

Abstract
BACKGROUND & AIMS:
Acute pancreatitis is characterized by premature intracellular activation of digestive proteases within pancreatic acini and a consecutive systemic inflammatory response. We investigated how these processes interact during severe pancreatitis in mice.
METHODS:
Pancreatitis was induced in C57Bl/6 wild-type (control), cathepsin B (CTSB)- knockout, and cathepsin L-knockout mice by partial pancreatic duct ligation with supramaximal caerulein injection, or by repetitive supramaximal caerulein injections alone. Immune cells that infiltrated the pancreas were characterized by immunofluorescence detection of Ly6g, CD206, and CD68. Macrophages were isolated from bone marrow and incubated with bovine trypsinogen or isolated acinar cells; the macrophages were then transferred into pancreatitis control or cathepsin-knockout mice. Activities of proteases and nuclear factor (NF)-κB were determined using fluorogenic substrates and trypsin activity was blocked by nafamostat. Cytokine levels were measured using a cytometric bead array. We performed immunohistochemical analyses to detect trypsinogen, CD206, and CD68 in human chronic pancreatitis (n = 13) and acute necrotizing pancreatitis (n = 15) specimens.
RESULTS:
Macrophages were the predominant immune cell population that migrated into the pancreas during induction of pancreatitis in control mice. CD68-positive macrophages were found to phagocytose acinar cell components, including zymogen-containing vesicles, in pancreata from mice with pancreatitis, as well as human necrotic pancreatic tissues. Trypsinogen became activated in macrophages cultured with purified trypsinogen or co-cultured with pancreatic acini and in pancreata of mice with pancreatitis; trypsinogen activation required macrophage endocytosis and expression and activity of CTSB, and was sensitive to pH. Activation of trypsinogen in macrophages resulted in translocation of NF-kB and production of inflammatory cytokines; mice without trypsinogen activation (CTSB-knockout mice) in macrophages developed less severe pancreatitis compared with control mice. Transfer of macrophage from control mice to CTSB-knockout mice increased the severity of pancreatitis. Inhibition of trypsin activity in macrophages prevented translocation of NF-κB and production of inflammatory cytokines.
CONCLUSIONS:
Studying pancreatitis in mice, we found activation of digestive proteases to occur not only in acinar cells but also in macrophages that infiltrate pancreatic tissue. Activation of the proteases in macrophage occurs during endocytosis of zymogen-containing vesicles, and depends on pH and CTSB. This process involves macrophage activation via NF-κB-translocation, and contributes to systemic inflammation and severity of pancreatitis.
Keywords: Pancreatic Inflammation, Mechanisms, Immune Response, Mouse Model
Graphical Abstract
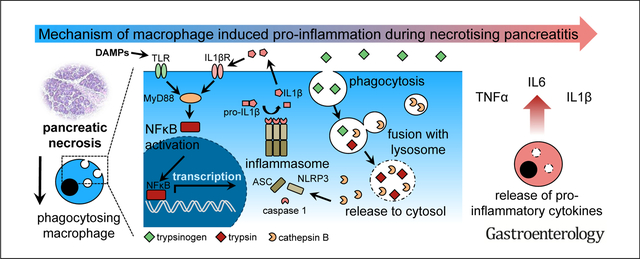
Acute pancreatitis is common and of increasing incidence in Western countries.1 The majority of cases suffer from a mild form of the disease but approximately 20% of patients develop severe pancreatitis, associated with pancreatic necrosis, systemic inflammation, and organ failure.2 An overwhelming systemic immune reaction, the so-called systemic inflammatory response syndrome, accounts for persistent multiorgan failure and seems to be responsible for the majority of systemic complications and mortality.3 The triggering events of this immune reaction and the pathophysiological mechanism that determine severity of the disease are still poorly understood.4
Self-digestion by its own proteases is considered to be a trigger of pancreatitis. Under pathologic conditions, the digestive serine protease trypsinogen is converted to active trypsin by the lysosomal hydrolase cathepsin B (CTSB) within the acinar cells.5–7 Premature intracellular protease activation is then followed by acinar cell death, which is accompanied by a pro-inflammatory response and leads to a prominent translocation of leukocytes. Especially cells of the innate immune system migrate into the injured organ.8 These cells are the first cells that reach the pancreas.9,10 This immune reaction boosts local damage and results in an increase in severity of disease.10,11 The importance of the protease/anti-protease balance for the development of pancreatitis is supported by the observation that germline mutations that increase the susceptibility toward developing pancreatitis mostly affect genes of the protease/anti-protease system, such as cationictrypsinogen (PRSS1), chymotrypsin-C (CTRC) or the trypsin-inhibitor SPINK1.12–16 How the intracellular activation of digestive proteases is linked to local and systemic inflammatory reaction is still being debated. On the one hand, acinar cells under experimental conditions activate nuclear factor ĸB,17 a key transcription factor for the proinflammatory response, but still do so in animals in which T7-trypsinogen, a major mouse-isoform of trypsinogen, has been deleted.18 On the other hand, acinar cell that undergo autodigestion and necrosis release damage-associated molecular patterns (DAMPs) such as free DNA, histones, or free adenosine triphosphate (ATP), which are recognized by immune cell receptors inducing a proinflammatory reaction and activation of the inflammasome pathway.19 The main task of infiltrating immune cells in the pancreas is thought to be defensive and includes the removal of cellular debris and necrosis. Most of the clearance is achieved through the action of macrophages that not only phagocytose cellular debris,20 but whose involvement in the subsequent immune reaction is profoundly influenced by what cellular components they are exposed to and which ones they ingest.21 For example, their clearance of apoptotic cellular waste involves largely noninflammatory pathways whereby components of necrotic cells can trigger a pro-inflammatory response.21 In the present study we found that, in areas of pancreatic necrosis, macrophages ingest zymogen-containing vesicles from damaged acinar cells that they convert these zymogens to active proteases in a CTSB-dependent manner and that the intramacrophage activation of trypsinogen is an important driver of local and systemic inflammation and disease severity.
