

SUMMARY
Group II introns are a class of retroelements that invade DNA through a copy-and-paste mechanism known as retrotransposition. Their coordinated activities occur within a complex that includes a maturase protein, which promotes splicing through an unknown mechanism. The mechanism of splice site exchange within the RNA active site during catalysis also remains unclear. We determined two cryo-EM structures at 3.6 Å resolution of a group II intron reverse splicing into DNA. These structures reveal that the branch-site domain VI helix swings 90°, enabling substrate exchange during DNA integration. The maturase assists catalysis through a transient RNA-protein contact with domain VI that positions the branch-site adenosine for lariat formation during forward splicing. These findings provide the first direct evidence for the role the maturase plays during group II intron catalysis. The domain VI dynamics closely parallel spliceosomal branch-site helix movement and provide strong evidence for a retroelement origin of the spliceosome.
In Brief
The structural basis for group II intron retrotransposition into DNA reveals mechanistic similarities with the spliceosome, supporting a shared evolutionary origin of these two processes
Graphical Abstract
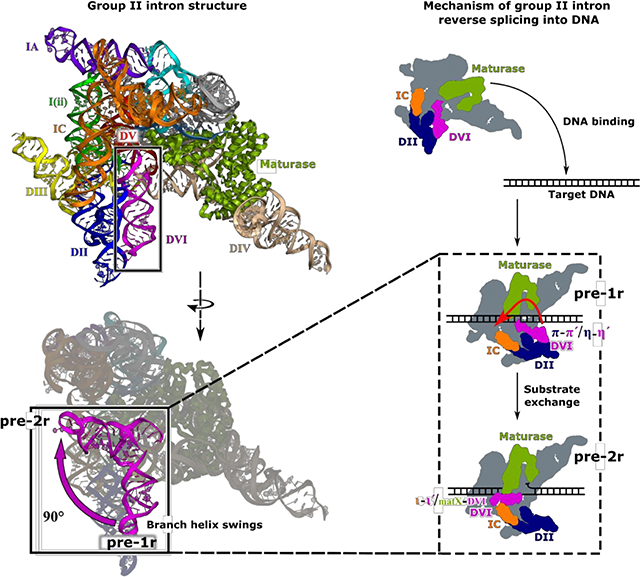
INTRODUCTION
Retroelements are selfish genetic elements that use an RNA intermediate to replicate within genomes through a copy-and-paste mechanism known as retrotransposition (Zimmerly et al., 1995a; Zimmerly et al., 1995b). Retroelements have successfully colonized eukaryotic DNA and now comprise ~45% of mammalian genomes (Deininger and Batzer, 2002). Retrotransposition occurs at high frequency in germ cells and in some somatic tissues, including the brain (Muotri et al., 2005). New retrotransposition events can lead to gene disruption and disease (Deininger and Batzer, 2002), such as the formation of cancer through disruption of tumor suppressor genes. Retroelements also have global effects on gene expression through insertion directly upstream of transcription promoters (Deininger and Batzer, 2002). As a result, they are thought to have had a major impact on the evolution and speciation of primates and other mammals (Ricci et al., 2018).
Group II introns are a class of retroelements that are hypothesized to be ancestral to the spliceosome and mammalian LINE elements (Eickbush, 1999). Group II introns have retained the capabilities of both systems; i.e. they can function as catalytic RNAs (ribozymes) through self-splicing reactions (Lambowitz and Zimmerly, 2011) and act as retroelements through insertion into double-stranded DNA (dsDNA). To function as a retroelement, a group II intron must first undergo the forward splicing reaction and excise itself from pre-messenger RNA (pre-mRNA) containing exons via a two-metal-ion mechanism (Toor et al., 2008). In this initial splicing reaction, a protein known as the maturase binds to the intron RNA and promotes its excision and exon ligation through formation of the 2′−5′ phosphodiester bond known as the lariat (Figure 1) (Wank et al., 1999). The excised intron lariat and its bound maturase protein constitute a retroelement complex that is able to engage in retrotransposition. The maturase is a multi-functional protein that contains reverse transcriptase (RT) and endonuclease (En) domains (Zimmerly et al., 2001). Group II intron retroelements are able to replicate through integration into dsDNA in a process known as target-primed reverse transcription (TPRT), in which both the RT and En domains of the maturase are required (Zimmerly et al., 1995b) (Figure S1). In this process, the group II intron retroelement complex first binds to the target DNA and partially melts the DNA duplex through both RNA and protein mediated interactions. Partial disruption of the DNA double helix exposes the intron binding sequences (IBS 1 to 3) found within the DNA and allows for the formation of Watson-Crick pairing interactions with the exon-binding sequences (EBS 1 to 3) within the intron RNA. These pairing interactions confer sequence specificity that determines the site of DNA integration. Once bound, the RNA reverse splices into the sense (top) strand of the DNA (Zimmerly et al., 1995a) via two sequential transesterification reactions (Figure 1). These reactions represent the reversal of the forward splicing reaction (Roitzsch et al., 2010); thus in the first step of reverse splicing, the 3′-OH of the intron RNA attacks the scissile phosphate of the top strand of a DNA target (Figure 1). This results in covalent attachment of the 3′ end of the RNA to the 3′ DNA. For the second transesterification step to occur, the first step products must be removed from the active site, followed by their replacement with the second step substrates through an unknown mechanism. Once this exchange is complete the second step can begin, with the newly created 3′-OH at the site of DNA cleavage engaging in nucleophilic attack upon the 2′−5′ phosphodiester bond of the lariat (Figure 1). Upon cleavage of the lariat bond, the intron is fully reverse spliced with the 5′ and 3′ ends of the RNA being connected to the DNA. The En domain of the maturase then cuts the antisense (bottom) strand of the DNA to create a free 3′-OH that is used as a primer for cDNA synthesis by the RT domain. Following the completion of cDNA synthesis, host DNA repair systems heal the nick in the bottom strand and replace the RNA in the top strand with DNA. The end result of this process is that a new DNA copy of the group II intron has been integrated into the genome via an RNA intermediate.
Figure 1.

Group II intron RNP complex formation and the mechanism of reverse splicing. The intron RNA first binds to the maturase protein and undergoes forward splicing, which consists of two sequential transesterification reactions. This results in exicision of the group II intron RNA from the flanking exons in the pre-mRNA. Upon the completion of forward splicing, the group II intron is competent to engage in reverse splicing reactions into a dsDNA target (sequence shown with intron binding sequences (IBS) labeled). The first stage of retrotransposition is reverse splicing of the intron RNA into the top strand of DNA.
The RNA component of a group II intron is a ribozyme that has a conserved secondary structure with six domains (Lambowitz and Zimmerly, 2011) (Figure S2). Domain I (DI) contains the EBS1 to EBS3 (Costa et al., 2000; Jacquier and Michel, 1987) used to target the group II intron to a specific DNA sequence. Domain IV (DIV) contains the open reading frame (ORF) for the maturase protein and provides the main binding site for the RT domain to form the fully assembled group II intron ribonucleoprotein (RNP) complex (Wank et al., 1999). Domain V (DV) harbors the active site of the ribozyme (Boulanger et al., 1995; Schmidt et al., 1996) and coordinates two catalytic magnesium ions (M1 and M2) in a binding pocket composed of the phosphate backbones of a conserved two-nucleotide (nt) bulge and an AGC catalytic triad (Toor et al., 2008). DV is homologous to the U2/U6 snRNA pairing that forms the active site of the spliceosome (Bertram et al., 2017; Galej et al., 2016; Hang et al., 2015). There is structural homology in the active site architecture of both complexes and they share an identical catalytic mechanism (Fica et al., 2013). In addition, the spliceosome contains the highly conserved Prp8 protein, which is an inactive RT that assists in active site formation (Galej et al., 2016; Hang et al., 2015). Prp8 also exhibits structural and sequence homology to the group II intron maturase protein (Galej et al., 2013). The 3′ end of the group II intron RNA encodes domain VI (DVI), which contains a bulged adenosine (A860) that forms the 2′−5′ lariat bond (Lambowitz and Zimmerly, 2011). This lariat bond is required for reverse splicing into a DNA substrate (Roitzsch et al., 2010) during the initial stages of retrotransposition. DVI exhibits conservation with the branch-site helix of the spliceosome that is responsible for forming the lariat during RNA splicing (Robart et al., 2014).
The current work addresses the following three questions pertaining to group II intron biology: 1) What is the mechanism of splice site substrate exchange between the two steps of RNA splicing? 2) How does the associated maturase protein promote forward splicing through binding to the intron RNA? 3) What is the molecular mechanism of reverse splicing into dsDNA? To address these questions, we determined structures of a thermostable group II intron from Thermosynechococcus elongatus (T.el.) (Mohr et al., 2010) in the process of DNA strand invasion. This group II intron exhibits high in vitro retrotransposition activity when incubated with a dsDNA target (Figure S3A). In addition, the thermostability of this complex makes it more amenable to high-resolution cryo-EM structure determination. The resulting structures reveal large-scale conformational rearrangements that provide detailed insight into the mechanism of retrotransposition and RNA splicing. These dynamics also have strong parallels with the spliceosome.
