

Abstract
Nanotechnology offers new solutions for the development of cancer therapeutics that display improved efficacy and safety. Although several nanotherapeutics have received clinical approval, the most promising nanotechnology applications for patients still lie ahead. Nanoparticles display unique transport, biological, optical, magnetic, electronic, and thermal properties that are not apparent on the molecular or macroscale, and can be utilized for therapeutic purposes. These characteristics arise because nanoparticles are in the same size range as the wavelength of light and display large surface area to volume ratios. The large size of nanoparticles compared to conventional chemotherapeutic agents or biological macromolecule drugs also enables incorporation of several supportive components in addition to active pharmaceutical ingredients. These components can facilitate solubilization, protection from degradation, sustained release, immunoevasion, tissue penetration, imaging, targeting, and triggered activation. Nanoparticles are also processed differently in the body compared to conventional drugs. Specifically, nanoparticles display unique hemodynamic properties and biodistribution profiles. Notably, the interactions that occur at the bio-nano interface can be exploited for improved drug delivery. This review discusses successful clinically approved cancer nanodrugs as well as promising candidates in the pipeline. These nanotherapeutics are categorized according to whether they predominantly exploit multifunctionality, unique electromagnetic properties, or distinct transport characteristics in the body. Moreover, future directions in nanomedicine such as companion diagnostics, strategies for modifying the microenvironment, spatiotemporal nanoparticle transitions, and the use of extracellular vesicles for drug delivery are also explored.
Keywords: drug delivery, extracellular vesicle, multifunctional, nanomedicine, nanoparticle
Graphical Abstract
1. Introduction
Definitions of nanotechnology vary in different countries, which has implications for the clinical approval of nanodrugs [1]. Nevertheless, the common denominator among various definitions of nanotechnology is the use of constructs with nanoscale dimensions (Box 1). There are three distinct advantages that arise from applying nanotechnology to the treatment of disease. The first advantage is that several functional components can be incorporated into nanotherapeutics as they are much larger than most conventional drugs. Each component can be tailored to contribute in different ways to overall therapeutic activity and safety. The second advantage is that materials on the nanoscale display unique electromagnetic properties, which can be exploited for therapeutic purposes. The third benefit of using nanotechnology in medicine is an increased ability to differentiate between pathological and normal tissues. This ability is based on exploiting distinct interactions and processing pathways in the body that are unique to nanoparticles. Notably, site-specific delivery of systemically injected drugs can be improved by taking advantage of bio-nano interactions.
Box 1 |. Definitions of Nanotechnology.
Technology that fulfills the following criteria: i) has components in the nanosize range, ii) is man-made, and iii) has properties that arise due to nanoscale dimensions (Mauro Ferrari, Chief Executive Officer and President of the Houston Methodist Research Institute in the United States) [211]
Work at the atomic, molecular, and supramolecular levels in order to understand and create materials, devices and systems with fundamentally new properties and functions because of their small structure (Robert Langer, Institute Professor at the Massachusetts Institute of Technology in the United States) [211]
Toolbox that provides nanometer-sized building blocks for the tailoring of new materials, devices, and systems (Jackie Ying, Professor, Agency for Science, Technology and Research in Singapore) [211]
Science, engineering, and technology conducted at the nanoscale, which is about 1–100 nm (informal definition by the United States National Nanotechnology Initiative) [212]
The use of tiny structures - less than 1,000 nm across - that are designed to have specific properties (informal definition by the European Medicines Agency) [213]
The branch of technology that deals with dimensions and tolerances of less than 100 nm, especially the manipulation of individual atoms and molecules (Oxford Dictionaries) [214]
In 1995, Doxil (liposomal doxorubicin) became the first nanoparticle-based drug for cancer treatment to receive clinical approval in the United States [2]. Since then, several other nanodrugs have entered the market for treatment of various diseases. These nanotherapeutics include topical, local, and systemically administered organic and inorganic particles. Clinically approved nanotherapeutics are often categorized based on material composition, belonging to the lipid, polymer, protein, or metal-based nanoparticle category [3]. However, an alternative grouping of nanotherapeutics is based on the above-mentioned principles by which materials on the nanoscale can improve therapeutic efficacy. This type of categorization gives a more complete understanding of the diverse benefits of nanotechnology in medicine. Table 1 summarizes the advantages of nanotherapeutics using examples of clinical-stage nanoparticles for cancer therapy. For a more comprehensive list of nanoparticles in the clinic, please refer to reviews by Wicki et al [4] and Anselmo et al [3]. Although certain entries in Table 1 could fall into several categories, it is clear that the full potential of nanotechnology in medicine still lies ahead as nanodrugs that exploit a comprehensive set of nanoscale benefits have yet to reach the clinic. In addition to introducing a new system for the classification of therapeutic nanoparticles, the focus of this review is to discuss clinically available cancer nanodrugs. Some promising future opportunities in the field of cancer nanomedicine are also reviewed in the closing section.
Table 1 |.
Advantages of nanotechnology in medicine with examples of clinical-stage cancer nanotherapeutics
| Name | Composition | Cancer type | Status | Refs |
|---|---|---|---|---|
| MULTIFUNCTIONALITY | ||||
| Solubilization/Sustained Release | ||||
| Abraxane | Albumin-bound paclitaxel | Breast, lung, and pancreatic cancer | Approved in the US (2005) | [27] |
| Genexol-PM | Polymeric nanoparticles with paclitaxel | Breast, lung, and ovarian cancer | Approved in Korea (2007) | [11] |
| Lipusu | Liposomal paclitaxel | Breast, lung, and ovarian cancer | Approved in China (2006) | [12] |
| Marqibo | Liposomal vincristine sulfate | Acute lymphoblastic leukemia | Approved in the US (2012) | [67] |
| Protection from degradation | ||||
| Atu027 | Liposomal small interfering RNA (siRNA) | Advanced or metastatic pancreatic cancer | Phase I/II completed (2016) | [27] |
| ALN-VSP02 | Liposomal siRNA | Solid tumors with liver involvement | Phase I completed (2011) | [44] |
| DCR-MYC | Lipid nanoparticle with Dicer-substrate siRNA | Advanced solid tumors | Phase I/II terminated (2016) | [46] |
| MRX34 | Liposomal micro RNA (miRNA) | Advanced cancers | Phase I terminated (2016) | [215] |
| Immunoevasion | ||||
| Doxil | Liposomal doxorubicin (pegylated) | HIV-related Kaposi sarcoma, ovarian cancer, and multiple myeloma | Approved in the US (1995) | [27] |
| Onivyde | Liposoml irinotecan (pegylated) | Advanced pancreatic cancer | Approved in the US (2015) | [54] |
| Combination therapy | ||||
| Vyxeos | Liposomal cytarabine and daunorubicin | High-risk acute myeloid leukemia | Approved in the US (2017) | [27] |
| Targeted therapy | ||||
| MM310 | Ephrin type-A receptor 2-targeted liposomal docetaxel | Solid tumors | Phase I ongoing | [82] |
| Triggered activation | ||||
| CX-2009 | CD166 probodymaytansinoid conjugate | Solid tumors | Phase I/II ongoing | [107] |
| CX-072 | Antiprogrammed death-ligand 1 (PD-L1) Probody | Solid tumors and lymphoma | Phase I/II ongoing | [105] |
| ThermoDox | Thermosensitive liposomal doxorubicin | Hepatocellular carcinoma | Phase III completed (2016) | [108] |
| UNIQUE ELECTROMAGNETIC PROPERTIES | ||||
| NanoTherm | Iron oxide NP | Brain tumors | Approved in Europe (2011) | [27] |
| AuroShell | Gold nanoshells | Prostate cancer | Phase I ongoing | [113] |
| UNIQUE TRANSPORT PROPERTIES | ||||
| Enhanced permeability and retention (EPR) effect | ||||
| SMANCS | Polymer-neocarzinostatin conjugate | Liver and renal cancer | Approved in Japan (1993) | [27] |
| DaunoXome | Liposomal daunorubicin | HIV-associated Kaposi’s sarcoma | Approved in the US (1996) | [65] |
| Myocet | Liposomal doxorubicin | Metastatic breast cancer | Approved in Europe/Canada (2000) | [66] |
| MEPACT | Liposomal muramyl tripeptide phophatidyl-ethanolamine | Osteosarcoma | Approved in Europe (2009) | [68] |
| Hemodynamics | ||||
| Nanoparticle generator | Porous silicon microparticle with polymeric doxorubicin | Breast cancer lung metastasis | Planning of phase I | [91, 161] |
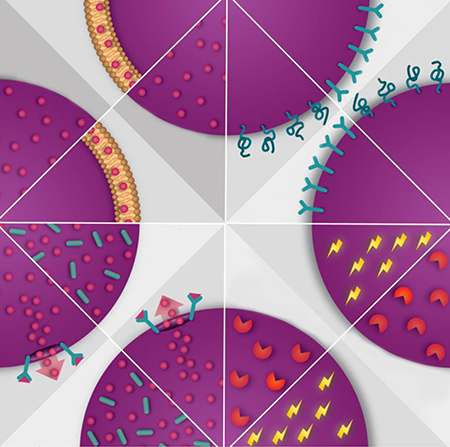
2. Exploiting multifunctionality
Nanomedicine enables the design of therapeutics with multiple functional elements. Namely, in addition to an active pharmaceutical ingredient, several other supportive components can be included due to the large size of nanoparticles compared to small molecules. These supportive components can aid in drug protection, site-specific activation, cellular uptake, and imaging. Figure 1 shows a summary of the various multifunctional properties of nanotherapeutics.
Figure 1 |. Multifunctional properties of nanotherapeutics.

2.1. Solubilization and sustained drug release
The simplest form of multifunctionality in nanomedicine is the addition of materials that enable drug solubilization. Several chemotherapeutic agents are poorly water soluble, which necessitates the use of toxic solvents for administration. For example, docetaxel and paclitaxel are administered with polysorbate 80 (Tween 80) and polyethoxylated castor oil (Kolliphor EL)/dehydrated ethanol, respectively [5], Accordingly, corticosteroids and antihistamines are prescribed to cancer patients in order to avoid the side effects of these solvents [6]. Nanomedicine offers a safer alternative to harmful formulations. For instance, Abraxane is an albumin-based paclitaxel-containing nanoparticle that has been approved by the United Stated Food and Drug Administration (FDA) for the treatment of several types of cancer [7]. Albumin is suitable for nanomedicine applications as it is the most abundant protein in the blood and serves as a carrier for endogenous hydrophobic molecules [8]. Clinical studies have shown that Abraxane administered at a 50% higher paclitaxel dose than the conventional formulation results in fewer and less severe cases of neutropenia, a common side effect of therapy [9]. Moreover, Abraxane can be administered over a shorter infusion time (30 min) than paclitaxel due to less risk of hypersensitivity reactions [10]. It is thought that the enhanced safety profile of Abraxane is potentially due to improved pharmacokinetics and the absence of Kolliphor EL [9]. Other examples of avoiding side effects from toxic solvents is the use of paclitaxel-containing polymeric micelles composed of polyethylene glycol (PEG) and poly-DL-lactic acid copolymers (Genexol-PM, approved in Korea [11]) and liposomes (Lipusu, approved in China [12]). Although nanomedicine usually enables higher drug doses to be administered, studies investigating optimal nanoparticle dosing, scheduling, and infusion rates are lacking. Additionally, due to the heterogeneity of nanotherapeutics the development of standardized methods for dose calculations is challenging. Notably, for breast cancer, Abraxane was shown to outperform solvent-based paclitaxel when administered every three weeks [9], while weekly injection schedules have revealed contradictory results in regards to the superiority of albuminbound paclitaxel. Specifically, one study demonstrated that Abraxane was superior to solventbased paclitaxel in regards to treatment response [13], while another study demonstrated a trend toward longer progression-free survival and less side effects with solvent-based paclitaxel [14]. The latter study suggests that nanoparticle-based therapy would primarily be advantageous for maintaining drug concentrations within the therapeutic window when doses are administered less frequently. However, it is worth noting that in this study, Bevacizumab was also administered in both treatment arms [14]. It is possible that Bevacizumab has different effects on free and nanoparticle-bound paclitaxel, especially as intratumoral accumulation of Abraxane is more dependent on vasculature properties. In fact, preclinical studies have demonstrated that antiangiogenic therapy lowers intratumoral drug accumulation [15].
Another important consideration for nanotherapeutics is high drug loading capacity to reduce exposure to excess materials that lack therapeutic efficacy. In particular, high levels of drug loading can be achieved with nanocrystals [16] or therapeutic agents that self-assemble into nanoparticles [17]. In addition to improving solubility, nanoparticles enable sustained drug release, which permits drug concentrations to stay within the therapeutic window instead of fluctuating between toxic and sub-therapeutic levels, which is usually the case for small molecules [18]. In a mouse study where liposomes with various drug release kinetics were compared, slower release rates, similar to that of clinically approved therapeutics, were found to lead to increased drug accumulation in tumors and improved therapeutic efficacy [19]. Notably, slower release kinetics also led to increased drug deposition in cutaneous tissues [19]. However, preclinical studies have also demonstrated that therapeutic efficacy is compromised with excessively slow release rates, as cancer cells have minimal exposure to bioavailable drugs [20].
