

Abstract
HER2-positive luminal B breast cancer (BC), a subset of the luminal B subtype, is ER-positive and HER2-positive BC which is approximately 10% of all BC. However, HER2-positive luminal B BC has received less attention and is less represented in previous molecular analyses than other subtypes. Hence, it is important to elucidate the molecular biology of HER2-positive luminal B BC to stratify patients in a way that allows them to receive their respective optimal treatment. We performed molecular profiling using targeted next-generation sequencing on 94 HER2-positive luminal B BC to identify its molecular characteristics. A total of 134 somatic nonsynonymous mutations, including 131 nonsynonymous single nucleotide variants and three coding insertions/deletions were identified in 30 genes of 75 samples. PIK3CA was most frequently mutated (38/94, 40.4%), followed by TP53 (31/94, 33.0%), and others were detected at lower frequencies. Recurrent germline mutations of MLH1 V384D were found in 13.8% (13/94), with a significantly high TP53 mutations rate. The frequency of MLH1 V384D germline mutation in individuals with HER2-positive luminal B BC was significantly higher than that observed in the controls. All 13 cases were classified as microsatellite stable tumors. Tumor mutation burdens (TMB) were not significantly different between MLH1 V384D carrier and wild type. The concordant results of microsatellite instability (MSI) and TMB suggest that the haploinsufficiency of MLH1 plays a role as a tumor predisposition factor rather than a direct oncogenic driver. Our study identified, for the first time, that MLH1 V384D germline variant is frequently detected in HER2-positive luminal B BC. MLH1 V384D germline variant may not only contribute to gastrointestinal cancer predisposition but may also contribute to BC in East Asians.
Subject terms: Breast cancer, Cancer genomics, Medical genomics
Introduction
Breast cancers (BC) are incredibly heterogeneous diseases in terms of the mutations, structural variations, and copy number aberrations they harbor. Genomic and molecular profiling analyses have extensively advanced our understanding of BC biology and have increased the elucidation of its five intrinsic molecular subtypes (luminal A, luminal B, HER2-enriched, basal-like, and normal breast like) by genome-wide expression analyses1,2. In intrinsic molecular subtypes, luminal B subtype is clinically and molecularly heterogeneous group. HER2-positive luminal B group, a subset of the luminal B subtype is ER-positive and HER2-positive BC, which is approximately 10% of all BC3,4. However, HER2-positive luminal B BC has received less attention and been less represented in previous molecular analyses than other subtypes. HER2-positive luminal B positive BC has been shown to be a heterogenous group5. Hormone receptor and HER2 co-expression may partially account for heterogeneity in the HER2-positive luminal B positive group. HER2 and ER pathways are strictly related in a bi-directional way6. HER2 signaling causes endocrine resistance, and ER modulates the response, not only to chemotherapy, but also to HER2 targeted therapy6, which is a major challenge in the treatment of HER2-positive luminal B BC. Therefore, it is important to elucidate the molecular biology of HER2-positive luminal B BC to stratify patients so that each can receive the optimal treatment.
More recently, using multi-omics profiling, analysis of a Korean BC cohort enriched with younger, premenopausal patients was found to harbor significant molecular differences from the TCGA cohort enriched with Western post-menopausal patients7. Korean BC cohort had significantly higher proportion of the HER2-positive luminal B BC subtype than TCGA cohort (16.1 vs 5.4%). Furthermore, there are also differences in molecular features and tumor immune microenvironments between Asian and Western BC.
Here we report a study in which molecular profiling is performed using targeted next-generation sequencing (NGS) of 94 HER2-positive luminal B BCs to identify the molecular characteristics of HER2-positive luminal B BC in East Asians.
Results
Clinicopathologic characteristics of 94 HER2-positive luminal B breast cancer patients
A total of 94 HER2-positive luminal B BC samples (KF) were analyzed in this study. The clinicopathologic characteristics are presented in Table 1 and Supplementary Table 1. The median age at diagnosis was 49 years (range, 25–75). Of patients, 52 (55.3%) were premenopausal, and 41 (43.6%) were post-menopausal. The most common histologic type was invasive ductal carcinoma (95.7%). Of these 94 patients, 72 (76.6%) received adjuvant trastuzumab, 80 (85.1%) received adjuvant chemotherapy, 72 (76.6%) received hormonal therapy, and 77 (81.9%) received radiation therapy. Eight of the 94 patients (8.5%) showed recurrence. The median time to recur after curative resection was 16.5 months and range was from 2 to 53 months.
Table 1.