RESULTS
Purification of a Group II Intron in Complex with a Target dsDNA
The reconstituted group II intron-maturase complex was incubated with desthiobiotin-labeled dsDNA containing the target IBS required for insertion. The RNA reverse splices into the top strand of the DNA to yield a mixture of retrotransposition intermediates. These intermediates correspond to the stage immediately prior to (pre-1r) and succeeding (pre-2r) the first step of reverse splicing, as well as fully reverse spliced intron RNA (post-2r) (Figure S3A). This mixture of reaction states is consistent with biochemical evidence showing that group II intron catalysis is reversible (Chin and Pyle, 1995). In the specific case of the T.el4h group II intron complex, this reversibility results in an equilibrium between the forward and reverse reactions. The complex was subsequently purified using an avidin column (Figure S3B). The purified sample was vitrified on cryo-EM grids for structure determination (Figure S4). Only the pre-1r and pre-2r states resulted in high-resolution 3D reconstructions. The additional 3D classes indicated disordered density in the active site as well as for DVI and thus were not investigated further. The pre-1r complex is bound to the DNA and in a state immediately prior to the first step of reverse splicing. In this structure, only the top strand of DNA is observed bound to the group II intron as the antisense strand is disordered after melting of the duplex. In the pre-2r complex, the target DNA has been cleaved and is covalently attached to the 3′ end of the intron RNA. The post-2r complex, which is observed biochemically in the sample (Figure S3A), was not identified as any of the 3D classes. It is likely that the post-2r complex was captured in one of the 3D classes that only yielded a low-resolution 3D reconstruction.
Cryo-EM Structures of Two States of the Group II Intron-Maturase Complex
Here we present the cryo-EM structures, each at 3.6 Å global resolution, of the T.el4h group II intron at two different stages of integration into a dsDNA target (Figure 2 and Table S1). The resolution of both maps approaches 3.0 Å for the large central portion of the complex (Figure S4D and Movie S1). The 2D class averages and resulting 3D reconstructions indicated orientation bias so a tilt specimen data collection strategy (Tan et al., 2017) was required to obtain more uniform and higher resolution density to allow modeling of individual nucleotides. In both states, the active sites of the RT domain and the group II intron ribozyme face a common DNA-binding cavity that is consistent with their interdependent roles during retrotransposition. In the pre-1r state, the intron RNA is Watson-Crick paired to the top strand of the DNA through the EBS-IBS interactions. The RT thumb/X domain of the maturase protein is also tightly bound to the RNA/DNA duplex. The 3′-OH of the RNA is coordinated to the catalytic M1 metal ion and is positioned directly above the scissile phosphate of the DNA, poised to undergo the first step of reverse splicing (Figure 3A). In the pre-2r state, the 3′ end of the RNA has been removed from the active site and is covalently attached to the 3′ DNA (Figure 3B). In this state, the lariat bond between U1 and A860 is positioned in the active site in preparation for the second step of reverse splicing. A broader inspection of the pre-1r and pre-2r structures reveals a large 90° swinging movement of the branch-site helix DVI (Figure 2 and Movie S2), which contains the bulged adenosine/lariat bond.
Figure 2.

Overall structure of the T.el4h group II intron. (A) A segmented low-pass filtered map of the group II intron in the pre-1r state is shown with DVI (red), the maturase protein (yellow), the target DNA (purple), and rest of the RNA (blue) highlighted. The structural model of T.el4h in the pre-1r state is also shown. Domains II-VI of the RNA are labeled DII to DVI and the maturase is labeled RT. DVI (boxed) adopts a downward conformation before DNA integration. RNA secondary structure diagrams show only the core domains of the pre-1r and pre-2r states. These diagrams approximate the domain organization evident from the cryo-EM structures. Intron RNA domains are labeled with Roman numerals and important tertiary interactions are indicated by Greek letters. (B) Similar segmented low-pass filtered map of the group II intron retroelement complex in the pre-2r state. The structural model indicates a 90° swinging of DVI.
Figure 3.

Active site architecture of the group II intron in the pre-1r and pre-2r states. (A) In the pre-1r state, the 3′-OH of C866 is positioned directly above the scissile phosphate (yellow) of the sense strand of DNA and is poised to initiate the first step of reverse splicing. C866 is held in place through hydrogen bonding between several conserved nucleotides, including those involved in the γ-γ′ interaction and the bulged adenosine (A860) pairing with G864. This structure represents the architecture of the active site immediately before the first step of reverse splicing. Density corresponding to the pre-1r state is shown to indicate the fit of the model to the cryo-EM data. (B) In the pre-2r state, the 3′ DNA nucleotide is covalently attached to C866 of the RNA. Between the pre-1r and pre-2r states, DVI swings 90° to remove the 3′ splice site from the active site. In addition, the dynamics of DVI help place the lariat bond in the active site for the second step of reverse splicing. An additional highly coordinated metal ion (M5) is bound to the two-nt bulge in the pre-2r state. This metal ion likely helps to stabilize the two-nt bulge in this alternate conformation. Density for the 3′ DNA nucleotide is only visible at a lower contour level. (C) Metal ion coordination of catalytic M1 in both the pre-1r and pre-2r states. M1 loses coordinating RNA ligands as reverse splicing proceeds.
Active Site Architecture for Reverse Splicing
In the pre-1r state, the scissile phosphate of the DNA is in the active site within DV and in close proximity to the 3′-OH nucleophile from the last nucleotide (C866) of the RNA. C866 is held in position for nucleophilic attack upon the scissile phosphate of the DNA target through Watson-Crick pairing with G479 to form the γ-γ′ interaction (Jacquier and Michel, 1990) (Figure 3A). At this stage of reverse splicing, strong density is only observed for the M1 catalytic metal ion, with M2 not being visible (Figure 3A). In transitioning to the pre-2r state, the target DNA is cleaved and the 3′ end of the RNA is covalently attached to the 3′ DNA at the site of DNA cleavage through a transesterification mechanism (Figure 2B and3B). Upon the completion of DNA cleavage and covalent attachment of the RNA, the metal ion M1 loses two out of five coordinating ligands (Figure 3C). The coordination between M1 and the scissile phosphate is lost due to stereochemical inversion during the transesterification reaction (Padgett et al., 1994). In addition, the 3′-OH of C866 now forms a phosphodiester bond with the DNA at the site of cleavage and therefore is unavailable to coordinate M1. The loss of these interactions with M1 is associated with an expansion of the catalytic metal ion binding pocket as well as a conformational rearrangement of the conserved two-nt bulge of DV (Figure S5).
The dynamic nature of the two-nt bulge has a significant impact upon the overall architecture of DV. The distal tetraloop of DV is anchored by the ζ-ζ′ interaction (Costa and Michel, 1995), which remains static between the two states (Figure S5). With ζ -ζ′ being fixed in space, the basal region of DV containing the RNA active site undergoes a 4.3 Å downward shift in the transition from the pre-1r to the pre-2r state (Movie S3). This overall expansion of DV exceeds the one base-pair register of 3.4 Å. The M1 catalytic metal ion undergoes a larger translation of 8 Å downwards as the basal stem of DV expands. To accommodate these shifts, several key, long-range interactions involving DV either rearrange or disengage. For example, DV is positioned within the scaffold of DI via minor groove interactions with two tetraloops to form the κ-κ′ (Boudvillain and Pyle, 1998) and μ-μ′ (Fedorova and Pyle, 2008) contacts. These tetraloops slide within the minor groove of DV during its expansion, shifting the corresponding receptor helix by one base-pair register (Movie S3). Therefore, the structural constraints imposed by κ-κ′ and μ-μ′ serve to guide the dynamics of DV. Our structures provide the first direct evidence of major conformational dynamics in the overall topology of active site DV.
Maturase Protein Promotes Substrate Exchange in the Ribozyme Active Site
A detailed comparison of the two structures reveals significant alterations in tertiary contacts that are associated with catalysis. The function of these conformational dynamics is to facilitate movement of the lariat bond between different stages of reverse splicing. Prior to DNA integration, the lariat bond is positioned ~20 Å away from the active site and DVI is engaged via two tetraloop-receptor interactions known as π-π′ and η-η′ (Robart et al., 2014) (Figure 2A and4A). After the initial covalent attachment of the RNA to the DNA, the branch-site helix DVI swings 90° (Figure 2B and4B), thus placing the lariat bond in the active site for the second step of reverse splicing (Figure 3C). In the resulting pre-2r structure in which π-π′ and η-η′ are disrupted, DVI is held in a horizontal position through interactions with a basic patch located in the maturase thumb/X domain (matX-DVI interaction), and the ι-ι′ interaction (Li et al., 2011) located within the IC stem of DI (Figure 2B and4C).