2.2. Protection from degradation
Another supportive function of nanoparticles is to provide protection from enzymatic and mechanical degradation of drugs. This property is particularly important when it comes to drugs that are sensitive to degradation, such as various gene-silencing agents, including antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), and microRNAs (miRNAs). Gene-silencing agents provide new ways to modulate disease-associated signaling pathways that are difficult to target using contemporary approaches. However, the blood, extracellular space, and lysosome contain high levels of ribonucleases and deoxyribonucleases that rapidly destroy oligonucleotides [21–23]. Therefore, chemical modifications and drug delivery systems are necessary to ensure the stability of these therapeutic agents [24]. Moreover, the negative charge and relatively large size of oligonucleotides prevent efficient cellular internalization [25]. In addition to protecting oligonucleotides from enzymes in the circulation and extracellular space, nanoparticles can aid in cellular uptake and lysosomal escape [24]. Notably, in vitro-in vivo correlations of therapeutic efficacy are difficult to make as most cell culture models lack key aspects of the biological environment. In a study investigating the ability of in vitro assays to predict the effect of hepatocellular siRNA nanodelivery in animal models, it was found that cell type and siRNA entrapment efficiency played a role, while nanoparticle size and zeta potential did not [26]. In particular, freshly isolated primary hepatocytes were more predictive of in vivo gene silencing efficiency compared to cancer cells. Interestingly, the use of HeLa cervical cancer cells lead to more predictive results than Hepa1–6 liver cancer cells. Overall, there is a need to evaluate and optimize in vitro assays for improving translation of nanodrugs.
Currently, there are several clinical trials for oligonucleotide-containing nanoparticles, and one has received clinical approval [27, 28]. A few chemically modified ASOs have also reached the market. For instance, fomivirsen, mipomersen, nusinersen, and eteplirsen are clinically approved in the United States for the treatment of cytomegalovirus retinitis (intraocular injection), homozygous familial hypercholesterolemia (subcutaneous injection), spinal muscular atrophy (intrathecal injection), and Duchenne muscular dystrophy (subcutaneous injection) respectively [29, 30]. A major challenge for the clinical translation of gene silencing agents is safety. In a pilot study of an intravenously injected lipid-conjugated ASO (Imetelstat) for myelofibrosis, 18% of patients experienced thrombocytopenia, which lead to intracranial hemorrhage and death in one patient [31]. Similarly, a phase II clinical trial of Imetelstat for pediatric brain tumors was prematurely terminated due to two deaths caused by thrombocytopenia-induced intratumoral hemorrhage [32]. However, platelet deficiency does not seem to be a class effect of ASOs [33] and may be due to specific chemical modifications that lead to binding to platelet-specific glycoprotein-VI receptors [34].
In 2016, the field of RNA interference (RNAi) faced a major setback as the development of revusiran, a promising siRNA-N-acetylgalactosamine (GalNAc) ligand conjugate for hereditary transthyretin amyloidosis with cardiomyopathy, was discontinued due to higher mortality than the placebo group in a phase III trial [35]. Potential reasons for increased mortality in response to revusiran are off-target effects, unexpected on-target effects, immunological activation, and poor study design in regards to statistical analysis. Mice studies have demonstrated that siRNAs can induce vascular and immune effects through activation of Toll-like receptor 3 (TLR3) [36], which is a sensor for foreign double-stranded RNA [37]. However, chemical modifications, such as those used for revusiran [38], have been shown to prevent TLR activation [39]. Moreover, design algorithms were used in the development of revusiran in an attempt to avoid off-target effects [35]. It is possible that on-target effects arising from suppression of wild type transthyretin could lead to side effects, as this protein plays an important role in several physiological processes. For instance, thransthyretin is a carrier for thyroid proteins and retinol-binding proteins in the blood, protects pancreatic β-cells from apoptosis, and displays proteolytic activity against amyloid β, apolipoprotein A-I, and amidated neuropeptide Y [40]. With several oligonucleotide nanomedicines in clinical trials, it remains to be seen whether toxicity will be common for this type of drugs or limited to specific therapeutic agents/formulations. In fact, encouraging safety results were recently reported for a phase III trial of patisiran [41], a liposome-based version of revusiran [42], which became the first siRNA-based therapy to receive clinical approval [28], marking a major breakthrough in this field. In regards to cancer treatments, RNAi nanotherapeutics are in earlier stages of clinical development. Specifically, lipid, cyclodextrin, and bacterial minicells have been utilized in phase I/II clinical trials for delivery of siRNAs and miRNAs [43]. Although the treatments were generally well tolerated with some mild inflammatory reactions, a small number of grade 3–4 adverse events [44–46] and one potential therapy-related death due to liver failure [44] were reported. The most favorable outcome of these trials was stable disease with some partial responses [43]. Accordingly, the clinical development of several siRNA nanotherapeutics for cancer, such as the lipid nanoparticle DCR-MYC, has been discontinued due to insufficient patient outcomes [47]. On the contrary to hereditary conditions that are caused by a single genetic defect, most cancers have several genome abnormalities that together contribute to malignancy. Therefore, clinical trials should be designed as combination therapies, as siRNA-based treatments for cancer are unlikely to be effective unless coupled with other therapeutic agents. Another promising application of nanomedicine is gene editing based on delivery of components of clustered regularly interspaced short palindromic repeats (CRISPR)/associated protein 9 (CRISPR/Cas9) technology. Although genome editing therapeutic agents are in early stages of development, nanoparticles offer an alternative to viral vectors, which pose various safety concerns [48].
2.3. Immunoevasion
Another example of multifunctionality in nanomedicine is the use of components that facilitate immunoevasion. Common strategies for avoiding immunological recognition and clearance of nanoparticles include surface modifications with anti-fouling polymers [49], self peptides [50], and cell coatings [51, 52]. A clinically approved strategy for decreasing nanoparticle uptake by the immune system relies on the stealth polymer, PEG. Nanoparticle pegylation creates a steric barrier that reduces binding of plasma opsonins and prevents interactions with cells [53]. Clinically approved examples of pegylated nanoparticles include Doxil [2] and Onivyde [54], which are liposomal formulations of chemotherapy drugs. In many cases, the altered pharmacokinetics of drugs as a result of nanodelivery reduces side effect. In fact, Doxil was approved for multiple myeloma based an equivalent efficacy but improved safety compared to free doxorubicin [2]. However, it is important to note that while nanotherapeutics generally reduce drug toxicity, they may cause new types of side effects to arise. For instance, although Doxil decreases the risk of cardiotoxicity, the occurrence of palmar-plantar erythrodysesthesia increases [55].
Notably, pegylation does not necessarily result in substantial improvements in tumor accumulation, although this strategy dramatically prolongs circulation times [56, 57]. The PEG dilemma is based on the inability of this polymer to specifically prevent interactions with immune cells, leading to decreased nanoparticle uptake in cancer cells as well [58]. Additionally, pegylation predominantly delays immunological clearance as opposed to preventing this process, as evidenced by studies demonstrating that phospholipid-PEG conjugates gradually detach from lipid nanoparticles in the circulation [59, 60]. Furthermore, in some patients, pegylated nanoparticles have been found to activate the complement system, which can increase immunological removal and cause hypersensitivity reactions [61, 62]. Frequent infusion reactions have also occurred when Doxil was administered over a 1 h period, causing longer infusion times (4 h) to be used [63]. In animal models, repeated injections have resulted in the production of PEG antibodies that are responsible for the accelerated blood clearance (ABC) phenomenon, in which pegylated nanoparticles are rapidly cleared by the immune system, paradoxically leading to much shorter circulation times [64]. However, it is questionable whether the ABC phenomenon is clinically relevant, as excessively high doses of pegylated liposomes have been used in animal studies. Notably, several clinically approved liposomes for cancer therapy are nonpegylated, including DaunoXome [65], Myocet [66], Marqibo [67], and MEPACT [68]. Taken together, more effective and specific strategies are necessary to avoid immunological clearance of nanoparticles. Efficient immunoevasion will likely require the concurrent use of microenvironmental modification strategies and innovative nanoparticle design approaches that avoid immunological triggers and incorporate markers of self, such as CD47 [50]. Various strategies for priming the innate immune system for reduced nanoparticle uptake will be discussed in the future directions section of this review. A major exception to the aforementioned approaches is immunotherapy, where the ability of nanoparticles to activate the immune system is advantageous. In fact, a few nanovaccines for the treatment of cancer have already reached clinical trials. For instance, a liposome-based vaccine that incorporates patient-specific cancer cell membrane proteins as well as interleukin-2 (IL-2) has shown promise for inducing sustained tumor-specific T cell responses in patients with lymphoma [69]. Indeed, one of the main advantages of nanoscale vaccines is the ability to incorporate multiple components, such as various antigens and adjuvants, in the same platform. Additionally, nanoparticles enable the use of safe and broadly applicable messenger RNA (mRNA)-based vaccines by providing protection from degradation and inducing cellular internalization. For example, in a small cohort of melanoma patients, antigen-specific T-cell responses occurred after systemic treatment with a lipid-based mRNA nanovaccine designed to target dendritic cells [70]. Alternatively, dendritic cells can be exposed to antigen-loaded nanoparticles ex vivo prior to injection into the body as cell-based vaccines [71].
2.4. Combination therapy
Combination treatment regimens are considered standard of care for most cancer types [72]. In fact, concurrent targeting of multiple malignant processes or different elements of the same process usually leads to improved outcomes [73]. Studies have also demonstrated that it is important to consider drug ratios to obtain optimal therapeutic synergy [74]. A major advantage of nanomedicine is the ability to deliver several drugs in the same nanoparticle. Indeed, it is easier to achieve ideal intratumoral and intracellular drug ratios by co-encapsulation in nanoparticles than taking into account the pharmacokinetics of individual therapeutic agents. In 2017, Vyxeos became the first nanomedicine for combination therapy to enter the market. This nanodrug is a liposomal formulation of cytarabine and daunorubicin [75] approved for the treatment of high-risk acute myeloid leukemia (AML). Vyxeos has a cytarabine to daunorubicin molar ratio of 5:1, as this ratio was found to display the highest synergistic therapeutic activity in cell culture and animal studies [76]. Importantly, it was also shown that the ratio was maintained for prolonged periods of time in the bone marrow [76]. In a phase III trial for secondary AML, patients treated with Vyxeos had a median overall survival of 9.56 months compared to 5.95 months in patients administered with a combination of free cytarabine and daunorubicin (standard of care) [77]. In addition to delivery of chemotherapy cocktails, it is likely that nanomedicine will be beneficial for successful implementation of several other therapeutic combinations. Furthermore, theranostic nanoparticles that contain both therapeutic and imaging agents could facilitate disease staging, treatment selection, response assessment, and recurrence detection [78]. Nanoparticle-mediated combination therapy could also be useful for increasing drug penetration depth in tumors. In fact, it is essential that drugs effectively penetrate tumor tissue to avoid subpopulations of cancer cells surviving and causing tumor recurrence [79]. Nanoparticles that incorporate proteolytic enzymes that break down the dense extracellular matrix (ECM) of tumors have been developed for improved intratumoral drug distribution [80, 81]. However, nanoparticles that contain permeation enhancers have not yet reached the clinical stage and are likely to display side effects-induced by proteolytic enzymes.
2.5. Targeting.
Nanoparticle targeting is based on surface conjugation of molecular targeting ligands. This strategy enables the design of drug carriers that recognize biomolecules that are associated with specific disease conditions. Such nanoparticles are usually designed to bind to molecules that are overexpressed on the surface of tumor vasculature or cancer cells. Targeted nanoparticles have not yet reached the market but several are currently undergoing clinical trials. For instance, MM310 is an ephrin type-A receptor 2 (EphA2)-targeted docetaxel-containing liposome that is in phase I clinical trials for patients with solid tumors [82]. Notably, EphA2 is present on the surface of multiple cells in the tumor microenvironment, including cancer cells, stromal cells, and endothelial cells [83]. The field of targeted nanodelivery underwent a major setback in 2016, when the promising nanodrug BIND-014 failed in a phase II clinical trial for the treatment of cervical and head-and-neck cancers, followed by the bankruptcy of Bind Therapeutics [84]. BIND-014 is a docetaxel-containing polymeric nanoparticle with surface ligands for prostate-specific membrane antigen (PSMA) [85], which is expressed on prostate cancer cells and on the vasculature of many other solid tumors [86]. Another commonly exploited target on tumor blood vessels is the αVβ3 integrin receptor [87], while nanoparticle surface ligands for the transferrin receptor [88] and folate receptors [89] have frequently been utilized for cancer cell targeting. Notably, molecular recognition of cancer cells does not increase nanoparticle passage from the circulatory system into the tumor interstitium, but rather promotes retention after extravasation has occurred. In addition to enhanced retention, one of the main benefits of cancer cell targeting is increased cellular internalization due to ligand-mediated endocytosis. In fact, even untargeted nanoparticles can substantially aid in the cellular uptake of therapeutic agents through various forms of endocytosis [90]. Nanoparticle-mediated internalization can also circumvent drug efflux pumps, thereby overcoming drug resistance [91].