Material and Methods
Isolation of Pancreatic Acini
Acini were isolated from mouse pancreas by collagenase digestion under sterile conditions, as previously reported.6 For details see Supplementary Materials and Methods.
Isolation of Bone Marrow-derived Macrophages
Bone marrow-derived macrophages (BMDM) were isolated from the femur and tibia of mice. A detailed protocol is provided in the Supplementary Materials.
Biochemical Assays
Serum amylase and different protease activities were measured as previously reported using substrates R110-Ile-Pro-Arg for trypsin, Suc-Ala-Ala-Pro-Phe-AMC for chymotrypsin, R110-Phe-Arg for CTSL, and AMC-Arg2 for CTSB.22 For details on in vivo imaging of proteases see Supplementary Materials.
Induction of Pancreatitis in Mice
All animal experiments were performed after prior approval by the Institutional Animal Care committee. C57Bl/6 mice were obtained from Charles River (Sulzfeld, Germany), CTSB−/−, cathepsin L (CTSL)−/−, and nucleotide-binding oligomerization domain-like receptor family pyrin domain- containing 3 (NLRP3)−/− mice were maintained in our animal facility; all mice strains are bred with a C57Bl/6 background.
Histology, Immunohistochemistry, and Immunofluorescence
Paraffin sections were used for H&E staining and Masson-Goldner-trichrome staining as previously reported.23 For details see Supplementary Materials.
Human Pancreatic Samples
Human chronic pancreatitis tissue was collected in the context of the ChroPac trial (ISRCTN38973832; http://www.isrctn.com/ISRCTN38973832).24 Necrotic tissue was harvested during endoscopic necrosectomy under the ethics committee approval for the ProZyt trial.
Statistical Analysis
All data are expressed as means ± standard error of the mean from at least 5 animals or experiments. Statistical analysis was performed using SigmaPlot and SigmaStat. Unpaired 2-tailed Student’s t-test or Rank-Sum test were used. Differences were considered significant for P < .05.
Results
Migration of Macrophages in Acute Pancreatitis
To study the pancreatic infiltrate we used 2 different models of acute pancreatitis: caerulein-induced pancreatitis and partial duct ligation to study extensive necrosis and exocrine tissue replacement by fibrosis after 14 days, as shown by H&E staining (Figure 1A).23
Figure 1.

Severe acute pancreatitis was induced by duct ligation and supramaximal caerulein stimulation. Infiltrating immune cells of the innate immune system were stained in pancreatic tissue (A) and quantified by cell counting (B). Time course and amount of neutrophil infiltration (Ly6g) and M1 macrophage (CD68) infiltration in the severe model of acute pancreatitis was compared with the mild form of the disease. H&E staining of the pancreas illustrating the severity of disease. The inflammatory infiltrate correlates with the amount of necrotic areas and the decrease of unaffected healthy exocrine tissue (C). The number of M2 macrophages (CD206) rises with increasing fibrosis (C). Three days after duct ligation the shape and volume of macrophages is significantly altered and increased in CD68-positive macrophages (D). Large CD68-positive macrophages display co-localization with pancreatic trypsinogen (E), indicating phagocytosis of dying acini or cellular waste. Neutrophils did not show any co-localization with trypsinogen (F). Asterisks (*) indicate significant differences with P < .05 (n = 5–8).
Staining of Ly6g illustrates the infiltration of granulocytes in pancreatic tissue. CD68 and CD206 are markers for macrophages, whereas CD68 is an indicator of the proinflammatory M1-phenotype and CD206 is only expressed on alternatively activated M2 macrophages. In contrast to the mild model of pancreatitis, the infiltration in severe necrotizing pancreatitis was dominated by CD68-positive macrophages (Figure 1A). Quantification of infiltrating cells per microscopic field revealed that CD68-positive macrophages represented the majority of infiltrating leukocytes in the ligation model of pancreatitis (Figure 1B), corresponding with the severity of the model. The number of infiltrating Ly6g-positive granulocytes did not differ between the pancreatitis models (Figure 1B). Supplementary Figure 1A and 1B show representative examples for the necrosis/fibrosis sequence in the ligation pancreatitis model. Alternatively, activated M2 macrophages were found increased at later time points (7 days and 14 days after duct ligation), which corresponded to the development of fibrosis. Whereas the number of M1 macrophages corresponded to the extent of necrosis at earlier time points (Figure 1B and 1C). Not only the number and phenotype of macrophages but also their morphology differed between models and over time (Figure 1D). Especially in necrotic areas, the size of infiltrating cells was significantly increased (Figure 1D).
It is well established that macrophages are responsible for clearance of necrotic areas by phagocytosing cellular debris.20 Co-labeling for trypsinogen, which is exclusively expressed by acinar cells, and CD68 suggested that macrophages phagocytose pancreatic enzymes or enzyme- containing vesicles from injured acinar cells within necrotic areas (Figure 1E). This phenomenon could only be observed in macrophages, where punctate vesicles contained trypsinogen, but not in other inflammatory cells (Figure 1F). Co-localization with chymotrypsin, another pancreatic serine-protease, was found in CD68-positive cells (Supplementary Figure 1C). CD68 positive M1 macrophages are mainly detected within necrotic areas, whereas CD206- positive cells are found in non-necrotic parts of the pancreas. Interestingly, we did not detect co-localization of trypsinogen within CD206-positive cells even 14 days postligation. Only few CD68-positive cells stained positive for trypsinogen later in the disease course (Supplementary Figure 1D–F). These data indicate that M1 macrophages in necrotic tissue areas of the pancreas ingest zymogen- containing components of acinar cells.