Figure 4.

Dynamic tertiary interactions hold DVI in two conformations. (A) In the pre-1r state, DVI (magenta) is held in place through two tetraloop-receptor interactions with DII (blue) named π-π′ and η-η′. (B) Superposition of DVI in both the pre-1r (magenta) and pre-2r (pink) states. DVI swings approximately 90° in the transition between stages of reverse splicing. (C) In the pre-2r state, DVI is held in a horizontal conformation through interactions with a basic cleft of the RT thumb/X domain and ι-ι′. Several conserved basic and polar residues reside in the thumb/X domain of the RT. They form a positively charged surface that captures DVI as it swings upward. A detailed view of ι-ι′ is shown to highlight the hydrogen bonds (dashed lines) that contribute to the interaction. This unusual interaction occurs between the minor grooves of DVI and the IC stem from DI.
To probe the importance of the matX-DVI contact, amino acid residues lining this RNA-protein interface were mutated and tested using a forward splicing assay. A forward splicing assay, rather than a reverse splicing assay, is necessary because the intron RNA must first splice in the presence of the maturase protein before it is competent to engage in subsequent reverse splicing reactions with DNA. Any mutations that completely inhibit forward splicing will not yield retroelement complexes competent for retrotransposition. Therefore, it is not possible to utilize reverse splicing as an assay to test the effects of these mutations in the protein. Three different maturase mutants (RT-1, RT-2, and RT-3) were tested for forward splicing activity. The protein has two patches (patches 1 and 2) of four amino acid residues each that comprise the interface that contacts the DVI helix. RT-1 (R357E / T358G / R405E / R406E) and RT-2 (T360G / H407D / K410D / K418D) have mutated surfaces corresponding to each patch, while RT-3 contains all the mutations from RT-1 and RT-2. A comparison of the forward splicing assays for WT and the RT mutants shows that mutagenesis of this region in the protein results in no visible formation of spliced lariat (Figure S6A). To verify that these mutations did not disrupt the overall structural fold of the maturase protein, these mutants were tested for both RT activity and RNA binding (Figure S6B, C, and D). Of the three mutants, only RT-1 retained intron RNA binding activity and was able to produce cDNA, as evidenced by a primer extension assay. Therefore, the RT-1 mutation retains the overall fold of the protein structure, while the RT-2 and RT-3 mutations severely disrupt the structure, abrogating RNA binding and cDNA synthesis. These data suggest the RT-1 mutation is specifically disrupting the matX-DVI interaction. This indicates that the matX-DVI interaction is essential for positioning the branched adenosine for nucleophilic attack on the 5′ splice site in the first step of forward splicing, as well as holding the lariat bond in the active site for the second step of reverse splicing. This provides biochemical evidence for a dynamic role for DVI during catalysis. In contrast to the severe effects observed in the RT-1 mutant, disruption of the ι-ι′ interaction does not preclude lariat formation (Figure S7), suggesting that the RNA-protein contact is more important for positioning DVI in this conformation.
Comparison to Previously Determined Crystal Structures
Previous crystal structures of the RNA component of group IIB and IIC introns (Robart et al., 2014; Toor et al., 2008) did not show DV or DVI conformational dynamics; however, there were hints of dynamics within these previous structures. For example, the pre-catalytic state of the IIB intron lacked density for the basal stem of DVI, indicating conformational heterogeneity within the crystal lattice (Robart et al., 2014). In addition, a base flipping event was seen during in crystallo hydrolytic splicing of the IIC intron that suggested DV was engaged in structural changes associated with catalysis (Marcia and Pyle, 2012). It is also likely that the tightly packed nature of the crystal lattice prevents the RNA from sampling the large-scale conformational rearrangements observed in our study. The swinging of DVI into the pre-2r state would likely not be accommodated within the confines of a crystal lattice.
A previously reported crystal structure suggested a shifting of base pairs surrounding the bulged adenosine in DVI during reverse splicing (Costa et al., 2016). We see no evidence of such a conformational change in our cryo-EM structures. The specimen used for structure determination by Costa et al. (2016) did not contain the maturase protein and was an artificial, chimeric hybrid of two different species of introns. Specifically, DVI from a different group II intron was attached to another IIC intron. This construct design is likely to have a negative effect upon the capture of large-scale conformational changes due to the fact that DVI coevolves with the rest of the intron RNA to form tertiary interactions (Toor et al., 2001). For example, tetraloop receptor interactions have defined partner sequences for forming contacts (Fiore and Nesbitt, 2013), which may not be preserved when artificially joining two species. As a result, this could preclude the visualization of the tertiary interactions necessary for DVI dynamics. The lack of the maturase could also be problematic as it is essential for stabilizing DVI in an alternative conformation as observed in our study.
DISCUSSION
DV and DVI Dynamics Associated with Substrate Exchange
Group II intron splicing requires two sequential transesterification reactions to be catalyzed in a single active site. The mechanism of substrate exchange between the two steps of group II intron splicing was previously unknown. We hypothesize that the observed 90° movement of DVI is initiated by conformational rearrangements that propagate outward from the active site in DV, which would couple catalysis with substrate exchange. Significant differences can be seen in the binding pocket for the two catalytic metal ions at each stage of reverse splicing. Specifically, the attachment of the 3′ end of the RNA to the DNA target is associated with an expansion of the metal ion binding pocket, which concurrently results in a lengthwise expansion of DV during the integration process (Figure S5 and Movie S3). This expansion has the potential to pull on the directly attached junction between DV and DVI (J5/6), thereby causing the adjacent DVI helix to detach from DII and swing into the alternate conformation.
The immediate consequence of the downward movement of the DV basal stem is a shift in the attached J5/6 linker sequence separating DV and DVI. In the pre-1r state, U826 and A827 from J5/6 insert into the minor groove at the base of DIV to form a newly identified tertiary interaction, which we have labeled φ-φ′ (Figure 5). The downward motion of DV is associated with the disengagement of φ-φ′ and the disruption of π-π′ and η-η′ in DVI. We observe another interaction in the pre-1r state, ψ-ψ′, which consists of a base triple between A401 from the conserved I(i) loop in DI and the U799-G816 base pair that is located directly underneath the two-nt bulge of DV (Figure 5). Disruption of this base triple through an A401G mutation completely inhibits lariat formation, highlighting the importance of this interaction for proper active site formation (Figure S7). The downward shift of DV occurs simultaneously with the disengagement of the ψ-ψ′ interaction, which lead to a rearrangement of the I(i) loop. This motion is transmitted to the proximal 5′ end because of its covalent attachment to DVI via the lariat bond. Therefore, DVI is pulled by two different regions – namely J5/6 and the 5′ end – which could favor the transition into the alternate conformation observed in the pre-2r state (Figure 5). Taken together, the data suggest a mechanism in which conformational changes in the catalytic DV propagate outward through a network of dynamic tertiary interactions. This results in the 90° swinging action observed in DVI, which facilitates substrate exchange during the reverse splicing process.
Figure 5.

Model of dynamic tertiary interactions that mediate active site rearrangement. (A) In the pre-1r state, both φ-φ′ and ψ-ψ′ are engaged. As the two-nt bulge rearranges between steps of catalysis, these two dynamic tertiary interactions are disrupted. These dynamics are associated with the 90° swinging motion observed for DVI. This motion is responsible for placing the lariat bond substrate in the active site for the second step of reverse splicing. The RNA domains are labeled with Roman numerals. Dynamic motion is indicated with black arrows and the M1 metal ions is shown as an orange sphere. (B) In the pre-1r state, both φ-φ′ and ψ-ψ′ are engaged. Black arrows indicate movement between steps of reverse splicing. In the pre-1r state two nucleotides from J5/6 (U826 and A827) are observed inserting into the minor groove of the basal stem of DIV. With φ-φ′ engaged, DVI exists in the down conformation. A conserved nucleotide from the I(i) loop (A401) is also observed participating in a base triple with U799 and G816 from DV to form ψ-ψ′. In the pre-1r state, the 5′ end of the RNA has a sharp kink and the bulged adenosine (A860) forms a non-canonical A-G base pair with G864. In the pre-2r state, both φ-φ′ and ψ- ψ′ are disengaged. As a result of the expansion of DV, φ-φ′ is forced to disengage in the pre-2r state, allowing J5/6 to act as a hinge that permits the 90° swinging motion of DVI. Due to the rearrangement of the two-nucleotide bulge, ψ-ψ′ also disengages. Density corresponding to the nucleotides involved in ψ-ψ′ are shown for both states to emphasize the disengagement of the interaction. The disruption of ψ-ψ′ results in a large conformational rearrangement of the I(i) loop and the 5′ end to accommodate the swinging of DVI. Backbone RMSD values are shown.
