

Abstract
Copy number variants (CNVs) play an important role in human disease and population diversity. Advancements in technology have allowed for the analysis of CNVs in thousands of individuals with disease in addition to thousands of controls. These studies have identified rare CNVs associated with neuropsychiatric diseases such as autism, schizophrenia, and intellectual disability. In addition, copy number polymorphisms (CNPs) are present at higher frequencies in the population, show high diversity in copy number, sequence, and structure, and have been associated with multiple phenotypes, primarily related to immune or environmental response. However, the landscape of copy number variation still remains largely unexplored, especially for smaller CNVs and those embedded within complex regions of the human genome. An integrated approach including characterization of single nucleotide variants and CNVs in a large number of individuals with disease and normal genomes holds the promise of thoroughly elucidating the genetic basis of human disease and diversity.
Keywords: copy number variant, segmental duplication, complex disease, microdeletions, microduplications, array-CGH
INTRODUCTION
Recent studies have indicated that copy number variants (CNVs) are widespread in the human genome and are a significant source of human genetic variation accounting for disease and population diversity. Advances in whole-genome technologies, including array comparative genomic hybridization (CGH), single nucleotide polymorphism (SNP) microarrays, and genome sequencing, have enabled the discovery and characterization of variants that are intermediate between large chromosomal aberrations (>1 Mbp) and smaller insertions or deletions (1–50 bp) (Figure 1). These intermediate-sized variants, essentially deletions and duplications in the human genome, are called CNVs. Between any two individuals the number of basepair differences due to CNVs is >100-fold higher compared with SNPs (72). Current advances have also led to large-scale association studies for common diseases to find not only common copy number polymorphisms (CNPs) but rare CNVs as well. Two distinct models of CNV disease association have emerged. The first model involves CNPs that occur at a population frequency of >1% but often exists in multicopy number states ranging from 0 to 30 copies per diploid genome (118). For these, variation in copy, content, or structure of sequences is important for understanding disease etiology. The second model involves CNVs that are individually rare (<1% frequency) but typically involve larger chromosomal segments (>100 kbp) and exist in fewer copy number states (segmental monosomy/hemizygous or trisomy). These CNVs are under strong selection pressure; their frequency in the population is largely contributed by de novo events, and they can only persist for a few generations. In this review, we explore the current state of knowledge in the study of rare and common CNVs in normal human populations in relation to human diseases, methodologies to study CNV disease associations, and future directions for research in understanding of the genetic etiology of complex diseases.
Figure 1.

Size and frequency of major categories of genetic variants. Different sized genetic variants as a function of frequency are shown. Overall, single nucleotide polymorphisms (SNPs) occur at a higher frequency and can be assayed by high throughput SNP genotyping. Copy number variants are intermediate-sized variants (>50 bp) and can be assayed by SNP microarrays or array comparative genomic hybridization. Large chromosomal aberrations are rarer, large, and microscopically visible after G-banding and are often associated with major congenital abnormalities (e.g., Down syndrome associated with trisomy 21).
RARE COPY NUMBER VARIANTS IN NORMAL POPULATIONS
Studies on copy number variation have been largely restricted to screening disease cohorts for rare CNVs. Such studies by themselves cannot necessarily distinguish variants that are truly pathogenic versus those that occur at a low frequency in the general population but are generally benign. Few studies are available documenting the CNV landscape in the general population. Itsara and colleagues analyzed 2,500 individuals by exploiting available Illumina-SNP microarray data collected from individuals with either no known disease or relatively mild phenotypic traits (53). Based on the assumption that these individuals were representative of the general population, the study provided some important insights into the size and frequency of CNVs. Focusing on only the largest events, the study found three to seven variants per person with a total of about 540 kbp of copy number variable DNA per person. Approximately 65% to 80% of individuals carry a CNV that is at least 100 kbp in size, five to ten percent of individuals harbor a CNV at least 500 kbp, and an astonishing one percent of individuals carry a large CNV of at least 1 Mbp in size. It is important to note that variants at the larger size range (>500 kbp), if observed as a de novo event within a gene-rich region, would usually be considered pathogenic in a clinical diagnostic setting (83). On an individual basis, approximately 71% of individual CNVs greater than 100 kbp are rare, i.e., seen in far less than one percent of the population, and events greater than 500 kbp were even rarer and observed in only one individual. This study also established the basic tenet of CNV studies: Rare CNVs harbor more genes than common CNVs and, particularly, homozygous deletions are significantly gene poor compared with the genome-wide average (p = 4.7 × 10−4). However, the authors also note that their estimate of size-wise CNV burden might be biased against smaller events due to the sensitivity of the array. In a follow-up analysis examining the new mutation frequency of large CNVs among children with mild to moderate asthma (54), the authors estimated conservatively that between 8,000 and 25,000 basepairs of genomic DNA were added or lost per transmission (compared with ∼100 basepairs changed by point mutations) and large CNVs were in the aggregate under relatively strong selection (s = 0.16). Thus, although CNVs were individually rare but collectively common (for example, eight percent of normal individuals carry an essentially private mutation greater than 500 kbp), most of these events would be eliminated relatively quickly as a result of purifying selection. These data argue that the impact of CNVs on human health must be significant.
GENOMIC DISORDERS AND VARIABLE NEURODEVELOPMENTAL PHENOTYPES
Classically, children with specific neurodevelopmental disorders ascertained by typical constellation of clinical features were tested by fluorescent in situ hybridization (FISH) or molecular karyotyping. Almost two decades of research resulted in delineating specific genomic regions to these syndromes, termed genomic disorders, such as Charcot-Marie-Tooth disease, Smith-Magenis syndrome, Williams syndrome, and DiGeorge syndrome (Figure 2). A large fraction of these variants arise by a nonallelic homologous recombination (NAHR) between high-identity segmental duplications. These genomic disorders are highly penetrant, recurrent, and can occur on different haplotype backgrounds in multiple unrelated individuals in a relatively short period of time, and they are under strong negative selection. The frequency of these rearrangements is thought to be partly dictated by the size and sequence identity of the flanking segmental duplications (Figure 3); the 22q11.2 deletion is the most common genomic disorder in children with developmental delay and intellectual disabilities. For at least 10 known genomic disorders, inversion variants or polymorphisms associate with directly orientating segmental duplications leading to NAHR (5, 6, 33, 36, 37, 93, 139).
Figure 2.
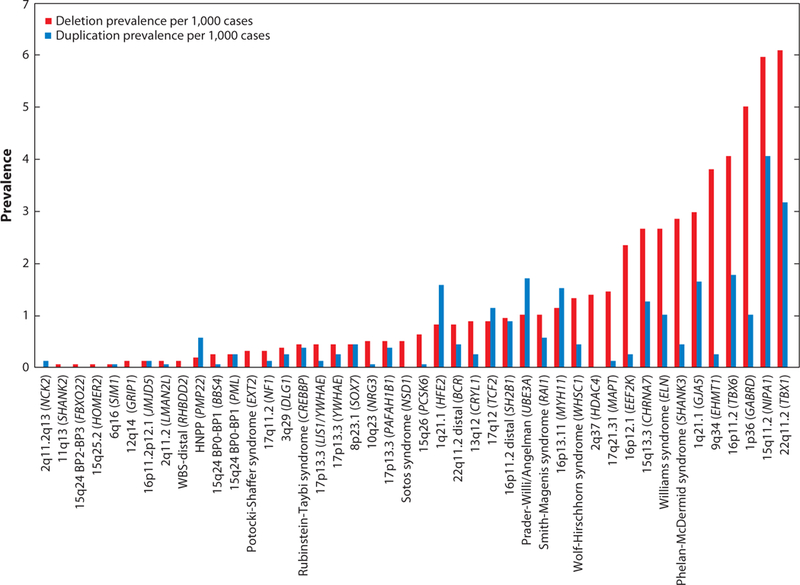
Prevalence of genomic disorders in individuals with developmental delay. The prevalence of rare deletions and duplications associated with neurodevelopmental disorders, also termed genomic disorders, is shown. The data were generated from an analysis of 15,767 individuals assessed for developmental delay and associated phenotypes (20a). Note that the prevalence of the deletion is higher compared with reciprocal duplications for most of the disorders. Also note that nonrecurrent rearrangements are generally rarer in frequency compared with recurrent segmental duplication-mediated rearrangements. The candidate genes within each of these genomic disorder regions are depicted within the parenthesis.