Clinicopathologic characteristics of 94 HER2-positive luminal B breast cancer patients’.
| Characteristics | No. of patients (%) |
|---|---|
| Age (years) | |
| Median (range) | 49 (25–75) |
| Menopausal status | |
| Pre-menopause | 52 (55.3) |
| Post-menopause | 41 (43.6) |
| N/A | 1 (1.1) |
| Histologic type | |
| IDC | 90 (95.7) |
| ILC | 1 (1.1) |
| Mucinous carcinoma | 2 (2.1) |
| Micropapillary carcinoma | 1 (1.1) |
| T-stage | |
| 1 | 43 (45.7) |
| 2 | 48 (51.1) |
| 3 | 3 (3.2) |
| N stage | |
| 0 | 50 (53.8) |
| 1 | 28 (30.1) |
| 2 | 12 (12.9) |
| 3 | 3 (3.2) |
| N/A | 1 (1.1) |
| Stage | |
| I | 32 (34.0) |
| II | 45 (47.9) |
| III | 16 (17.1) |
| IV | 1 (1.1) |
| Histologic grade | |
| I | 2 (2.1) |
| II | 33 (35.1) |
| III | 59 (62.8) |
| Herceptin Tx | |
| Yes | 72 (76.6) |
| No | 17 (18.1) |
| NA | 5 (5.3) |
| Chemo Tx | |
| Yes | 80 (85.1) |
| No | 9 (9.6) |
| NA | 5 (5.3) |
| Hormonal Tx | |
| Yes | 72 (76.6) |
| No | 17 (18.1) |
| NA | 5 (5.3) |
| Radiation Tx | |
| Yes | 77 (81.9) |
| No | 12 (12.8) |
| NA | 5 (5.3) |
| Recur | |
| Yes | 8 (8.5) |
| No | 81 (86.2) |
| NA | 5 (5.3) |
Genomic profiling in HER2-positive luminal B breast cancer
We sequenced 170 cancer-related genes with FFPE tumor tissue of 94 HER2-positive luminal B BC. Targeted sequencing summary is shown in Supplementary Table 4. A total of 134 somatic nonsynonymous mutations, including 131 nonsynonymous single nucleotide variants (nsSNVs) and three coding insertions/deletions (indels), were identified in 30 genes of 75 samples. A full list of variants is shown in Supplementary Table 5. Figure 1 depicts sequence variants of 17 genes detected in more than two samples. The most frequently mutated was PIK3CA (38/94, 40.4%), followed by TP53 (31/94, 33.0%), with the others detected at lower frequencies. All PIK3CA variants were missense, and they occurred mostly in two hotspots (34/38, 89.5%) located in the helical and kinase domains (exon 9 and exon 20), resulting in activation of the gene with constant auto-phosphorylation. The majority of TP53 mutations were missense (27/31, 87.1%), and the remaining were truncating. The nature of tumor suppressor was reflected by the fact that the loci of the variants were scattered. When compared with The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC), the frequency of PIK3CA mutations was higher in KF (40.4%) than TCGA (25.0%) and METABRIC (23.8%). If variants with low allele frequency (AF) (<5%) were included, the difference achieved statistical significance (p = 0.01). On the other hand, the frequency of TP53 was significantly lower (p = 0.01) in KF (33.0%) than TCGA (42.3%) and METABRIC (54%) (Supplementary Fig. 1). We checked total read depth (TR) and variant allele frequency (VAF) of the TCGA and METABRIC BC data set. The median VAF was 33.7, 36.2, and 16.1 for TCGA, METABRIC, and KF, respectively, while the median TR was 72x, 176x, and 752x (Supplementary Fig. 2). VAF and TR were significantly different among three groups (p < 2.2e-16). Since we sequenced relatively high depth with median mean depth 1578x, we found more subclonal events that might have been missed in previous studies. Among the variants with recurrent positions shared by more than two samples, others than PIK3CA and TP53, were mostly “uncertain significance” of ClinVar annotation. CDH1 p.V832M, CHEK2 p.H371Y, and ERBB2 p.V747L were “likely pathogenic” or “pathogenic” in ClinVar annotation. The others were annotated as “uncertain significance” in ClinVar. Unfortunately, genes frequently mutated in HER2-positive luminal B BC in TCGA and METABRIC (GATA3, ARID1A, and KMT2C) were not included in our gene panels. Their mutation frequency could not be assessed.
Figure 1.

Recurrently mutated genes in HER2-positive luminal B BC. Each column represents a case. The top panel shows tumor mutation burden. The middle panel shows MLH1 V384D germline mutation status: dark purple, mutant type; light purple, wild type. The bottom panel shows distribution of mutations. Four mutation types are distinguished by different colors. The right panel represents the mutation frequency in the 94 samples.