It is noteworthy that both DV and its U2/U6 homologue have exhibited a static overall topology during catalysis in all previous structures of the group II intron and the spliceosome, respectively. However, rearrangements of the catalytic triplex in the active site have been observed in crystal structures of the group IIB intron (Chan et al., 2018), which may be a prelude to the DV and DVI dynamics observed in our work. We suggest that the analogous U2/U6 pairing or a spliceosomal protein component may also initiate large-scale conformational dynamics involving the branch-site helix in the spliceosome.
Mechanism of Reverse Splicing into DNA
Based on the distinct pre-1r and pre-2r structures observed in the current work, we propose a mechanism of retrotransposition that accounts for the integration of the RNA into the top strand of the DNA target (Figure 6). The group II intron-maturase complex first binds to the target DNA and partially melts the duplex, allowing the RNA to Watson-Crick pair with the DNA in a sequence-specific manner. In the pre-1r state, DVI is engaged with DII via the π-π′ and η-η′ interactions and positions the 3′-OH of the intron RNA to engage in nucleophilic attack at the target site, catalyzed by magnesium ions M1 and M2 in the active site. As a result, the 3′ end of the RNA is covalently attached to the 5′ end of the newly cleaved DNA. In preparation for the second step of reverse splicing, π-π′ and η-η′ are disrupted, thus allowing DVI to swing 90° and become stabilized by the matX-DVI and ι-ι′ interactions. DVI rearrangement places the 2′−5′ phosphodiester bond of the lariat in the catalytic core. The 3′-OH of the 5′ DNA then engages in nucleophilic attack on the 2′−5′ phosphodiester bond, resulting in cleavage of the lariat and covalent attachment of the 5′ end of the RNA to the DNA target.
Figure 6.

Model of DNA integration and active site substrate exchange. In the initial stage of integration, the dsDNA is partially melted and the RNA selects the appropriate insertion site through Watson-Crick pairing with the DNA (pre-1r). Once the dsDNA is bound, the 3′-OH of the RNA is positioned in the active site to attack the scissile phosphate bond (red arrow). This results in the 3′ end of the RNA being covalently attached to the 3′ DNA (post-1r). After the first step of reverse splicing, the rearrangement of DV translates outward causing DVI to swing by 90° (black arrow). DVI is stabilized in its alternate conformation by both the ι-ι′ and matX-DVI interactions (pre-2r). In this new conformation of DVI, the lariat bond is presented to the active site for nucleophilic attack by the 3′-OH of the 5′ strand of DNA (red arrow). The end result of this mechanism is that the intron RNA is covalently attached to dsDNA at both its 5′ and 3′ ends (post-2r). Newly formed covalent bonds between the RNA and DNA are highlighted as red circles.
We hypothesize that after the intron is fully reverse spliced into the DNA, additional conformational rearrangements will occur to coordinate the activities of the En and RT domains of the maturase protein. Density corresponding to the En domain was absent in the pre-1r and pre-2r states. However, this domain must bind to the DNA after the intron is fully reverse spliced in order to cleave the bottom strand. Future work to capture the subsequent stages of retrotransposition will likely give insight into these outstanding questions.
Mechanism of Maturase-Promoted RNA Splicing
For over 20 years, the maturase has been known to promote splicing by binding to the intron RNA; however, the precise mechanism of how this protein promotes group II intron ribozyme catalysis has remained unknown. The pre-1r and pre-2r structures show that the maturase protein promotes splicing by forming an essential contact with DVI of the intron RNA (matX-DVI). Stabilization of DVI in this alternate conformation by the maturase is required for the first step of forward splicing. The maturase helps to position the adenosine nucleophile directly over the 5′ splice site, which contradicts the prevailing view that the maturase merely provides a chaperone-like function for the proper overall folding of the RNA structure.
The matX-DVI interaction has parallels to the RNase H-like domain of Prp8 interacting with the spliceosomal branch-site helix. A conserved beta-finger structure from the RNase H-like domain interacts with the branch-site helix to properly position the adenosine for lariat formation in the spliceosome (Figure 7A). In addition, Prp8 contacts with the 5′ exon sequence (Galej et al., 2016) in a manner similar to that seen with the maturase binding to the DNA target. Therefore, Prp8 functions in an analogous manner in the spliceosome to promote the branching reaction.
Figure 7.

Group II intron and the spliceosome share mechanistic dynamics. (A) In group II introns the thumb/x domain of the maturase protein promotes splicing by binding to DVI of the RNA and stabilizing the bulged adenosine in the active site to facilitate lariat formation. In the spliceosomal C complex (PDB Accession Code 5LJ5), the RNaseH-like domain of prp8 plays a similar role in binding and transitioning the homologous branch helix through the different stages of splicing (Galej et al., 2016). (B) The DVI helix of the group II intron undergoes a 90° swinging motion between the pre-1r and pre-2r stages of DNA integration. Identical dynamics are observed in the spliceosome in the transition between the C complex (PDB Accession Code 5LJ5) (Galej et al., 2016) and the post-catalytic P (post-P) complex (PDB Accession Code 6QDV) (Fica et al., 2019). The post-P complex and the C complex are structurally homologous to the pre-1r and pre-2r states, respectively. This conserved dynamic motion supports the hypothesis that the spliceosome has evolutionary origins as a group II intron-like retroelement.
Comparison with Branch-Site Helix Dynamics in the Spliceosome
The conformational dynamics seen for DVI in the group II intron are almost identical to those seen for the branch-site helix in the spliceosome (Figure 7B). Specifically, the branch-site helix in the spliceosome undergoes a 90° degree swinging movement in the transition between the C and P stages of catalysis (Fica et al., 2019; Galej et al., 2016). In both systems, this conformational rearrangement accomplishes the exchange of the two splice site substrates between the different stages of catalysis. These mechanistic parallels suggest that this mechanism of substrate exchange first originated in a group II intron-like ancestor and was retained during the evolutionary progression toward the spliceosome.
The changes in the topology of DV may also occur in the homologous U2/U6 snRNA pairing in the spliceosome. The structural basis for the nucleobase conservation of the catalytic triad and the two-nt bulge in both systems cannot be explained solely by the phosphate backbone forming a pocket for metal ion binding. We hypothesize that there may be transient nucleobase interactions that have not been entirely captured in previous crystal structures. There has been some evidence of subtle conformational dynamics involving DV, however the current study indicates that this domain may be involved in more significant topological rearrangements associated with catalysis than previously expected. It is possible that DV could play a role in communicating the state of the active site to the adjacent DVI. Further verification of the importance of DV dynamics would likely require structure determination of the group II intron at different stages of reverse splicing.
Retroelement Origins of the Spliceosome
We hypothesize that these observed branch-site helix dynamics first originated in a group II intron ancestor, with the spliceosomal descendants retaining this movement as a mechanism of substrate exchange. The conservation of conformational dynamics between the two systems is striking and is one of the strongest pieces of functional evidence for a direct evolutionary relationship between the two systems. In addition, the spliceosome contains the highly conserved Prp8 protein, which exhibits extensive structural homology to the group II intron maturase protein (Galej et al., 2013). Given the structural and mechanistic similarities between the two systems, the spliceosome likely evolved from a former selfish retroelement that evolved a new role in pre-mRNA splicing. A retroelement origin for the spliceosome may also explain the proliferation of introns in higher eukaryotes to such an extent that they now comprise >25% of the genome in mammals. The spliceosome has been found to engage in reverse splicing reactions into mRNAs in vitro (Tseng and Cheng, 2008), which may be indicative of its former function as a retroelement. It has been hypothesized that reverse splicing could be followed by conversion into a cDNA copy (Lee and Stevens, 2016), which we suggest may be synthesized by a LINE element RT. The newly synthesized cDNA would subsequently integrate into the genome through homologous recombination. Yet another possibility is that the spliceosome may have retained a low level of activity for direct insertion into DNA via reverse splicing in a manner similar to that seen in the TPRT mechanism. This hypothesis can be tested by challenging the spliceosome with different DNA substrates to identify any vestigial activity responsible for intron proliferation.
STAR Methods
CONTACT FOR REAGENTS AND RESOURSE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Navtej Toor (ntoor@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Two strains of E. coli were used in this study. DH5α cells acquired from Thermo Fisher Scientific were used to prepared plasmid for in vitro transcription. These cells were grown in Luria-Bertani (LB) broth overnight at 37°C and recovered using centrifugation at 5000 RCF. To express all recombinant protein constructs, Rosetta 2 (DE3) cells acquired from New England Biolabs were used. For all protein purification, the cells were grown in LB broth at 37°C to an optical density of 0.6–0.8 and then induced with 1 mM IPTG. After induction, the cells were grown at 22°C for 48 hours. The cells were then harvested using centrifugation.