Macrophages Phagocytosing Digestive Zymogens From Injured Acinar Cells Undergo Activation
To investigate the role of phagocytosing macrophages, we performed co-incubation experiments using bone marrow-derived macrophages and freshly prepared acinar cells. Pure macrophages in culture are shown in Figure 2A. Isolated acini were stimulated with 1 μmol/L cholecystokinin (CCK) before co-incubation. Staining of trypsinogen and CD68 allowed discrimination between acinar cells and macrophages (Figure 2A) and showed that macrophages co-localize with zymogens or zymogen-containing vesicles from damaged acinar cells in vitro.
Figure 2.

BMDM were co-incubated with CCK-stimulated acini. Staining of CD68 marked macrophages, whereas trypsinogen was used as a marker for acini (A). Phagocytosis of acinar cell compartments is demonstrated by intra-macrophage trypsinogen staining (arrow). Six hours after co-incubation the transcription factor NFĸBp65 showed nuclear translocation in macrophages compared with untreated controls (B). Cytokine secretion of macrophages was significantly increased after 6 hours of co-incubation, as seen by secretion IL6, TNFα, and MCP-1. Acini alone showed a significantly lower secretion of these cytokines (C). Treatment of macrophages with LPS served as control. Furthermore, macrophage morphology changed during co-incubation (D-E). CD69, an early activation marker belonging to the C-type lectin domain family, is increased on macrophages co-incubated with acini or isolated zymogen granules (F). Asterisks (*) indicate significant differences with P < .05 (n = 6); # indicate a significant difference to all other conditions.
Staining macrophages for nuclear factor ĸB p65 (NFĸBp65) revealed a cytoplasmic distribution in unstimulated control cells, whereas 6 hours of co-incubation with CCK-stimulated acini induced a translocation of NFĸBp65 into the nucleus of CD68-positive macrophages (Figure 2B). In the same setting, co-incubation of macrophages with CCK-stimulated acini resulted in a significant increase of cytokine secretion, here shown for the proinflammatory cytokines interleukin-6 (IL6) and tumor necrosis factor α (TNFα), as well as the chemokine monocyte chemoattractant protein-1 (MCP-1) (Figure 2C). Lipopolysaccharide (LPS)-stimulated macrophages served as controls. Acini were also able to secrete cytokines, but the majority of cytokine release originated from macrophages (Figure 2C). When morphology of macrophages was analyzed for cell size (Figure 2D), co-incubation with acini in vitro resulted in a significant change in macrophage morphology, resembling the results of tissue sections (Figure 1D). Macrophages developed a spreading phenotype and changed their morphology in forward side scatter on fluorescence-activated cell sorter analysis (Figure 2E). Macrophages are activated upon co-incubation with acini or isolated zymogen granules (Figure 2F), which are engulfed by macrophages (Supplementary Figure 2A–C). These data indicate that co-incubation of macrophages with acinar cells results in activation of macrophages and a differentiation to a pro-inflammatory M1 phenotype.
Macrophages Phagocytose Zymogen-containing Vesicles and Activate Trypsinogen Intracellularly in a CTSB-dependent Manner
The detection of trypsin activity within phagocytosing macrophages raised the question whether activated trypsin was engulfed or whether macrophages activate trypsinogen intracellularly. We therefore isolated macrophages from bone marrow of C57Bl/6 mice and co-incubated them with bovine trypsinogen in a concentration of 10 μg/mL (see Supplementary Figure 2D). Labelling for trypsinogen and CD68 indicated that trypsinogen is internalized by macrophages (Figure 3A) and then converted to active trypsin intracellularly (Figure 3B). Intracellular trypsin activity in macrophages could be blocked completely by 50 μmol/L nafamostate, a potent digestive serine protease inhibitor, by bafilomycin-A1, an inhibitor of V-ATPases that neutralizes the usually acidic intravesicular pH, and by CA074me, a cell permeant CTSB inhibitor. Both substances led to a pronounced reduction in CTSB and trypsin activity in macrophages (Figure 3B). Furthermore, in vivo imaging of isolated macrophages co-incubated with trypsinogen revealed active trypsin inside macrophages (Figure 3C). Final evidence that trypsinogen undergoes activation in a CTSB-dependent manner within phagocytosing macrophages came from experiments using CTSB- and CTSL-deleted mouse macrophages incubated with bovine trypsinogen. Trypsin activity was significantly reduced in macrophages from CTSB−/− mice while it was increased in macrophages from CTSL−/− mice (Figure 3D), which is in accordance with the counteracting role of CTSL to CTSB.25 Immunofluorescence staining for CTSB and trypsinogen in macrophages incubated with bovine trypsinogen revealed a punctate localization of both enzymes in a vesicular compartment of macrophages (Figure 3E). These experiments show that macrophages activate phagocytosed trypsinogen intracellularly.
Figure 3.

BMDM were co-incubated with bovine trypsinogen over a time period of 6 hours. Immunofluorescence staining of trypsinogen revealed an intracellular membrane confined punctiform localization within CD68-positive macrophages after 6 hours (A). Activity measurements in cell lysates of macrophages proved a significant increase of trypsin activity in macrophages co-incubated with trypsinogen. Nafamostate, CA074me, or bafilomycin-A1 and lysosomal acidification were able to completely block trypsin activity (B), but only CA074me and bafilomycin-A1 caused a significant decrease in CTSB activity (B). Visual detection of active trypsin by the fluorogenic substrate R110-CBZ-Ile-Pro-Arg demonstrated intracellular located trypsin activity within macrophages (C). Comparison of WT macrophages with CTSB or CTSL-deficient macrophages showed significantly decreased trypsin activity in CTSB-deleted macrophages and increased activity in CTSL-deleted cells (D). Immunofluorescence staining indicates co-localization of phagocytosed trypsinogen and macrophage CTSB (E). Asterisks (#) indicate significant differences with P < .05 (n = 4–6).