Frequent MLH1 mutations in HER2-positive luminal B breast cancer
Although there is a slight difference in mutation frequencies of PIK3CA and TP53, we obtained similar mutation profiles as the previous large-scale studies. To identify other novel findings within our data, we focused on rare germline mutations. The probable germline mutations could not be perfectly filtered out in clinical cancer genome analysis, since matched normal samples usually are not examined. Under the same condition, rare germline mutations with clinical significance could be included in the present study. Therefore, we reviewed variants with minor allele frequency (MAF) between 1–5% in gnomAD EAS, which was excluded to filter out putative germline polymorphism. Interestingly, recurrent MLH1 mutations on chr3:37067240 (p.V384D) were identified in 13 samples (prevalence: 13.8%, MAF: 0.069). Thirteen HER2-positive luminal B BC tissues revealed a MLH1 V384D with median 45.09% VAF (range: 31.8–60.12). The median of depth of coverage in the region is 1273 (range: 69–2121). The MAFs of that position were 0.003 and 0.036 in gnomAD All and EAS, respectively. We checked MAF of MLH1 V384D in non-cancer Korean database of 1100 individuals (http://152.99.75.168/KRGDB/menuPages/intro.jsp). The frequency was 0.035, which is almost identical to gnomAD EAS and about one half the value of HER2-positive luminal B BC. The frequency of the MLH1 V384D mutation in individuals with HER2-positive luminal B BC was significantly higher than that observed in the controls (Fig. 2). We compared mutational frequencies of MLH1 V384D with those of HER2-negative luminal type BC and triple negative type BC (in-house data, not published). The frequency was 8.2% (19/233) in HER2-negative luminal type and 4.8% (5/104) in triple negative type. We have no available data of HER2-positive luminal B BC, the frequency of MLH1 V384D for that group could not be assessed. MLH1 V384D mutation might be associated with ER positivity, further study is needed to check its relationship with HER2 positivity.
Figure 2.
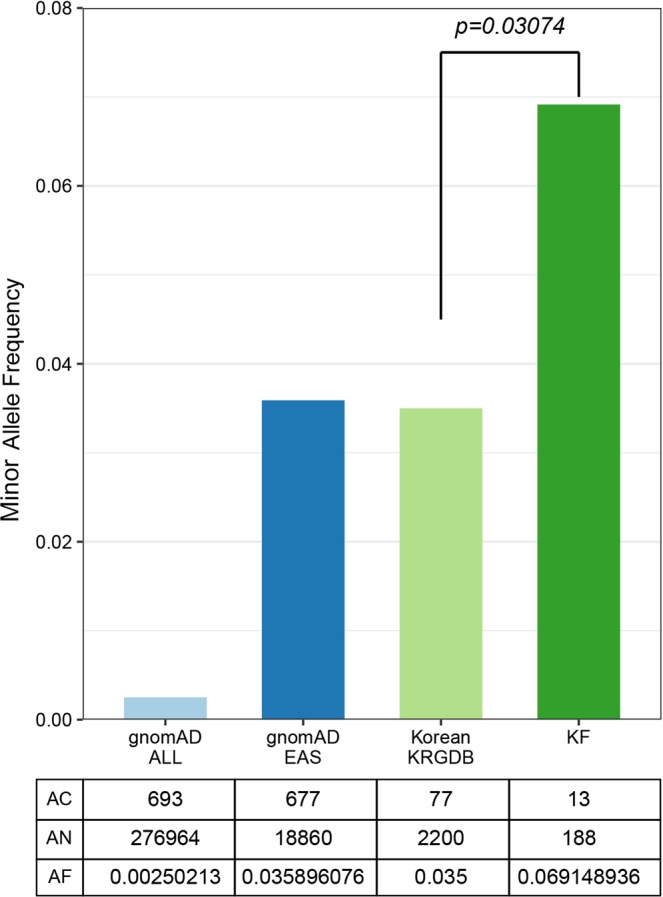
Different minor allele frequencies among four populations. Bar plot compares minor allele frequency of HER2-positive luminal B BC with three non-cancer populations, gnomAD all, East Asian (EAS), non-cancer Korean (KRGDB) and present study (KF). The bottom table represents each allele number (AN), allele count (AC), and allele frequency (AF).
Validation for MLH1 V384D mutation
To check whether MLH1 V384D was a germline or somatic, we performed conventional Sanger sequencing in each tumor and matched normal breast tissues. The variant of c.1151T > A (V384D) was detected in all 13 pairs of tumor and normal samples (Fig. 3a). We confirmed these MLH1 V384D mutations are germline heterozygous mutation.
Figure 3.

Representative results of Sanger sequencing, MSI and immunohistochemical analysis of MLH1 expression of a germline MLH1 V384D mutant case. (a) Sanger sequencing of MLH1 V384D for tumor and matched normal revealed that it is germline heterozygous variant. (b) MSI profiles with five Bethesda markers in the same case demonstrated MSS. (c) Representative cases with focal loss of MLH1 expression in a mutant case and intact MLH1 expression in a wild-type case. Abbreviations: T = tumor tissue; N = normal tissue.
Microsatellite instability (MSI) analysis was performed in all 13 cases. None of the cases revealed instability at any of the five loci analyzed, and they were classified as microsatellite stable tumor (MSS, 0/5) (Fig. 3b). To evaluate the functional loss of MLH1 protein, we performed immunohistochemical analysis for MLH1 protein. In three of 13 cases with MHL1 mutation, both carcinoma and adjacent normal breast tissue showed an area of focal loss, while all 81 cases without MLH1 mutation showed uniform positive staining (Fig. 3c).