METHOD DETAILS
Plasmid cloning
The WT, RT-1, RT-2, and RT-3 6xHis-MBP-T.el4h maturase genes were synthesized (Genscript) and cloned into a pET15b vector using the NdeI and BamHI restriction sites. A single G275D mutation was made in all constructs to restore the conserved YADD active site of the RT. The cloned plasmids were then transformed into Rosetta 2 cells (NEB). The WT T.el4h intron gene was synthesized (Genscript) containing a single C236T mutation to restore the conserved δ-δ′ and the DIV ORF was removed. The WT T.el4h intron gene was cloned into a pUC57 plasmid using the EcoRV restriction site. The WT intron construct was used for retroelement assembly and structural studies and contained an 18-nt 5′ exon and a 9-nt 3′ exon followed by a HindIII cut site. The cloned intron RNA plasmid was transformed into DH5α cells. The target DNA primers were synthesized by IDT, with the desthiobiotin tag included only for the avidin purification through integration into the DNA.
Sense DNA target sequence.:
GATAGAGATTTTCCCAGGGTTGGCCGAGCGGATGAGGCAGCGAAC
Antisense DNA target sequence:
GTTCGCTGCCTCATCCGCTCGGCCAACCCTGGGAAAATCTCTATC
T.el4h RT purification
A 100 mL overnight culture of 6X-His-MBP-T.el4h was prepared in LB containing carbenicillin. 20 mL of the overnight culture was diluted into 2 L of LB containing carbenicillin. The cells were grown to an optical density of 0.8 and then induced with 1 mM IPTG. The cells were incubated at 22°C with shaking for 48 hrs. Cells were harvested by centrifugation at 5,000 rpm for 10 minutes at 4°C and the cell pellet was resuspended in 50 mL of lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM KCl, 2 M urea, 10 mM imidazole, 5 mM 2-mercaptoethanol, and PMSF). The cells were lysed using a probe sonicator at 60% amplitude for a total of 80 seconds. The lysate was cleared of cell debris through centrifugation at 12,000 rpm for 45 minutes at 4°C. The resulting supernatant was transferred to a clean tube with 2 mL of Ni-NTA (Qiagen). The mixture was allowed to batch bind for 1 hr at 4°C. The resin was collected in an empty Bio-Rad gravity purification column. The resin was first washed with 5 column volumes of lysis buffer followed by 5 column volumes of a high salt wash buffer (20 mM Tris-HCl pH 7.5, 1.5 M KCl, 2 M urea, 10 mM imidazole, 5 mM 2-mercaptoethanol). The resin was re-equilibrated to 500 mM KCl by rinsing with 5 column volumes of lysis buffer supplemented with 10% glycerol. The bound protein then underwent a refolding protocol on-column by stepwise reduction in urea. The protein was eluted from the column using buffer containing 250 mM imidazole. The recovered protein was exchanged into filtration buffer (20 mM Tris-HCl pH 7.5, 500 mM KCl, 5 mM 2-mercaptoethanol, 10% glycerol) using a 50 kDa cut-off filter (EMD-Millipore). The protein-containing solution was rinsed a total of six times with 14 mL of filtration buffer. For the last buffer exchange, the protein solution was concentrated to 500 μL and brought to 50% glycerol for long term storage at −80°C.
In vitro RNA transcription
T.el4h plasmid was linearized using an engineered HindIII restriction site. In vitro transcription reactions were prepared in a 1 mL total volume. 40 μg of linear DNA template was added to transcription buffer containing: 50 mM Tris-HCl pH 7.5, 17.5 mM MgCl2, 5 mM DTT, 2 mM spermidine, 0.05% Triton X-100, and 2 mM of each NTP. T7 polymerase was added to initiate in vitro transcription. Thermophilic inorganic pyrophosphatase was also included to minimize pyrophosphate precipitate. The reaction was placed at 37°C for 3 hrs. Turbo DNase (20 units) was then added along with 12 μL of 100 mM CaCl2 and allowed to react for 1 hr at 37°C. Proteinase K (200 μg) was then added at 37°C for 1 hr. The mixture was centrifuged to remove any precipitate. The supernatant was filtered through a 0.2 μm filter followed by a 100 kDa cutoff filter. The RNA containing solution was buffer exchanged a total of 7 times with 14 mL of filtration buffer (5 mM cacodylate pH 6.5 and 10 mM MgCl2). For the last buffer exchange, the RNA was concentrated to approximately 10 mg/mL.
In vitro reverse splicing assay
To perform reverse splicing experiments, the target DNA sense strand was 5΄ radiolabeled with 32P. 50 pmol of DNA primer were mixed with 50 pmol of [γ- 32P] ATP, 5 μL of reaction buffer, and 20 units of T4 polynucleotide kinase (New England BioLabs) in a total reaction volume of 50 μL. The solution was incubated for 30 minutes at 37°C. The mixture was then passed through a desalting gel filtration column to capture the unreacted [γ−32P] ATP. The labelled primers were ethanol precipitated using NH4OAc and linear polyacrylamide as a carrier. The resulting pellet was resuspended in water. To analyze RNA insertion, radiolabeled sense DNA was annealed to cold antisense DNA by heating the mixture to 90°C for 2 minutes and allowing the solution to cool at room temperature for 15 minutes. T.el4h intron RNA was then spliced with T.el4h maturase protein in 10 mM MgCl2, 40 mM Tris-HCl pH 7.5, 500 mM NH4Cl, and 5 mM DTT for 5 minutes at 50°C. Annealed target DNA was added along with 200 μM dNTPs and allowed to react at 50°C for an additional 5 minutes. The samples were then quenched by phenol/chloroform extraction and the nucleic acids were recovered by performing an ethanol precipitation with linear polyacrylamide as a carrier. The pellets were resuspended in formamide and analyzed on a denaturing 4% 19:1 acrylamide:bis-acrylamide PAGE gel with 8 M urea. The gels were imaged using a Bio-Rad phosphorimager.
RNP purification
To selectively purify group II intron bound to dsDNA, insertion into desthiobiotin-labeled DNA was used to capture active complexes. To assemble the retroelement complex for purification, a 5 mL solution containing 500 μg of T.el4h and 1 mg of T.el4h RT in 40 mM Tris-HCl pH 7.5, 500 mM NH4Cl, 10 mM MgCl2, and 5 mM DTT was prepared. The solution was placed at 50°C for 5 minutes and then 10 μg of pre-annealed, desthiobiotin-labeled DNA target was added. The solution was placed back at 50°C for an additional 5 minutes. The resulting mixture was then added to 300 uL of soft-link avidin resin and allowed to bind for 1 hour. The supernatant was then removed and the resin was washed with 5 mL of wash buffer (40 mM Tris-HCl pH 7.5, 300 mM NH4Cl, 10 mM MgCl2, and 5 mM DTT) a total of 6 times. The bound complex was eluted with elution buffer (40 mM Tris-HCl pH 7.5, 300 mM NH4Cl, 10 mM MgCl2, 5 mM DTT, and saturated biotin). The eluent was recovered and concentrated using a 30 kDa molecular weight cut-off filter to a concentration of 100 ng/μL.
EM sample preparation and data collection
For cryo-EM analysis of the T.el4h group II intron, 2.5 μl of RNP sample was loaded onto plasma cleaned UltrAuFoil R1.2/1.3 300-mesh grids (Quantifoil). The grids were blotted before being plunged into liquid ethane cooled by liquid nitrogen using a manual plunger under >80% humidity at 4 °C. Leginon (Suloway et al., 2005) was used for data collection. Micrographs were recorded in a Titan Krios microscope (FEI) operating at 300 kV (Figure S4), equipped with a K2 Summit direct electron detector (Gatan) operating in counting mode. A tilt-specimen data collection strategy was employed to address the preferred sample orientation issue (Tan et al., 2017). In total, 998, 736, 967 and 741 micrographs were collected at 0°, 10°, 20° and 30° tilt angles respectively. A nominal magnification of 39,000x and a defocus range of −1.0 μm to −3.0 μm was used for imaging the sample, giving an effective pixel size of 0.79 Å at the specimen level. Each micrograph was dose-fractionated to 80 frames under a dose rate of 4.55 e-/pixel/s, with a total exposure time of 8 s and a frame exposure time of 0.1 s, resulting in a total dose of ~58e-/Å2.