Co-incubation of BMDM with freshly prepared and CCK- stimulated acini (rather than trypsinogen) again resulted in intracellular localization of trypsinogen as well as amylase within macrophages. In addition, a co-localization of CTSB with trypsinogen was observed (Figure 4A). Intracellular localization of protease activity showed a membrane-confined punctate localization for active trypsin and active chymotrypsin (Figure 4B). Furthermore, fluorescent-labeled zymogen granules showed trypsin activity visualized by R110-Ile-Pro-Arg cleavage (Supplementary Figure 2A). Western blotting of macrophage lysates confirmed the intracellular presence of trypsinogen and amylase (Figure 4C). Activity of trypsin in macrophages after coincubation with acini was significantly increased. Even when macrophages were co-incubated with acini isolated from CTSB−/− mice, we observed a significant increase in trypsin activity (Figure 4D), indicating that macrophage CTSB, rather than acinar cell CTSB, drives trypsin activation. When we used macrophages isolated from CTSB−/− mice we found no increase in trypsin activity after co-incubation with acini from wild-type (WT) or CTSB−/− mice. Both nafamostate and CA074me blocked trypsin activity after coincubation (Figure 4E), indicating that intracellular trypsin activation in macrophages is entirely dependent on the presence and activity of CTSB. Amylase activity was measured in macrophages as a marker of phagocytosis of acinar cell components. Uptake was not impaired by nafamostate pretreatment, whereas pretreatment of macrophages with CA074me resulted in an increase in intracellular amylase activity (Figure 4E), indicating that inhibition of CTSB leads to impaired degradation of amylase. To investigate whether phagocytosis is the mechanism of uptake for pancreatic zymogens, we used cytochalasin-B to block phagocytosis by inhibition of the network formation of actin filaments.26 Cytochalasin-B greatly reduced trypsin, chymotrypsin, and amylase activity in macrophages exposed to either bovine trypsinogen or acinar cells (Figure 4F). The activity of CTSB was not affected by cytochalasin-B treatment (Figure 4F).
Figure 4.

BMDM were co-incubated with freshly prepared acini stimulated with 1 μmol/L CCK. Co-incubation of macrophages with acini led to intracellular localization of trypsinogen and pancreatic amylase in CD68-positive macrophages (A). Further- more, co-localization of trypsinogen and CTSB was observed (A). Live-cell imaging with fluorochrome substrates for trypsin (green fluorescent ± DAPI [4’,6-diamidino-2-phenylindole]) or chymotrypsin (blue fluorescent) demonstrated an intracellular punctiform localization of active pancreatic enzymes within macrophages (B). Western blotting of macrophage lysates stain positive for trypsinogen when co-incubated with acini or trypsinogen, but only macrophages co-incubated with acini showed a band for pancreatic amylase (C). Macrophages from WT animals revealed significantly increased trypsin activity when coincubated with acini, even with acini of CTSB−/− mice, in contrast to macrophages of CTSB-deleted mice, which showed no increase in trypsin activity if co-incubated with WT or CTSB-deleted acini (D). Inhibition of serine proteases by nafamostate completely abolished trypsin activity. Similarly, inhibition of CTSB activity by CA074me reduced intracellular trypsin activity in macrophages but the uptake of acinar cell proteins, as demonstrated by amylase activity, was not reduced by the nafamostate or CA074me (E). Treatment of macrophages with cytochalasin-B reduced the uptake of acinar cell proteins as well as trypsinogen, resulting in decreased trypsin activity and decreased amylase or chymotrypsin activity in macrophages. CTSB activity was not affected by cytochalasin-B (F). Asterisks (*) indicate significant differences with P < .05 (n = 4–6).
Blockade of Digestive Protease Activation Within Macrophages Reduces Pro-inflammatory Signaling and Cytokine Secretion
Macrophages engulf cellular debris from injured acini and incorporate pancreatic zymogens and zymogen-containing vesicles. Phagocytosed zymogens, especially trypsinogen, undergo activation in a CTSB-dependent manner within macrophages. We therefore investigated the effect of this intracellular protease activation on macrophages.
Translocation of NFĸBp65 into the nucleus indicates pro-inflammatory differentiation of macrophages (M1). Co-incubation of BMDM with CCK-stimulated acini resulted in a nuclear redistribution of NFĸBp65. This translocation was blocked by nafamostate (Figure 5A), indicating that trypsin activity is involved in this process. Untreated macrophages served as negative controls while LPS treatment served as positive controls (Figure 5A). Both nafamostate and bafilomycin-A1 abolished intracellular trypsin activity without affecting phagocytosis, as shown by an unimpaired amylase uptake (Figure 5B). Cytokine release of macrophages co-incubated with acini was determined by cytometric bead array. Treatment with nafamostate significantly reduced secretion of pro-inflammatory cytokines such as IL6, MCP-1, and TNFα. Treatment with bafilomycin-A1 had a similar effect, with the exception of TNFα (Figure 5C). The effect of bafilomycin-A1 on TNFα secretion in macrophages has been described previously.27 Comparing the cytokine release of macrophages from WT mice with CTSB−/− cells, we found significantly reduced IL6, TNFα, and MCP-1 secretion after co-incubation with acini (Figure 5D). This is not a generalized defect because CTSB−/− macrophages responded to LPS adequately and in the same manner as WT macrophages (Supplementary Figure 2E). Transcriptome analysis of macrophages was performed 6 hours after co-incubation with acini. Transcriptional alteration of NFĸB-related genes was affected by pre-treatment with nafamostate, blocking intracellular trypsin activity and proving a trypsin-mediated effect on NFĸB-signaling. In particular, NFĸB2 and Rela were significantly decreased (Student’s t-test of fold change) (Figure 5E and 5F).
Figure 5.