There are several potential reasons for disappointing outcomes with targeted nanotherapeutics, including increased immunological recognition and clearance due to the presence of surface ligands. Moreover, targeting molecules enlarge the size of nanoparticles, which could lead to decreased levels of extravasation and intratumoral penetration. Another problematic factor is the binding-site-barrier, which occurs when ligands bind with high affinity to target molecules, thereby preventing further diffusion throughout the tumor [92]. Therefore, it is important to balance binding affinity and diffusion ability when designing targeted nanoparticles. Strategies to achieve this balance include the use of fast-penetrating nanoparticles, low-affinity ligands, stimuli-triggered drug release, and ligand unveiling strategies [93, 94]. Moreover, targeting approaches should be tailored based on tumor types. For example, the stroma content of tumors is likely to have a major impact on which cells in the tumor microenvironment would be ideal targets [93]. Accordingly, an understanding of the tissue and cell surface distribution of molecular targets as well as the effect of ligand-receptor binding on nanoparticle diffusion and cellular internalization will be critical for successful clinical translation of targeted nanoparticles. An additional challenge of targeted delivery is the protein corona that forms around the surface of nanoparticles upon exposure to biological fluids [95]. This layer of biomolecules can mask surface ligands and prevent target recognition [96]. Moreover, protein binding to the nanoparticle surface can lead to conformational changes, which could induce an immunological response [97]. Additionally, the biomolecular layer can affect biodistribution, nanoparticle degradation, and drug release. A potential solution to the corona problem is surface modification of nanoparticles to promote binding of specific plasma proteins that dictate preferential transport properties [98]. For instance, one study showed that polymeric nanoparticles coated with polysorbate 80 attracted apolipoprotein binding, which mediated blood brain barrier crossing [99]. Additionally, carbon nanotube-polymer complexes have been surface engineered to display binding pockets for specific proteins [100]. Similarly, libraries of hydrogel nanoparticles with distinct biomolecular absorption properties can be used to dictate the composition of the protein corona [101]. Although studies evaluating interactions between nanoparticles and biomolecules following plasma incubation can be informative, studies that in a systematic manner address the impact of nanoparticle characteristics on pharmacokinetics are essential for improving tumor-targeting. Coupling in vivo studies with mathematical modeling of biomolecule-nanoparticle interactions [102] will be valuable for establishing a general framework for optimizing nanoparticle design. Moreover, in situ measurements of the hydrodynamic radius of nanoparticles using fluorine-19 diffusion-ordered nuclear magnetic resonance spectroscopy may provide dynamic protein absorption measurements in vivo [103]. Additionally, although proteomic studies evaluating the composition of the protein corona are frequently reported, visualization of the biomolecular layer could provide important structural insight. Taken together, an increased understanding of interactions that occur at the bio-nano interface will be critical for the success of targeted delivery.
2.6. Triggered activation
Nanomedicine also enables the triggered activation or release of therapeutic agents in the tumor. There are several stimuli in the tumor microenvironment that can be used as triggers, such as low pH, high levels of glutathione, and elevated amounts of enzymes. Additionally, exogenous stimuli, such as heat, ultrasound, and magnetic/electric fields, can be applied to externally accessible tumors to induce drug release from nanomaterials that are responsive to specific energy sources. Clinical-stage examples of stimuli-responsive therapeutics include Probodies, which are antibodies that are activated by tumor-associated proteases, such as urokinase-type plasminogen activator, membrane-type serine protease 1, legumain, and various matrix metalloproteinases (MMPs) [104]. The antigen-binding site of Probodies is masked by a peptide that is cleaved in the tumor microenvironment, thereby reducing antibody binding to normal cells. For instance, CX-072 is a anti-programmed death-ligand 1 (PD-L1) Probody in phase I/II clinical trials for patients with solid tumors and lymphoma [105]. This Probody has the potential to decrease the side effects of anti-PD-L1 therapy, including the risk of autoimmune-like conditions [104, 106]. Another example is CX-2009, an anti-CD166 probody-maytansinoid DM4 conjugate that is undergoing phase I/II clinical trials for solid tumors [107]. CD166 is an antibody that is upregulated in tumors, while DM4 is a cytotoxic agent. Although Probodies and antibody-drug conjugates are multifunctional nanosized therapeutics, they are not generally categorized as nanomedicine. Nevertheless, similarly to nanodrugs, Probodies have the potential to reduce side effects and enable the use of higher doses of therapeutic antibodies. A clinical stage example of a nanoparticle strategy that utilizes an external trigger for drug release is ThermoDox, which is a thermosensitive liposome that releases doxorubicin in response to temperature elevations above 39 °C [108]. In a Phase III trial of ThermoDox coupled with radiofrequency-induced heating for hepatocellular carcinoma, the primary endpoint of progression-free survival was not reached [108]. Potential reasons for the failure of this trial include cancer cell resistance to doxorubicin, inadequate dose of doxorubicin, inappropriate primary endpoints, and unexpected anticancer activity in the control group that received radiofrequency ablation (RFA) without ThermoDox [108]. Additionally, the lack of a standardized procedure for RFA is likely to have impacted the results of this study. In fact, retrospective analysis of patients that received RFA for at least 45 min did show a substantial improvement in progression-free survival [108]. This study highlights the importance of selecting appropriate drug candidates, drug doses, diseases, and treatment procedures in order to gain a clear understanding of the benefits of the nanoscale component of the treatment strategy.
3. Exploiting electromagnetic properties
Inorganic nanoparticles can convert energy from external sources into heat, which can be utilized for therapeutic purposes. A major advantage of these strategies is that the external energy source, such as near-infrared light, radiofrequency waves, and magnetic fields, can usually be localized to a specific area in the body in order to reduce damage to healthy tissues. In fact, nanoparticle-based thermal therapy can cause intratumoral temperatures to rise above 70 °C [109], resulting in cancer cell death. Most of the therapies that have shown promise in the preclinical and clinical setting are based on local injection of nanoparticles. However, it is important to develop systemically administered therapies, as the majority of cancer deaths are caused by metastatic lesions that are impossible to individually target through local interventions. Studies have shown that high concentrations of nanoparticles are necessary to achieve hyperthermia (> 42 °C) or thermal ablation (> 50 °C) [110]. Therefore, heat-generation capacities and biodistribution profiles should be optimized to potentially achieve therapeutic efficacy through systemic injections.
The only example of a clinically approved treatment strategy that utilizes the unique electromagnetic properties of nanoparticles for thermal ablation is NanoTherm. NanoTherm is an iron oxide nanoparticle-based suspension, which is approved in Europe for the treatment of patients with brain tumors [111]. Iron oxide nanoparticles are administered through local infusion into the brain, after which patients are exposed to an alternating magnetic field that causes heat-induced tumor ablation. Studies have shown that this treatment combined with radiotherapy has few side effects and extends overall survival compared to conventional treatment options [111]. In addition to the clinical utilization of magnetic nanoparticles, clinical trials are underway to assess the safety and efficacy of laser-heated gold nanoshells (AuroShell) in prostate cancer patients [112, 113]. In the preclinical setting, near-infrared light [114, 115] and radiofrequency irradiation [116] have been applied to heat gold and carbon nanoparticles for cancer therapy. Besides tumor ablation, less dramatic nanoparticle-induced temperature elevations have been used to enhance intratumoral permeability for improved drug delivery [117]. However, the limited tissue penetration depth of near-infrared light brings into question the utility of this strategy for clinical applications. Although radio waves can easily penetrate through the body, there is controversy surrounding the contribution of gold nanoparticles to radiofrequency heating. In fact, one study reported that electrolytes in nanoparticle suspensions are responsible for radio wave-induced temperature elevations [118].
4. Exploiting mass transport characteristics
The circulatory system is responsible for the transport of nutrients, oxygen, waste products, cells, and endocrine factors throughout the body. The blood circulation is also exploited for the delivery of intravenous or oral drugs, which account for the largest proportion of therapeutic agents. The characteristics of the vascular system differ based on developmental stage, organ type, and disease condition. Indeed, properties such as vessel diameter, blood flow velocity, and vascular permeability vary depending on the physiological function of the tissue. For example, the capillaries in the lungs are narrower than those in other organs to ensure that red blood cells come in close contact with alveolar spaces, enabling efficient gas exchange [119]. Conventional drugs are often smaller than 1 nm and can efficiently diffuse throughout most tissues in the body regardless of organ-specific vasculature characteristics. Although widespread diffusion permits exposure of diseased cells to drugs, dose limitations are necessary due to damage inflicted on healthy cells. Drug doses that are intended to avoid adverse side effects are in many cases insufficient to treat disease. Therefore, there is a need to develop strategies for site-specific drug delivery. Compared to small molecules, nanoparticle transport in the body is more dependent on vasculature properties. For instance, widespread diffusion of nanoparticles in tissues throughout the body rarely takes place, as nanoparticles are too large to pass through the vascular wall. Moreover, studies have indicated that the cutoff size for renal clearance of rigid particles is 5.5 nm [120]. A large portion of nanoparticles in the circulation are sequestered by the liver due to vascular fenestrations and resident macrophages that are responsible for the clearance of pathogens and cellular debris, which are often in the nanosize range [121, 122]. The dependence of nanoparticle transport on vasculature properties can be exploited for tissue-specific delivery to avoid complete liver clearance. Accordingly, the term transport oncophysics was coined to denote a different way of viewing cancer, which enables the design of drug delivery strategies that take advantage of the unique transport properties of tumors [123–125]. The physics component indicates that physics is required to understand the movement of nanoparticles within and between various biological compartments in the body.
The growth of a tumor beyond 2–3 mm in diameter requires the recruitment of a vasculature network to ensure efficient gas, nutrient, and waste exchange [126]. The characteristics of tumor blood vessels differ substantially from healthy vasculature as the neovascularization process (tumor-associated angiogenesis) is forced to take place in a short period of time to accommodate rapid tumor growth [127]. In particular, tumors often display a disorganized vasculature network and individual blood vessels exhibit immature properties, such as endothelial fenestrations. Drug delivery systems can be designed to exploit the unique characteristics of cancer blood vessels to increase drug accumulation in tumors. The most well known phenomenon that relates to the utilization of abnormal tumor vasculature properties for drug delivery purposes is the enhanced permeability and retention (EPR) effect. Another emerging drug delivery strategy that exploits differences between normal and tumor blood vessels is based on hemodynamics.
4.1. EPR effect
The EPR effect, which was first described in 1986 [128, 129], entails the increased accumulation of nanosized matter in tumors primarily due to vascular fenestrations (up to ~500 nm [130]) and poor lymphatic drainage. Specifically, immature vasculature has reduced pericyte coverage and less endothelial tight junctions, causing gaps in the vessel wall. Additionally, a lack of functional lymphatic vessels, abnormal blood flow patterns, and a dense ECM have been proposed as explanations for enhanced retention of nanoparticles in the tumor microenvironment after extravasation [131, 132]. In particular, it is likely that the characteristic intratumoral increase in ECM components [133] leads to adhesive interactions with nanoparticles. Fibronectin and collagen have both been referred to as extracellular glue due to the capacity of these proteins to bind a wide variety of molecules [134, 135]. In fact, collagen is derived from the Greek word ‘kolla’, which means glue [135]. In addition to the biomolecular adhesive properties of ECM components, it is probable that the structural organization of the protein fiber network will entrap nanoparticles. Another reason for accumulation of nanoparticles in tumors is interactions with cells in the tumor microenvironment. For instance, it has been shown that tumor-associated macrophages can promote intratumoral uptake of nanotherapeutics [136]. The beneficial effects of many clinically approved nanoparticles, especially liposomes [137] and polymeric nanoparticles [138], have been attributed to the EPR effect. In the preclinical setting, liposomal delivery has been found to lead to a several fold increase in intratumoral drug accumulation compared to administration of free drugs [139–141]. Although there is evidence of the EPR effect in humans, considerable heterogeneity exists in the prevalence of this phenomenon among individuals [142]. For example, the accumulation of liposomes in tumors varied from 0–3.5% of the injected dose in patients with head and neck, breast, and lung cancer [143]. Notably, two out of 17 [143] and two out of 22 [144] cancer patients exhibited undetectable levels of intratumoral liposomes, suggesting that the EPR effect may be entirely absent in certain cases. Moreover, there was an inverse correlation between tumor size and liposome accumulation/kg tumor tissue [143]. This observation may be due to large tumors having more necrotic areas that contribute to tumor size but lack functional vasculature. Indeed, in animal studies, larger tumors have been associated with higher levels of necrosis and less liposome uptake normalized by tumor weight [145]. In another patient study, tumor vasculature density, measured by CD31 staining, was found to correlate with liposome accumulation [146]. In addition to liposomes, the EPR effect has also been observed in patient studies with other types of nanoparticles. For instance, polymeric nanoparticles were shown to display increased deposition in tumors compared to adjacent noncancerous tissues in nine out of nine cancer patients [147]. Notably, preferential tumor accumulation of nanoparticles has been observed for both primary [143, 144, 146–148] and metastatic [144, 149] cancer patients. Analysis of 17 clinical studies revealed that the EPR effect was most pronounced in pancreatic, colon, breast, and stomach cancers, which displayed more than ten times higher intratumoral nanoparticle levels compared to those in normal tissue [150]. In contrast, head and neck cancer and melanoma patients displayed a twofold increase in preferential tumor accumulation [150]. In addition to interpatient variability in regards to the EPR effect, it is probable that high levels of intratumoral heterogeneity exist. In fact, in one study, nanoparticle levels differed substantially (four-fold) in two samples taken from different locations of a stomach tumor [151]. Mathematical modeling [152] and experimental studies [79] have demonstrated that such variations in spatial drug distribution can lead to the formation of drug resistance, especially in cases in which cancer cells have limited ability to migrate. The amount of variation observed in the distribution of nanoparticles in these small cohorts of cancer patients is a strong indicator that the EPR effect in humans is more heterogeneous than what has been observed in the preclinical setting. In fact, the characteristics of tumors in animal models and patients often differ substantially in regards to growth rate, size relative to body mass, and features of the microenvironment, all of which are likely to influence the neovascularization process, which serves as the underlying cause of the EPR effect. In particular, cancer cell injections in animals result in tumors that lack typical tumor-stroma interactions, which play a major role vascularization and nanodelivery. Taken together, the lack of animal models that accurately recapitulate the EPR effect as well as inter-patient and intra-patient variability is likely to play a major role in the clinical failure of several nanotherapeutics.