Figure 3.
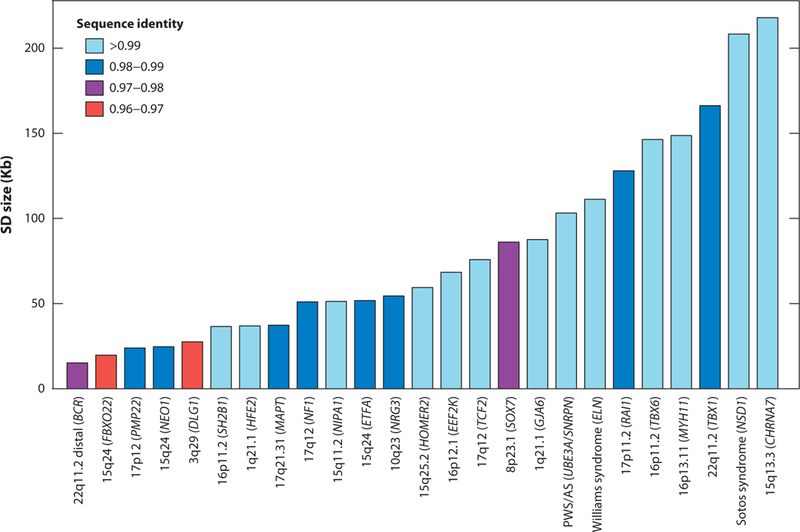
Sequence identity and size of segmental duplications mediating disease-associated genomic rearrangements. Properties of the segmental duplications participating in nonallelic homologous recombination (NAHR) events are shown. Note that direct orientation of segmental duplications is also a general requirement and individuals possessing such architecture are predisposed for an NAHR event. Operationally, segmental duplications are defined as large blocks with greater than 10 kbp of repeat sequences with >95% sequence identity.
Bert de Vries and colleagues estimated the diagnostic significance of whole-genome array CGH using bacterial artificial chromosome clones for a general population of patients with intellectual disability and developmental delay (24). This study identified 10% of cases with a large (size ranging from 500 kbp to 12 Mbp) de novo CNV—a greater than twofold diagnostic yield compared with conventional Giemsa-banded karyotyping. Alternatively, applying a targeted methodology to discover CNVs at NAHR rearrangement sites termed genomic hotspots, Sharp and colleagues identified a series of novel rearrangements in individuals with developmental delay (107). In the next few years, hotspot-mediated rearrangements were not only associated with intellectual disability and developmental delay but were also implicated in other complex diseases such as autism, schizophrenia, bipolar disorder, amyotrophic lateral sclerosis, attention deficit hyperactivity disorder (ADHD), and Tourette syndrome. Excluding syndromic rearrangements on chromosomes 17q21.31 (61, 107, 111), 15q24 (110), and 17q23.3 (9), many recurrent CNVs are associated with variable penetrance and expressivity. For example, the 1.5 Mbp deletion on 15q13.3 was initially identified in individuals with developmental delay (109). Further studies showed that the same chromosomal change was enriched in cases with autism (95) and schizophrenia (19, 117) and, in fact, accounts for approximately one percent of cases with idiopathic generalized epilepsy (46). Similarly, the ∼1.6 Mbp microdeletion on 1q21.1 was enriched in individuals with developmental delay (82), autism (120), schizophrenia (19, 117), and cardiac defects (43). Further, microdeletions on chromosome 17q12 were first discovered in cognitively normal children with renal cysts and maturity-onset diabetes of the young (RCAD syndrome) (81). The associated phenotypes were further extended to include developmental delay, brain malformation, seizures (89), schizophrenia, and autism (86). Another example is a microdeletion on 3q29 initially observed in a large set of cases with developmental delay (8, 14, 25), which was also recently reported in individuals with schizophrenia (88). Thus, the newly identified CNVs transcend phenotypic boundaries but cause concerns of ascertainment bias and resolution of clinical assessment. It was recently observed that more than one CNV (or the two-hit model) can contribute to severe developmental delay and often is responsible for phenotypic variability associated with genomic disorders. Interestingly, the cooccurring CNVs were large (>500 kbp) and contained several genes, were rare in frequency, and were sometimes also associated with a known genomic disorder—as observed in cases with a 16p12.1 microdeletion (39). These observations indicate potential involvement of one or more dosage-sensitive genes within the CNV interval in early developmental pathways.
Despite the importance of genomic hotspots, a larger fraction of CNV neurocognitive disease burden is contributed by rearrangements generated by nonrecurrent mechanisms such as microhomology/microsatellite-mediated break-induced repair, nonhomologous end joining or fork stalling, and template switching mechanisms (reviewed in 44). These rearrangements do occur at a relatively lower frequency and may be caused by different mutational mechanisms, including replication-based rearrangement and nonhomologous end joining. Unlike genomic hotspots where the de novo mutation frequency is elevated, assessing the pathogenic significance of these types of events has been problematic, requiring the screening of many thousands of patients and controls to establish pathogenicity. Examples include duplications involving PLP1 in Pelizaeus-Merzbacher disease (66), PMP22 in Charcot-Marie-Tooth disease (137), and LMNB1 in autosomal dominant leukodystrophy (94). Several nonrecurrent deletions and duplications have been reported in autism and schizophrenia, including ASTN2 and NRXN1 disruptions in both autism (76) and schizophrenia (60, 126), SHANK2, SHANK3, and PTCHD1 CNVs in autism (76), and MYT1L and CNTND2 (126) disruptions in schizophrenia. Recently, based on the hypothesis that rare CNVs smaller than 500 kbp that reside in regions other than NAHR hotspots may contribute to the etiology of schizophrenia, Vacic and colleagues performed a large-scale genome-wide scan in 8,290 patients with schizophrenia and 7,431 matched controls (124). A 362-kbp microduplication on 7q36.3 overlapping or upstream of the vasoactive intestinal peptide receptor gene, VIPR2, was detected in 2.5% of the cases compared with only 0.03% controls. Further, functional studies also suggested altered cyclic-AMP signaling in the cultured lymphocytes of patients compared with controls.
CHALLENGES IN THE DISCOVERY AND DIAGNOSIS OF DISORDERS ASSOCIATED WITH RARE CNVS
The study of rare CNVs has several associated challenges. First, discovery of rare variants requires screening several thousands of well-ascertained cases and matched controls. The need for having an ethnicity-matched control population for comparing the frequency of identified variant cannot be overstated. For example, it is known now that segmental duplication architecture is stratified in different ethnic populations and can predispose or protect individuals for large disease-associated CNVs. Such stratified regions in the genome have been identified for CNVs for at least half a dozen regions, including 17q21.31, 16p12.1, and 7q11.23 (reviewed in 38). Second, although there is an exponential increase in the throughput of processing samples for CNVs, the uniformity of diagnosis at all participating clinical centers, resolution of assessment, and objective assessment for severity and variability of a particular clinical feature has often been of concern. Resolution of clinical assessment and a clinical acumen for subtle features are exceptionally important in the diagnosis of early-onset or variable phenotype. For example, clinical diagnosis of patients with duplication syndromes is often missed because of the subtle manifestation of most of the phenotypes, unlike, for example, Smith-Magenis syndrome associated with the 17p11.2 deletion, where sleep disturbance, craniofacial features, overt behaviors, and obesity are the key constellation of features (28a, 39a). In contrast, the reciprocal duplication of 17p11.2 manifests with variable features ranging from craniofacial abnormalities and growth retardation to idiopathic autism features and ADHD (100).