Data processing
A total of 3442 movie stacks were drift corrected and electron-dose weighted using MotionCor2 (Zheng et al., 2017). The average defocus level of each aligned micrograph (without dose weighting) was determined with GCTF (Zhang, 2016). 3,368 micrographs with minimum drift and charging were selected for further data processing. Nearly 10,000 particle images were manually selected and subjected to 2D classification with Relion (Kimanius et al., 2016; Scheres, 2012). From these images, a 3D ab initio model was obtained in cryoSPARC (Punjani et al., 2017). Projections of this model were then used as templates for auto-picking, which yielded 416,000 particles from non-dose-weighted micrographs with Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/). The defocus level of each particle image was determined with GCTF. The particle images were extracted from dose-weighted micrographs with 4-fold binning and were subjected to one round of 2D classification. Each of the 401,889 selected particles were recentered and re-extracted without binning. These particles were 3D refined to yield a density map with a global resolution of 3.6 Å according to the gold-standard FSC 0.143 criteria. This map was further improved to 3.1 Å resolution with re-boxed polished particle images (particle images after motion correction and radiation dose weighting). These particles were 3D classified with a spherical mask into 10 classes without image alignment. The particle images belonging to two distinct conformational states (pre-1r (73,086 particles) and pre-2r (87,706 particles)) were 3D refined to yield maps with final global resolutions at 3.6 Å. Local resolution maps were created using ResMap (Swint-Kruse and Brown, 2005) and displayed using UCSF Chimera (Pettersen et al., 2004).
Model building and structure refinement
All modelling was performed in COOT (Emsley and Cowtan, 2004) with the RCrane plugin (Keating and Pyle, 2012) used for the RNA component. The coordinates were refined in real space using PHENIX (Adams et al., 2010; Afonine et al., 2018) and COOT. Appropriate protein and RNA secondary structure restraints were imposed during refinement. The secondary structure was used as a guide for the placement of the helices within the density. The quality of the density allowed for the differentiation of purines and pyrimidines, with large amino acids side chains being visible. To initiate modelling of the T.el4h RT protein, the coordinates from the IIC RT known as GsI-IIC (PDB accession code 6AR1) (Stamos et al., 2017), were rigid body fit into the density. To improve the protein model, a map was generated using local alignment focusing on the RT density. This resulted in a map at 3.1 Å for the RT component of the complex. Using the locally refined map and the 6AR1 model, the T.el4h RT model was built into the density using COOT. The RT coordinates were then rigid body fit into the final 3.6 Å maps and refined in real space using PHENIX. All software was compiled by SBGrid (Morin et al., 2013).
RT activity assay
Primer extension was performed on Oceanobacillus iheyensis RNA to determine the reverse transcriptase activity of the WT and mutant maturase proteins. Primer was first 5′ end-labelled by mixing the following ingredients in a tube: 500 ng primer, 10 μCi of γ−32P-ATP (10 μCi/μl; 3000 Ci/mmol), 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 5 mM dithiothreitol, 1 μl of T4 polynucleotide kinase (10 units/μl) and H2O for a total volume of 50 μl. This was incubated at 37°C for 30 minutes and heat denatured at 60°C for 20 minutes. The resulting solution was run through a Sephadex G-50 column to remove unincorporated nucleotides, and ethanol precipitated with 4M NH4OAc. The pellet was washed with 1 ml of ice-cold 75% ethanol and resuspended in 15 μl of H2O. The primer was annealed to RNA by mixing 1 μl of primer (10,000 cpm) and 5 μl (100 ng/μl) of RNA and heating to 65°C for 10 min followed by cooling on ice for 5 min. 2 μl of 5X first strand buffer (250 mM Tris-HCl (pH 8.3), 375 mM KCl and 15 mM MgCl2), 1 μl of 0.1 M DTT, and 1 μl 10 mM dNTPs were added, mixed, and heated at 25°C for 2 min. Three μl of WT or mutant maturase protein was added and incubated at 55°C for a further 60 min. Reverse transcriptase was inactivated by phenol/chloroform extraction. Seven μl of formamide loading dye was added to the sample and 1,000 cpm were resolved on a 8% polyacrylamide (19:1 acrylamide:bisacrylamide ratio)/8 M urea gel.
Gel filtration coelution assay
In order to assess intron RNA/maturase protein complex formation, a gel filtration coelution assay was performed. Intron RNA was incubated with maturase protein at 50°C for 10 minutes in a buffered solution containing 5 mM MgCl2, 0.5 M NH4Cl, 40 mM Tris-HCl (pH 7.5), and 5 mM dithiothreitol. The resulting solution was injected onto a HiLoad 16/60 Superdex 200 prep grade column equilibrated with running buffer (5 mM MgCl2, 0.5 M NH4Cl, 40 mM Tris-HCl (pH 7.5), and 2 mM dithiothreitol) at a flow rate of 1 mL/min. The progress of the elution was monitored by UV absorbance at 260 nm. The eluent was fractionated into 1.5 mL aliquots. The fraction containing the assembled complex (Frac 38) was collected and analyzed for the presence of intron RNA and maturase protein using SDS-PAGE gel electrophoresis. The RNA and protein bands were visualized using a coomassie blue stain.
In vitro forward splicing assay
The constructs used for the in vitro forward splicing assays contained WT T.el4h sequence with the DIV ORF removed and a 250-nucleotide 5′ exon and 150-nucleotide 3′ exon. The Δι-ι′ mutant also contained a G94A and G116A mutation. The Δψ-ψ′ construct also contained a single A401G mutation. These constructs were cloned into the pUC57 plasmid. Plasmid was linearized using HindIII and used for in vitro transcription with T7 RNA polymerase. Radiolabeled transcripts were prepared using 10 μCi [α−32P]UTP (3,000 Ci mmol−1 ), 0.5 mM UTP, 1 mM other NTPs, and 10 mM MgCl2. Transcripts were gel purified on a 4% polyacrylamide (19:1)/8 M urea gel, RNA was recovered by diffusion into 300 mM NaCl, 0.01% SDS, 1 mM EDTA. Forward splicing experiments were performed for 1 min at 50 °C in a splicing buffer containing 5 mM MgCl2, 0.5 M NH4Cl, 40 mM Tris-HCl (pH 7.5) and 5 mM dithiothreitol. Reactions were stopped by phenol/chloroform extraction. Splicing products were resolved using a denaturing 4% polyacrylamide (19:1)/8 M urea gels.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA concentrations were determined using a Nanodrop spectrophotometer (Thermo-Fisher). Maturase protein concentrations were determined using an SDS-PAGE gel with a concentration gradient of BSA (Thermo-Fisher). Per-residue backbone RMSD values were calculated using superposed models in UCSF Chimera (Figures 5 and S5). All statistical validation performed on the two deposited models (6ME0 and 6MEC) was done using PHENIX (Table S1). Forward splicing assays were performed in triplicate with only one representative being shown (Figures S6A and S7).
DATA AND SOFTWARE AVAILABILITY
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-9105 (pre-1r) and EMD-9106 (pre-2r). The coordinates of the atomic models have been deposited in the Protein Data Bank under accession codes 6ME0 (pre-1r) and 6MEC (pre-2r).
Supplementary Material
Movie 1. Active site domain V of the pre-1r state shown in density, Related to Figure 2.
Figure S5. Conformational rearrangements observed in DV between the pre-1r (red) and pre-2r states (blue), Related to Figure 5. The ζ′ loop remains static between the two steps of DNA insertion. A rearrangement of the conserved two-nucleotide bulge is associated with a conformational change in DV that ultimately leads to a downward expansion of the basal stem of this domain. The bulge is highly dynamic during DNA integration and shifts the active site metal M1 downward by 8 Å. Backbone RMSD values are shown for DV residues.
Figure S6. Characterization of the matX-DVI interaction, Related to Figure 4. (A) A urea-PAGE gel analyzing maturase-promoted forward splicing of T.el4h RNA. Lariat only forms when intron RNA is incubated with WT maturase protein. (B) Primer extension experiment using WT maturase as well as RT mutants. Both WT and RT-1 maturase show cDNA synthesis. (C) A cartoon representation of a gel filtration chromatogram. To evaluate RNA/maturase binding, a co-elution experiment was performed by determining the composition of the RNP-containing fraction 38 for all samples. (D) An SDS-PAGE gel analyzing fraction 38 for WT maturase, RNA only, RNA/WT maturase, RNA/RT-1 maturase, RNA/RT-2 maturase, and RNA/RT-3 maturase samples. Only RNA/WT maturase, and RNA/RT-1 maturase show coelution of both RNA and protein. RT-1 (R357E / T358G / R405E / R406E) and RT-2 (T360G / H407D / K410D / K418D) have mutated surfaces corresponding to each patch, while RT-3 contains all the mutations from RT-1 and RT-2.