MLH1 V384D in patients with 13 HER2-positive luminal B breast cancers
The 13 patients included 12 females and one male. Of them, six patients (46.2%) were premenopausal and six (46.2%) were postmenopausal woman. The detailed clinical and pathologic findings are summarized in Supplementary Table 6. Histologic types were invasive ductal carcinoma of no special type in 12 cases and mucinous carcinoma in one case. Tumor stage was IA in four cases, stage IIA in five cases, and stage IIB in four cases by AJCC eighth edition. None of the patients had any recurrence. Table 2 shows a comparison between the clinicopathologic and molecular data of the 13 patients with the MLH1 V384D germline mutation and those of the 81 patients with wild MLH1. The two groups were statistically similar with regard to age, menopausal status, T stage, N stage, AJCC stage, histologic grade, PIK3CA mutation status, and recurrence. The frequency of TP53 mutation in the MLH1 V384D germline mutation group was significantly higher than that of the wild MLH1 group (p = 0.03). Tumor mutation burdens (TMB) were not significantly different between the two groups, which is concordant with the result of MSI analysis.
Table 2.
The clinicopathologic data of 13 patients with MLH1 V384D germline mutation compared with 81 patients with wild MLH1.
| MLH1 wild | MLH1 V384D | p-value | |
|---|---|---|---|
| (N = 81, 86.2%) | (N = 13, 13.8%) | ||
| Age | 48 (25–74) | 52 (42–71) | 0.145 |
| Menopausal status | 0.134 | ||
| Pre | 46 (56.8%) | 6 (46.2%) | |
| Post | 35 (43.2%) | 6 (46.2%) | |
| N/A | 0 | 1 (7.7%) | |
| T_stage | 0.517 | ||
| 1 | 37 (45.7%) | 6 (46.2%) | |
| 2 | 42 (51.9%) | 6 (46.2%) | |
| 3 | 2 (2.5%) | 1 (7.7%) | |
| N_stage | 0.496 | ||
| 0 | 43 (53.1%) | 8 (61.5%) | |
| 1 | 23 (28.4%) | 5 (38.5%) | |
| 2 | 12 (14.8%) | 0 | |
| 3 | 3 (3.7%) | 0 | |
| Stage | 0.262 | ||
| 1 | 28 (34.6%) | 4 (30.8%) | |
| 2 | 36 (44.4%) | 9 (69.2%) | |
| 3 | 16 (19.8%) | 0 | |
| 4 | 1 (1.2%) | 0 | |
| Histologic grade | 0.188 | ||
| 1 | 1 (1.2%) | 1 (7.7%) | |
| 2 | 30 (37.0%) | 3 (23.1%) | |
| 3 | 50 (61.7%) | 9 (69.2%) | |
| PIK3CA mutation | 0.774 | ||
| Not identified | 46 (56.8%) | 8 (61.5%) | |
| Present | 35 (43.2%) | 5 (38.5%) | |
| TP53 mutation | 0.027 | ||
| Not identified | 58 (71.6%) | 5 (38.5%) | |
| Present | 23 (28.4%) | 8 (61.5%) | |
| Recurrence | 0.651 | ||
| No | 68 (84.0%) | 13 (100%) | |
| Yes | 8 (9.9%) | 0 | |
| N/A | 5 (6.2%) | 0 |
Discussion
The present study presents molecular profiling in 94 HER2-positive luminal B BCs using a targeted next-generation sequencing assay. We sequenced 170 cancer-related genes, and variants were shown to be enriched in PIK3CA and TP53. Recurrent germline mutations of MLH1 were found in 13.8%, with a significantly high TP53 mutations rate.
The two top-ranked genes were PIK3CA and TP53, and variants in the other genes exhibited long tail distribution. These results are consistent with those of previous studies on molecular analysis of patients with BC8,9. The prevalence of PIK3CA mutations has been variously reported in accordance with the different biologic subtypes of BC, which tends to be more common in hormone receptor positive BC (35%) than HER2 positive BC (22–31%) and triple negative BC (8.3%)8,9. When compared with TCGA and METABRIC HER2-positive luminal B BC, the PIK3CA mutation frequency was higher in KF population. But considering VAF, our results included subclonal events with low VAF, which might be missed in the TCGA and METABRIC. This is supported by a recent research that also revealed a comparably high mutation rate of PIK3CA (22.1%, 23/104) in ultradepth sequencing of triple-negative BC10. PIK3CA mutant group are associated with low chemosensitivity and resistance to anti-HER2 therapy in metastatic HER2-positive luminal B BC11,12. TP53 mutations are associated with triple-negative BC group, more aggressive subtypes and resistance to chemotherapies13. In a recent study, the significance of TP53 as a prognostic marker in BC was different in a molecular subgroup14. They reported that TP53 mutations were independent poor prognostic marker in luminal B, HER2-enriched groups14. In our study, MLH1 mutant group had significantly high TP53 mutation rate. Although no recurrence identified in MLH1 mutant group, it is necessary to confirm the relationship of MLH1 mutation and prognosis in a larger sample group.