Another example is the ∼400 kbp duplication on 15q13.3 containing CHRNA7. Although smaller (114) as well as larger deletions encompassing CHRNA7 have been associated with a variety of neurodevelopmental disorders (46, 95, 109, 125), the smaller reciprocal duplication of CHRNA7 is of unknown significance (119). Although deep phenotyping can recover patients with subclinical features mostly associated with duplications, overall the frequency of patients with deletions is usually higher than that of duplications. It is noteworthy that a deletion not only causes haploinsufficiency for dosage-sensitive genes within the genomic interval but also unmasks recessive alleles within the region. A good example is hearing loss associated with patients with Smith-Magenis syndrome due to recessive mutations in MYO15A (69). Thus, the genome is likely more tolerant for duplications than for deletions. Analysis of recombination products using microsatellite repeats and SNPs within and flanking the rearranged chromosomal segments suggests that recombination between segmental duplications can occur in three ways: intrachromatid, interchromatid, and interchromosomal (121). Although all the three mechanisms can generate deletions, only the latter two can result in duplications. This mechanism also provides physical evidence for increased frequency of deletions compared with duplications.
Clinical DNA microarrays are now routinely used for molecular testing of children with intellectual disability/developmental delay phenotypes. Although analysis of large disease cohorts has essentially helped identify novel, rare deletions and duplications, this advance also led to the discovery of variants ranging in population frequency between one percent and five percent, which are deemed of unknown clinical significance. Conventionally, variants are considered pathogenic if they arise de novo in the proband, are rare (<1%) in frequency, and are not observed in unaffected siblings or ethnically matched controls. Depending on the array platform and design of probes, the diagnostic yield of cytogenetic laboratories, i.e., the proportion of referred cases with a pathogenic CNV, ranges between 5% and 20%. Although the standard microarray design consists of relatively uniform distribution of probes throughout the genome, other versions have also been in vogue with high probe density targeting coding exons, specific candidate genes, or genomic hotspot regions, in addition to a uniform density of probes in the genomic backbone (83).
DE NOVO AND INHERITED BASIS OF RARE COPY NUMBER VARIANTS IN COMMON COMPLEX DISEASES
The extreme genetic heterogeneity of common complex diseases, including autism and schizophrenia, and the high de novo mutation rate have hindered linkage studies of inherited susceptibility loci. Initial discoveries of CNVs in complex diseases were based on locus-specific case-control association models in which deletions and duplications for a particular region of the genome were expected to be more represented in cases than healthy controls. The discovery of CNVs in autism and schizophrenia was also based on a new mutation model where only de novo variants were considered pathogenic (105, 132). Two pioneering studies exemplify this model. Sebat and colleagues utilized the representational oligonucleotide microarray analysis CNV detection methodology in 165 families affected with autism and compared with 99 control families. They identified 7.2% (14/195) enrichment for de novo variants in cases compared with 1.02% (2/196) in unaffected controls (p = 0.0005). This study also observed a higher incidence of deletions in cases with autism when both CNVs in the controls were duplications. Similarly, Walsh and colleagues tested the total genome-wide mutational burden in individuals with schizophrenia compared with controls (127). They studied 150 individuals with schizophrenia and compared them with 268 ancestry-matched controls and identified that, compared with only five percent frequency in controls, CNVs of recent origin accounted for 15% of adult onset (p = 0.0008) and 20% of early onset schizophrenia (p = 0.0001). Pathway analysis showed that these mutations disrupted genes involved in neuronal signaling networks including glutamate and neuregulin pathways.
Some studies did not find enrichment for large, rare CNVs in cases compared with controls, as described by Walsh and colleagues (40, 52, 91, 112). For example, Shi and colleagues analyzed 155 cases with schizophrenia and 187 matched controls—all recruited from the Han Chinese population in Shanghai. No significant enrichment was observed for rare CNVs (>100 kbp) in the case cohort compared with controls in this study. No increase in rare CNV (>500 kbp) burden was also reported by Ikeda and colleagues (52) after analyzing 575 patients with schizophrenia and 564 control subjects from Japan. Similarly, Glessner and colleagues (40) analyzed CNV data from 977 cases with schizophrenia and 2,000 healthy controls of European ancestry and then followed up the positive findings in another set of 758 cases of schizophrenia and 1,485 controls. They were unable to replicate the previously reported overrepresentation of rare CNVs affecting many genes in schizophrenia compared with controls. Need and colleagues (91) also tested the large CNV enrichment in 1,013 cases with schizophrenia and 1,084 matched controls. They could not provide strong support for the hypothesis that schizophrenia patients have a significantly greater load of large (>100 kbp), rare CNVs. It is important to note that such observations could be due to differences in sample quality, cell line artifacts, probe resolution, GC content, lack of genotype information, subphenotype characterization and clinical heterogeneity, age of onset of disease, and platform-specific biases. In all of these studies, previously reported loci, including 1q21.1, 15q11.2, 15q13.3, and 22q11.2 and NRXN1 deletions, were supported, although at a nominal significance.
In a family-based study of 359 individuals with schizophrenia, Xu and colleagues observed an eightfold increase in de novo CNVs in cases with sporadic schizophrenia compared with controls (132). In comparison, rare inherited CNVs were only modestly enriched (1.5-fold compared with controls) in sporadic cases. The same group also found that although rare de novo CNVs were not enriched compared with sporadic cases, individually rare inherited CNVs were more frequent in multiplex families with schizophrenia (133). This observation replicated a similar result obtained by Marshall and colleagues wherein higher rates of de novo variants were detected in families with single affected cases with autism spectrum disorder (ASD) compared with families with two or more affected siblings (76). Searching for common inherited factors segregating in a recessive fashion to result in autism, Morrow and colleagues recruited 104 families, including 79 simplex and 25 multiplex families (87). A large proportion of these families had a history of parental consanguinity with cousin marriages (88/104, 19 multiplex and 69 simplex). Using homozygosity mapping, homozygous deletions involving genes associated with synaptic functions such as DIA1, PCDH10, ROBO2, NHE9, and SCN7A were identified. Overall, the frequency of de novo CNVs segregating with ASD was null (0%) in consanguineous multiplex families (0/42) and 1.9% in consanguineous simplex families (1/52 patients). This estimate is considerably lower than that reported by Sebat and colleagues (105) for nonconsanguineous families, suggesting an increased role for inherited factors in autism families with shared ancestry.
Considering the impact of de novo CNVs and the inherited basis of complex disease, two studies exemplified the genetic basis of simplex and multiplex autism. Zhao and colleagues analyzed autism risk in multiplex families from the Autism Genetic Resource Exchange (AGRE) and found strong evidence for dominant transmission to male offspring (138). They put forth a model of autism risk in which the families fall into two categories: those in which the autism risk is low, as in the majority of families, and those in which the risk is high. Under this model, sporadic autism occurs because of a spontaneous mutation with high penetrance, particularly in males from low-risk families, and multiplex autism occurs in high-risk families in which the susceptible-allele carrying mother has low or no penetrance, and inheritance of this allele occurs in a dominant fashion with male-specific penetrance. Itsara and colleagues proposed an alternate model that unifies all these observations by analyzing 717 multiplex pedigrees from the AGRE cohort for de novo CNVs (54). They found that the de novo CNVs within multiplex autism families segregated disproportionately to affected siblings over unaffected siblings. In fact, there was a fourfold enrichment of de novo CNVs in the affected cases of multiplex autism compared with unaffected siblings. Under their model, simplex cases are enriched for high risk, causative mutations and therefore have a significantly increased frequency of new mutations compared with the general population. In contrast, multiplex autism cases are not enriched for causative mutations but carry some inherited predisposition for disease, and hence the overall rate of de novo CNVs within multiplex pedigrees is not any different from the general population. However, when de novo CNVs arise within multiplex autism pedigrees, they occur on an already sensitized genetic background for disease and hence de novo CNVs segregate disproportionately to affected versus unaffected siblings. Thus, although rare, causative variants may cause sporadic disease such as autism, in multiplex families autism is a result of multiple cooccurring mutations. The inherited predisposition in many instances is not sufficient enough to reach a threshold of full-blown disease. Girirajan and colleagues proposed the necessity in some families for a second mutational hit to manifest severe neuropsychiatric disease (39). It is therefore plausible that many of these CNVs by themselves are not fully penetrant, but in concert with other genetic factors they would provide a molecular etiology for the genetic complexity of these diseases.