Figure S7. Maturase-promoted forward splicing assay, Related to Figures 4 and 5. Maturase-promoted forward splicing was performed to compare WT RNA with Δι- ι′ (G94A/G116A) and Δψ- ψ′ (A401G) mutants. The Δι- ι′ mutant shows a decrease in lariat formation when compared the WT RNA. The Δψ- ψ′ mutant leads to a complete inhibition of lariat formation indicating its importance for proper active site formation during the first step of forward splicing. The final two lanes contain WT RNA spliced with the RT-1 maturase mutant. When compared to the Δψ- ψ′ mutant RNA, the severity of the first step defect is identical. However, the ι- ι′ mutant is still competent for lariat formation, suggesting that the matX-DVI interaction is essential to position the bulged adenosine in the active site for the first step of forward splicing or the second step of reverse splicing.
Table S1. Cryo-EM data collection and refinement statistics, Related to Figure 2.
Movie 2. Large-scale dynamics of DVI between the pre-1r and pre-2r states of the group II intron, Related to Figures 2 and 4.
Movie 3. Active site domain V dynamics associated with splicing, Related to Figure 5. The κ-κ′ interaction (grey) and the μ-μ′ (yellow) are shown sliding within the minor groove of DV.
Figure S1. Mechanism for group II intron target-primed reverse transcription (TPRT), Related to Figure 1. A group II intron retroelement complex is made up of lariat RNA and its associated maturase protein. To initiate retrotransposition, the retroelement complex first binds to the target DNA. The RNA then catalyzes reverse splicing into the sense strand through two sequential transesterification reactions. Once the RNA has been covalently attached to the DNA, the endonuclease (En) domain of the maturase cleaves the antisense strand of DNA and the reverse transcriptase (RT) begins synthesis of a cDNA copy of the RNA. The RNA is then degraded and the cDNA copy is fully integrated into the genome through host repair mechanisms. The structural intermediates captured in these cryo-EM experiments have been shaded.
Figure S2. RNA secondary structure of T.el4h, Related to Figure 2. An RNA secondary structure of the T.el4h construct used in the cryo-EM experiments. The RNA contains six conserved domains labeled with Roman numerals. The exon binding sequences are labeled (EBS 1-3). Tertiary interactions are labeled with Greek symbols. The dsDNA target used in these studies is shown and the intron binding sequences are labeled (IBS 1-3).
Figure S3. Reverse splicing activity assay of a group II intron and purification scheme of the assembled RNP complex, Related to Figures 1 and 2. (A) A polyacrylamide gel showing the reverse splicing activity of the T.el4h group II intron. Bands corresponding to the pre-1r, pre-2r, and post-2r states are visible. (B) Workflow of retroelement purification. Desthiobiotin-labeled dsDNA was incubated with the retroelement complex. Once the RNA was bound to the DNA, it was purified from unbound RNP particles by mixing with avidin resin. For simplicity, only the fully integrated complex is shown bound to avidin; however, all bound states of this complex will be captured using this method. The DNA-bound complexes were then eluted using a buffer saturated with biotin. For more information see the Materials and Methods.
Figure S4. Cryo-EM data processing, Related to Figures 2 and 3. (A) An example micrograph of T.el4h frozen on UltrAuFoil R1.2/1.3 300-mesh grids (Quantifoil). The image was acquired at a nominal magnification of 39,000x. (B) 2D class averages of T.el4h. (C) Data processing workflow and structure determination of the T.el4h group II intron. After 3D classification, two distinct conformational states were identified (pre-1r and pre-2r). All particles forming these two states were combined into two separate data sets and further refined. The additional 3D classes were not further processed owing to disordered density in the active site and for DVI. The overall global resolution of each of the resulting 3D reconstructions was 3.6 Å. See Materials and Methods for further details on the cryo-EM data processing. (D) Local resolution maps of the pre-1r and pre-2r cryo-EM maps (generated using UCSF Chimera). (E) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of pre-1r and pre-2r. Curves calculated between two half maps of pre-1r (red), between the model and the full map of pre-1r used for model refinement (green), between the two half maps of pre-2r (blue), and between the model and the full map of pre-2r for model refinement (magenta). (F) Euler angle distribution plot of pre-1r (left) and pre-2r (right) showing the relative orientation of the particles used in the final 3D reconstruction. Three 3D FSC isosurfaces at a threshold cutoff of 0.5 in three axial orientations describing the directional resolution (isotropy) of the refined maps are displayed to the right of each Euler plot.
Highlights.
Mechanism of group II intron reverse splicing into DNA
Mechanism of splice site exchange during group II intron splicing
Mechanistic contribution of maturase protein to group II intron splicing
Conserved branch-site helix dynamics between the group II intron and spliceosome
ACKNOWLEDGEMENTS
We thank Youngmin Jeon for grid preparation and Bill Anderson for help with the electron microscopes. We also thank Mandy Janssen and Timothy Booth for both grid preparation and preliminary cryo-EM data collection. We thank Kevin Corbett for assistance with protein modeling. We thank Geoffrey Haack for assistance with video editing.
Funding: This work was supported by NIH grants 1R01GM123275 awarded to N.T., 1R01GM033050 awarded to T.S.B., and NIH DP5 OD021396 and U54GM103368 awarded to D.L.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, and Lührmann R (2017). Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542, 318–323. [DOI] [PubMed] [Google Scholar]
- Boudvillain M, and Pyle AM (1998). Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron self-splicing: a NAIM analysis. EMBO J 17, 7091–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger SC, Belcher SM, Schmidt U, Dib-Hajj SD, Schmidt T, and Perlman PS (1995). Studies of point mutants define three essential paired nucleotides in the domain 5 substructure of a group II intron. Mol Cell Biol 15, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RT, Peters JK, Robart AR, Wiryaman T, Rajashankar KR, and Toor N (2018). Structural basis for the second step of group II intron splicing. Nat Commun 9, 4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, and Pyle A (1995). Branch-point attack in group II introns is a highly reversible transesterification, providing a potential proofreading mechanism for 5’-splice site selection. RNA 1, 391–406. [PMC free article] [PubMed] [Google Scholar]
- Costa M, and Michel F (1995). Frequent use of the same tertiary motif by self-folding RNAs. EMBO J 14, 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Michel F, and Westhof E (2000). A three-dimensional perspective on exon binding by a group II self-splicing intron. EMBO J 19, 5007–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Walbott H, Monachello D, Westhof E, and Michel F (2016). Crystal structures of a group II intron lariat primed for reverse splicing. Science 354, 1118. [DOI] [PubMed] [Google Scholar]
- Deininger PL, and Batzer MA (2002). Mammalian retroelements. Genome Res 12, 1455–1465. [DOI] [PubMed] [Google Scholar]
- Eickbush TH (1999). Mobile introns: retrohoming by complete reverse splicing. Curr Biol 9, R11–14. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fedorova O, and Pyle AM (2008). A conserved element that stabilizes the group II intron active site. RNA 14, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Oubridge C, Wilkinson ME, Newman AJ, and Nagai K (2019). A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science 363, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, Dai Q, Staley JP, and Piccirilli JA (2013). RNA catalyses nuclear pre-mRNA splicing. Nature 503, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore JL, and Nesbitt DJ (2013). An RNA folding motif: GNRA tetraloop-receptor interactions. Q Rev Biophys 46, 223–264. [DOI] [PubMed] [Google Scholar]
- Galej WP, Oubridge C, Newman AJ, and Nagai K (2013). Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature 493, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Wilkinson ME, Fica SM, Oubridge C, Newman AJ, and Nagai K (2016). Cryo-EM structure of the spliceosome immediately after branching. Nature 537, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J, Wan R, Yan C, and Shi Y (2015). Structural basis of pre-mRNA splicing. Science 349, 1191–1198. [DOI] [PubMed] [Google Scholar]
- Jacquier A, and Michel F (1987). Multiple exon-binding sites in class II self-splicing introns. Cell 50, 17–29. [DOI] [PubMed] [Google Scholar]
- Jacquier A, and Michel F (1990). Base-pairing interactions involving the 5’ and 3’-terminal nucleotides of group II self-splicing introns. J Mol Biol 213, 437–447. [DOI] [PubMed] [Google Scholar]
- Keating KS, and Pyle AM (2012). RCrane: semi-automated RNA model building. Acta Crystallogr D Biol Crystallogr 68, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres S, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. In bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, and Zimmerly S (2011). Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol 3, a003616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, and Stevens SW (2016). Spliceosomal intronogenesis. Proc Natl Acad Sci U S A 113, 6514–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CF, Costa M, and Michel F (2011). Linking the branchpoint helix to a newly found receptor allows lariat formation by a group II intron. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcia M, and Pyle AM (2012). Visualizing group II intron catalysis through the stages of splicing. Cell 151, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr G, Ghanem E, and Lambowitz AM (2010). Mechanisms used for genomic proliferation by thermophilic group II introns. PLoS Biol 8, e1000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, and Gage FH (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435, 903–910. [DOI] [PubMed] [Google Scholar]
- Padgett RA, Podar M, Boulanger SC, and Perlman PS (1994). The stereochemical course of group II intron self-splicing. Science 266, 1685–1688. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Ricci M, Peona V, Guichard E, Taccioli C, and Boattini A (2018). Transposable Elements Activity is Positively Related to Rate of Speciation in Mammals. J Mol Evol 86, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robart AR, Chan RT, Peters JK, Rajashankar KR, and Toor N (2014). Crystal structure of a eukaryotic group II intron lariat. Nature 514, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitzsch M, Fedorova O, and Pyle AM (2010). The 2’-OH group at the group II intron terminus acts as a proton shuttle. Nat Chem Biol 6, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U, Podar M, Stahl U, and Perlman PS (1996). Mutations of the two-nucleotide bulge of D5 of a group II intron block splicing in vitro and in vivo: phenotypes and suppressor mutations. RNA 2, 1161–1172. [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Lentzsch AM, and Lambowitz AM (2017). Structure of a Thermostable Group II Intron Reverse Transcriptase with Template-Primer and Its Functional and Evolutionary Implications. Mol Cell 68, 926–939.e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Swint-Kruse L, and Brown CS (2005). Resmap: automated representation of macromolecular interfaces as two-dimensional networks. Bioinformatics 21, 3327–3328. [DOI] [PubMed] [Google Scholar]
- Tan YZ, Baldwin PR, Davis JH, Williamson JR, Potter CS, Carragher B, and Lyumkis D (2017). Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Hausner G, and Zimmerly S (2001). Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA 7, 1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Keating KS, Taylor SD, and Pyle AM (2008). Crystal structure of a self-spliced group II intron. Science 320, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, and Cheng SC (2008). Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science 320, 1782–1784. [DOI] [PubMed] [Google Scholar]
- Wank H, SanFilippo J, Singh RN, Matsuura M, and Lambowitz AM (1999). A reverse transcriptase/maturase promotes splicing by binding at its own coding segment in a group II intron RNA. Mol Cell 4, 239–250. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Eskes R, Yang J, Perlman PS, and Lambowitz AM (1995a). A group II intron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell 83, 529–538. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Guo H, Perlman PS, and Lambowitz AM (1995b). Group II intron mobility occurs by target DNA-primed reverse transcription. Cell 82, 545–554. [DOI] [PubMed] [Google Scholar]
- Zimmerly S, Hausner G, and Wu X (2001). Phylogenetic relationships among group II intron ORFs. Nucleic Acids Res 29, 1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1. Active site domain V of the pre-1r state shown in density, Related to Figure 2.
