

Abstract
See “Deletion of IκBα activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A,” by Neuhöfer P, Liang S, Einwächter H, et al, on page 192; and “Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice,” by Huang H, Liu Y, Daniluk J, et al, on page 202.
The nuclear factor (NF)-κB inducible transcription factors are critical regulators of cell behavior both in health and disease.1–7 NF-κB mediates the expression of hundreds of genes that play key roles in inflammation, immunity, cell survival and proliferation, and is induced by a great variety of stresses. There are 5 NF-κB family members in mammals: RelA/p65, RelB, c-Rel, p50 (derived from its precursor p105/NF-κB1), and p52. NF-κB proteins bind to their cognate DNA sites as homo- or heterodimers, such as the prototypical p50-p65, and activate (in most cases) or negatively regulate target gene transcription. In most “resting” cells, NF-κB is kept inactive in the cytoplasm through sequestration in complexes with the inhibitor of NF-κB (IκB) proteins, such as IκBα and IκBβ. The sequestration prevents NF-κB migration into the nucleus, its’ binding to DNA, and transcriptional activation. Typically, in response to an inducing stimulus the IκBs are phosphorylated on specific Ser residues by IκB kinases (IKKs), which results in IκB ubiquitylation and proteasome-mediated degradation, allowing NF-κB nuclear translocation. The IKK complex is composed of 2 related kinases, IKK1 (or IKKα) and IKK2 (or IKKβ), and a scaffolding protein NEMO/IKKγ, which is critical for IKK activation. IKK activation involves recruitment of adaptor proteins and multiple ubiquitination events. The main (“canonical”) pathway, which is triggered by proinflammatory cytokines, for example, tumor necrosis factor-α, is primarily mediated by IKK2, and results in activation of p50-p65 and induction of genes regulating inflammation and cell survival. Transcriptional activity of NF-κB dimers is also regulated by their posttranslational modification, for example, C-terminal phosphorylation of p65. Thus, NF-κB activation is mediated and regulated at 3 levels: IKK, IκB, and the NF-κB dimers.
Inflammation and parenchymal cell death are key pathologic responses of pancreatitis and determinants of disease severity.8 Although the mechanisms initiating pancreatitis are not clear, the disease is believed to originate in injured acinar cells. In most patients, the ensuing inflammatory response and pancreatic damage ultimately resolve, but in severe cases unrelenting systemic inflammatory response syndrome leads to multiple organ (especially lung) failure, a major cause of mortality in acute pancreatitis.8 Uncontrolled inflammation also contributes to parenchymal necrosis, greater amounts of which bode a worse prognosis.
Dramatic increases in cytokines and chemokines were found in 1990s in both human and experimental pancreatitis.9 It was further shown that injured acinar cells themselves produce and release a variety of inflammatory mediators,10,11 followed by recruitment of inflammatory cells into the pancreas and systemic inflammation.12 Prompted by these results, an early pancreatic NF-κB activation was found in the “classical” cerulein model of acute pancreatitis and the ex vivo model of CCK-hyperstimulated acinar cells.13,14 Since then, NF-κB activation in pancreatitis has been studied in >250 publications15; it occurs in all experimental models, involves p50-p65 activation (i.e., the canonical pathway) mediated by degradation of both IκBα and IκBβ, and is associated with induction of cytokines and other inflammatory mediators. Mechanisms mediating NF-κB activation in acinar cells involve cytosolic Ca2+ increase and novel PKC isoforms.15 The p50-p65 and p50-p50 dimers are the only activated NF-κB complexes shown in pancreatitis.