METHODS TO STUDY RARE COPY NUMBER VARIANT DISEASE ASSOCIATIONS
The locus and mutational heterogeneity of CNVs have created difficulties in traditional data analysis, and as a result new disease-association methods and disease models have been proposed to explain prevalence of complex neurocognitive and neurobehavioral diseases (Figure 4). Glessner and colleagues, for example, resorted to identifying perturbed gene pathways based on CNVs on different loci and determining if specific biological pathways were enriched in cases with schizophrenia (40). This approach identified a significant enrichment of synaptic transmission genes in the cases compared with controls. A pathway-based approach was also utilized in a study of ADHD by Elia and colleagues in a set of 335 cases and 2,026 unrelated healthy individuals (28). In addition to genes important for synaptic transmission and central nervous system development, genes previously implicated in autism, schizophrenia, and Tourette syndrome, such as AUTS2, A2BP1, CNTNAP2, and IMMP2L, were identified.
Figure 4.

Methods for associating rare copy number variants (CNVs) to neurodevelopmental disease. (a) Pathogenicity has been classically associated with a de novo or new mutation model. Pathogenic variants are expected to be strongly selected and the prevalence of these CNVs is essentially maintained by de novo occurrence. (b) Case-control association study to infer pathogenicity for a CNV. Locus-specific CNV frequency is compared in cases and controls under the assumption that the pathogenic CNV is enriched in cases that manifest the disease. This comparison is only valid when both the cohorts are matched for age, sex, and ethnicity and assayed on the comparable CNV detection platform. (c) Sliding window or segment-based approach to identify pathogenic regions in the genome. Such analysis can identify a specific genic region or a locus enriched in cases compared with controls. (d) Size-wise comparison of CNV data as a function of frequency is a good estimate of selective pressure on CNVs. This method provides an estimate of the odds ratio for a particular sized variant. (e) Pathway-based analysis for assessing pathogenicity of the individually rare but collectively common CNVs. This model is generally applicable in the study of complex neuropsychiatric disease wherein related genes are thought to interact in a common neurological pathway. An altered homeostatic state resulting in disease is inferred when two or more genes within the same pathway are disrupted. (f) The global CNV rate and gene disruptions as a function of pathogenic association. The total number of rare CNVs and the number of genes disrupted by deletion or duplication can also be considered for testing pathogenicity. Such a method was recently utilized by Pinto and colleagues in a large-scale study of individuals with autism (98).
Another measure of selective pressure acting on individuals with a disease and healthy controls is to perform size-wise comparison of frequency of CNVs. Such analysis can further be utilized to generate the odds of observing a CNV of a particular size in individuals with disease compared with controls and would also be a better model for comparing CNV data from disparate phenotypes or subphenotypes. Not conforming to candidate gene or genomic region based associations, Glessner and colleagues analyzed two independent cohorts of cases and controls to identify CNVs conferring susceptibility to ASD (41). They applied a segment-based or sliding-window scoring approach that scans the entire genome in overlapping windows for consecutive probes with more frequent copy number changes in cases compared with controls. This approach will identify CNV regions (CNVRs) and, using a Fisher’s exact test or permutations, p values for significant CNVR enrichment in cases compared with controls can be deduced. Alternatively, a gene-based scoring approach that examines CNVs within regions of the genes for specific enrichment in cases compared with controls can be applied. Using this approach, Glessner and colleagues identified seven genes within or surrounding CNVs with an increased frequency in cases compared with controls. Interestingly, these genes, including UBE3A, PARK2, RFWD2, and FBXO40, are involved in the ubiquitin pathway, thereby suggesting a novel pathogenesis for ASD (41). The gene-based sliding window approach is advantageous because the CNVs targeting different parts of the gene but falling within the same segment would have been missed by a segment-based approach.
A more functionally relevant model is to focus on variants disrupting genes. A comparison of the number of genes affected by CNVs in individuals with disease compared with controls is also a good measure of contribution of CNVs to pathogenicity. Such analysis has been performed by Walsh and colleagues for schizophrenia (127) and more recently by Pinto and colleagues (98). Because association of individual, rare CNVs often has insufficient power to discriminate benign variants from disease-causing variants, Pinto and colleagues sought to find whether genes and CNVs previously associated with ASD and intellectual disability were enriched in cases compared with controls. Using this criterion, they defined three gene sets: an ASD-implicated list consisting of those genes strongly implicated in autism and also observed in cases with intellectual disability, genes implicated in intellectual disability but not ASD, and ASD candidate genes from previously reported studies. Although they found a higher proportion of cases with rare CNVs overlapping ASD-implicated and intellectual disability genes, there was no enrichment for the ASD candidate genes. The authors further applied pathway analysis and identified a significant enrichment for GTPase/Ras signaling, cellular proliferation, synaptic transmission, and glutaminergic pathways. These analyses also suggest potential pathways and genes specific to a particular manifestation, such as idiopathic autism or Asperger syndrome, or indicate the culmination of genes and pathways common to intellectual disability and ASD.
PROPERTIES OF COPY NUMBER POLYMORPHISMS
Although the CNVs associated with neurocognitive disease are rare, CNVs can, on occasion, rise to high frequency in human populations. These variants are generally termed as CNPs. Two types of CNPs may be distinguished: those that simply represent a gain or loss of a particular segment of DNA, called biallelic CNPs, and those where the underlying sequence can exist as a series of whole integers, termed multicopy CNPs. Surveys of CNPs in the general population have identified large numbers of these variants in the human genome and have informed our understanding of the genomic mutational landscape (16, 18, 22, 47, 51, 56, 78, 80, 106, 108). CNPs are not distributed uniformly in the genome but are enriched in segmental duplications (7). Multiple studies have estimated a fourfold to tenfold enrichment of CNPs within regions of segmental duplications (21, 70, 101, 108, 131). Many of the same mechanisms that lead to the formation of rare pathogenic CNVs likely contribute to patterns of CNPs. Recent efforts in CNV breakpoint sequencing have revealed that microhomology of sequence at the breakpoints, NAHR, and L1 retrotransposition are the most common mechanisms leading to the formation of CNVs (17, 57). Therefore, CNPs in segmental duplication-rich regions likely have a higher mutation/recurrence rate compared with CNPs in unique regions of the genome because of the propensity of segmental duplications to drive NAHR. Not surprisingly, almost all cases of multicopy CNPs correspond to regions previously identified as segmental duplications (7). This observation is supported by studies of structural variation in nonhuman primates. Specifically, significant overlap between CNV loci in humans and nonhuman primates has been observed, and these loci are highly enriched for segmental duplications in the compared species (65, 97). It is highly unlikely that these CNVs are ancestral polymorphisms; instead, the enrichment for segmental duplications in these shared CNV loci supports a higher rate of recurrent CNVs in segmental duplication-rich regions. An interesting aspect with respect to human evolution has been the apparent acceleration of duplication in the common ancestor of humans and African great apes (75). Taken together, these studies of segmental duplications demonstrate that these regions of the genome are highly dynamic.
CNPs can overlap genes (18, 51, 56, 101, 106), and CNPs are enriched for coding sequences (21). Genes overlapped by CNPs are enriched for immune and environmental response pathways (18, 21, 101). This enrichment suggests that CNPs are candidates for local, adaptive selection in human populations, which may be indicated by large differences in frequency between populations. Several analyses have identified such CNPs with potential clinical relevance. For example, individuals of African ancestry tend to have more copies of CCL3L1 than individuals of European ancestry (42). CCL3L1 encodes a chemokine ligand, which binds to the coreceptor for HIV. There is controversial evidence for a role of copy number variation of this locus in HIV infection (11, 34, 42, 45, 123). A deletion in APOBEC3B, which functions in innate immunity, is most frequent in East Asian individuals (58). Other population differentiated CNPs overlap UGT2B17 (78), which is involved in steroid metabolism, OCLN (13), which is required for hepatitis C viral entry, and a taste receptor cluster on chromosome 12 (13). Several of these genes are located in segmental duplications, and much, but not all, of the association of CNPs with genes can be accounted for by the enrichment of genes in segmental duplications (21). Furthermore, several genes within segmental duplications show strong evidence of positive selection (55, 99), and these genes are highly copy number variable in humans (4, 118). The importance of CNPs in segmental duplications is underscored by the fact that the most copy number variable genes in the human genome are in segmental duplications (4, 70, 108, 118) and by the finding that CNPs in segmental duplications are more likely to have frequency differences across populations compared with CNPs in unique regions of the genome (13). Therefore, CNPs, especially those within segmental duplications, represent an important class of human genetic diversity with potential implications for human diseases.