4.2. Tumor hemodynamics
The disorganized structure of tumor vasculature leads to blood flow abnormalities. The flow of fluid through a tube can be expressed as the shear rate, which is calculated based on the tube diameter and fluid velocity. In general, the shear rates that occur in tumor blood vessels are lower (< 100 s−1) than those in normal blood vasculature (> 100 s−1) [153, 154]. Accordingly, a complementary strategy to utilizing the EPR effect for improved tumor drug delivery is taking advantage of hemodynamics. Specifically, drug carriers can be designed to preferentially attach to the vasculature in blood flow conditions with low shear rates (Fig. 2). The two requisites for particle attachment is proximity to the vessel wall during flow and sufficient adhesive interactions with the endothelium. The size and shape of drug carriers can be tailored to meet both of these criteria. For instance, discoidal microparticles display a tendency to drift laterally against the wall, while spherical particles follow the streamline [155]. Additionally, disc-shaped microparticles have a much larger parallel surface area that can adhere to the vessel wall [153, 156]. Therefore, it is not surprising that platelets display a discoidal morphology as hemostasis is dependent on binding of these cell fragments to the endothelium [157]. Notably, although the vast majority of drug delivery systems are spherical, discoidal particles appear to be superior in regards to attaching to the vasculature. Importantly, the diameter and height of disc-shaped particles can be fine-tuned to obtain different vasculature binding patterns in normal and tumor tissues. In fact, in normal vasculature with higher shear rates, strong hydrodynamic forces cause adhered particles to dislodge from the endothelium [153, 156]. On the contrary, reduced shear rates in tumor blood vessels enable permanent adhesion of microparticles to the vasculature. In essence, strong particle binding to the endothelium is promoted by low shear rates, while high shear rates lead to transient particle adhesion. Hemodynamic targeting approaches for cancer therapy rely on identifying optimal ratios between particle adhesion and hydrodynamic forces. Mathematical modeling and animal studies have demonstrated that discoidal microparticle-based drug delivery systems can be designed to preferentially bind to tumor vasculature, resulting in increased tumor accumulation and improved therapeutic efficacy compared to spherical drug carriers [91, 153, 158, 159]. For instance, in a mouse model of breast cancer, tumor accumulation of discoidal silicon microparticles was fivefold higher than that of spherical particles with the same diameter [158]. Similarly, in a mouse model of melanoma lung metastasis, the accumulation of disc-shaped microparticles in tumor tissue was ten times higher than that of spherical nanoparticles [159]. Although certain dimensions of these particles are in the micron size range, some dimensions are nano-sized and the particles have other nanotechnological features, such as nanoscale pores. Other non-spherically shaped particles, such as rods, have also outperformed spheres in binding to the endothelium of target organs [160]. An important consideration for the development of nanotehrapeutics with unconventional shapes is potential manufacturing challenges. While lithography-based techniques can be used for large-scale fabrication of silicon [161] and polymeric [162] particles of various shapes and sizes, this may prove less feasible for other materials, such as lipids.
Figure 2 |. Hemodynamic-based tumor targeting.

Spherical nanoparticles follow the streamline (upper panel), while discoidal microparticles flow close to the vessel wall (lower panel). Proximity to the endothelium and a large parallel surface area that can attach to the vessel wall make disc-shaped particles ideal for vascular adhesion. Unique hemodynamics in normal and tumor blood vessels lead to different patterns of particle adhesion. Specifically, the disorganized vasculature network of tumors leads to lower shear rates and permanent attachment of discoidal microparticles
Notably, patients have also been shown to display disorganized tumor vasculature characteristics [163]. In fact, an intravital microscopy study demonstrated that vasculature shear rates of human tumors are lower than those observed in animal models [163], highlighting the promising potential of hemodynamic targeting in the clinical setting. Clinical trials that utilize drug delivery strategies based on fluid dynamics are currently being planned for the treatment of breast cancer lung metastasis based on promising preclinical results [91, 161].
5. Future Directions
The use of nanocarriers has led to major improvements in site-specific drug delivery compared to administration of free drugs. These improvements range from a 10 to 100-fold increase in intratumoral drug accumulation [139, 141, 164, 165]. Despite major advancements in the biodistribution of drugs, less than 1% of systemically injected nanoparticles typically reach the tumor [57]. Over 90% of an injected nanoparticle dose usually ends up in the liver and spleen, as these organs are responsible for the clearance of nanosized cell debris and pathogens, which are often indistinguishable from nanotherapeutics [121, 122]. Moreover, the reasons for failed clinical trials in nanomedicine are largely unknown and are likely to be a combination of multiple complex factors [166]. Accordingly, nanomedicine still faces major challenge (Table 2) that necessitates the development of additional therapeutic strategies to enhance tumor accumulation. Such approaches include engineered extracellular vesicles, drug delivery vehicles that undergo spatiotemporal transitions, strategies to prime the microenvironment, and patient stratification according to tumor properties.
Table 2 |.
Limitations of nanotherapeutics
| Type | Limitations |
|---|---|
| Clinically approved | |
| Non-pegylated nanoparticles | • Rapid clearance by the immune system |
| • Low tumor accumulation | |
| Pegylated liposomes | • Polyethylene glycol (PEG) dilemma (reduced interactions with cancer cells) |
| • Transient protection from immunological clearance | |
| • Potential activation of the complement system | |
| • Accelerated blood clearance (ABC) phenomenon (antibody-mediated clearance following repeated injections) | |
| Iron oxide nanoparticles | • Require local injection to achieve sufficient quantities for thermal ablation |
| • Unknown effects of long-term accumulation in the body | |
| Preclinical/Clinical trials | |
| RNA interference nanoparticles | • Complete protection required due to susceptibility of RNA to degradation |
| • Potential side effects | |
| • Endosomal escape strategies necessary | |
| • siRNA-based therapeutics are unlikely to be effective as monotherapy | |
| • Toxicity of cationic nanoparticles that result in higher loading efficiency | |
| DNA, mRNA nanoparticles | • Problematic nanoparticle encapsulation due to large size |
| • Immune responses in intracellular compartments | |
| • Endosomal escape strategies necessary | |
| Immunotherapeutic nanoparticles | • Unspecific immune activation |
| • Costly personalized approaches | |
| Nanoparticles with permeation enhancers | • Side effects |
| • Risk of increased metastasis | |
| Targeted nanoparticles | • Masking of targeting ligands by the protein corona |
| • Binding-site-barrier (ligands bind with high affinity to target molecules, thereby preventing further diffusion throughout the tumor) | |
| • Decreased extravasation and intratumoral penetration due to increased size (targeting ligands on the surface) | |
| • Potentially increased immunoactivation (targeting ligands on the surface) | |
| Triggered-release strategies | • Tumors need to be accessible if using external energy triggers |
| • Rarely completely specific to tumor tissue if using triggers in the microenvironment | |
| Gold nanoparticles | • Limited tissue penetration depth of infrared light |
| • Unknown effects of long-term accumulation in the body | |
| Non-spherical nanoparticles | • Manufacturing challenges |
| • Material limitations | |
| Transitional/multistage nanoparticles | • Complex manufacturing |
| • Complex characterization | |
| • Regulatory challenges |
5.1. Engineered extracellular vesicles
Extracellular vesicles are released by all cells and play a central role in cell communication by serving as endogenous carriers for biomolecules over both short and long distances [167]. Genetic or chemical engineering approaches can transform extracellular vesicles to drug delivery vehicles [168, 169]. The complexity of natural vesicle membranes is challenging to replicate in the synthetic setting and provides unique functional properties. For instance, it was recently shown that extracellular vesicles carrying gene silencing agents against oncogenic KRAS display improved therapeutic efficacy in pancreatic cancer models compared to liposomes [170]. Notably, it was postulated that the presence of the transmembrane protein CD47 was a major contributing factor for increased anticancer activity, as this protein reduces immunological clearance [170]. The use of extracellular vesicles as drug delivery vehicles is promising due to the abundance of surface proteins that can be exploited for tissue-specific targeting. However, successful implementation of these biological nanoparticles for drug delivery will be dependent on the identification and isolation of vesicles that display favorable biomolecular profiles. Another approach that also exploits biological membranes for drug delivery is the use of particles coated with cell membranes, including those derived from leukocytes [51] and erythrocytes [52]. On the contrary to engineered extracellular vesicles, membrane-coated particles are not confined to spherical shapes [51]. The major challenge for clinical translation of extracellular vesicles and other biological membrane-based therapeutics is the development of scalable cost-effective production methods [171, 172]. Indeed, the most common method for the isolation of vesicles is ultracentrifugation, which is expensive, labor-intensive, and difficult to scale-up for clinical-grade manufacturing [173]. Additionally, batch-to-batch consistency, purity, integrity, and recovery are low with this isolation method, necessitating the use of alternative production techniques for engineered extracellular vesicles. Despite these challenges, extracellular vesicles for immunotherapy entered clinical trials more then ten years ago. Specifically, in 2005, the results from metastatic melanoma and non-small cell lung cancer phase I clinical trials were published [174, 175]. These trials revealed that tumor antigen-loaded exosomes derived from autologous dendritic cells were safe and in a few cases lead to a minor or partial therapeutic response. Accordingly, further research is necessary to identify the most effective ways of utilizing extracellular vesicles for immunotherapy. There are also several other promising nanotechnology-based strategies for inducing anticancer immune responses. These approaches are more than a decade away from clinical translation and have been extensively reviewed elsewhere [176–178].
5.2. Spatiotemporal nanoparticle transitions
In addition to the incorporation of multiple functional elements, drug delivery systems can be designed to display transitional properties in a spatiotemporal manner. Namely, the characteristics of nanoparticles change with time as they move from one compartment in the body to another. In fact, nanoparticle properties that are beneficial in some biological settings are unfavorable in others. For example, while pegylation provides an immunoevasive function in the circulation, this surface modification reduces cellular uptake in the tumor microenvironment. Multistage or transitional drug delivery systems that display adjustable stability, size, and surface properties can be developed to overcome such challenges [179]. The multistage vector is a drug delivery system that consists of several components that are active at various stages in the drug delivery process [161, 180]. The first stage is a silicon microparticle that is designed to navigate in the circulatory system and bind to inflamed vasculature. The microparticle then releases a second stage, which consists of nanoparticles that display optimal properties for transport within the tumor interstitium. The final stage comprises a therapeutic molecule that binds to an intracellular target. The multistage vector is a versatile platform that has previously been used for the delivery of chemotherapeutic agents [91, 181], anti-inflammatory drugs [182], gene-silencing agents [183–185], and therapeutic proteins [186] encapsulated in various nanocarriers. Recently, a new multistage platform based on an injectable nanoparticle generator was developed [91]. This generator is an intravenously administered discoidal silicon microparticle that spontaneously produces and releases polymeric doxorubicin nanoparticles into the tumor microenvironment. Specifically, nanoparticles are formed by self-assembly of polymeric doxorubicin strands loaded inside the microparticle. The nanoparticles are then internalized by cancer cells through endocytosis, thereby overcoming drug efflux pumps [91]. Doxorubicin is released inside acidic endosomes following the cleavage of a pH-sensitive linker. In a breast cancer lung metastasis model, the survival time doubled in mice administered with the nanoparticle generator compared to those treated with a liposomal formulation containing the same dose of doxorubicin [91]. Other examples of multistage drug delivery system are 80–100 nm nanoparticles that in response to tumor enzymes or acidity release smaller 5–10 nm nanoparticles that can more easily diffuse throughout the tumor [187–189]. An alternative approach to the sequential release of nanoparticles is the design of multilayer nanodrugs. For instance, PEG layers can be shed in response to enzymes in the tumor microenvironment [190]. Additionally, the acidic environment of the tumor can be exploited to induce surface modifications that expose cell-penetrating [191] and nucleus-targeting peptides [192]. In conclusion, nanoproperty changes are necessary to ensure beneficial bio-nano interactions in different biological compartments. Although spatiotemporal transitions substantially improve drug delivery, it is important to note that complex nanotherapeutics face greater manufacturing and regulatory challenges. Nevertheless, large-scale production of the injectable nanoparticle generator based on photolithography and electrochemical etching according to the Current Good Manufacturing Practices (CGMPs) enforced by the United States FDA has been established [161]. Toxicity studies based on CGMP quality particles have been performed and an investigational new drug application for breast cancer lung metastases will be submitted within the next 12 months. Clinical approval of a wide range of multifunctional and transitional nanoparticles will require retraining of the pharmaceutical industry to perform cost-effective manufacturing of nanoparticles.