In most studies, pharmacologic NF-κB inhibition ameliorated the inflammatory response, necrosis, and other parameters of pancreatitis severity.15 However, the pharmacologic agents were largely nonspecific, such as antioxidants and proteasomal inhibitors. The first knockout study16 found amelioration of cerulein pancreatitis in mice deficient in p50/p105, with decreased tumor necrosis factor-α level and leukocyte accumulation. In accord with these data, adenoviral transfer of RelA/p65 to elevate NF-κB in pancreas resulted in neutrophil infiltration and pancreatic and lung injury.17 Thus, a consensus was building that NF-κB activation in pancreatitis is detrimental. The next significant development came in 2007 in 2 studies,18,19 which both used the Cre/loxP technology, but came to mutually opposing conclusions. In one of them,18 acinar-cell–specific, inducible expression of constitutively active (CA) IKK2 was sufficient to trigger severe pancreatitis in the transgenic mice, and also aggravated cerulein pancreatitis. Furthermore, acinar-cell–specific expression of dominant negative IKK2 significantly improved cerulein pancreatitis. These results confirmed and expanded the previous findings.15 In stark contrast, the study19 from Schmid’s group found aggravation of cerulein pancreatitis in mice with pancreas-specific ablation of rela gene (termed rela∆panc), with more parenchymal necrosis and more inflammation in pancreas and lung. The authors concluded that p65 protects acinar cells from death associated with inflammation. Their findings revealed a previously unappreciated complexity of NF-κB roles in pancreatitis. However, the results of these 2 studies18,19 created a paradox in that both activation and inhibition of NF-κB in acinar cells aggravated disease.
The 2 new studies20,21 in this issue of GASTROENTEROLOGY continue along these “parallel tracks”— one20 using mostly inhibition, and the other21 activation of the IKK/NF-κB pathway. Neuhöfer et al20 (the Schmid-Algül group) generated mice with pancreas specific ablation of IκBα (termed IκBa∆panc), with the rationale that this would lead to constitutive activation of p65. The IκBa∆panc mice, indeed, showed some basal NF-κB activation and small increases in pancreatic cytokines and chemokines, which did not cause any overt phenotype. But cerulein- and L-arginine–induced pancreatitis was ameliorated in IκBa∆panc mice, with less pancreatic and lung injury and improvements in serum amylase, pancreatic necrosis, and trypsin activity.20 To further prove that the protective effects of IκBα ablation were through p65, the authors crossed IκBa∆panc with rela∆panc mice and showed that amelioration of cerulein pancreatitis was lost in the double-knockout IκBa∆panc × rela∆panc mice. The results reinforce this group’s previous conclusion19 that acinar cell NF-κB activation is protective in acute pancreatitis. Microarray analysis implicated serine protease inhibitor 2A (Spi2A) in the protective effect of NF-κB, and lentiviral-mediated expression of Spi2A improved cerulein pancreatitis.20
By contrast, Huang et al21 (the Logsdon-Ji group) pursued the strategy of directly activating acinar cell NF-κB by developing several strains of transgenic mice. One strain was generated by crossing mice expressing “loxP-stop-loxP-p65” (LSL-p65) construct and mice with inducible Cre recombinase under a strong acinar-cell–specific promoter. LSL excision in the resultant LSL-p65/Cre mice, indeed, caused an increase in p65, but this was compensated by greatly upregulated IκBα (the IκBα gene expression is induced by NF-κB) and did not lead to any obvious phenotype. However, upon induction of cerulein pancreatitis the LSL-p65/Cre mice displayed greater pancreatic NF-κB activation, early and dramatic induction of proinflammatory cytokines, and more severe disease, compared with controls, that did not express transgenic p65 (eg, harboring Cre alone). Thus, in contrast with Neuhöfer et al,20 the high level of acinar cell p65 did not protect, but aggravated pancreatitis. Furthermore, repeated administration of modest amounts of cerulein, which did not cause pancreatic damage in control mice, resulted in chronic pancreatitis injury in LSL-p65/Cre mice, with persistent inflammation and fibrosis.21 The second strain of transgenic mice, LSL-IKK2/Cre, similarly allowed inducible, acinar-cell–specific expression of IKK2-CA. In these mice, there was no compensatory up-regulation of IκBα, and the induction of IKK2-CA resulted in spontaneous pancreatitis within 7 days. The pancreas damage and inflammation increased with longer times of IKK2-CA expression. Furthermore, the injury was worse in triple transgenic LSL-IKK2/LSL-p65/Cre mice that combined elevated levels of p65 and IKK2-CA expression. Within several days, these mice spontaneously developed severe inflammation as well as pancreatic fibrosis (in particular, increased α-smooth muscle actin). The results21 are in accord with the previous findings from Logsdon’s group17 and the results from Baumann et al18 on IKK2-CA and dominant negative IKK2 transgenic mice.