BMDM were co-incubated with CCK-stimulated acini for 6 hours. Immunofluorescence staining of NFĸBp65 showed translocation to the nucleus of LPS stimulated macrophages compared with untreated controls. Co-incubation of macrophages and acini resulted in nuclear translocation of NFĸBp65, while treatment with nafamostate prohibited nuclear translocation (A). Treatment with nafamostate as well as bafilomycin-A1 inhibited intracellular trypsin activity in macrophages, but did not affect phagocytosis, as demonstrated by amylase activity (B). After 6 hours of co-incubation, the cytokine release in the supernatant was measured (C). Blockade of protease activation with nafamostate or bafilomycin-A1 significantly reduced IL6-, MCP-1, and TNFα release. The same effect was observed in CTSB-deleted macrophages compared with WT cells (D). Transcriptome analysis showed increased expression of NFĸB-related genes in macrophages after co-incubation with acini, which is reduced upon nafamostate treatment for NFĸB2 and Rela (E–F). Asterisks (*) indicate significant differences with P < .05 (n = 4–6); # indicate a significant difference of untreated cells compared with treatment.
These data suggest that the pro-inflammatory phenotype of macrophages in pancreatitis is dependent on CTSB-mediated trypsinogen activation within phagocytosing macrophages.
Protease Activation Within Macrophages Acts as DAMP and Induces NLRP3 Inflammasome Activation
Pathway analysis of macrophage transcriptome data by Ingenuity Pathway Analysis shows a significant down-regulation of the toll-like receptor pathways, as well as the NFĸB pathway, after co-incubation with acini and trypsin inhibition by nafamostate (Figure 6A). MyD88 is the upstream regulator (P = 2.0E−6) that is affected most. Detailed analysis of the NFĸB pathway suggests a critical role of IL1β for the activation of NFĸB (Supplementary Figure 3). Secretion and maturation of IL1β depends on inflammasome activation and caspase-1.19,28 Immunohistochemistry of caspase-1 shows expression within infiltrating inflammatory cells but not in pancreatic acini 3 days after duct ligation (Figure 6B). Secretion of mature IL1β from macrophages in response to acini proves in vitro activation of the inflammasome; in contrast, CCK-stimulated acini did not secret IL1β. Blockade of macrophage cathepsins by Ca074me or E64d abolished IL1β secretion (Figure 6C). Genetic deletion of NLRP3, an essential part of the inflammasome complex, did not affect macrophage phagocytosis and intracellular protease activation upon co-incubation with WT acini (Figure 6D). NFĸB translocation into the nucleus (Figure 6E), as well as cytokine secretion of IL6, TNFα, and MCP-1, was reduced in the same way as after treatment with nafamostate (Figure 6F).
Figure 6.

Pathway analysis of transcriptome data was performed by using Ingenuity Pathway Analysis software. Major pathways that were induced or repressed in macrophages co-incubated with acini ± nafamostate reveal down-regulation of the toll-like receptor pathway, as well as the NFĸB-pathway (A). The major inducer of the NFĸB pathway via MyD88 was IL1β.54 IL-1β maturation depends on caspase-1, which is exclusively expressed in infiltrating inflammatory cells and not in acini during pancreatitis (B). In vitro assay of BMDM revealed secretion of IL1β into the medium 6 hours after co-incubation with acini, while treatment with 50 μmol/L CA074me or 20 μmol/L E64d abolished IL1β release. Acini by themselves were not able to release mature IL1β (C). BMDM from NLRP3−/− mice phagocytose in the same way as macrophages from WT mice, as shown by trypsin and amylase content (D). NFĸB translocation into the nucleus was reduced in NLRP3-deleted macrophages after co-incubation with acini (E), resulting in a decreased secretion IL6, TNFα, and MCP-1 (F).
Protease activation within macrophages results in IL1β maturation and secretion, which enhances the proinflammatory macrophage phenotype by acting on the IL1β receptor expressed on macrophages in an autocrine loop (Supplementary Figure 2C).
Adoptive Transfer of WT Macrophages Restores Trypsinogen Activation During Severe Necrotizing Pancreatitis in CTSB−/− Mice
As previously reported, significant numbers of macrophages migrate into the pancreas during the early phase of pancreatitis, and these infiltrating cells express considerable concentrations of CTSB (Supplementary Figure 4A). In fact, CTSB is the second most abundant protein in macrophages (data not shown). This would make it plausible that macrophages also contribute to intrapancreatic trypsinogen activation in vivo. We therefore induced severe pancreatitis in WT- and cathepsin B-deleted animals. Two days after duct ligation we performed an adoptive transfer of BDMB generated from WT or CTSB−/− mice (Figure 7A) and sacrificed animals 24 hours after adoptive transfer of macrophages. Staining for CTSB indicated the presence of CTSB in all cells of WT animals; however, in WT animals receiving CTSB−/− macrophage transfer, we detected CTSB-negative cells in the pancreas. In contrast, in CTSB−/− mice no signal for CTSB was detected and only CTSB−/− mice that had received WT macrophages displayed some CTSB-positive infiltrating cells in the pancreas (Figure 7B). We observed a prominent infiltration of F4/80-positive macrophages in all animals, independently of the adoptive transfer. H&E histology showed pancreatic damage and necrosis in all animals, but to a significantly lesser degree in CTSB-deficient animals (Figure 7B).
Figure 7.