METHODOLOGIES FOR ASSESSING COPY NUMBER POLYMORPHISMS
Surveys of CNPs have identified large numbers of these variants; however, in most cases the lists of variants do not show complete overlap (3). Many studies have identified and/or genotyped CNPs using microarray hybridization approaches. Array CGH involves hybridizing a sample of interest and a reference sample, each labeled with a different fluorescent dye, to microarrays of DNA probes covering regions of interest. Differences in hybridization between the two samples can indicate the presence of a copy number difference between the samples. High-density SNP arrays can also be employed for copy number measurement. In these experiments, deviations from the expected intensities of the two alleles across nearby SNPs are suggestive of a copy number change in a specific sample (22, 80). However, SNP microarray platforms tend to have a paucity of probes in duplication-rich regions of the genome and are unable to capture a large number of known CNPs (22, 29, 70). Around half of simple deletion variants are not well captured by even the highest-density SNP arrays, and this number increases when more complex multicopy variants within segmental duplication-rich regions are considered (22). Most SNP microarray and array CGH platforms are designed relative to the human genome reference sequence. However, recent work has led to the identification of insertions of sequences not in the reference genome assembly (56, 68, 128). In fact, many of these novel insertions are polymorphic in human populations and, thus, represent genetic variants that will be missed by using microarrays designed to the reference genome assembly (59). Recently, methods based on massively parallel sequencing data have been used to survey the landscape of copy number variation. CNVs can be identified as regions with an excess or a paucity of mapping sequence reads (3, 7, 136). In addition, paired-end reads that map abnormally compared with the distribution of insert sizes may be indicative of the presence of a CNV (49, 56, 63, 122). When applied to whole-genome sequence data, these approaches have revealed smaller CNVs than microarray-based studies (84) and can accurately assess the copy number of segmental duplications, which are often intractable with microarrays (4, 118). As whole-genome sequencing becomes feasible in large numbers of individuals, these methodologies hold promise for disentangling the role of CNVs in disease.
ASSOCIATION TESTING OF COPY NUMBER POLYMORPHISMS TO DISEASE
In order to test CNPs for association to disease, a diploid copy number is usually assigned to all subjects in the analysis. Many studies have genotyped CNPs based on clear separation hybridization values for individuals with distinct copy number genotypes (18, 22, 80). Integer copy numbers are assigned to each cluster of individuals based on population genetic assumptions (18, 80), SNP allele hybridization (SNP microarrays) (22, 80), or single channel intensity data (array CGH) (13, 59, 96). In order to perform association testing on CNPs with discrete integer copy numbers, the counts of diploid copy numbers can be compared between cases and controls (Figure 5). For CNPs with higher copy numbers, it is currently difficult or impossible to assign integer copy numbers, especially from microarray hybridization data. Such variants are often within segmental duplications and are often excluded from further analysis. Given the potential importance of CNPs in segmental duplications discussed above, it will be important to test these variants for association to disease. One approach to testing such CNPs for association to disease is to compare the distributions of copy numbers in cases and controls to look for enrichment of low or high copy number in the cases (Figure 5). However, this approach is susceptible to false positive results when there are small systematic differences between the processing of cases and controls (10). Read-depth copy number estimates from whole-genome sequence data have shown good concordance with FISH (4, 118). As whole-genome sequencing becomes feasible on large numbers of individuals, it will be possible to test CNPs in segmental duplications for association to disease using these methodologies.
Figure 5.

Methods for associating copy number polymorphisms (CNPs) to disease. (a) For a CNP with discrete copy number genotypes, the counts of each copy number genotype can be compared across cases and controls. (b) For a CNP where discrete copy number genotypes cannot be assigned, the distribution of copy numbers can be compared between cases and controls. (c) CNPs associated to disease can be identified indirectly through the association of a single nucleotide polymorphism in linkage disequilibrium.
In addition to direct measurements of copy number, CNPs have also been assessed using indirect linkage disequilibrium (LD) based approaches. LD occurs when nearby genetic variants are correlated because of human demographic history. LD exists between SNPs and some CNPs (16, 18, 47, 70, 80). After identifying common SNPs associated with disease from genome-wide association studies, one can look for variants that are in LD with the most associated SNPs. Several studies have identified CNPs associated with disease using this approach (18, 79, 115, 129). It is important to consider the properties of CNPs in high LD with SNPs before concluding that imputation of CNP genotypes from SNP data allows for comprehensive association testing of CNPs. Many studies of LD between SNPs and CNPs have focused on simple deletions and duplications (i.e., biallelic), which can be accurately assigned copy number genotypes. The majority of these biallelic CNPs are in high LD with SNPs (13, 18, 80). However, CNPs in segmental duplication-rich regions show less LD to SNPs (13, 70). The reduction of LD of CNPs in segmental duplications is not due to a lower SNP density in duplication-rich regions of the genome (13, 104) but may be explained by the presence of dispersed copies of duplicated CNPs or by recurrent mutation of CNPs in segmental duplications (13, 70, 104). Nevertheless, the lack of LD between CNPs in segmental duplications and SNPs implies that variants will be intractable using imputation-based methods and must be measured directly.
COPY NUMBER POLYMORPHISMS ASSOCIATED WITH HUMAN DISEASE
Numerous CNPs have been associated with complex human diseases (Table 1). Many of these associations are to immune-related phenotypes; however, this may be due to the fact that CNPs are enriched for immune-related genes, and these genes are considered candidates for these phenotypes. CNPs in the human leukocyte antigen (HLA) region have been associated with multiple diseases, including Crohn’s disease, rheumatoid arthritis, and type 1 diabetes (20). SNPs in the HLA region have been strongly associated with many diseases (113) and given the strong LD across this locus, it is not surprising that CNPs in this region also show association to disease. A large number of the CNPs associated with disease are in segmental duplications highlighting the role of these regions in disease, including association of copy number of the β-defensin locus on chromosome 8 with psoriasis and Crohn’s disease (32, 48) and low copy number of FCGR3B associated with lupus (30). Several studies of CNPs in segmental duplications have revealed the difficulty of conducting association studies with these variants. The lower copy number of CCL3L1 has been associated with susceptibility to HIV infection (42), but other studies have failed to replicate this association (34, 123). It is unclear whether this lack of reproducibility is an indication that the original result was spurious or was caused by the difficulty in accurately genotyping CNPs, especially segmental duplications like the CNP containing CCL3L1 (45). Followup analysis has also failed to replicate the association of β-defensin copy number with Crohn’s disease using different copy number genotyping methods (2, 32). Therefore, although CNPs in segmental duplications are likely important for human genetic diversity, accurate genotyping methods are required to test these variants for association to disease.
Table 1.