Interestingly, we found that 13.8% of patients carried the MLH1 V384D germline mutation of the 94 HER2-positive luminal B BC. MLH1 is a tumor suppressor gene involved in DNA mismatch repair (MMR)15. MLH1 germline mutations are known to cause Lynch syndrome16. The most common malignant tumors in Lynch syndrome are colorectal and endometrial carcinoma17. In addition to germline mutations, somatic mutations in MLH1 have been identified in colorectal and endometrial carcinomas18. Although the MLH1 mutation was generally known to occur in the gastrointestinal cancer susceptibility syndrome, several studies suggested that MLH1 mutation carriers have increased risk of various cancers, such as renal, pancreatic, urinary bladder, prostate, and female BC12,19–21. In particular, there was controversy as to whether the MMR deficiency increased BC22–26. One study prospectively stated that the risk of BC was increased about four-fold above that of the control group19. More recently, another study found 10 MLH1 germline mutations in 8085 unselected Chinese BC patients, including 965 HER2-positive luminal B BC patients27. However, there was a report of 25 BC susceptibility genes in 488 patients of a Western BC population, including 63 HER2-positive luminal B BC patients, which were not found to have a germline MLH1 mutation28.
MLH1 dimerizes with PMS2 to form MutL heterodimer, which plays an important role in the MMR system29. The V384D mutation is likely to impair the stability of the hMLH1 protein, which can eventually partially reduce the interaction between hMLH1 and hPMS230. Finally, it was demonstrated that the V384D mutation reduced MMR activity in vitro31. Therefore, the MLH1 V384D mutation is partly a loss of function variant. In our study, we found that the MLH1 V384D mutation is a heterozygous germline mutation.
A heterozygous germline mutation of tumor suppressor gene allele establishes a predisposition to develop cancer32. The mechanism to explain the heterozygous effect is haploinsufficiency32. These effects can be directly attributable to the reduction in gene dosage or can work in cooperation with other tumorigenesis or haploinsufficient events32. Heterozygous MSH2 mutant mice appear to be more susceptible to tumor formation than wild-type animals33–35. MSH2 haploinsufficiency might have phenotypic effects that could contribute to progression of cancers in HNPCC individuals33–35. That is why haploinsufficiency might increase the cancer risk in MLH1 V384D mutation carrier30.
In our study, the frequency of the MLH1 V384D germline mutation in the HER2-positive luminal B BC group was two times higher than the mutation rate in both East Asian and Korean patients. Few published studies in East Asia have been about MLH1 V384D germline mutation in sporadic cancer. Ohasawa et al.36 stated that MLH1 V384D mutant was detected in 6% of 670 colorectal cancer patients, but it was only detected in 1.5% of 322 patients in the control group in the Japanese population. Peng et al.37 reported that a MLH1 V384D mutant was significantly higher in a colorectal cancer group than in the 311 members of a control group (9.0% vs 0.3%) in the Chinese population. Recently, Chiu et al.38 reported that the MLH1 V384D allele was overrepresented in EGFR L858R-positive lung adenocarcinoma patients with epidermal growth tyrosine kinase inhibitor (EGFR-TKI) resistance. Therefore, ethnic differences are evident the frequency of MLH1 V384D mutation between East Asian and Western populations.
We also identified that the frequency of the TP53 mutation was significantly high in the MLH1 V384D germline mutation carrier. Several studies have demonstrated that malignancy arising in carriers of BRCA1/BRCA2 germline mutations showed the higher rate of TP53 mutations39,40. These observations suggest that the loss of p53 function may be a critical genetic event of tumorigenesis in background of a germline BRCA1 / BRCA2 mutation. Consistent with this theory, our results can be explained in this context.
Few studies have investigated the germline mutations in BC susceptibility genes27,28. Germline mutations particularly for BRCA1 mutations are significantly associated with triple-negative BC phenotype among the four molecular subgroups27, which indicated that triple-negative BC reveals high level of genomic instability and a more aggressive phenotype. On the other hand, HER2–positive BC exhibits fewer germline mutations than other molecular subgroups27. This is explained by the fact that HER2 is a potent driver gene that could initially induce tumorigenesis41. In our study, we did not perform these analyses in other BC molecular subgroups. It is yet unknown whether MLH1 V384D germline mutation was significantly different among other molecular subgroups. Therefore, additional studies for more diverse unselected BC are needed to establish this.
It is now well-known that MMR-deficient tumors exhibit a high tumor mutation burden (TMB). Several studies assessed microsatellite instability using TMB42,43. In our study, there was no significant different between MLH1 V384D mutant and the wild group with regard to TMB, and all members of the MLH1 V384D mutant group showed MSS in conventional MSI analysis. Cancers with MSI-H phenotype in Lynch syndrome develop when second hit mutation occurs in wild-type allele followed in addition to pre-existing germline mutation44. The concordant results of MSI analysis and TMB suggest that the haploinsufficiency of MLH1 plays a role as a tumor predisposition factor rather than a direct oncogenic driver.
In conclusion, our study identified, for the first time, MLH1 V384D germline variant, which is frequently detected in HER2-positive luminal B BC. MLH1 V384D germline variant may not only contribute to gastrointestinal cancer predisposition but may also contribute to BC in East Asians. Further investigation into the role of MLH1 V384D germline mutation in BC will be needed to identify the association between MMR genes and BC risk.