Adoptive-transfer of BMDM from WT mice into CTSB-deleted animals and vice versa was performed (A). Staining of CTSB showed infiltrating CTSB-positive cells in the pancreas of CTSB-deleted mice after adoptive transfer of WT macrophages, while adoptive transfer of CTSB-deleted macrophages in CTSB-deleted mice showed no signal (B). F4/80 staining illustrates macrophage infiltration in all animals to the same extent and H&E staining of the pancreas showed pancreatic damage and necrosis in all groups with a higher degree of severity in WT mice (B). Measurement of CTSB activity revealed a significantly higher activity in CTSB-deleted animals treated with WT macrophages, while WT mice displayed the highest activity (C). The adoptive transfer of WT macrophages restored trypsin activity in CTSB-deleted animals, and the additional transfer of WT macrophages in CTSB+/+ mice resulted in an increase in trypsinogen activation compared with the transfer of CTSB−/− macrophages (C). In addition to trypsin activity, serum amylase levels were also increased in CTSB−/− animals with CTSB+/+ macrophages, as well as serum levels of MCP-1 (D). Asterisks (*) indicate significant differences with P < .05 (n = 8–12). Human chronic pancreatitis and necrosectomy specimens were stained for CD68-positive macrophages. Chronic pancreatitis tissue showed in healthy parts a lower number of macrophages and an excessive infiltration of CD68-positive cells in parts of the section where acute damage was observed (E). A human necrosectomy specimen showed only detached areas with CD68-positive cells; for most parts of the tissue only ghosts of cells were detected (E). The highly infiltrated parts of chronic pancreatitis tissue were stained for trypsin and cD68. Here, we detected co-localization of trypsin in CD68-positive macrophages similar to that in mice (F).
CTSB activity in pancreatic homogenates was completely suppressed in CTSB−/− animals and only in animals that had received WT macrophages did we observed an increase in CTSB activity during pancreatitis (Figure 7C). Untreated animals without duct ligation pancreatitis served as control. In pancreatic homogenates from CTSB−/− mice, we detected only traces of active trypsin and even after induction of pancreatitis we did not detect a significant increase in trypsin activity when CTSB−/− macrophages were adoptively transferred. Only in animals that had received WT macrophages we found a significant increase in trypsin activity in pancreatic tissue homogenates (Figure 7C). In line with these results, in CTSB−/− animals adoptive transfer of WT macrophages significantly increased trypsinogen activation in WT animals compared with mice that had received CTSB−/− macrophages. These data confirm our findings from the in vitro studies that CTSB-containing macrophages contribute to intrapancreatic trypsinogen activation in the course of severe acute pancreatitis in vivo. The time course of trypsinogen activation in the duct ligation model showed elevated trypsin activity over 14 days, and we observed trypsinogen phagocytosing macrophages at all time points (Supplementary Figure 5). Macrophage-derived protease activation depended on the presence of necrosis. A much greater effect on disease severity and systemic inflammation was observed in CTSB−/− animals 3 days after duct ligation (Supplementary Figure 4B and 4C). The differences between CTSB−/− and CTSB+/+ animals were more pronounced in the severe model of pancreatitis compared with the mild caerulein-induced disease model.5 In a genetic model of chronic pancreatitis in Lamp2-deficient mice,29 we observed enlarged CD68-positive macrophages within the pancreas in conjunction with intracellular trypsinogen and CTSB co-localization (Supplementary Figure 4D).
It is well established that inflammatory response determines the severity of pancreatic damage, and infiltrating monocytes and neutrophils can directly induce acinar cell damage.10,11 Adoptive transfer of WT macrophages in CTSB-deleted animals resulted not only in an increase in serum amylase but also in serum MCP-1 in animals that had received WT macrophages (Figure 7D). Systemic inflammation thus depends on intra-macrophage trypsinogen activation, which may be the reason why CTSB−/− mice display less systemic inflammation.
Macrophages in Human Pancreatic Tissue
Paraffin sections of human pancreatic tissue from chronic pancreatitis patients, as well as necrotic tissue from endoscopic necrosectomies, were stained for CD68. Macrophage infiltration was observed in human chronic pancreatitis tissue with regional differences. Tissue samples from necrosectomy specimens contained only a low number of vital cells, but macrophage infiltration was also observed here (Figure 7E). Co-localization of trypsinogen and CD68 in human chronic pancreatitis samples proved that macrophages phagocytose trypsinogen in humans in the same way as in mouse models of pancreatitis (Figure 7F). In addition to CD68+ macrophages, CD206+ M2 macrophages were also detected in pancreatic tissue, but we never observed co-localization of trypsinogen in CD206+ cells (Supplementary Figure 6). Our data suggest that phagocytosing macrophages can actively contribute to the regulation of the immune response in the course of pancreatitis in mice and men.
Discussion
Macrophages are cells of the innate immune system that migrate into the pancreas within hours after the onset of pancreatitis and are involved in local injury via the release of cytokines such as TNFα.10,30,31 Pancreatitis is primarily a sterile inflammation and activation of immune cells in the early disease course is largely independent of infectious pathogens. The activation of immune cells and recruitment of leukocytes is thought to be mediated by the release of pro-inflammatory mediators and chemokines directly from acinar cells17,30 as well as via DAMPs (eg, free DNA, his-tones, or free ATP) activating toll-like-receptors and the inflammasome complex on immune cells.19,28 This DAMP-related activation of immune cells links inflammation directly to pancreatic damage. In this context, acinar damage and local inflammation form a causal loop activating each other in a chain reaction. Intracellular protease activation is known to be a hallmark of pancreatitis32 but is only indirectly linked to the inflammatory response. From T7 trypsinogen-deleted mice we have learned that NFĸB activation can occur independently of trypsinogen activation.18 So far, most investigations have either focused on acini or on inflammatory cells, while the present project addresses the cross-talk between intracellular protease activation and inflammation.