5.3. Priming the microenvironment
A complementary approach to developing innovative nanoparticles for treatment of disease is the design of strategies aimed at targeting the microenvironment. Such approaches have generally focused on modifying tumor characteristics, such as the vasculature and ECM, for improved nanodelivery [193]. Indeed, there are several barriers in the tumor microenvironment that prevent effective nanomedicine delivery. These obstacles include uneven blood flow distribution, interstitial fluid pressure, a dense ECM, and a large population of stromal cells [93]. In animal models, antiangiogenic and angiogenic agents have been administered to normalize cancer vasculature for improved nanoparticle accumulation [194]. Other studies have utilized hyperthermia to enhance nanoparticle permeability across the tumor endothelium [117]. Furthermore, degradation of modification of the ECM can decompress cancer vasculature and improve intratumoral diffusion of nanoparticles [93]. The tumor microenvironment can also be restructured through the inactivation or destruction of cancer-associated fibroblasts [93]. In the clinical setting, angiotensin inhibitors, which alter the vasculature and ECM of tumors [195], have been explored for improved delivery of chemotherapeutic agents. The findings from such clinical trials are inconclusive as retrospective studies have indicated promising results [196, 197], while other trials have failed to show any improvement [198]. It is worth noting that the effect of tumor microenvironment alterations on nanoparticle permeation may differ substantially from that of small molecules, as nanodrugs display unique transport characteristics. Moreover, a potential drawback of increased vascular permeability and reduced ECM thickness is the promotion of cancer cell invasiveness. In addition to modifying the tumor microenvironment for improved drug delivery, healthy organs such as the liver and spleen can be targeted to alter nanoparticle biodistribution [199]. For example, pretreatment of mice with gadolinium chloride was found to deactivate Kupffer cells in the liver, leading to increased accumulation of quantum dots in tumors [200]. Saturation of the innate immune system with decoy liposomes prior to injection of therapeutic iron oxide nanoparticles also demonstrated promise for improving tumor homing [201]. Additionally, preconditioning of the mononuclear phagocyte system with the clinically approved antimalarial agent chloroquine was shown to reduce nanoparticle uptake in the liver and improve intratumoral accumulation in a mouse model [202]. This pretreatment strategy is a simple, broadly applicable, and easily implementable approach to potentially increase site-specific nanodelivery in the clinical setting [203]. Based on common biodsitribution trends [121, 122], a 1% redirection of nanoparticles from the mononuclear phagocyte system to the tumor would be sufficient to double intratumoral drug levels. Assuming that there is a correlation between tumor drug concentration and anticancer efficacy, a minor redistribution of nanoparticles could be a determining factor between life and death. Namely, small changes in liver and spleen accumulation could have major implications for therapeutic efficacy. The clinical translation of strategies to modify the mononuclear phagocyte system for improved tumor delivery of nanoparticles should be easily implementable in certain cases, as agents such as chloroquine that deactivate the innate immune system are already on the market and have shown acceptable safety profiles when combined with chemotherapeutic agents [204]. However, such strategies could also impact tumor-associated macrophages, which could have favorable or unfavorable effects depending on the tumor type [202]. Notably, the translation of therapeutic strategies to modify the tumor microenvironment has limited relevance unless animal models resemble clinical pathobiology. Accordingly, the suitability of tumor models should be assessed by comparing gene expression, histological features, and heterogeneity to biobanks of patient tumors [166]. In general, genetically engineered and patient-derived xenograft models tend to most accurately recapitulate the tumor microenvironment of patients [166].
5.4. Companion diagnostics
Another approach to improve the performance of nanotherapeutics is to stratify patients according to tumor characteristics, such as vasculature density, vessel permeability, blood flow velocity, pressure gradients, and ECM density. Methods for patient stratification can be based on biomarker profiles or imaging. For instance, in animal models, it was shown that the ratio of MMP-9 to tissue inhibitor of metalloproteinase 1 (TIMP-1) can serve as a serum biomarker of nanoparticle accumulation in tumors [205]. Moreover, the content of capillary-wall collagen has been shown to be a biophysical marker of nanoparticle extravasation from tumor vasculature [206]. Additionally, several angiogenesis biomarkers have been reported, including circulating cells, proteins, gene expression profiles, and functional parameters [207]. For instance, higher levels of circulating endothelial cells and endothelial cell progenitors are indicative of increased angiogenesis [208]. In the clinical setting, it has been demonstrated that imaging techniques can be used to predict intratumoral nanoparticle accumulation and therapeutic efficacy. For example, the uptake of iron nanoparticles in patients with advanced solid tumors was quantified by magnetic resonance imaging prior to treatment with Onivyde (irinotecan liposomes) [209]. Notably, intratumoral levels of iron nanoparticles were predictive of therapeutic responses to the liposomes [209]. Additionally, in metastatic breast cancer patients treated with radiolabeled doxorubicin-containing liposomes, tumor accumulation quantified by positron emission tomography was shown to correlate with treatment outcome [210]. This example highlights the value of theranostic nanoparticles that provide information about the success of drug delivery in addition to having therapeutic effects. Taken together, companion diagnostics present a promising tool for identifying patients that are likely to respond to nanotherapeutics. Future applications also include further pre-selection of patients according to the most suitable nanoparticle properties, such as size, shape, and charge. In order for companion diagnostics to achieve clinical relevance in nanomedicine, a decision-making framework needs to be created. The generation of such a framework requires improved tools and databases to assess tumor heterogeneity, especially in regards to parameters that dictate nanoparticle delivery to tumors. Moreover, the focus should be shifted from formulation-driven development of nanotherapeutics to design criteria that are based on treating specific tumor types [166].
6. Conclusions
The reasons for the failures of clinical trials to reach proposed objectives are invariably multifold, complex, and interwoven with each other. Unfortunately, it is the norm rather than the exception that therapeutic agents (chemo-, biological, or nanotechnological) prove to be very effective and cell-selective in targeting certain cancer cell populations in vitro, and even in suitable animal models, yet fail dramatically in clinical trials. It stands to reason that the biodistribution of the agent may be a fundamental factor for these failures, with insufficient concentrations being realized at target sites, and unwanted concentrations elsewhere causing dose-limiting toxicities. The biodistribution of therapeutic agents is largely controlled by the ability of the drug to penetrate across biological barriers, especially in the evolving forms they present in the course of carcinogenesis. The strategy of adding targeting moieties to therapeutic nanoparticles to increase their localization specificity has not yielded clinically approved drugs, to date, despite 30 years of attempts by many laboratories and pharmaceutical companies. This pitfall is related to that the fact that the addition of molecular targeting agents increases recognition specificity, but at the cost of much greater difficulties in addressing biological barriers. This review highlights critical obstacles in nanomedicine and the necessity to develop strategies for addressing them in a sequential fashion for improved outcomes in clinical trials. The various benefits and characteristics of nanoparticles summarized in this review form the knowledge basis from which further strategies can be deployed to improve cancer therapeutics. Specifically, the major advantages of nanotherapeutics are multifunctionality, unique electromagnetic characteristics, and distinct transport properties. Although many of these benefits have been individually implemented in the clinical setting, successful future nanotherapeutics will likely integrate a wider range of valuable nanoscale characteristics combined with strategies to modify the microenvironment. In regards to clinical oncology, nanomedicine has the potential to substantially improve the treatment of aggressive diseases, such as triple negative breast cancer and pancreatic cancer. Specifically, the microenvironment of pancreatic tumors poses unique challenges due to poor vascularization and dense stroma, requiring a multipronged approach to treatment. In the case of metastatic breast cancer, nanoparticle-based treatments show promise for overcoming drug resistance and achieving site-specific delivery through approaches such as hemodynamic targeting. Although nanomedicine offers new and improved solutions for the treatment of cancer, there is a need for an improved understanding of bio-nano interactions in the body, as this interface is a determining factor of the success of nanotherapeutics. Accordingly, systematic studies that evaluate the impact of nanoparticle characteristics on biomolecular interactions and pharmacokinetics will be critical for the design of superior treatments. Furthermore, the commercialization process for nanotherapeutics is challenging due to a lack of industry experience in large-scale clinical-grade manufacturing. Indeed, pharmaceutical production facilities are primarily specialized in small molecule and antibody production. In conclusion, an increase in industry knowledge of scalable nanoparticle synthesis coupled with the design of transitional multifunctional nanoparticles, microenvironmental priming strategies, and companion diagnostics is likely to radically transform cancer treatment.
Acknowledgements
Mauro Ferrari is the Ernest Cockrell Jr. Presidential Distinguished Chair at the Houston Methodist Research Institute. This work was supported by the National Cancer Institute Physical Sciences-Oncology Network of the National Institutes of Health, under award number U54CA210181. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program, under award number W81XWH-17-1-0389. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. Additionally, this work was supported by intramural funding from Mayo Clinic. The authors thank Dr. Piotr Godzinski, Director of the Office of Cancer Nanotechnology Research at the National Cancer Institute, for reviewing the manuscript and providing valuable comments.
Author Biographies

Mauro Ferrari, Ph.D. is the President and CEO of Houston Methodist Research Institute, where he holds the Ernest Cockrell Jr. Presidential Distinguished Chair. He also serves as Executive Vice President of the Houston Methodist Hospital System and Senior Associate Dean and Professor of Medicine at Weill Cornell Medical College.

Joy Wolfram, Ph.D. is an Assistant Professor of Medicine at the Mayo Clinic, where she leads the Nanomedicine and Extracellular Vesicles Laboratory. Her main research interests include cancer and tissue injury and her goal is to bring new nanomedicines with increased therapeutic efficacy and safety to the clinic.
Footnotes
Declaration of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Boverhof DR, Bramante CM, Butala JH, Clancy SF, Lafranconi M, West J, Gordon SC, Regul. Toxicol. Pharmacol, 73 (2015) 137–150. [DOI] [PubMed] [Google Scholar]
- [2].Barenholz Y, Control J Release, 160 (2012) 117–134. [DOI] [PubMed] [Google Scholar]
- [3].Anselmo AC, Mitragotri S, Bioengineering & Translational Medicine, 1 (2016) 10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wicki A, Witzigmann D, Balasubramanian V, Huwyler J, Control J Release, 200 (2015) 138–157. [DOI] [PubMed] [Google Scholar]
- [5].ten Tije AJ, Verweij J, Loos WJ, Sparreboom A, Clin. Pharmacokinet, 42 (2003) 665685. [DOI] [PubMed] [Google Scholar]
- [6].Yardley DA, Control J Release, 170 (2013) 365–372. [DOI] [PubMed] [Google Scholar]
- [7].Hawkins MJ, Soon-Shiong P, Desai N, Adv Drug Deliv Rev, 60 (2008) 876–885. [DOI] [PubMed] [Google Scholar]
- [8].Fanali G, di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P, Mol. Aspects Med, 33 (2012) 209–290. [DOI] [PubMed] [Google Scholar]
- [9].Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J, J. Clin. Oncol, 23 (2005) 7794–7803. [DOI] [PubMed] [Google Scholar]
- [10].Stinchcombe TE, Nanomedicine (Lond), 2 (2007) 415–423. [DOI] [PubMed] [Google Scholar]
- [11].Hwang L, Douglas C, Nam D, Trieu V, Eur. J. Cancer, 50 (2014) e70. [Google Scholar]
- [12].Koudelka S, Turanek J, Control J Release, 163 (2012) 322–334. [DOI] [PubMed] [Google Scholar]
- [13].Untch M, Jackisch C, Schneeweiss A, Conrad B, Aktas B, Denkert C, Eidtmann H, Wiebringhaus H, Kummel S, Hilfrich J, Warm M, Paepke S, Just M, Hanusch C, Hackmann J, Blohmer JU, Clemens M, Darb-Esfahani S, Schmitt WD, Dan Costa S, Gerber B, Engels K, Nekljudova V, Loibl S, von Minckwitz G, German Breast G, Arbeitsgemeinschaft Gynakologische Onkologie-Breast I, Lancet Oncol, 17 (2016) 345–356. [DOI] [PubMed] [Google Scholar]
- [14].Rugo HS, Barry WT, Moreno-Aspitia A, Lyss AP, Cirrincione C, Leung E, Mayer EL, Naughton M, Toppmeyer D, Carey LA, Perez EA, Hudis C, Winer EP, J. Clin. Oncol, 33 (2015) 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, Hendrikse NH, Eriksson J, Windhorst AD, Postmus PE, Verheul HM, Serne EH, Lammertsma AA, Smit EF, Cancer Cell, 21 (2012) 82–91. [DOI] [PubMed] [Google Scholar]
- [16].Liu F, Park JY, Zhang Y, Conwell C, Liu Y, Bathula SR, Huang L, J. Pharm. Sci, 99 (2010) 3542–3551. [DOI] [PubMed] [Google Scholar]
- [17].Shen J, Wolfram J, Ferrari M, Shen H, Mater Today (Kidlington), 20 (2017) 95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ediriwickrema A, Saltzman WM, ACS Biomater Sci Eng, 1 (2015) 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Charrois GJ, Allen TM, Biochim. Biophys. Acta, 1663 (2004) 167–177. [DOI] [PubMed] [Google Scholar]
- [20].Johnston MJ, Semple SC, Klimuk SK, Edwards K, Eisenhardt ML, Leng EC, Karlsson G, Yanko D, Cullis PR, Biochim. Biophys. Acta, 1758 (2006) 55–64. [DOI] [PubMed] [Google Scholar]
- [21].Koczera P, Martin L, Marx G, Schuerholz T, Int J Mol Sci, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nadano D, Yasuda T, Kishi K, Clin. Chem, 39 (1993) 448–452. [PubMed] [Google Scholar]
- [23].Weissmann G, New Engl. J. Med, 273 (1965) 1084–1090. [DOI] [PubMed] [Google Scholar]
- [24].Shen H, Sun T, Ferrari M, Cancer Gene Ther, 19 (2012) 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Molinaro R, Wolfram J, Federico C, Cilurzo F, Di Marzio L, Ventura CA, Carafa M, Celia C, Fresta M, Expert opinion on drug delivery, 10 (2013) 1653–1668. [DOI] [PubMed] [Google Scholar]
- [26].Whitehead KA, Matthews J, Chang PH, Niroui F, Dorkin JR, Severgnini M, Anderson DG, ACS nano, 6 (2012) 6922–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nat. Rev. Cancer, 17 (2017) 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hoy SM, Drugs, (2018). [Google Scholar]
- [29].Chi X, Gatti P, Papoian T, Drug Discov. Today, 22 (2017) 823–833. [DOI] [PubMed] [Google Scholar]
- [30].Aartsma-Rus A, Krieg AM, Nucleic Acid Ther, 27 (2017) 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, Laborde RR, Wassie E, Schimek L, Hanson CA, Gangat N, Wang X, Pardanani A, Engl N J. Med, 373 (2015) 908–919. [DOI] [PubMed] [Google Scholar]
- [32].Salloum R, Hummel TR, Kumar SS, Dorris K, Li S, Lin T, Daryani VM, Stewart CF, Miles L, Poussaint TY, Stevenson C, Goldman S, Dhall G, Packer R, Fisher P, Pollack IF, Fouladi M, Boyett J, Drissi R, Neurooncol J, 129 (2016) 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Crooke ST, Baker BF, Kwoh TJ, Cheng W, Schulz DJ, Xia S, Salgado N, Bui HH, Hart CE, Burel SA, Younis HS, Geary RS, Henry SP, Bhanot S, Mol. Ther, 24 (2016) 1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Flierl U, Nero TL, Lim B, Arthur JF, Yao Y, Jung SM, Gitz E, Pollitt AY, Zaldivia MT, Jandrot-Perrus M, Schafer A, Nieswandt B, Andrews RK, Parker MW, Gardiner EE, Peter K, J. Exp. Med, 212 (2015) 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Garber K, Nat Biotech, 34 (2016) 1213–1214. [DOI] [PubMed] [Google Scholar]
- [36].Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J, Nature, 452 (2008) 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Alexopoulou L, Holt AC, Medzhitov R, Flavell RA, Nature, 413 (2001) 732–738. [DOI] [PubMed] [Google Scholar]
- [38].Khvorova A, Engl N J. Med, 376 (2017) 4–7. [DOI] [PubMed] [Google Scholar]
- [39].Broering R, Real CI, John MJ, Jahn-Hofmann K, Ickenstein LM, Kleinehr K, Paul A, Gibbert K, Dittmer U, Gerken G, Schlaak JF, Int. Immunol, 26 (2014) 35–46. [DOI] [PubMed] [Google Scholar]
- [40].Prapunpoj P, Leelawatwattana L, FEBS J, 276 (2009) 5330–5341. [DOI] [PubMed] [Google Scholar]
- [41].BusinessWire, http://www.businesswire.com/news/home/20170920005628/en/AlnylamSanofi-Report-Positive-Topline-Results-APOLLO, (2017).