Figure S5. Conformational rearrangements observed in DV between the pre-1r (red) and pre-2r states (blue), Related to Figure 5. The ζ′ loop remains static between the two steps of DNA insertion. A rearrangement of the conserved two-nucleotide bulge is associated with a conformational change in DV that ultimately leads to a downward expansion of the basal stem of this domain. The bulge is highly dynamic during DNA integration and shifts the active site metal M1 downward by 8 Å. Backbone RMSD values are shown for DV residues.
Figure S6. Characterization of the matX-DVI interaction, Related to Figure 4. (A) A urea-PAGE gel analyzing maturase-promoted forward splicing of T.el4h RNA. Lariat only forms when intron RNA is incubated with WT maturase protein. (B) Primer extension experiment using WT maturase as well as RT mutants. Both WT and RT-1 maturase show cDNA synthesis. (C) A cartoon representation of a gel filtration chromatogram. To evaluate RNA/maturase binding, a co-elution experiment was performed by determining the composition of the RNP-containing fraction 38 for all samples. (D) An SDS-PAGE gel analyzing fraction 38 for WT maturase, RNA only, RNA/WT maturase, RNA/RT-1 maturase, RNA/RT-2 maturase, and RNA/RT-3 maturase samples. Only RNA/WT maturase, and RNA/RT-1 maturase show coelution of both RNA and protein. RT-1 (R357E / T358G / R405E / R406E) and RT-2 (T360G / H407D / K410D / K418D) have mutated surfaces corresponding to each patch, while RT-3 contains all the mutations from RT-1 and RT-2.
Figure S7. Maturase-promoted forward splicing assay, Related to Figures 4 and 5. Maturase-promoted forward splicing was performed to compare WT RNA with Δι- ι′ (G94A/G116A) and Δψ- ψ′ (A401G) mutants. The Δι- ι′ mutant shows a decrease in lariat formation when compared the WT RNA. The Δψ- ψ′ mutant leads to a complete inhibition of lariat formation indicating its importance for proper active site formation during the first step of forward splicing. The final two lanes contain WT RNA spliced with the RT-1 maturase mutant. When compared to the Δψ- ψ′ mutant RNA, the severity of the first step defect is identical. However, the ι- ι′ mutant is still competent for lariat formation, suggesting that the matX-DVI interaction is essential to position the bulged adenosine in the active site for the first step of forward splicing or the second step of reverse splicing.
Table S1. Cryo-EM data collection and refinement statistics, Related to Figure 2.
Movie 2. Large-scale dynamics of DVI between the pre-1r and pre-2r states of the group II intron, Related to Figures 2 and 4.
Movie 3. Active site domain V dynamics associated with splicing, Related to Figure 5. The κ-κ′ interaction (grey) and the μ-μ′ (yellow) are shown sliding within the minor groove of DV.
Figure S1. Mechanism for group II intron target-primed reverse transcription (TPRT), Related to Figure 1. A group II intron retroelement complex is made up of lariat RNA and its associated maturase protein. To initiate retrotransposition, the retroelement complex first binds to the target DNA. The RNA then catalyzes reverse splicing into the sense strand through two sequential transesterification reactions. Once the RNA has been covalently attached to the DNA, the endonuclease (En) domain of the maturase cleaves the antisense strand of DNA and the reverse transcriptase (RT) begins synthesis of a cDNA copy of the RNA. The RNA is then degraded and the cDNA copy is fully integrated into the genome through host repair mechanisms. The structural intermediates captured in these cryo-EM experiments have been shaded.
Figure S2. RNA secondary structure of T.el4h, Related to Figure 2. An RNA secondary structure of the T.el4h construct used in the cryo-EM experiments. The RNA contains six conserved domains labeled with Roman numerals. The exon binding sequences are labeled (EBS 1-3). Tertiary interactions are labeled with Greek symbols. The dsDNA target used in these studies is shown and the intron binding sequences are labeled (IBS 1-3).
Figure S3. Reverse splicing activity assay of a group II intron and purification scheme of the assembled RNP complex, Related to Figures 1 and 2. (A) A polyacrylamide gel showing the reverse splicing activity of the T.el4h group II intron. Bands corresponding to the pre-1r, pre-2r, and post-2r states are visible. (B) Workflow of retroelement purification. Desthiobiotin-labeled dsDNA was incubated with the retroelement complex. Once the RNA was bound to the DNA, it was purified from unbound RNP particles by mixing with avidin resin. For simplicity, only the fully integrated complex is shown bound to avidin; however, all bound states of this complex will be captured using this method. The DNA-bound complexes were then eluted using a buffer saturated with biotin. For more information see the Materials and Methods.
Figure S4. Cryo-EM data processing, Related to Figures 2 and 3. (A) An example micrograph of T.el4h frozen on UltrAuFoil R1.2/1.3 300-mesh grids (Quantifoil). The image was acquired at a nominal magnification of 39,000x. (B) 2D class averages of T.el4h. (C) Data processing workflow and structure determination of the T.el4h group II intron. After 3D classification, two distinct conformational states were identified (pre-1r and pre-2r). All particles forming these two states were combined into two separate data sets and further refined. The additional 3D classes were not further processed owing to disordered density in the active site and for DVI. The overall global resolution of each of the resulting 3D reconstructions was 3.6 Å. See Materials and Methods for further details on the cryo-EM data processing. (D) Local resolution maps of the pre-1r and pre-2r cryo-EM maps (generated using UCSF Chimera). (E) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of pre-1r and pre-2r. Curves calculated between two half maps of pre-1r (red), between the model and the full map of pre-1r used for model refinement (green), between the two half maps of pre-2r (blue), and between the model and the full map of pre-2r for model refinement (magenta). (F) Euler angle distribution plot of pre-1r (left) and pre-2r (right) showing the relative orientation of the particles used in the final 3D reconstruction. Three 3D FSC isosurfaces at a threshold cutoff of 0.5 in three axial orientations describing the directional resolution (isotropy) of the refined maps are displayed to the right of each Euler plot.
Data Availability Statement
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-9105 (pre-1r) and EMD-9106 (pre-2r). The coordinates of the atomic models have been deposited in the Protein Data Bank under accession codes 6ME0 (pre-1r) and 6MEC (pre-2r).