The 2 studies20,21 perpetuate the paradox that activation of IKK2 or p65 leads to both aggravation (even induction) and amelioration of pancreatitis. However, this paradox can be resolved, at least in part, by realizing that acinar cell NF-κB activation, indeed, triggers both pro- and anti-inflammatory pathways (Figure 1). A major pro-inflammatory mechanism, emphasized in Ref. 21, is the initial release of cytokines and other mediators induced by NF-κB. The opposite effect of NF-κB19,20 is protection against acinar cell necroptosis that can limit pancreatic inflammation through as-yet unclear mechanisms. These mechanisms may involve limiting inflammasome activation by factors released from damaged or necrotic cells.22 Of note, the role of inflammasome in pancreatitis is just beginning to be explored.23 The protective effect of NF-κB could also be through limiting interleukin (IL)-1β processing and secretion.24 In this regard, it would be instructive to compare changes in the pancreatic and circulating levels of IL-1β between the transgenic NF-κB strains at different stages of cerulein pancreatitis.
Figure 1.
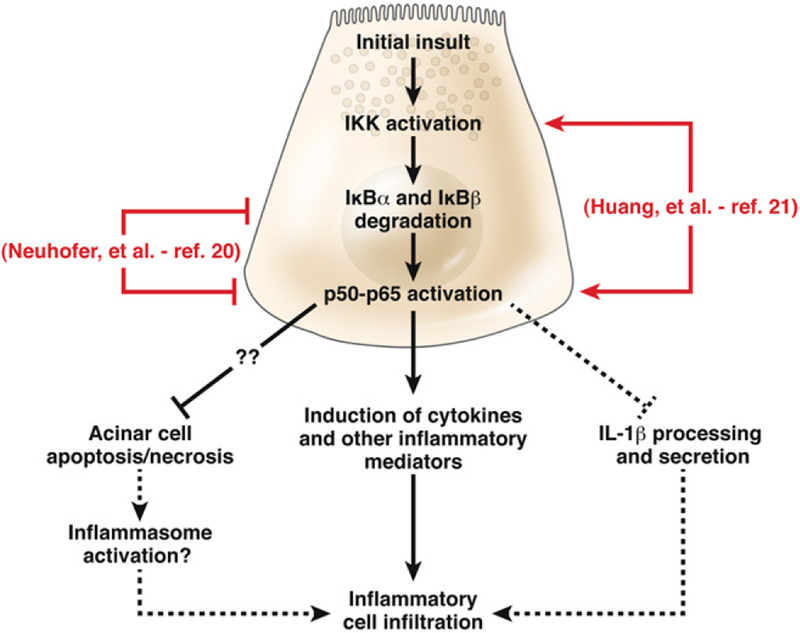
Pathways through which acinar cell NF-κB activation regulates inflammation in pancreatitis. A major proinflammatory mechanism is the induction of cytokines and other mediators, as emphasized by Huang et al.21 In contrast, Neuhöfer et al20 emphasize the protective effect of NF-κB against acinar cell necroptosis, limiting pancreatic inflammation. The mechanisms underlying this prosurvival effect of NF-κB are not clear but may involve PAP1,19 Spi2A,20 Bcl-xL,35 and other proteins. One mechanism whereby acinar cell necrosis may promote pancreatic inflammation is through activation of the inflammasome.22,23 The protective effect of NF-κB activation could also be through limiting IL-1β processing and secretion.24
The balance between these—as well as other yet undefined—mechanisms determines the extent, pattern, and kinetics of the inflammatory response, thus “integrating” the various effects of NF-κB activation. The higher level of NF-κB activity in transgenic mice generated in Huang et al21 clearly favors the proinflammatory “arm”; and increased local inflammation promotes acinar cell necrosis,25,26 which may overpower the protective, prosurvival effect of NF-κB. It is not so clear whether pancreatitis-induced inflammation is less or more in the IκBa∆panc mice, as the authors themselves state.20 In addition, the different approaches targeting IKK2, IκBα and p65 in20,21 may have nonoverlapping effects (see below).