Macrophages are characterized by a high plasticity. Differentiation of macrophages changes their role in the pathways of inflammation. Alternatively activated macrophages (M2) have mainly an anti-inflammatory profile promoting wound healing or fibrosis,33,34 whereas classically activated macrophages (M1) are characterized by a prominent pro-inflammatory phenotype and play a critical role for host defenses against pathogens.33 In pancreatitis, the role of macrophages appears to change during the different disease stages. In the chronic phase of the disease mainly alternatively activated M2 macrophages are detected in the pancreas and involved in fibrosis formation via stimulation of pancreatic stellate cells.23,34 In acute pancreatitis, macrophages infiltrate into the pancreas, polarize into the pro-inflammatory M1 phenotype, and drive additional tissue damage.10,23 Our results show a positive correlation between the extent of pancreatic damage and the number of infiltrating macrophages, whereas for neutrophils no such correlation existed. Because the major task of neutrophils is bacterial clearance by respiratory burst in pancreatitis,35 a primarily sterile inflammation, the infiltration of neutrophils is more or less an unspecific response, although their ability to form neutrophil extracellular traps within small pancreatic ducts appears to contribute to disease progression.36 In contrast to neutrophils, monocytes and macrophages can fulfill multiple functions depending on their polarization.33 One of these functions is the elimination of apoptotic cells, necrotic tissue, and cellular debris.20 Removal of apoptotic or necrotic cells is a crucial function of macrophages and necessary for maintaining tissue homeostasis and regeneration.37 We could show that CD68-positive macrophages are attracted to necrotic areas in the pancreas where they phagocytose cellular debris. Cells that undergo apoptosis are rapidly removed without inducing a pro-inflammatory response. Necrotic cell death, the major form of cell death during severe forms of pancreatitis,38 leads to a pro-inflammatory response of macrophages and can contribute to disease severity.21,37 We observed a strong pro-inflammatory reaction of macrophages exposed to CCK-stimulated acini, leading to nuclear translocation of NFĸBp65 and the release of high concentrations of pro-inflammatory cytokines. Thus, macrophages trigger a pro-inflammatory reaction during acute pancreatitis. The pro-inflammatory phenotype (M1) depends on necrotic acinar cell death, the lack of DAMP signaling leads to an alternative macrophage activation (M2 phenotype) that is associated with fibrosis induction3.4 The balance between M1 and M2 macrophages correlates to the necrosis-fibrosis sequence, which underlines the crucial role of macrophages during acute and chronic pancreatitis.23,34,39
The pancreas synthesizes and secretes a multitude of digestive enzymes and, in contrast to other organs, the macrophages of the pancreas could phagocytose large amounts of zymogens when they ingest injured acini or their components. During phagosome maturation, fusion with lysosomes is a critical event for the degradation of phagocytosed content.40 In pancreatitis, CTSB (the major activator of trypsinogen5,6) and pancreatic digestive proteases undergo co-localization within the same compartment, not only in acinar cells, but apparently also in macrophages. Here we show that trypsinogen undergoes activation in macrophages. This protease activation follows the same pathway as in acinar cells and depends on the presence of CTSB and an acidic pH in the vesicular compartment. Moreover, in mouse models of acute pancreatitis, we could restore protease activation in CTSB-deleted mice by adoptive transfer of CTSB-expressing macrophages and demonstrated their migration into the inflamed pancreas. This means that pancreatic protease activation is not restricted to acinar cells. Protease activation is not only induced in acinar cells by transmigrating leukocytes,10 it also arises within phagosomes of macrophages. It is well established that the rise in intrapancreatic trypsin activity during pancreatitis follows a biphasic pattern.5 Intracellular protease activation in acinar cells in the early phase of the disease begins in a zymogen granule-enriched subcellular fraction,6,32 whereas the second peak of trypsin activity (after hours, rather than minutes) localizes to a lighter, non-zymogen fraction.6 While it is technically impossible to distinguish the cellular source of trypsin-containing subcellular vesicles, our results make it likely that the second peak of protease activation during pancreatitis originates from macrophages, rather than other types of leukocytes5 or acinar cells. Prior studies on macrophages during pancreatitis demonstrated a decreased protease activation in mice after macrophage depletion with clodronate-containing liposomes.10 Severity of disease depends on macrophage-derived cytokine secretion, such as TNFα,31 which is responsible for intra-acinar protease activation and necrosis.10 Our data show that the second, macrophage-driven peak of intrapancreatic trypsin activity has an effect on disease severity. While previous studies have suggested that the initial, intra-acinar cell trypsin activation does not affect the local or systemic inflammatory response,18 our results indicate that trypsinogen activation within macrophages greatly increases inflammation. The nuclear translocation of NFĸBp65, as well as the pro-inflammatory release of cytokines from macrophages or their differentiation to the M1-phenotype, could be abolished by trypsin inhibition (nafamostate), pH neutralization (bafylomycin-A1), or by deletion or inhibition of CTSB. The regulation of immune responses via serine proteases is not unheard of. During cystic fibrosis, cleavage of the phosphatidylserine receptor by polymorphonuclear leukocyte (PMN) elastase results in impaired macrophage-mediated tissue clearance and ongoing inflammation.41 In vitro experiments using isolated macrophages demonstrated a pro-inflammatory effect of serine proteases that could be prevented by protease inhibitors.42 Activation of the phosphatidylserine receptor was found to be involved in the anti-inflammatory response of macrophages43,44 and the recognition of apoptotic cells.43,45 Another crucial mechanism for macrophage activation is the formation of the inflammasome complex that leads to secretion of IL1β. It is well known that phagosomal rupture and the release of multiple cathepsins into the cytosol induces formation of the inflammasome complex.46 On the other hand, active trypsin destabilizes cathepsin-containing compartments like phagolysosomes.47 Taken together, active trypsin acts as a destabilizing agent on lysosomes that induces inflammasome activation and IL1β release. Macrophages express the IL1β receptor and react in an autocrine pro-inflammatory loop on IL1β stimulation with NFĸB translocation to the nucleus.48 Thus, intra-macrophage protease activation can result in an autocrine loop in macrophage activation that additionally enhances the pro-inflammatory response (Supplementary Figure 7). Even if, according to animal data, blockage of the IL1β pathway could be a therapeutic option for the treatment of severe acute pancreatitis in man, similar to TNFα blockage, we would like to propose a word of caution: It is well known that treating animals with a IL1 receptor agonist (IL1ra) reduces systemic inflammation, and serum levels of IL6 and TNFα, as well as pancreatic damage.49 Furthermore, genetic deletion of IL1β50 or components of the inflammasome complex reduce disease severity and systemic inflammation.19,28,51 However, all those experimental treatments were given prophylactically and we do not have data on the effect of IL1β inhibition in fully established systemic inflammatory response syndrome. Thus, pancreatic inflammation appears to be a self-sustaining mechanism driven by acinar cells and macrophages. Also, in human chronic pancreatitis samples or necrosectomy specimens, macrophage infiltration and ingestion of trypsinogen seem to be a relevant characteristic.