Selected copy number polymorphisms associated with complex diseases
| Gene | Disease/trait | Variant type | Associated allele | Reference |
|---|---|---|---|---|
| DEFB4, DEFB103, DEFB104 | Psoriasis | Amplificationa,b | High copy number | (48) |
| DEFB4, DEFB103, DEFB104 | Crohn’s disease | Amplificationa,b | Low copy number | (2, 32) |
| CCL3L1 | HIV/AIDS | Amplificationa,b | Low copy number | (34, 42, 45, 123) |
| C4 | Lupus | Amplificationa,b | Low copy number | (135) |
| FCGR3B | Glomerulonephritis in Lupus patients | Amplificationa,b | Low copy number | (1, 30) |
| FCGR3B | Lupus | Amplificationa,b | Low copy number | (30) |
| IRGM | Crohn’s disease | Upstream deletion | Deletion | (79) |
| CFHR1, CFHR3 | Age-related macular degeneration | Deletiona | No deletion | (50, 116) |
| CYP2D6 | Reduced drug metabolism | Deletiona | Deletion | (35) |
| RHD | Rh-negative blood group | Deletiona | Deletion | (15) |
| OPN1LW, OPN1MW | Color blindness | Deletion | Deletion | (27, 90) |
| LPA | Coronary heart disease | Amplificationa,b | Low copy number | (64) |
| SMN2 | Severity of spinal muscular atrophy | Amplificationa,b | Low copy number | (130) |
| AZFc region | Spermatogenetic failure | Deletiona | Deletion | (102) |
| CYP21A2 | Congenital adrenal hyperplasia | Amplificationa,b | Two copies/chrom | (62) |
| UGT2B17 | Osteoporosis | Deletionb | No deletion | (134) |
| UGT2B17 | Graft-versus-host disease | Deletionb | Deletion | (77) |
| LCE3B, LCE3C | Psoriasis | Deletion | Deletion | (23) |
| NEGR1 | Obesity | Upstream deletion | Deletion | (129) |
| NBPF23 | Neuroblastoma | Deletiona,b | Deletion | (26) |
| TSPAN8 | Type 2 diabetes | Amplificationa,b | Low copy number | (20) |
| HLA | Crohn’s disease, rheumatoid arthritis, type 1 diabetes | Multiple CNVsa | Various | (20) |
| GPRC5B | Obesity | Upstream deletion | Deletion | (115) |
Multicopy CNP, more than three diploid copy numbers observed in the population.
CNP is in a segmental duplication.
Interestingly, CNPs can be modifiers of rare disease-causing mutations. For example, spinal muscular atrophy (SMA) can be caused by homozygous deletion of SMN1 (67). SMN1 only differs by eight basepair differences from SMN2, which is a highly identical paralog duplicated in close proximity (67). A silent paralogous sequence variant in SMN2 leads to skipping of exon 7; therefore, the presence of SMN2 cannot completely compensate for the SMN1 deletion in SMA patients (71, 85). However, the higher copy number of SMN2 in SMA patients can reduce the severity of disease. Specifically, individuals with the most severe form of SMA usually die before the age of two and have only one to two copies of SMN2 (12, 31, 73). In contrast, individuals with the mildest form of SMA often have three or more copies of SMN2 and survive into adulthood (12, 31, 73). The relationship between the copy number of SMN1 and SMN2 suggests that CNPs may interact with rare pathogenic variants and lead to phenotypic variability. Because the sequence identity of segmental duplications containing SMN1 and SMN2 is so high, the paralogs cannot be distinguished with microarrays. Thus, microarray hybridization experiments only allow for the determination of the total copy number of SMN genes. However, to know an individual’s SMA status, it is critical to know the sequence of specific paralogous sequence variants that differ between SMN1 and SMN2 (31). Copy number variation of the segmental duplications containing the SMN genes points to the importance of the sequence content of copy number variable regions as well as absolute copy number (Figure 6). In addition, recurrent CNVs can have slightly different breakpoints that cannot be resolved by microarrays but may have important phenotypic consequences (56) (Figure 6). Massively parallel sequencing data has the potential to uncover the effects of sequence content in addition to copy number. Paralogous sequence variants can be cataloged and sequencing read-depth over these positions allows for the determination of paralog-specific copy number (118). Furthermore, there are new methods to obtain breakpoint resolution from sequencing read data (59, 63, 84). Such methods and sequencing data on more individuals will allow for the CNP landscape to be comprehensively tested for association to disease.
Figure 6.

The importance of assessing copy number and sequence content. A simplified diagram of the survival of motor neuron (SMN) locus is depicted based on the reference genome assembly; however, it is known that this region is highly variable and can exist in multiple configurations (103). Copy number variation at this locus is likely mediated by many pairs of paralogous segmental duplications (arrows). Spinal muscular atrophy is caused by homozygous deletion of SMN1. The phenotypic effects of this rare copy number variant are modified by a copy number polymorphism encompassing SMN2, a highly identical paralog of SMN1, which differs from SMN1 by a splice site variant that leads to skipping of exon 7. Different structures of the SMN locus are diagrammed with the expected result of hybridizing individuals with these structures against an individual with four total SMN copies (the reference assembly configuration). Note that array CGH can be used to estimate total copy of the SMN genes, and the sequence content of these copies is critical for phenotypic outcome.
FUTURE DIRECTIONS
CNVs are abundant and impactful but can be difficult to discover and genotype accurately. Their importance in human disease has become increasingly apparent over the past five years. We now appreciate that at least 15% of human neurodevelopmental disease is due to rare and large copy number changes that result in local dosage imbalance for dozens of genes. Other large CNVs, both inherited and de novo, have been implicated in the etiology of autism, schizophrenia, kidney dysfunction, and congenital heart disease. Surprisingly, studies of the general population suggest that although such alleles are rare, collectively they are quite common and under strong purifying selection. These features mean that a significant fraction of the human population carries an unbalanced genome. Such individuals may be sensitized for the effect of another variant that could potentially interact with these CNVs in a digenic manner. The cooccurrence of multiple, rare CNVs has been used to explain the comorbidity and variable expressivity associated with particular variants in cases of severe developmental delay. There is circumstantial evidence that the full complement of both CNVs and SNPs may be important for understanding genetic diseases more broadly (92). It is clear that investigations into the genetic basis of disease without consideration of CNVs will miss an important component of the heritability (74).
Despite numerous advances, the landscape of copy number variation still remains largely unexplored especially for CNVs below the routine detection limit of SNP microarrays (20–30 kbp). Genome sequencing and CNV characterization of a large number of normal genomes will be required in order to interpret potentially pathogenic CNVs. Moreover, the role of multicopy CNVs and the genes therein warrant further exploration. Such variants are the most likely to recurrently mutate, the least likely to be tagged by a flanking SNP, and will vary not only in copy but also in content, as both functional genes and nonfunctional pseudogenes of near-perfect sequence identity are gained and lost. Although next-generation sequencing will open these genes to more accurate genotyping, disease association will depend on a fundamental understanding of the genetic diversity of these regions at the sequence level. If the past decade was a boon for CNV discovery, the next should focus on integrating all classes of genetic variation (SNPs, CNVs, and indels) into a more complete model of human disease.
SUMMARY POINTS.
Rare CNVs contribute significantly to the human neuropsychiatric disease burden.
Several of these rare variants are individually rare but collectively common even in the normal populations but are enriched in individuals with the disease.
Rare variants are under tremendous selective pressure and hence their frequency in the population is contributed by de novo or new mutations.
Considerable phenotypic variability has been uncovered for each of the rare pathogenic variants and can be explained by a two-hit model for complex disease
CNPs are enriched in segmental duplications making these variants challenging to assess; however, CNPs are important contributors to human genetic diversity and disease.
CNPs can contain genes that are enriched for immune and environmental response functions, and several CNPs have been associated to human phenotypes.
In addition to differing in copy number, segmental duplications can differ in copy, sequence content, and structure.
FUTURE ISSUES.
Availability of high quality sequences will allow for the genotyping of not just copy numbers but also sequence content and structure of complex regions of the human genome.
To fully explore the relationship of CNVs with disease, a comprehensive analysis of genetic diversity in these loci from different population of normal individuals is needed.
There is a need for an objective and complete phenotypic assessment of individuals with disease for comprehensive genotype-phenotype correlation studies.
Integration of all genetic variation, including rare and common CNVs as well as single nucleotide variants, is important to study complex disease.
Functional analysis of CNVs using model organisms or cell-culture strategies are the important next step in providing evidence for causality of the identified variants.
Development of novel treatment strategies from CNV studies.