Materials and Methods
Patients
This cohort study with retrospective data evaluated clinical, histological, and immunohistochemical charcteristics of all patients with histologically confirmed 94 HER2-positive luminal B BC (Supplementary Table 1). The cohort was composed of Korean non-related individuals. The samples were obtained from patients who underwent surgical resection for primary BC at the Konkuk University Medical Center from January 2012 to December 2016. All patients provided informed consent before surgical resection. Study approval was obtained from the Institutional Review Board of Konkuk University Medical Center (KUH1210049) and all experiments were performed in accordance with the approved guidelines and regulations.
Medical records and H&E-stained sections were reviewed to obtain clinicopathologic information. The following histopathologic variables of the invasive carcinomas were determined: histologic subtype, T stage, N stage, AJCC stage, Bloom-Richardson histologic grade. Samples with tumor cellularity >50% were included in this study.
Targeted Next-generation sequencing
Using the Custom Cancer Panel (Agilent Technologies, Inc., Santa Clara, California, USA) after DNA isolation from formalin-fixed, paraffin-embedded (FFPE) samples, we sequenced 170 cancer-related genes (Supplementary Table 2) to identify genetic mutations in 94 samples from HER2-positive luminal B BC patients. A dsDNA HS Assay Kit (Life Technologies, Grand Island, NY) was used to quantify DNA samples by Qubit 2.0. Sequencing libraries were prepared with SureSelectXT Library Prep Kit (Agilent Technologies, Inc., Santa Clara, California, USA) as previously decribed45. In brief, 200 ng of genomic DNA from the FFPE samples was fragmented by Covaris E220 instrument (Covaris, Woburn, MA) and then subjected to end repair, A-Tailing, and adapter ligation. Agencourt AMPure XP beads (Beckman Coulter, Beverly, MA) were used to remove unligated adaptors. The resulting libraries were PCR-amplified and purified with Agencourt AMPure XP beads. Captured libraries were PCR-amplified using Illumina p5 and p7 primers and purified with Agencourt AMPure XP beads. Library was quantified using a KAPA Library Quantification Kit (KAPA Biosystems), and its fragment size was analyzed by Bioanalyzer 2100 (Agilent Technologies, Cedar Creek, TX). Once ready, libraries were sequenced on Illumina HiSeq2500 platforms (Illumina, San Diego, CA).
Identification of somatic mutations
FASTQ file quality control was performed using FastQC v0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) software. The adapter sequences were removed by cutadapt v1.9.1.46. Sequencing reads were mapped to Human Genome version 19 (hg19) using the Burrows-Wheeler Aligner47. Poorly mapped reads that had mapping quality (MAPQ) below 20 were removed using Samtools v.1.3.1.48. Local realignment around indels and base quality score recalibration were applied with the Genome Analysis Toolkit (GATK 3.4.0)49. Duplicated reads were discarded using Picard MarkDuplicates v2.2.4 (https://broadinstitute.github.io/picard/). The MuTect2 algorithm was used to identify somatic mutations, including single nucleotide variants (SNVs), small insertions, and deletions (INDELs)50. False positive variant calls that originated from oxoG artifacts were excluded. All the variants were annotated using SnpEff & SnpSift v4.3,51 VEP52 and oncotator53. To identify a high-confidence list of putative somatic mutations, the following filtering steps were applied: (1) total allele count >=50 and variant allele frequency (VAF) > = 5%; (2) minor allele frequency (MAF) < 1% in gnomAD54 all and east Asian (EAS), and 1000 genome project55; (3) nonsynonymous SNVs or indels in coding regions; (4) exclude variants with “benign” or “likely-benign” of ClinVar clinical significance value. Copy number variations (CNVs) were detected using MuTect2 with default parameters. Fifty non-cancer samples (in-house data) were used as the panel of normal.
Comparing with public data
Mutation frequencies of candidate genes were compared with those of the public database. We downloaded the clinical and mutation profile data of two major large-scale study, “Breast Invasive Carcinoma (TCGA, Nature 2012)”56 and “Breast Cancer (METABRIC, Nature 2012 & Nat Commun 2016)”57 from cBioPortal. (http://www.cbioportal.org). HER2-positive luminal B BC was selected by the “Positive” value of “ER Status” and “HER2 Status” in the clinical data from samples with both clinical and mutational data available. Fifty-two of 825 TCGA (6.3%) and 100 of 2368 METABRIC (4.2%) met the criteria. Extracted variants were annotated using VEP and further filtered by the same criteria used to identify the somatic mutations as descripted above.
Validation of MLH1 mutation by Sanger sequencing in tumor and matched normal samples
We performed Sanger sequencing of matched normal tissue to demonstrate whether MLH1 mutations found in targeted sequencing were germline alterations as previously descripted58. Amplification was performed in a total volume of 20 μL containing 100ng-1μg of DNA from formalin-fixed and paraffin-embedded tissue using Maxime PCR PreMix (iNtRON Biotechnology, Korea). The primer sequence is shown in Supplementary Table 3. The PCR products were purified and analyzed using an ABI3730XL DNA analyzer (Appied Biosystem, Foster City, CA) for Sanger sequencing to validate the presence of each mutation.