The complexities of the IKK/IκB/NF-κB system, in particular both pro- and anti-inflammatory effects, are discussed in numerous reviews that appeared in 2012 to commemorate 25 years since the discovery of NF-κB (eg, a special issue of Immunological Reviews4–7). The following are some of the aspects relevant to20,21 (1) both the extent and the mode of NF-κB manipulation can have pro-foundly different consequences, as shown in the liver and intestine, and in particular on mice with NEMO ablation (essentially complete blockade of NF-κB) versus IKK2 ablation (less NF-κB inhibition) versus manipulating IκB (variable extent of NF-κB inhibition).1,2,6 In this regard, it will be interesting to assess the effects of NEMO ablation on experimental pancreatitis. (2) Modulating the NF-κB pathway has very different effects on the acute versus chronic inflammation and tissue damage, as shown by IKK2 ablation in intestinal epithelial cells.27,28 That is why it would be important to carefully examine the time-course of the effects of manipulating NF-κB in pancreatitis, in particular at the early, acute stage versus the repair/resolution stage. Equally important is elucidation of the role of NF-κB activation in inflammatory cells, which is often opposite to that in epithelial (ie, acinar) cells. Myeloid-cell specific NF-κB inhibition has been utilized for this purpose in other organs (eg, Eckmann et al28) and was shown in a chronic pancreatitis model to decrease overall inflammation and ameliorate disease.29 (3) Manipulating the IKK/IκB/NF-κB system at 1 level can perturb pathways that would not be affected by modulation at other levels. For example, the IKKs have targets beyond the NF-κB pathway, and some of their effects are independent of IKK kinase activity.1–3 Such pathways would not be affected by modulating “lower” levels, namely, p65. On the other hand, p65 ablation affects pathways involving p65 posttranslational modifications and interactions with other transcription factors1–5 (such as p53, shown to play a role in pancreatitis30). The absence of p65 can be compensated by other NF-κB dimers,5 which obviously does not occur when manipulating the IKKs.
One objective of Neuhöfer et al20 was to explore the interrelations between intra-acinar NF-κB and trypsinogen activation, both early events in pancreatitis. Findings from different studies17,31–33 show that intra-acinar trypsin does not affect NF-κB; however, whether NF-κB activation in acinar cells can or cannot directly modulate trypsin activity is not quite clear. One way of testing this would be to measure agonist (eg, CCK)-induced trypsin activity in acinar cells isolated from NF-κB transgenic mice,20,21 to bypass the effects of infiltrating inflammatory cells on trypsinogen activation in vivo.26,34 In our studies (unpublished observations), the blockade of NF-κB activation in p50−/− p65+/− acinar cells had no effect on CCK-induced trypsin activity, indicating there is no direct connection between the 2 events.
A common theme in the reviews on IKK/IκB/NF-κB1–7 is that this system is strikingly “context-dependent” and its’ fine-tuning will require exhaustive characterization of the underlying mechanisms. The studies in this issue of GASTROENTEROLOGY20,21 expand our knowledge of NF-κB in pancreatitis, but also underscore both its complexity and the gaps in our understanding of this pathway. Building on these results, elucidation of the many and diverse “trades” of NF-κB will clarify its role in the pathogenesis of pancreatitis and, hopefully, harness the therapeutic potential of this system for treatment of pancreatitis.