- [42].Garber K, Nat Biotech, 33 (2015) 577–577. [DOI] [PubMed] [Google Scholar]
- [43].Barata P, Sood AK, Hong DS, Cancer Treat. Rev, 50 (2016) 35–47. [DOI] [PubMed] [Google Scholar]
- [44].Cervantes A, Alsina M, Tabernero J, Infante JR, LoRusso P, Shapiro G, Paz-Ares LG, Falzone R, Hill J, Cehelsky J, White A, Toudjarska I, Bumcrot D, Meyers R, Hinkle G, Svrzikapa N, Sah DW, Vaishnaw A, Gollob J, Burris HA, J. Clin. Oncol, 29 (2011) 3025–3025. [Google Scholar]
- [45].Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, Keil O, Lange C, Giese K, Kaufmann J, Khan M, Drevs J, J. Clin. Oncol, 32 (2014) 4141–4148. [DOI] [PubMed] [Google Scholar]
- [46].Tolcher AW, Papadopoulos KP, Patnaik A, Rasco DW, Martinez D, Wood DL, Fielman B, Sharma M, Janisch LA, Brown BD, Bhargava P, Ratain MJ, J. Clin. Oncol, 33 (2015) 11006–11006. [Google Scholar]
- [47].Whitfield JR, Beaulieu ME, Soucek L, Front Cell Dev Biol, 5 (2017) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, Leong KW, Chem. Rev, 117 (2017) 9874–9906. [DOI] [PubMed] [Google Scholar]
- [49].Amoozgar Z, Yeo Y, Wiley Interdiscip Rev Nanomed Nanobiotechnol, 4 (2012) 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE, Science, 339 (2013) 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E, Nature nanotechnology, 8 (2013) 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc. Natl. Acad. Sci. U. S. A, 108 (2011) 10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pasut G, Veronese FM, Control J Release, 161 (2012) 461–472. [DOI] [PubMed] [Google Scholar]
- [54].Lamb YN, Scott LJ, Drugs, 77 (2017) 785–792. [DOI] [PubMed] [Google Scholar]
- [55].Soloman R, Gabizon AA, Clin. Lymphoma Myeloma, 8 (2008) 21–32. [DOI] [PubMed] [Google Scholar]
- [56].Knop K, Hoogenboom R, Fischer D, Schubert US, Angew. Chem. Int. Ed. Engl, 49 (2010) 6288–6308. [DOI] [PubMed] [Google Scholar]
- [57].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nature Reviews Materials, 1 (2016) 16014. [Google Scholar]
- [58].Hatakeyama H, Akita H, Harashima H, Biol. Pharm. Bull, 36 (2013) 892–899. [DOI] [PubMed] [Google Scholar]
- [59].Pasut G, Paolino D, Celia C, Mero A, Joseph AS, Wolfram J, Cosco D, Schiavon O, Shen H, Fresta M, Control J Release, 199 (2015) 106–113. [DOI] [PubMed] [Google Scholar]
- [60].Parr MJ, Ansell SM, Choi LS, Cullis PR, Biochim. Biophys. Acta, 1195 (1994) 21–30. [DOI] [PubMed] [Google Scholar]
- [61].Chanan-Khan A, Szebeni J, Savay S, Liebes L, Rafique NM, Alving CR, Muggia FM, Ann. Oncol, 14 (2003) 1430–1437. [DOI] [PubMed] [Google Scholar]
- [62].Moghimi SM, Andersen AJ, Hashemi SH, Lettiero B, Ahmadvand D, Hunter AC, Andresen TL, Hamad I, Szebeni J, Control J Release, 146 (2010) 175–181. [DOI] [PubMed] [Google Scholar]
- [63].Marina NM, Cochrane D, Harney E, Zomorodi K, Blaney S, Winick N, Bernstein M, Link MP, Clin. Cancer. Res, 8 (2002) 413–418. [PubMed] [Google Scholar]
- [64].Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H, Control J Release, 88 (2003) 3542. [DOI] [PubMed] [Google Scholar]
- [65].J Int Assoc Physicians AIDS Care, 2 (1996) 50–51. [PubMed] [Google Scholar]
- [66].Batist G, Barton J, Chaikin P, Swenson C, Welles L, Expert Opin. Pharmacother, 3 (2002) 1739–1751. [DOI] [PubMed] [Google Scholar]
- [67].Deitcher OR, Glaspy J, Gonzalez R, Sato T, Bedikian AY, Segarini K, Silverman J, Deitcher SR, Clin. Lymphoma Myeloma Leuk, 14 (2014) 197–202. [DOI] [PubMed] [Google Scholar]
- [68].Frampton JE, Paediatr. Drugs, 12 (2010) 141–153. [DOI] [PubMed] [Google Scholar]
- [69].Neelapu SS, Baskar S, Gause BL, Kobrin CB, Watson TM, Frye AR, Pennington R, Harvey L, Jaffe ES, Robb RJ, Popescu MC, Kwak LW, Clin. Cancer. Res, 10 (2004) 83098317. [DOI] [PubMed] [Google Scholar]
- [70].Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, Sahin U, Nature, 534 (2016) 396–401. [DOI] [PubMed] [Google Scholar]
- [71].Xia X, Mai J, Xu R, Perez JE, Guevara ML, Shen Q, Mu C, Tung HY, Corry DB, Evans SE, Liu X, Ferrari M, Zhang Z, Li XC, Wang RF, Shen H, Cell reports, 11 (2015) 957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lane D, Nat Biotech, 24 (2006) 163–164. [DOI] [PubMed] [Google Scholar]
- [73].Alexis F, Mol. Ther, 22 (2014) 1239–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Blanco E, Sangai T, Wu S, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, Ferrari M, Mol. Ther, 22 (2014) 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dicko A, Kwak S, Frazier AA, Mayer LD, Liboiron BD, Int. J. Pharm, 391 (2010) 248–259. [DOI] [PubMed] [Google Scholar]
- [76].Tardi P, Johnstone S, Harasym N, Xie S, Harasym T, Zisman N, Harvie P, Bermudes D, Mayer L, Leuk. Res, 33 (2009) 129–139. [DOI] [PubMed] [Google Scholar]
- [77].Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR, Stone RM, Bixby DL, Kolitz JE, Schiller GJ, Wieduwilt MJ, Ryan DH, Hoering A, Chiarella M, Louie AC, Medeiros BC, J. Clin. Oncol, 34 (2016) 7000–7000. [Google Scholar]
- [78].Ryu JH, Lee S, Son S, Kim SH, Leary JF, Choi K, Kwon IC, Control J Release, 190 (2014) 477–484. [DOI] [PubMed] [Google Scholar]
- [79].Minchinton AI, Tannock IF, Nat. Rev. Cancer, 6 (2006) 583–592. [DOI] [PubMed] [Google Scholar]
- [80].Zhou H, Fan Z, Deng J, Lemons PK, Arhontoulis DC, Bowne WB, Cheng H, Nano Lett, 16 (2016) 3268–3277. [DOI] [PubMed] [Google Scholar]
- [81].Villegas MR, Baeza A, Vallet-Regi M, ACS Appl Mater Interfaces, 7 (2015) 24075–24081. [DOI] [PubMed] [Google Scholar]
- [82].U.S. National Library of Medicine, ClinicalTrials.gov,, https://clinicaltrials.gov/ct2/show/NCT03076372 (2017). [DOI] [PubMed]
- [83].Pasquale EB, Nat. Rev. Cancer, 10 (2010) 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ledford H, Nature, 533 (2016) 304–305. [DOI] [PubMed] [Google Scholar]
- [85].Von Hoff DD, Mita MM, Ramanathan RK, Weiss GJ, Mita AC, LoRusso PM, Burris HA 3rd, Hart LL, Low SC, Parsons DM, Zale SE, Summa JM, Youssoufian H, Sachdev JC, Clin. Cancer. Res, 22 (2016) 3157–3163. [DOI] [PubMed] [Google Scholar]
- [86].Chang SS, O’Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB, Clin. Cancer. Res, 5 (1999) 2674–2681. [PubMed] [Google Scholar]
- [87].Danhier F, Le Breton A, Preat V, Mol. Pharm, 9 (2012) 2961–2973. [DOI] [PubMed] [Google Scholar]
- [88].Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML, Biochim. Biophys. Acta, 1820 (2012) 291–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Garcia-Bennett A, Nees M, Fadeel B, Biochem. Pharmacol, 81 (2011) 976–984. [DOI] [PubMed] [Google Scholar]
- [90].Iversen TG, Skotland T, Sandvig K, Nano Today, 6 (2011) 176–185. [DOI] [PubMed] [Google Scholar]
- [91].Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, Shen J, Liu H, Hu Z, Chen L, Huang Y, Koay E, Huang Y, Liu J, Ensor JE, Blanco E, Liu X, Ferrari M, Shen H, Nat. Biotechnol, 34 (2016) 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Allen TM, Nat. Rev. Cancer, 2 (2002) 750–763. [DOI] [PubMed] [Google Scholar]
- [93].Chauhan VP, Jain RK, Nature materials, 12 (2013) 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Harris TJ, von Maltzahn G, Lord ME, Park JH, Agrawal A, Min DH, Sailor MJ, Bhatia SN, Small, 4 (2008) 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA, Proc. Natl. Acad. Sci. U. S. A, 105 (2008) 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Aberg C, Mahon E, Dawson KA, Nature nanotechnology, 8 (2013) 137–143. [DOI] [PubMed] [Google Scholar]
- [97].Lundqvist M, Sethson I, Jonsson BH, Langmuir, 20 (2004) 10639–10647. [DOI] [PubMed] [Google Scholar]
- [98].Mahon E, Salvati A, Baldelli Bombelli F, Lynch I, Dawson KA, Control J Release, 161 (2012) 164–174. [DOI] [PubMed] [Google Scholar]
- [99].Kreuter J, Shamenkov D, Petrov V, Ramge P, Cychutek K, Koch-Brandt C, Alyautdin R, Drug Target J, 10 (2002) 317–325. [DOI] [PubMed] [Google Scholar]
- [100].Zhang J, Landry MP, Barone PW, Kim JH, Lin S, Ulissi ZW, Lin D, Mu B, Boghossian AA, Hilmer AJ, Rwei A, Hinckley AC, Kruss S, Shandell MA, Nair N, Blake S, Sen F, Sen S, Croy RG, Li D, Yum K, Ahn JH, Jin H, Heller DA, Essigmann JM, Blankschtein D, Strano MS, Nature nanotechnology, 8 (2013) 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].O’Brien J, Shea KJ, Acc. Chem. Res, 49 (2016) 1200–1210. [DOI] [PubMed] [Google Scholar]
- [102].Xia XR, Monteiro-Riviere NA, Riviere JE, Nature nanotechnology, 5 (2010) 671–675. [DOI] [PubMed] [Google Scholar]
- [103].Carril M, Padro D, Del Pino P, Carrillo-Carrion C, Gallego M, Parak WJ, Nature communications, 8 (2017) 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Pauthner M, Yeung J, Ullman C, Bakker J, Wurch T, Reichert JM, Lund-Johansen F, Bradbury AR, Carter PJ, Melis JP, MAbs, 8 (2016) 617–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].U.S. National,, https://clinicaltrials.gov/ct2/show/NCT03013491 (2017). Library of Medicine, ClinicalTrials.gov
- [106].Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, Wolchok JD, Ann. Oncol, 27 (2016) 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].U.S. National Library of Medicine, ClinicalTrials.gov,, https://clinicaltrials.gov/ct2/show/NCT03149549 (2017). [DOI] [PubMed]
- [108].Dou Y, Hynynen K, Allen C, Control J Release, 249 (2017) 63–73. [DOI] [PubMed] [Google Scholar]
- [109].Johannsen M, Thiesen B, Jordan A, Taymoorian K, Gneveckow U, Waldofner N, Scholz R, Koch M, Lein M, Jung K, Loening SA, Prostate, 64 (2005) 283–292. [DOI] [PubMed] [Google Scholar]
- [110].Cervadoro A, Giverso C, Pande R, Sarangi S, Preziosi L, Wosik J, Brazdeikis A, Decuzzi P, PLoS One, 8 (2013) e57332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Maier-Hauff K, Ulrich F, Nestler D, Niehoff H, Wust P, Thiesen B, Orawa H, Budach V, Jordan A, Neurooncol J, 103 (2011) 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Stern JM, Kibanov Solomonov VV, Sazykina E, Schwartz JA, Gad SC, Goodrich GP, Int. J. Toxicol, 35 (2016) 38–46. [DOI] [PubMed] [Google Scholar]