Molecular findings for microsatellite instability (MSI)
We performed an MSI analysis on paraffin-embedded tissues to evaluate their MSI status. The MSI status of the tumor samples was determined using the five-marker Bethesda panel (BAT25, BAT26, D5S346, D2S123 and D17S250)59. Polymerase chain reaction (PCR) products were run on an Qsep 100 DNA fragment analyzer (Bioptic Inc., Taiwan) and analyzed using Qsep 100 viewer (Bioptic Inc., Taiwan). Microsatellite instability was defined by the presence of different sized alleles in tumor DNA compared with the matched normal DNA sample. We classified the results into microsatellite instability-high (MSI-H), microsatellite instability–low (MSI-L) and microsatellite stable (MSS) in tumors according to Bethesda guidelines60.
Immunohistochemical analysis
Immunohistochemical analysis for MLH1 protein was performed in 13 cases using the automated XT iVIEW DAB V.1 procedure on the BenchMark XT (Ventana, Tucson, AZ, USA), with incubation with MLH1 primary antibody (1:100, ES05, Dako) at 37 °C for 32 minutes. This was followed by a standard Ventana signal amplification and counterstaining with hematoxylin. Slides were mounted and examined by light microscopy. Only nuclear staining was considered for interpretation of data.
Statistical analysis
The Pearson chi-squared test and the Fisher’s exact test were used for the analysis. The time to recurrence-free survival was defined from the day of first surgery until recurrence. All tests were two-sided, with P < 0.05 considered as statistically significant. All statistical analyses were performed using the SPSS software (SPSS Inc., Chicago IL, USA).
Supplementary information
Acknowledgements
This work was supported the Post-Genome Technology Development Program (10053582, Development of Cancer Panel Companion Diagnostics System Based on NGS for the Personalized Medicine against Cancer) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea).
Author Contributions
W.S. Kim, H.Y. Park and S.E. Lee designed the experiments. S.E. Lee collected clinical samples and information. K.-Y. Kim, J.-H. Park, H. Roh, H.Y. Park, and S.E. Lee performed experiments and analyzed data. H.Y. Park and S.E. Lee interpreted results. The manuscript was drafted by H.Y. Park, S.E. Lee and edited by W.S. Kim. H.S. Lee provided expert consultation. All authors read and approved the final manuscript.
Data Availability
Please contact the corresponding author for all data requests.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ha Young Park, Email: pmint00@naver.com.
Wan-Seop Kim, Email: wskim@kuh.ac.kr.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-47439-3.
References
- 1.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader, N. et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. Journal of the National Cancer Institute106 (2014). [DOI] [PMC free article] [PubMed]
- 4.Parise CA, Caggiano V. Breast Cancer Survival Defined by the ER/PR/HER2 Subtypes and a Surrogate Classification according to Tumor Grade and Immunohistochemical Biomarkers. Journal of cancer epidemiology. 2014;2014:469251. doi: 10.1155/2014/469251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu VS, Kanaya N, Lo C, Mortimer J, Chen S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? The Journal of steroid biochemistry and molecular biology. 2015;153:45–53. doi: 10.1016/j.jsbmb.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine reviews. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kan Z, et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nature communications. 2018;9:1725. doi: 10.1038/s41467-018-04129-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saal LH, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer research. 2005;65:2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 9.Stemke-Hale K, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mark Kriegsmann VE, et al. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget. 2014;5:9952–9965. doi: 10.18632/oncotarget.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loibl S, et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Annals of oncology: official journal of the European Society for Medical Oncology. 2016;27:1519–1525. doi: 10.1093/annonc/mdw197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loibl S, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:3212–3220. doi: 10.1200/JCO.2014.55.7876. [DOI] [PubMed] [Google Scholar]
- 13.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silwal-Pandit L, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res. 2014;20:3569–3580. doi: 10.1158/1078-0432.CCR-13-2943. [DOI] [PubMed] [Google Scholar]
- 15.Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annual review of genetics. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- 16.Lynch HT, Smyrk T. An update on Lynch syndrome. Current opinion in oncology. 1998;10:349–356. doi: 10.1097/00001622-199807000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Vasen HF, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62:812–823. doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haraldsdottir S, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147:1308–1316 e1301. doi: 10.1053/j.gastro.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Win AK, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:958–964. doi: 10.1200/JCO.2011.39.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrow E, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clinical genetics. 2009;75:141–149. doi: 10.1111/j.1399-0004.2008.01125.x. [DOI] [PubMed] [Google Scholar]
- 21.Engel C, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:4409–4415. doi: 10.1200/JCO.2012.43.2278. [DOI] [PubMed] [Google Scholar]