Copy number variant (CNV): an imbalance of genomic sequence (>50 bp) that alters the diploid status of a particular locus
Comparative genomic hybridization (CGH): a method where the DNA from two individuals are labeled with different fluorescent dyes and hybridized to a microarray
Copy number polymorphism (CNP): copy number variant present at greater than one percent frequency in a population and mostly occurring in multiple copy number states
Genomic disorder: rare copy number variants associated with neurodevelopmental disease
Nonallelic homologous recombination (NAHR): unequal crossing over between nonallelic but high identity sequences, such as segmental duplications or Alu repeats, that generates recurrent rearrangements
Segmental duplication: a large block (>10 kbp) of interspersed duplicated sequences with >95% sequence identity constituting five to six percent of the genome
De novo variants: variants observed only in the proband and not transmitted or inherited from the parents
ASD: autism spectrum disorder
ACKNOWLEDGMENTS
This work was supported by NIH HD065285 to E.E.E. and Ruth L. Kirschstein National Research Service Award (NRSA) fellowship (F32HG006070) to CDC. E.E.E. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, et al. 2006. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439:851–55 [DOI] [PubMed] [Google Scholar]
- 2.Aldhous MC, Abu Bakar S, Prescott NJ, Palla R, Soo K, et al. 2010. Measurement methods and accuracy in copy number variation: failure to replicate associations of β-defensin copy number with Crohn’s disease. Hum. Mol. Genet 19:4930–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alkan C, Coe BP, Eichler EE. 2011. Genome structural variation discovery and genotyping. Nat. Rev. Genet 12:363–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, et al. 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet 41:1061–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, et al. 2010. A large, complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat. Genet 42:745–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonacci F, Kidd JM, Marques-Bonet T, Ventura M, Siswara P, et al. 2009. Characterization of six human disease-associated inversion polymorphisms. Hum. Mol. Genet 18:2555–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, et al. 2002. Recent segmental duplications in the human genome. Science 297:1003–7 [DOI] [PubMed] [Google Scholar]
- 8.Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, et al. 2008. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol. Cytogenet 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, et al. 2010. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am. J. Hum. Genet 86:454–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, et al. 2008. A robust statistical method for case-control association testing with copy number variation. Nat. Genet 40:1245–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya T, Stanton J, Kim EY, Kunstman KJ, Phair JP, et al. 2009. Reply to: “CCL3L1 and HIV/AIDS susceptibility.” Nat. Med 15:1112–15 [DOI] [PubMed] [Google Scholar]
- 12.Biros I, Forrest S. 1999. Spinal muscular atrophy: untangling the knot? J. Med. Genet 36:1–8 [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell CD, Sampas N, Tsalenko A, Sudmant PH, Kidd JM, et al. 2011. Population-genetic properties of differentiated human copy-number polymorphisms. Am. J. Hum. Genet 88:317–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clayton-Smith J, Giblin C, Smith RA, Dunn C, Willatt L. 2010. Familial 3q29 microdeletion syndrome providing further evidence of involvement of the 3q29 region in bipolar disorder. Clin. Dysmorphol 19:128–32 [DOI] [PubMed] [Google Scholar]
- 15.Colin Y, Cherif-Zahar B, Le Van Kim C, Raynal V, Van Huffel V, Cartron JP. 1991. Genetic basis of the RhD-positive and RhD-negative blood group polymorphism as determined by Southern analysis. Blood 78:2747–52 [PubMed] [Google Scholar]
- 16.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. 2006. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet 38:75–81 [DOI] [PubMed] [Google Scholar]
- 17.Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, et al. 2010. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet 42:385–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464:704–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium IS. 2008. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455:237–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium TWTCC. 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464:713–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, et al. 2011. A copy number variation morbidity map of developmental delay. Nat. Genet 10.1038/ng.909 [DOI] [PMC free article] [PubMed]
- 21.Cooper GM, Nickerson DA, Eichler EE. 2007. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet 39:S22–29 [DOI] [PubMed] [Google Scholar]
- 22.Cooper GM, Zerr T, Kidd JM, Eichler EE, Nickerson DA. 2008. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat. Genet 40:1199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Cid R, Riveira-Munoz E, Zeeuwen PL, Robarge J, Liao W, et al. 2009. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet 41:211–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, et al. 2005. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet 77:606–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Digilio MC, Bernardini L, Mingarelli R, Capolino R, Capalbo A, et al. 2009. 3q29 microdeletion: a mental retardation disorder unassociated with a recognizable phenotype in two mother-daughter pairs. Am. J. Med. Genet. A 149A:1777–81 [DOI] [PubMed] [Google Scholar]
- 26.Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, et al. 2009. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459:987–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drummond-Borg M, Deeb SS, Motulsky AG. 1989. Molecular patterns of X chromosome-linked color vision genes among 134 men of European ancestry. Proc. Natl. Acad. Sci. USA 86:983–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elia J, Gai X, Xie HM, Perin JC, Geiger E, et al. 2010. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 15:637–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Elsea SH, Girirajan S. 2008. Smith-Magenis syndrome. Eur. J. Hum. Genet 16(4):412–21 [DOI] [PubMed] [Google Scholar]
- 29.Estivill X, Cheung J, Pujana MA, Nakabayashi K, Scherer SW, Tsui LC. 2002. Chromosomal regions containing high-density and ambiguously mapped putative single nucleotide polymorphisms (SNPs) correlate with segmental duplications in the human genome. Hum. Mol. Genet 11:1987–95 [DOI] [PubMed] [Google Scholar]
- 30.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, et al. 2007. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet 39:721–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. 2002. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet 70:358–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, et al. 2006. A chromosome 8 gene-cluster polymorphism with low human β-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet 79:439–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feuk L 2010. Inversion variants in the human genome: role in disease and genome architecture. Genome Med 2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Field SF, Howson JM, Maier LM, Walker S, Walker NM, et al. 2009. Experimental aspects of copy number variant assays at CCL3L1. Nat. Med 15:1115–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaedigk A, Blum M, Gaedigk R, Eichelbaum M, Meyer UA. 1991. Deletion of the entire cytochrome P450 CYP2D6 gene as a cause of impaired drug metabolism in poor metabolizers of the debrisoquine/sparteine polymorphism. Am. J. Hum. Genet 48:943–50 [PMC free article] [PubMed] [Google Scholar]
- 36.Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, et al. 2001. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am. J. Hum. Genet 68:874–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, et al. 2003. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum. Mol. Genet 12:849–58 [DOI] [PubMed] [Google Scholar]
- 38.Girirajan S, Eichler EE. 2010. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet 19:R176–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, et al. 2010. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet 42:203–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39a.Girirajan S, Vlangos CN, Szomju BB, Edelman E, Trevors CD, et al. 2006. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet. Med 8(7):417–27 [DOI] [PubMed] [Google Scholar]
- 40.Glessner JT, Reilly MP, Kim CE, Takahashi N, Albano A, et al. 2010. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA 107:10584–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. 2009. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459:569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, et al. 2005. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–40 [DOI] [PubMed] [Google Scholar]
- 43.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, et al. 2009. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet 41:931–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009. Mechanisms of change in gene copy number. Nat. Rev. Genet 10:551–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He W, Kulkarni H, Castiblanco J, Shimizu C, Aluyen U, et al. 2009. Reply to: “Experimental aspects of copy number variant assays at CCL3L1.” Nat. Med 15:1117–20 [DOI] [PubMed] [Google Scholar]
- 46.Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, et al. 2009. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet 41:160–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. 2006. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet 38:82–85 [DOI] [PubMed] [Google Scholar]
- 48.Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, et al. 2008. Psoriasis is associated with increased β-defensin genomic copy number. Nat. Genet 40:23–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. 2009. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res 19:1270–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. 2006. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet 38:1173–77 [DOI] [PubMed] [Google Scholar]
- 51.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, et al. 2004. Detection of large-scale variation in the human genome. Nat. Genet 36:949–51 [DOI] [PubMed] [Google Scholar]
- 52.Ikeda M, Aleksic B, Kirov G, Kinoshita Y, Yamanouchi Y, et al. 2010. Copy number variation in schizophrenia in the Japanese population. Biol. Psychiatry 67:283–86 [DOI] [PubMed] [Google Scholar]
- 53.Itsara A, Cooper GM, Baker C, Girirajan S, Li J, et al. 2009. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet 84:148–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, et al. 2010. De novo rates and selection of large copy number variation. Genome Res 20:1469–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, et al. 2001. Positive selection of a gene family during the emergence of humans and African apes. Nature 413:514–19 [DOI] [PubMed] [Google Scholar]
- 56.Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453:56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, et al. 2010. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143:837–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. 2007. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet 3:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, et al. 2010. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods 7:365–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kirov G, Rujescu D, Ingason A, Collier DA, O’Donovan MC, Owen MJ. 2009. Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr. Bull 35:851–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, et al. 2006. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet 38:999–1001 [DOI] [PubMed] [Google Scholar]