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012;26:203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007;8:49–62. [DOI] [PubMed] [Google Scholar]
- 3.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol 2009;19:404–413. [DOI] [PubMed] [Google Scholar]
- 4.Hinz M, Arslan SÇ, Scheidereit C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol Rev 2012;246: 59–76. [DOI] [PubMed] [Google Scholar]
- 5.Smale ST. Dimer-specific regulatory mechanisms within the NF-κB family of transcription factors. Immunol Rev 2012;246:193–204. [DOI] [PubMed] [Google Scholar]
- 6.Pasparakis M. Role of NF-κB in epithelial biology. Immunol Rev 2012;246:346–358. [DOI] [PubMed] [Google Scholar]
- 7.DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev 2012;246:379–400. [DOI] [PubMed] [Google Scholar]
- 8.Pandol SJ, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. Gastroenterology 2007;132:1127–1151. Erratum: ibid, 133:1056. [DOI] [PubMed] [Google Scholar]
- 9.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg 1998;175:76–83. [DOI] [PubMed] [Google Scholar]
- 10.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest 1997;100:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grady T, Liang P, Ernst SA, et al. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 1997;113:1966–1975. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia M, Brady M, Shokuhi S, et al. Inflammatory mediators in acute pancreatitis. J Pathol 2000;190:117–125. [DOI] [PubMed] [Google Scholar]
- 13.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 1998;275:G1402–1414. [DOI] [PubMed] [Google Scholar]
- 14.Steinle AU, Weidenbach H, Wagner M, et al. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999;116:420–430. [DOI] [PubMed] [Google Scholar]
- 15.Rakonczay Z Jr., Hegyi P, Takacs T, et al. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut 2008; 57:259–267. [DOI] [PubMed] [Google Scholar]
- 16.Altavilla D, Famulari C, Passaniti M, et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab Invest 2003;83:1723–1732. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Ji B, Han B, et al. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 2002;122:448–457. [DOI] [PubMed] [Google Scholar]
- 18.Baumann B, Wagner M, Aleksic T, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest 2007;117:1502–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Algul H, Treiber M, Lesina M, et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest 2007;117:1490–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neuhöfer P, Liang S, Einwächter H, et al. Deletion of IκBα activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A. Gastroenterology 2013;144:192 201. [DOI] [PubMed] [Google Scholar]
- 21.Huang H, Liu Y, Daniluk J, et al. Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 2013;144:202 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strowig T, Henao-Mejia J, Elinav E, et al. Inflammasomes in health and disease. Nature 2012;481:278–286. [DOI] [PubMed] [Google Scholar]
- 23.Hoque R, Sohail M, Malik A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011;141:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007;130:918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology 1996;111:1081–1091. [DOI] [PubMed] [Google Scholar]
- 26.Sendler M, Dummer A, Weiss FU, et al. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 2012. April 5 [Epub ahead of print]. [DOI] [PubMed]
- 27.Chen LW, Egan L, Li ZW, et al. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med 2003;9:575–581. [DOI] [PubMed] [Google Scholar]
- 28.Eckmann L, Nebelsiek T, Fingerle AA, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci U S A 2008;105:15058–15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treiber M, Neuhöfer P, Anetsberger E, et al. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology 2011;141:1473–1485. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura Y, Do JH, Yuan J, et al. Inflammatory cells regulate p53 and caspases in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 2010;298:G92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tando Y, Algül H, Schneider G, et al. Induction of IkappaB-kinase by cholecystokinin is mediated by trypsinogen activation in rat pancreatic lobules. Digestion 2002;66:237–245. [DOI] [PubMed] [Google Scholar]
- 32.Ji B, Gaiser S, Chen X, et al. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J Biol Chem 2009;284:17488–17498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dawra R, Sah RP, Dudeja V, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 2011;141: 2210–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002; 122:974–984. [DOI] [PubMed] [Google Scholar]
- 35.Sung KF, Odinokova IV, Mareninova OA, et al. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp Cell Res 2009; 315:1975–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]