in refractory SE. Therefore, the duration of the SE experienced by the rats in this study was consistent with clinically defined refractory SE and the observed effects of MDZ on the refractory SE was consistent with the use of MDZ in the treatment of refractory SE in human patients. From this perspective, it is not surprising that we observed efficacy of this treatment paradigm at delayed treatment times. It should be noted that despite having been very heavily sedated by this dose, the rats were not completely anaesthetized.
Neuroprotection by MDZ when administered with different delays after seizure onset
Despite the clear effects of MDZ on electrographic SE, we did not observe a robust effect on neuronal death, as measured with FJB, at any of the three different delay times of treatment. When neuronal loss from DFP-induced SE was analyzed for each delay time (i.e., 30, 60, and 120 min), MDZ administration had no detectable effect on FJB-measured neuronal loss. Presumably, “necrotic” neuronal loss occurred progressively during the 30, 60, and 120 min periods of SE before the MDZ-induced suppression of seizure activity. The significant difference in neuronal loss measured with FJB when the data from the three different delay times were grouped together was likely due to the larger group sizes. This result from the grouped data regarding neuroprotection supports our hypothetical proposal regarding our observation of efficacious seizure suppression at prolonged delay times (i.e., up to 120 min between seizure onset and administration of MDZ). Although MDZ did provide neuroprotection when all of the data were grouped together, the lack of effect of MDZ on neuronal loss at individual delays and the small effect of MDZ when these data were grouped together emphasize the need to focus on new treatments that target the molecular pathways that would reduce seizure-induced neuronal death when administered after electrographic SE has been suppressed with drugs like MDZ. These data are consistent with other reports that have shown little or no effect of MDZ on neuronal death in delayed treatment (≥ 40 min) of NA-induced SE 13,39,40. In contrast, treatment with MDZ at 10-20 min appears to be effective at reducing neuronal death 40,46. Our analysis of the change in power in the gamma band for up to 20 hr from the time of treatment suggests that the observed neuronal damage may be affected by the reemergence of epileptiform activity hours after treatment, which is similar to what has been previously shown by Wu et al 40.
The NIH CounterACT Neurotherapeutics Screening (CNS) Program
This work was supported by the NIH CounterACT Neurotherapeutics Screening (CNS) Program, which is funded through an interagency agreement (AOD16024–001-01000) between the NIH Office of the Director (OD) and the U.S. Army Medical Research Institute of Chemical Defense under the oversight of the Chemical Countermeasures Research Program (CCRP) within the Office of Biodefense Research (OBRS) at the National Institute of Allergy and Infectious Diseases (NIAID/NIH). The primary goal of the CNS Program is to provide a screening assay available to any participant who is interested in generating data towards application for grant funding from the CounterACT program. As such, the scope of the work here is limited to the development of a medium-throughput assay that somewhat models a real-life exposure scenario, and not open to traditional academic scientific endeavor. Indeed, these experiments raise numerous basic science questions, such as the identity of FJB labeled neurons, the time course of neuronal death, what effect multiple dosing paradigms (repeated treatment) might have on the seizure activity, and others; however, such studies are beyond the scope of this screening program. The attempt to mimic real-life exposure scenarios in the design of the assay were delayed treatment times, use of antidotes (for military personnel or first responders, pre-treatment prior to anticipated exposure is a legitimate scenario) and one-time dosing of a benzodiazepine approximately equal to two auto-injectors carried by military personnel or stocked in the US Dept. of Health and Human Services Strategic National Stockpile CHEMPACK Program.
Conclusions
Although it has long been stated that benzodiazepines lose their efficacy with increasing time after onset on SE, we found a robust effect of MDZ on DFP-induced SE when tested at delays of 30, 60 and even 120 min. This apparent discrepancy with previous results may reflect the nature of the models, the benzodiazepines under study, and/or the methods of seizure analysis; however, the present data are consistent with several other studies that have observed some form of benzodiazepine efficacy at longer delay times, particularly in models of organophosphate-induced SE. While the delayed MDZ intervention was quite effective at reducing seizures during DFP-induced SE, it only had barely detectable effects on neuronal death, as assessed with FJB. These data also demonstrate that OP-induced SE persists and slowly re-emerges after single treatments with MDZ. This model of delayed treatment will hopefully be useful in the development of new therapeutic interventions that could enhance the anti-seizure efficacy of MDZ or produce a more robust reduction in neuronal death.
Key Bullet Points.
MDZ-induced suppression of DFP-induced seizures was equivalent in magnitude regardless of treatment delay (i.e., seizure duration).
A small but significant reduction in neuronal death was detected for MDZ treated rats only when data from the entire cohort was combined.
These data support the need for the development of additional neuroprotective therapies to mitigate the effects of OP exposure.