- 62.Koppens PF, Hoogenboezem T, Degenhart HJ. 2002. Duplication of the CYP21A2 gene complicates mutation analysis of steroid 21-hydroxylase deficiency: characteristics of three unusual haplotypes. Hum. Genet 111:405–10 [DOI] [PubMed] [Google Scholar]
- 63.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318:420–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kraft HG, Lingenhel A, Kochl S, Hoppichler F, Kronenberg F, et al. 1996. Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol 16:713–19 [DOI] [PubMed] [Google Scholar]
- 65.Lee AS, Gutierrez-Arcelus M, Perry GH, Vallender EJ, Johnson WE, et al. 2008. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum. Mol. Genet 17:1127–36 [DOI] [PubMed] [Google Scholar]
- 66.Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131:1235–47 [DOI] [PubMed] [Google Scholar]
- 67.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, et al. 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–65 [DOI] [PubMed] [Google Scholar]
- 68.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, et al. 2001. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum. Genet 109:535–41 [DOI] [PubMed] [Google Scholar]
- 70.Locke DP, Sharp AJ, McCarroll SA, McGrath SD, Newman TL, et al. 2006. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet 79:275–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lorson CL, Hahnen E, Androphy EJ, Wirth B. 1999. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 96:6307–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lupski JR. 2007. Genomic rearrangements and sporadic disease. Nat. Genet 39:S43–47 [DOI] [PubMed] [Google Scholar]
- 73.Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, et al. 2002. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med 4:20–26 [DOI] [PubMed] [Google Scholar]
- 74.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. 2009. Finding the missing heritability of complex diseases. Nature 461:747–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, et al. 2009. A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457:877–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, et al. 2008. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet 82:477–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCarroll SA, Bradner JE, Turpeinen H, Volin L, Martin PJ, et al. 2009. Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nat. Genet 41:1341–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, et al. 2006. Common deletion polymorphisms in the human genome. Nat. Genet 38:86–92 [DOI] [PubMed] [Google Scholar]
- 79.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, et al. 2008. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet 40:1107–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, et al. 2008. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet 40:1166–74 [DOI] [PubMed] [Google Scholar]
- 81.Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, et al. 2007. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet 81:1057–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, et al. 2008. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med 359:1685–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, et al. 2010. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet 86:749–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, et al. 1999. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet 8:1177–83 [DOI] [PubMed] [Google Scholar]
- 86.Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, et al. 2010. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet 87:618–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, et al. 2008. Identifying autism loci and genes by tracing recent shared ancestry. Science 321:218–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mulle JG, Dodd AF, McGrath JA, Wolyniec PS, Mitchell AA, et al. 2010. Microdeletions of 3q29 confer high risk for schizophrenia. Am. J. Hum. Genet 87:229–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagamani SC, Erez A, Shen J, Li C, Roeder E, et al. 2010. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet 18:278–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. 1986. Molecular genetics of inherited variation in human color vision. Science 232:203–10 [DOI] [PubMed] [Google Scholar]
- 91.Need AC, Ge D, Weale ME, Maia J, Feng S, et al. 2009. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet 5:e1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, et al. 2011. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet 585–89 [DOI] [PMC free article] [PubMed]
- 93.Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, et al. 2001. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat. Genet 29:321–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, et al. 2006. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet 38:1114–23 [DOI] [PubMed] [Google Scholar]
- 95.Pagnamenta AT, Wing K, Sadighi Akha E, Knight SJ, Bolte S, et al. 2009. A 15q13.3 microdeletion segregating with autism. Eur. J. Hum. Genet 17:687–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perry GH, Ben-Dor A, Tsalenko A, Sampas N, Rodriguez-Revenga L, et al. 2008. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet 82:685–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, et al. 2006. Hotspots for copy number variation in chimpanzees and humans. Proc. Natl. Acad. Sci. USA 103:8006–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, et al. 2010. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466:368–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Popesco MC, Maclaren EJ, Hopkins J, Dumas L, Cox M, et al. 2006. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science 313:1304–7 [DOI] [PubMed] [Google Scholar]
- 100.Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, et al. 2007. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet 80:633–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, et al. 2006. Global variation in copy number in the human genome. Nature 444:444–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Repping S, Skaletsky H, Brown L, van Daalen SK, Korver CM, et al. 2003. Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat. Genet 35:247–51 [DOI] [PubMed] [Google Scholar]
- 103.Schmutz J, Martin J, Terry A, Couronne O, Grimwood J, et al. 2004. The DNA sequence and comparative analysis of human chromosome 5. Nature 431:268–74 [DOI] [PubMed] [Google Scholar]
- 104.Schrider DR, Hahn MW. 2010. Lower linkage disequilibrium at CNVs is due to both recurrent mutation and transposing duplications. Mol. Biol. Evol 27:103–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. 2007. Strong association of de novo copy number mutations with autism. Science 316:445–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, et al. 2004. Large-scale copy number polymorphism in the human genome. Science 305:525–28 [DOI] [PubMed] [Google Scholar]
- 107.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, et al. 2006. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet 38:1038–42 [DOI] [PubMed] [Google Scholar]
- 108.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, et al. 2005. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet 77:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, et al. 2008. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet 40:322–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, et al. 2007. Characterization of a recurrent 15q24 microdeletion syndrome. Hum. Mol. Genet 16:567–72 [DOI] [PubMed] [Google Scholar]
- 111.Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, et al. 2006. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet 38:1032–37 [DOI] [PubMed] [Google Scholar]
- 112.Shi YY, He G, Zhang Z, Tang W, Zhang J Jr., et al. 2008. A study of rare structural variants in schizophrenia patients and normal controls from Chinese Han population. Mol. Psychiatry 13:911–13 [DOI] [PubMed] [Google Scholar]
- 113.Shiina T, Inoko H, Kulski JK. 2004. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 64:631–49 [DOI] [PubMed] [Google Scholar]
- 114.Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, et al. 2009. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat. Genet 41:1269–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, et al. 2010. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet 42:937–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spencer KL, Hauser MA, Olson LM, Schmidt S, Scott WK, et al. 2008. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum. Mol. Genet 17:971–77 [DOI] [PubMed] [Google Scholar]
- 117.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, et al. 2008. Large recurrent microdeletions associated with schizophrenia. Nature 455:232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, et al. 2010. Diversity of human copy number variation and multicopy genes. Science 330:641–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, et al. 2010. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum. Mutat 31:840–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, et al. 2007. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet 39:319–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, et al. 2008. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet 40:90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, et al. 2005. Fine-scale structural variation of the human genome. Nat. Genet 37:727–32 [DOI] [PubMed] [Google Scholar]
- 123.Urban TJ, Weintrob AC, Fellay J, Colombo S, Shianna KV, et al. 2009. CCL3L1 and HIV/AIDS susceptibility. Nat. Med 15:1110–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, et al. 2011. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, et al. 2009. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J. Med. Genet 46:511–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vrijenhoek T, Buizer-Voskamp JE, van der Stelt I, Strengman E, Sabatti C, et al. 2008. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet 83:504–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, et al. 2008. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320:539–43 [DOI] [PubMed] [Google Scholar]
- 128.Wang J, Wang W, Li R, Li Y, Tian G, et al. 2008. The diploid genome sequence of an Asian individual. Nature 456:60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, et al. 2009. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet 41:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, et al. 1999. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet 64:1340–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, et al. 2007. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet 80:91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M. 2008. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet 40:880–85 [DOI] [PubMed] [Google Scholar]
- 133.Xu B, Woodroffe A, Rodriguez-Murillo L, Roos JL, van Rensburg EJ, et al. 2009. Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc. Natl. Acad. Sci. USA 106:16746–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang TL, Chen XD, Guo Y, Lei SF, Wang JT, et al. 2008. Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am. J. Hum. Genet 83:663–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, et al. 2007. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet 80:1037–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. 2009. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 19:1586–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. 2009. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet 41:849–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, et al. 2007. A unified genetic theory for sporadic and inherited autism. Proc. Natl. Acad. Sci. USA 104:12831–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, et al. 2008. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet 40:1076–83 [DOI] [PMC free article] [PubMed] [Google Scholar]