- [113].U.S. National Library of Medicine, ClinicalTrials.gov,, https://clinicaltrials.gov/ct2/show/NCT02680535 (2016). [DOI] [PubMed]
- [114].Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL, Proc. Natl. Acad. Sci. U. S. A, 100 (2003) 13549–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Chakravarty P, Marches R, Zimmerman NS, Swafford AD, Bajaj P, Musselman IH, Pantano P, Draper RK, Vitetta ES, Proc. Natl. Acad. Sci. U. S. A, 105 (2008) 8697–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Glazer ES, Zhu C, Massey KL, Thompson CS, Kaluarachchi WD, Hamir AN, Curley SA, Clin. Cancer. Res, 16 (2010) 5712–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kirui DK, Celia C, Molinaro R, Bansal SS, Cosco D, Fresta M, Shen H, Ferrari M, Advanced healthcare materials, 4 (2015) 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Li D, Jung YS, Tan S, Kim HK, Chory E, Geller DA, Colloid Interface Sci J, 358 (2011) 47–53. [DOI] [PubMed] [Google Scholar]
- [119].Nabors LK, Baumgartner WA Jr., Janke SJ, Rose JR, Wagner WW Jr., Capen RL, J Appl Physiol (1985), 94 (2003) 1634–1640. [DOI] [PubMed] [Google Scholar]
- [120].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV, Nat. Biotechnol, 25 (2007) 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H, Nano Today, 10 (2015) 487–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WC, Control J Release, (2016). [DOI] [PubMed] [Google Scholar]
- [123].Ferrari M, Trends Biotechnol, 28 (2010) 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Koay EJ, Ferrari M, Phys. Biol, 11 (2014) 060201. [DOI] [PubMed] [Google Scholar]
- [125].Michor F, Liphardt J, Ferrari M, Widom J, Nat. Rev. Cancer, 11 (2011) 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gimbrone MA Jr., Leapman SB, Cotran RS, Folkman J, J. Exp. Med, 136 (1972) 261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Siemann DW, Cancer Treat. Rev, 37 (2011) 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Matsumura Y, Maeda H, Cancer Res, 46 (1986) 6387–6392. [PubMed] [Google Scholar]
- [129].Gerlowski LE, Jain RK, Microvasc. Res, 31 (1986) 288–305. [DOI] [PubMed] [Google Scholar]
- [130].Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK, Cancer Res, 55 (1995) 3752–3756. [PubMed] [Google Scholar]
- [131].Fang J, Nakamura H, Maeda H, Adv Drug Deliv Rev, 63 (2011) 136–151. [DOI] [PubMed] [Google Scholar]
- [132].Torchilin V, Adv Drug Deliv Rev, 63 (2011) 131–135. [DOI] [PubMed] [Google Scholar]
- [133].Kalluri R, Zeisberg M, Nat. Rev. Cancer, 6 (2006) 392–401. [DOI] [PubMed] [Google Scholar]
- [134].Zollinger AJ, Smith ML, Matrix Biol, 60–61 (2017) 27–37. [DOI] [PubMed] [Google Scholar]
- [135].Snedeker JG, Gautieri A, Muscles Ligaments Tendons J, 4 (2014) 303–308. [PMC free article] [PubMed] [Google Scholar]
- [136].Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, Weissleder R, Nature communications, 6 (2015) 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Gentile E, Cilurzo F, Di Marzio L, Carafa M, Ventura CA, Wolfram J, Paolino D, Celia C, Future Oncology, 9 (2013) 1849–1859. [DOI] [PubMed] [Google Scholar]
- [138].Maeda H, Adv Drug Deliv Rev, 46 (2001) 169–185. [DOI] [PubMed] [Google Scholar]
- [139].Forssen EA, Coulter DM, Proffitt RT, Cancer Res, 52 (1992) 3255–3261. [PubMed] [Google Scholar]
- [140].Mayer LD, Bally MB, Cullis PR, Wilson SL, Emerman JT, Cancer Lett, 53 (1990) 183–190. [DOI] [PubMed] [Google Scholar]
- [141].Merian J, De Souza R, Dou Y, Ekdawi SN, Ravenelle F, Allen C, Int. J. Pharm, 488 (2015) 154–164. [DOI] [PubMed] [Google Scholar]
- [142].Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC, Cancer Res, 73 (2013) 2412–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, Stewart JS, Clin. Cancer. Res, 7 (2001) 243–254. [PubMed] [Google Scholar]
- [144].Presant CA, Proffitt RT, Turner AF, Williams LE, Winsor D, Werner JL, Kennedy P, Wiseman C, Gala K, McKenna RJ, et al. , Cancer, 62 (1988) 905–911. [DOI] [PubMed] [Google Scholar]
- [145].Harrington KJ, Rowlinson-Busza G, Syrigos KN, Abra RM, Uster PS, Peters AM, Stewart JS, Br. J. Cancer, 83 (2000) 684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Koukourakis MI, Koukouraki S, Giatromanolaki A, Archimandritis SC, Skarlatos J, Beroukas K, Bizakis JG, Retalis G, Karkavitsas N, Helidonis ES, J. Clin. Oncol, 17 (1999) 3512–3521. [DOI] [PubMed] [Google Scholar]
- [147].Clark AJ, Wiley DT, Zuckerman JE, Webster P, Chao J, Lin J, Yen Y, Davis ME, Proc. Natl. Acad. Sci. U. S. A, 113 (2016) 3850–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Presant CA, Blayney D, Proffitt RT, Turner AF, Williams LE, Nadel HI, Kennedy P, Wiseman C, Gala K, Crossley RJ, et al. , Lancet, 335 (1990) 1307–1309. [DOI] [PubMed] [Google Scholar]
- [149].Symon Z, Peyser A, Tzemach D, Lyass O, Sucher E, Shezen E, Gabizon A, Cancer, 86 (1999) 72–78. [PubMed] [Google Scholar]
- [150].Natfji AA, Ravishankar D, Osborn HMI, Greco F, J. Pharm. Sci, 106 (2017) 3179–3187. [DOI] [PubMed] [Google Scholar]
- [151].Boulikas T, Stathopoulos GP, Volakakis N, Vougiouka M, Anticancer Res, 25 (2005) 3031–3039. [PubMed] [Google Scholar]
- [152].Fu F, Nowak MA, Bonhoeffer S, PLoS Comput. Biol, 11 (2015) e1004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Adriani G, de Tullio MD, Ferrari M, Hussain F, Pascazio G, Liu X, Decuzzi P, Biomaterials, 33 (2012) 5504–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Sevick EM, Jain RK, Cancer Res, 49 (1989) 3513–3519. [PubMed] [Google Scholar]
- [155].Lee SY, Ferrari M, Decuzzi P, Nanotechnology, 20 (2009) 495101. [DOI] [PubMed] [Google Scholar]
- [156].Decuzzi P, Ferrari M, Biomaterials, 27 (2006) 5307–5314. [DOI] [PubMed] [Google Scholar]
- [157].Kuwahara M, Sugimoto M, Tsuji S, Matsui H, Mizuno T, Miyata S, Yoshioka A, Arterioscler. Thromb. Vasc. Biol, 22 (2002) 329–334. [DOI] [PubMed] [Google Scholar]
- [158].Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X, Adv. Funct. Mater, 22 (2012) 4225–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Mi Y, Mu C, Wolfram J, Deng Z, Hu TY, Liu X, Blanco E, Shen H, Ferrari M, Advanced healthcare materials, 5 (2016) 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S, Proc. Natl. Acad. Sci. U. S. A, 110 (2013) 10753–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wolfram J, Shen H, Ferrari M, Control J Release, 219 (2015) 406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Canelas DA, Herlihy KP, DeSimone JM, Wiley Interdiscip Rev Nanomed Nanobiotechnol, 1 (2009) 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Fisher DT, Muhitch JB, Kim M, Doyen KC, Bogner PN, Evans SS, Skitzki JJ, Nature communications, 7 (2016) 10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Suzuki R, Takizawa T, Kuwata Y, Mutoh M, Ishiguro N, Utoguchi N, Shinohara A, Eriguchi M, Yanagie H, Maruyama K, Int. J. Pharm, 346 (2008) 143–150. [DOI] [PubMed] [Google Scholar]
- [165].Chau Y, Dang NM, Tan FE, Langer R, J. Pharm. Sci, 95 (2006) 542–551. [DOI] [PubMed] [Google Scholar]
- [166].Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST, Adv Drug Deliv Rev, 108 (2017) 25–38. [DOI] [PubMed] [Google Scholar]
- [167].S ELA, Mager I, Breakefield XO, Wood MJ, Nat. Rev. Drug Discov, 12 (2013) 347–357. [DOI] [PubMed] [Google Scholar]
- [168].van der Meel R, Fens MH, Vader P, van Solinge WW, Eniola-Adefeso O, Schiffelers RM, Control J Release, 195 (2014) 72–85. [DOI] [PubMed] [Google Scholar]
- [169].Borrelli DA, Yankson K, Shukla N, Vilanilam G, Ticer T, Wolfram J, Control J Release, 273 (2018) 86–98. [DOI] [PubMed] [Google Scholar]
- [170].Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R, Nature, 546 (2017) 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Busatto S, Vilanilam G, Ticer T, Lin WL, Dickson DW, Shapiro S, Bergese P, Wolfram J, Cells, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach J-M, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, Bedina Zavec A, Benmoussa A, Berardi AC, Bergese P, Bielska E, Blenkiron C, BobisWozowicz S, Boilard E, Boireau W, Bongiovanni A, Borràs FE, Bosch S, Boulanger CM, Breakefield X, Breglio AM, Brennan MÁ, Brigstock DR, Brisson A, Broekman MLD, Bromberg JF, Bryl-Górecka P, Buch S, Buck AH, Burger D, Busatto S, Buschmann D, Bussolati B, Buzás EI, Byrd JB, Camussi G, Carter DRF, Caruso S, Chamley LW, Chang Y-T, Chen C, Chen S, Cheng L, Chin AR, Clayton A, Clerici SP, Cocks A, Cocucci E, Coffey RJ, Cordeiro-da-Silva A, Couch Y, Coumans FAW, Coyle B, Crescitelli R, Criado MF, D’Souza-Schorey C, Das S, Datta Chaudhuri A, de Candia P, De Santana EF, De Wever O, del Portillo HA, Demaret T, Deville S, Devitt A, Dhondt B, Di Vizio D, Dieterich LC, Dolo V, Dominguez Rubio AP, Dominici M, Dourado MR, Driedonks TAP, Duarte FV, Duncan HM, Eichenberger RM, Ekström K, El Andaloussi S, Elie-Caille C, Erdbrügger U, Falcón-Pérez JM, Fatima F, Fish JE, Flores-Bellver M, Försönits A, FreletBarrand A, Fricke F, Fuhrmann G, Gabrielsson S, Gámez-Valero A, Gardiner C, Gärtner K, Gaudin R, Gho YS, Giebel B, Gilbert C, Gimona M, Giusti I, Goberdhan DCI, Görgens A, Gorski SM, Greening DW, Gross JC, Gualerzi A, Gupta GN, Gustafson D, Handberg A, Haraszti RA, Harrison P, Hegyesi H, Hendrix A, Hill AF, Hochberg FH, Hoffmann KF, Holder B, Holthofer H, Hosseinkhani B, Hu G, Huang Y, Huber V, Hunt S, Ibrahim AG-E, Ikezu T, Inal JM, Isin M, Ivanova A, Jackson HK, Jacobsen S, Jay SM, Jayachandran M, Jenster G, Jiang L, Johnson SM, Jones JC, Jong A, Jovanovic-Talisman T, Jung S, Kalluri R, Kano S.