- 22.Boyd J, et al. Male breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast cancer research and treatment. 1999;53:87–91. doi: 10.1023/A:1006030116357. [DOI] [PubMed] [Google Scholar]
- 23.Scott RJ, et al. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. American journal of human genetics. 2001;68:118–127. doi: 10.1086/316942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shanley S, et al. Breast cancer immunohistochemistry can be useful in triage of some HNPCC families. Familial cancer. 2009;8:251–255. doi: 10.1007/s10689-008-9226-4. [DOI] [PubMed] [Google Scholar]
- 25.Jensen UB, et al. Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast cancer research and treatment. 2010;120:777–782. doi: 10.1007/s10549-009-0449-3. [DOI] [PubMed] [Google Scholar]
- 26.Walsh MD, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16:2214–2224. doi: 10.1158/1078-0432.CCR-09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun J, et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin Cancer Res. 2017;23:6113–6119. doi: 10.1158/1078-0432.CCR-16-3227. [DOI] [PubMed] [Google Scholar]
- 28.Tung N, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reyes GX, Schmidt TT, Kolodner RD, Hombauer H. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124:443–462. doi: 10.1007/s00412-015-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan Y, et al. Analysis of hMLH1 missense mutations in East Asian patients with suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2007;13:7515–7521. doi: 10.1158/1078-0432.CCR-07-1028. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi M, et al. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer research. 2007;67:4595–4604. doi: 10.1158/0008-5472.CAN-06-3509. [DOI] [PubMed] [Google Scholar]
- 32.Santarosa M, Ashworth A. Haploinsufficiency for tumour suppressor genes: when you don’t need to go all the way. Biochimica et biophysica acta. 2004;1654:105–122. doi: 10.1016/j.bbcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 33.de Wind N, Dekker M, van Rossum A, van der Valk M, te Riele H. Mouse models for hereditary nonpolyposis colorectal cancer. Cancer research. 1998;58:248–255. [PubMed] [Google Scholar]
- 34.DeWeese TL, et al. Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11915–11920. doi: 10.1073/pnas.95.20.11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouffler SD, Hofland N, Cox R, Fodde R. Evidence for Msh2 haploinsufficiency in mice revealed by MNU-induced sister-chromatid exchange analysis. British journal of cancer. 2000;83:1291–1294. doi: 10.1054/bjoc.2000.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohsawa T, et al. Colorectal cancer susceptibility associated with the hMLH1 V384D variant. Molecular medicine reports. 2009;2:887–891. doi: 10.3892/mmr_00000187. [DOI] [PubMed] [Google Scholar]
- 37.Peng, H. X. et al. Molecular analysis of MLH1 variants in Chinese sporadic colorectal cancer patients. Genetics and molecular research: GMR15 (2016). [DOI] [PubMed]
- 38.Chiu CH, et al. MLH1 V384D polymorphism associates with poor response to EGFR tyrosine kinase inhibitors in patients with EGFR L858R-positive lung adenocarcinoma. Oncotarget. 2015;6:8407–8417. doi: 10.18632/oncotarget.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips KA, et al. Frequency of p53 mutations in breast carcinomas from Ashkenazi Jewish carriers of BRCA1 mutations. Journal of the National Cancer Institute. 1999;91:469–473. doi: 10.1093/jnci/91.5.469. [DOI] [PubMed] [Google Scholar]
- 40.Smith PD, et al. Novel p53 mutants selected in BRCA-associated tumours which dissociate transformation suppression from other wild-type p53 functions. Oncogene. 1999;18:2451–2459. doi: 10.1038/sj.onc.1202565. [DOI] [PubMed] [Google Scholar]
- 41.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 42.Middha, S. et al. Hechtman. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precision Oncology (2017). [DOI] [PMC free article] [PubMed]
- 43.Stadler ZK, et al. Reliable Detection of Mismatch Repair Deficiency in Colorectal Cancers Using Mutational Load in Next-Generation Sequencing Panels. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2016;34:2141–2147. doi: 10.1200/JCO.2015.65.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24. doi: 10.1186/s13000-017-0613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang J, et al. GCC2-ALK as a targetable fusion in lung adenocarcinoma and its enduring clinical responses to ALK inhibitors. Lung Cancer. 2018;115:5–11. doi: 10.1016/j.lungcan.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 46.Martin Marcel. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 47.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics43, 11 10 11–33 (2013). [DOI] [PMC free article] [PubMed]
- 50.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLaren W, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos AH, et al. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Genomes Project C, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cancer GenomeAtlas, N. Comprehensive molecular portraits of human breast tumours. Nature490, 61–70 (2012). [DOI] [PMC free article] [PubMed]
- 57.Pereira B, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hyeon J, et al. Targeted deep sequencing of gastric marginal zone lymphoma identified alterations of TRAF3 and TNFAIP3 that were mutually exclusive for MALT1 rearrangement. Mod Pathol. 2018;31:1418–1428. doi: 10.1038/s41379-018-0064-0. [DOI] [PubMed] [Google Scholar]
- 59.Boland CR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer research. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 60.Umar A, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Please contact the corresponding author for all data requests.