Acknowledgements
We thank Peter Roper for regular updating of the QuoStatus software for automated SE analysis and Melissa Smolik for help collecting and assembling the data.
Funding
This work was supported by USAMRICD subcontract W81XWH-14-C-0119, W81XWH-16-C-0140 and W81XWH-18-C-0181.
Footnotes
Disclosures
F.E. Dudek has equity interest in and receives gifts (reduced cost for wireless equipment) and consultant fees from Epitel, Inc., which is a company that makes wireless recording devices. He has also received consulting fees from Neurotherapeutics Pharma, Rugen Holdings, and Neurona Therapeutics. The other authors declare no other financial interests.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
REFERENCES
- 1.Hulse EJ, Davies JO, Simpson AJ, et al. Respiratory complications of organophosphorus nerve agent and insecticide poisoning. Implications for respiratory and critical care. Am J Respir Crit Care Med 2014;190:1342–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lotti M Clinical toxicology of anticholinesterase agents in humans. Handbook of Pesticide Toxicology; 2001:1043–1085. [Google Scholar]
- 3.Karalliedde L, Senanayake N. Organophosphorus insecticide poisoning. Br J Anaesth 1989;63:736–750. [DOI] [PubMed] [Google Scholar]
- 4.Bajgar J Optimal choice of acetylcholinesterase reactivators for antidotal treatment of nerve agent intoxication. Acta Medica (Hradec Kralove) 2010;53:207–211. [DOI] [PubMed] [Google Scholar]
- 5.Jokanovic M, Stojiljkovic MP. Current understanding of the application of pyridinium oximes as cholinesterase reactivators in treatment of organophosphate poisoning. Eur J Pharmacol 2006;553:10–17. [DOI] [PubMed] [Google Scholar]
- 6.Leadbeater L, Inns RH, Rylands JM. Treatment of poisoning by soman. Fundam Appl Toxicol 1985;5:S225–231. [DOI] [PubMed] [Google Scholar]
- 7.Voicu VA, Bajgar J, Medvedovici A, et al. Pharmacokinetics and pharmacodynamics of some oximes and associated therapeutic consequences: a critical review. J Appl Toxicol 2010;30:719–729. [DOI] [PubMed] [Google Scholar]
- 8.Voicu VA, Thiermann H, Radulescu FS, et al. The toxicokinetics and toxicodynamics of organophosphonates versus the pharmacokinetics and pharmacodynamics of oxime antidotes: biological consequences. Basic Clin Pharmacol Toxicol 2010;106:73–85. [DOI] [PubMed] [Google Scholar]
- 9.Wetherell J, Hall T, Passingham S. Physostigmine and hyoscine improves protection against the lethal and incapacitating effects of nerve agent poisoning in the guinea-pig. Neurotoxicology 2002;23:341–349. [DOI] [PubMed] [Google Scholar]
- 10.Apland JP, Aroniadou-Anderjaska V, Figueiredo TH, et al. The limitations of diazepam as a treatment for nerve agent-induced seizures and neuropathology in rats: comparison with UBP302. J Pharmacol Exp Ther 2014;351:359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apland JP, Figueiredo TH, Qashu F, et al. Higher susceptibility of the ventral versus the dorsal hippocampus and the posteroventral versus anterodorsal amygdala to soman-induced neuropathology. Neurotoxicology 2010;31:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prager EM, Aroniadou-Anderjaska V, Almeida-Suhett CP, et al. The recovery of acetylcholinesterase activity and the progression of neuropathological and pathophysiological alterations in the rat basolateral amygdala after soman-induced status epilepticus: relation to anxiety-like behavior. Neuropharmacology 2014;81:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih TM, Duniho SM, McDonough JH. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol Appl Pharmacol 2003;188:69–80. [DOI] [PubMed] [Google Scholar]
- 14.Wasterlain CG, Fujikawa DG, Penix L, et al. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 1993;34 Suppl 1:S37–53. [DOI] [PubMed] [Google Scholar]
- 15.Filliat P, Coubard S, Pierard C, et al. Long-term behavioral consequences of soman poisoning in mice. Neurotoxicology 2007;28:508–519. [DOI] [PubMed] [Google Scholar]
- 16.Langston JL, Wright LK, Connis N, et al. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: increased startle reactivity and perseverative behavior. Pharmacol Biochem Behav 2012;100:382–391. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman A, Eisenkraft A, Finkelstein A, et al. A decade after the Tokyo sarin attack: a review of neurological follow-up of the victims. Mil Med 2007;172:607–610. [DOI] [PubMed] [Google Scholar]
- 18.Yanagisawa N, Morita H, Nakajima T. Sarin experiences in Japan: acute toxicity and long-term effects. J Neurol Sci 2006;249:76–85. [DOI] [PubMed] [Google Scholar]
- 19.Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631–637. [DOI] [PubMed] [Google Scholar]