-i., Kaur S, Kawamura Y, Keller ET, Khamari D, Khomyakova E, Khvorova A, Kierulf P, Kim KP, Kislinger T, Klingeborn M, Klinke DJ, Kornek M, Kosanović MM, Kovács ÁF, Krämer-Albers E-M, Krasemann S, Krause M, Kurochkin IV, Kusuma GD, Kuypers S, Laitinen S, Langevin SM, Languino LR, Lannigan J, Lässer C, Laurent LC, Lavieu G, Lázaro-Ibáñez E, Le Lay S, Lee M-S, Lee YXF, Lemos DS, Lenassi M, Leszczynska A, Li ITS, Liao K, Libregts SF, Ligeti E, Lim R, Lim SK, Linē A, Linnemannstöns K, Llorente A, Lombard CA, Lorenowicz MJ, Lörincz ÁM, Lötvall J, Lovett J, Lowry MC, Loyer X, Lu Q, Lukomska B, Lunavat TR, Maas SLN, Malhi H, Marcilla A, Mariani J, Mariscal J, Martens-Uzunova ES, Martin-Jaular L, Martinez MC, Martins VR, Mathieu M, Mathivanan S, Maugeri M, McGinnis LK, McVey MJ, Meckes DG, Meehan KL, Mertens I, Minciacchi VR, Möller A, Møller Jørgensen M, MoralesKastresana A, Morhayim J, Mullier F, Muraca M, Musante L, Mussack V, Muth DC, Myburgh KH, Najrana T, Nawaz M, Nazarenko I, Nejsum P, Neri C, Neri T, Nieuwland R, Nimrichter L, Nolan JP, Nolte-’t Hoen ENM, Noren Hooten N, O’Driscoll L, O’Grady T, O’Loghlen A, Ochiya T, Olivier M, Ortiz A, Ortiz LA, Osteikoetxea X, Ostegaard O, Ostrowski M, Park J, Pegtel DM, Peinado H, Perut F, Pfaffl MW, Phinney DG, Pieters BCH, Pink RC, Pisetsky DS, Pogge von Strandmann E, Polakovicova I, Poon IKH, Powell BH, Prada I, Pulliam L, Quesenberry P, Radeghieri A, Raffai RL, Raimondo S, Rak J, Ramirez MI, Raposo G, Rayyan MS, Regev-Rudzki N, Ricklefs FL, Robbins PD, Roberts DD, Rodrigues SC, Rohde E, Rome S, Rouschop KMA, Rughetti A, Russell AE, Saá P, Sahoo S, Salas-Huenuleo E, Sánchez C, Saugstad JA, Saul MJ, Schiffelers RM, Schneider R, Schøyen TH, Scott A, Shahaj E, Sharma S, Shatnyeva O, Shekari F, Shelke GV, Shetty AK, Shiba K, Siljander PRM, Silva AM, Skowronek A, Snyder OL, Soares RP, Sódar BW, Soekmadji C, Sotillo J, Stahl PD, Stoorvogel W, Stott SL, Strasser EF, Swift S, Tahara H, Tewari M, Timms K, Tiwari S, Tixeira R, Tkach M, Toh WS, Tomasini R, Torrecilhas AC, Tosar JP, Toxavidis V, Urbanelli L, Vader P, van Balkom BWM, van der Grein SG, Van Deun J, van Herwijnen MJC, Van Keuren-Jensen K, van Niel G, van Royen ME, van Wijnen AJ, Vasconcelos MH, Vechetti IJ, Veit TD, Vella LJ, Velot É, Verweij FJ, Vestad B, Viñas JL, Visnovitz T, Vukman KV, Wahlgren J, Watson DC, Wauben MHM, Weaver A, Webber JP, Weber V, Wehman AM, Weiss DJ, Welsh JA, Wendt S, Wheelock AM, Wiener Z, Witte L, Wolfram J, Xagorari A, Xander P, Xu J, Yan X, Yáñez-Mó M, Yin H, Yuana Y, Zappulli V, Zarubova J, Žėkas V, Zhang J.-y., Zhao Z, Zheng L, Zheutlin AR, Zickler AM, Zimmermann P, Zivkovic AM, Zocco D, Zuba-Surma EK, Journal of Extracellular Vesicles, 7 (2018) 1535750.30637094 [Google Scholar]
- [173].Gardiner C, Di Vizio D, Sahoo S, Thery C, Witwer KW, Wauben M, Hill AF, J Extracell Vesicles, 5 (2016) 32945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Escudier B, Dorval T, Chaput N, André F, Caby M-P, Novault S, Flament C, Leboulaire C, Borg C, Amigorena S, Boccaccio C, Bonnerot C, Dhellin O, Movassagh M, Piperno S, Robert C, Serra V, Valente N, Le Pecq J-B, Spatz A, Lantz O, Tursz T, Angevin E, Zitvogel L, Journal of Translational Medicine, 3 (2005) 10–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A, Hsu D-H, Le Pecq J-B, Lyerly HK, Journal of Translational Medicine, 3 (2005) 9–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Shen H, Sun T, Hoang HH, Burchfield JS, Hamilton GF, Mittendorf EA, Ferrari M, Semin. Immunol, 34 (2017) 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Jiang W, Yuan H, Chan CK, von Roemeling CA, Yan Z, Weissman IL, Kim BYS, Nat. Rev. Drug Discov, 16 (2017) 369–370. [DOI] [PubMed] [Google Scholar]
- [178].Hu X, Wu T, Bao Y, Zhang Z, Control J Release, 256 (2017) 26–45. [DOI] [PubMed] [Google Scholar]
- [179].Sun Q, Zhou Z, Qiu N, Shen Y, Adv. Mater, 29 (2017). [DOI] [PubMed] [Google Scholar]
- [180].Venuta A, Wolfram J, Shen H, Ferrari M, Journal of Materials Chemistry B, 5 (2017) 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Blanco E, Sangai T, Hsiao A, Ferrati S, Bai L, Liu X, Meric-Bernstam F, Ferrari M, Cancer Lett, 334 (2013) 245–252. [DOI] [PubMed] [Google Scholar]
- [182].Scavo MP, Gentile E, Wolfram J, Gu J, Barone M, Evangelopoulos M, Martinez JO, Liu X, Celia C, Tasciotti E, Vilar E, Shen H, Colloids Surf. B. Biointerfaces, 136 (2015) 694703. [DOI] [PubMed] [Google Scholar]
- [183].Shen J, Wu X, Lee Y, Wolfram J, Yang Z, Mao ZW, Ferrari M, Shen H, J Vis Exp, (2015) 52075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Zhang M, Xu R, Xia X, Yang Y, Gu J, Qin G, Liu X, Ferrari M, Shen H, Biomaterials, 35 (2014) 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Shen J, Xu R, Mai J, Kim HC, Guo X, Qin G, Yang Y, Wolfram J, Mu C, Xia X, Gu J, Liu X, Mao ZW, Ferrari M, Shen H, ACS nano, 7 (2013) 9867–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Scott B, Shen J, Nizzero S, Boom K, Persano S, Mi Y, Liu X, Zhao Y, Blanco E, Shen H, Ferrari M, Wolfram J, Pharmacol. Res, 111 (2016) 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popovic Z, Jain RK, Bawendi MG, Fukumura D, Proc. Natl. Acad. Sci. U. S. A, 108 (2011) 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Li HJ, Du JZ, Du XJ, Xu CF, Sun CY, Wang HX, Cao ZT, Yang XZ, Zhu YH, Nie S, Wang J, Proc. Natl. Acad. Sci. U. S. A, 113 (2016) 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Li HJ, Du JZ, Liu J, Du XJ, Shen S, Zhu YH, Wang X, Ye X, Nie S, Wang J, ACS nano, 10 (2016) 6753–6761. [DOI] [PubMed] [Google Scholar]
- [190].Zhu L, Wang T, Perche F, Taigind A, Torchilin VP, Proc. Natl. Acad. Sci. U. S. A, 110 (2013) 17047–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Xu X, Saw PE, Tao W, Li Y, Ji X, Yu M, Mahmoudi M, Rasmussen J, Ayyash D, Zhou Y, Farokhzad OC, Shi J, Nano Lett, 17 (2017) 4427–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Li L, Sun W, Zhong J, Yang Q, Zhu X, Zhou Z, Zhang Z, Huang Y, Adv. Funct. Mater, 25 (2015) 4101–4113. [Google Scholar]
- [193].Khalid A, Persano S, Shen H, Zhao Y, Blanco E, Ferrari M, Wolfram J, Expert opinion on drug delivery, (2016) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Jiang W, Huang Y, An Y, Kim BY, ACS nano, 9 (2015) 8689–8696. [DOI] [PubMed] [Google Scholar]
- [195].Stylianopoulos T, Jain RK, Proc. Natl. Acad. Sci. U. S. A, 110 (2013) 18632–18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Keizman D, Huang P, Eisenberger MA, Pili R, Kim JJ, Antonarakis ES, Hammers H, Carducci MA, Eur. J. Cancer, 47 (2011) 1955–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Wilop S, von Hobe S, Crysandt M, Esser A, Osieka R, Jost E, Cancer Res J Clin. Oncol, 135 (2009) 1429–1435. [DOI] [PubMed] [Google Scholar]
- [198].Nakai Y, Isayama H, Ijichi H, Sasaki T, Takahara N, Ito Y, Matsubara S, Uchino R, Yagioka H, Arizumi T, Hamada T, Miyabayashi K, Mizuno S, Yamamoto K, Kogure H, Yamamoto N, Hirano K, Sasahira N, Tateishi K, Tada M, Koike K, Invest. New Drugs, 31 (2013) 1294–1299. [DOI] [PubMed] [Google Scholar]
- [199].Samuelsson E, Shen H, Blanco E, Ferrari M, Wolfram J, Colloids Surf. B. Biointerfaces, 158 (2017) 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Diagaradjane P, Deorukhkar A, Gelovani JG, Maru DM, Krishnan S, ACS nano, 4 (2010) 4131–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Liu T, Choi H, Zhou R, Chen IW, Nano Today, 10 (2015) 11–21. [Google Scholar]
- [202].Wolfram J, Nizzero S, Liu H, Li F, Zhang G, Li Z, Shen H, Blanco E, Ferrari M, Sci. Rep, 7 (2017) 13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Pelt J, Busatto S, Ferrari M, Thompson EA, Mody K, Wolfram J, Pharmacol. Ther, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Montanari F, Lu M, Marcus S, Saran A, Malankar A, Mazumder A, Blood, 124 (2014) 5775. [Google Scholar]
- [205].Yokoi K, Tanei T, Godin B, van de Ven AL, Hanibuchi M, Matsunoki A, Alexander J, Ferrari M, Cancer Lett, 345 (2014) 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Yokoi K, Kojic M, Milosevic M, Tanei T, Ferrari M, Ziemys A, Cancer Res, 74 (2014) 4239–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Sessa C, Guibal A, Del Conte G, Ruegg C, Nat. Clin. Pract. Oncol, 5 (2008) 378–391. [DOI] [PubMed] [Google Scholar]
- [208].Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, Rawn K, Voskas D, Dumont DJ, Ben-David Y, Lawler J, Henkin J, Huber J, Hicklin DJ, D’Amato RJ, Kerbel RS, Cancer Cell, 7 (2005) 101–111. [DOI] [PubMed] [Google Scholar]
- [209].Ramanathan RK, Korn RL, Raghunand N, Sachdev JC, Newbold RG, Jameson G, Fetterly GJ, Prey J, Klinz SG, Kim J, Cain J, Hendriks BS, Drummond DC, Bayever E, Fitzgerald JB, Clin. Cancer. Res, (2017). [DOI] [PubMed] [Google Scholar]
- [210].Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX, LoRusso PM, Munster PN, Campbell K, Gaddy DF, Leonard SC, Geretti E, Blocker SJ, Kirpotin DB, Moyo V, Wickham TJ, Hendriks BS, Clin. Cancer. Res, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Theis T, Parr D, Binks P, Ying J, Drexler KE, Schepers E, Mullis K, Bai C, Boland JJ, Langer R, Dobson P, Rao CN, Ferrari M, Nature nanotechnology, 1 (2006) 8–10. [DOI] [PubMed] [Google Scholar]
- [212].U.S. National Nanotechnology Initiative, https://www.nano.gov/nanotech101/what/definition, (2017).
- [213].European Medicines Agency, http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000345.jsp&, (2017).
- [214].“nanotechnology”, https://www.oxforddictionaries.com, (2017).
- [215].Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, Smith S, Bader AG, Kim S, Hong DS, Invest. New Drugs, 35 (2017) 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]