- 20.Shorvon S Status epilepticus: its clinical features and treatment in children and adults. Cambridge University Press: Cambridge, England; 1994. [Google Scholar]
- 21.Treiman DM. Treatment of convulsive status epilepticus. Int Rev Neurobiol 2007;81:273–285. [DOI] [PubMed] [Google Scholar]
- 22.Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol 1988;101:267–275. [DOI] [PubMed] [Google Scholar]
- 23.Manno EM. Status epilepticus: current treatment strategies. Neurohospitalist 2011;1:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sirven JI, Waterhouse E. Management of status epilepticus. Am Fam Physician 2003;68:469–476. [PubMed] [Google Scholar]
- 25.Silbergleit R, Lowenstein D, Durkalski V, et al. RAMPART (Rapid Anticonvulsant Medication Prior to Arrival Trial): a double-blind randomized clinical trial of the efficacy of intramuscular midazolam versus intravenous lorazepam in the prehospital treatment of status epilepticus by paramedics. Epilepsia 2011;52 Suppl 8:45–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lockey AS. Emergency department drug therapy for status epilepticus in adults. Emerg Med J 2002;19:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonough JH, McMonagle JD, Shih TM. Time-dependent reduction in the anticonvulsant effectiveness of diazepam against soman-induced seizures in guinea pigs. Drug Chem Toxicol 2010;33:279–283. [DOI] [PubMed] [Google Scholar]
- 28.Shih T, McDonough JH Jr., Koplovitz I. Anticonvulsants for soman-induced seizure activity. J Biomed Sci 1999;6:86–96. [DOI] [PubMed] [Google Scholar]
- 29.Todorovic MS, Cowan ML, Balint CA, et al. Characterization of status epilepticus induced by two organophosphates in rats. Epilepsy Res 2012;101:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci 1997;17:7532–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogalski R, Rogalski A. Benzodiazepine selection in the management of status epilepticus: a review. Adv Emerg Nurs J 2015;37:83–94; quiz E82. [DOI] [PubMed] [Google Scholar]
- 32.McDonough JH, Jr., McMonagle J, Copeland T, et al. Comparative evaluation of benzodiazepines for control of soman-induced seizures. Arch Toxicol 1999;73:473–478. [DOI] [PubMed] [Google Scholar]
- 33.Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmkuhle MJ, Thomson KE, Scheerlinck P, et al. A simple quantitative method for analyzing electrographic status epilepticus in rats. J Neurophysiol 2009;101:1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White AM, Williams PA, Ferraro DJ, et al. Efficient unsupervised algorithms for the detection of seizures in continuous EEG recordings from rats after brain injury. J Neurosci Methods 2006;152:255–266. [DOI] [PubMed] [Google Scholar]
- 36.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000;874:123–130. [DOI] [PubMed] [Google Scholar]
- 37.Pouliot W, Bealer SL, Roach B, et al. A rodent model of human organophosphate exposure producing status epilepticus and neuropathology. Neurotoxicology 2016;56:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pouliot W, Bialer M, Hen N, et al. A comparative electrographic analysis of the effect of sec-butyl-propylacetamide on pharmacoresistant status epilepticus. Neuroscience 2013;231:145–156. [DOI] [PubMed] [Google Scholar]
- 39.Chapman S, Yaakov G, Egoz I, et al. Sarin-induced brain damage in rats is attenuated by delayed administration of midazolam. Neurotoxicology 2015;49:132–138. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, Kuruba R, Reddy DS. Midazolam-Resistant Seizures and Brain Injury after Acute Intoxication of Diisopropylfluorophosphate, an Organophosphate Pesticide and Surrogate for Nerve Agents. J Pharmacol Exp Ther 2018;367:302–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy SD, Reddy DS. Midazolam as an anticonvulsant antidote for organophosphate intoxication--A pharmacotherapeutic appraisal. Epilepsia 2015;56:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossetti AO, Lowenstein DH. Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol 2011;10:922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez A, Lantigua H, Lesch C, et al. High-dose midazolam infusion for refractory status epilepticus. Neurology 2014;82:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trinka E, Hofler J, Leitinger M, et al. Pharmacotherapy for Status Epilepticus. Drugs 2015;75:1499–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Acon-Chen C, Koenig JA, Smith GR, et al. Evaluation of acetylcholine, seizure activity and neuropathology following high-dose nerve agent exposure and delayed neuroprotective treatment drugs in freely moving rats. Toxicol Mech Methods 2016;26:378–388. [DOI] [PubMed] [Google Scholar]