Macrophages appear to play a crucial role in regulating the immune response and are the dominant immune cell population that migrates into the pancreas. In addition, some genetic models of chronic pancreatitis, such as Lamp2-deficient mice or IL1β-transgenic mice, support an important role of macrophages in the disease development.29,52 Activation and recruitment of macrophages was previously thought to be mediated by cytokine release of acinar cells.53 Recent data further suggested a critical role of DAMPs, which activate the inflammasome/caspase-1 complex.19,28 According to our study, a third mechanism of macrophage activation must be added - that induced by intramacrophage trypsinogen activation. This mechanism is unique to the pancreas, depends on the phagocytosis of cellular components of injured acini in necrotic tissue, depends on the activity of CTSB in macrophages (rather than in acinar cells), and lends itself to therapeutic interventions. Most importantly, targeting intra-macrophage trypsinogen activation would address disease severity at a time point when pancreatitis and tissue necrosis are fully established, rather than trying to treat an early event that most often has already passed when the patient is admitted to the emergency room. This option would clearly be attractive for therapeutic studies.
Supplementary Material
EDITOR’S NOTES.
BACKGROUND AND CONTEXT
Necrotizing acute pancreatitis induces an overwhelming systemic inflammatory response (SIRS), which is mediated by infiltrating phagocytosing macrophages. Persistent SIRS results in multiorgan failure.
NEW FINDINGS
Pancreatic protease activation, a hallmark of acute pancreatitis, is not restricted to acinar cells but phagocytosing macrophages are able to intracellularly activate trypsinogen to trypsin in a cathepsin B dependent manner. Active trypsin inside macrophages acts as DAMP and fuels systemic inflammation.
LIMITATIONS
Macrophage mediated protease activation is limited to a severe model of acute necrotising pancreatitis.
IMPACT
Intrapancreatic protease activation within macrophages is directly linked to NFĸB activation and the systemic inflammatory response determining severity of the disease. This allows therapeutic targeting of two causals mechanism in pancreatitis.
Funding
The Deutsche Forschungsgemeinschaft (DFG MA 4115/1–2/3, DFG SE 2702/2–1, GRK 1947;A3), the Federal Ministry of Education and Research (BMBF GANIMED 03IS2061A and BMBF 0314107, 01ZZ9603, 01ZZ0103, 01ZZ0403, 03ZIK012), and the European Union (EU-FP-7: EPC-TM), V-630-S-150-2012/132/ 133 and ESF/14-BM-A55-0045/16.
Abbreviations used in this paper:
- ATP
adenosine triphosphate
- BMDM
bone marrow-derived macrophages
- CCK
cholecystokinin
- CTSB
cathepsin B
- CTSL
cathepsin L
- DAMP
damage-associated molecular pattern
- IL
interleukin
- LPS
lipopolysaccharide
- MCP-1
monocyte chemoattractant protein-1
- NKĸBp65
nuclear factor ĸB p65
- NLRP3
nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3
- TNFα
tumor necrosis factor a
- WT
wild-type
Footnotes
Conflict of interest
The authors disclose no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2017.10.018.
References
- 1.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013; 144:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernández CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373. [DOI] [PubMed] [Google Scholar]
- 3.Working Group IAP/APA Acute Pancreatitis Guidelines. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013;13(Suppl 2): e1–e15. [DOI] [PubMed] [Google Scholar]
- 4.Mayerle J, Dummer A, Sendler M, et al. Differential roles of inflammatory cells in pancreatitis. J Gastroenterol Hepatol 2012;27(Suppl 2):47–51. [DOI] [PubMed] [Google Scholar]
- 5.Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 2000;106: 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sendler M, Maertin S, John D, et al. Cathepsin B activity initiates apoptosis via digestive protease activation in pancreatic acinar cells and experimental pancreatitis. J Biol Chem 2016;291:14717–14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirano T, Saluja A, Ramarao P, et al. Apical secretion of lysosomal enzymes in rabbit pancreas occurs via a secretagogue regulated pathway and is increased after pancreatic duct obstruction. J Clin Invest 1991;87: 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnekenburger J, Schick V, Krüger B, et al. The calcium binding protein S100A9 is essential for pancreatic leukocyte infiltration and induces disruption of cell-cell contacts. J Cell Physiol 2008;216:558–567. [DOI] [PubMed] [Google Scholar]
- 9.Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology 1996; 111:1081–1091. [DOI] [PubMed] [Google Scholar]
- 10.Sendler M, Dummer A, Weiss FU, et al. Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 2013;62:430–439. [DOI] [PubMed] [Google Scholar]
- 11.Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 2002;122:974–984. [DOI] [PubMed] [Google Scholar]
- 12.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141–145. [DOI] [PubMed] [Google Scholar]
- 13.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000; 25:213–216. [DOI] [PubMed] [Google Scholar]
- 14.Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008; 40:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keim V, Bauer N, Teich N, et al. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med 2001;111: 622–626. [DOI] [PubMed] [Google Scholar]
- 16.Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet 2012;44:1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol 1998;275: G1402–G1414. [DOI] [PubMed] [Google Scholar]
- 18.Dawra R, Sah RP, Dudeja V, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 2011;141:2210–2217.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoque R, Sohail M, Malik A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011;141: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poon IKH, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 2010;17:381–397. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol 2002;2:787–795. [DOI] [PubMed] [Google Scholar]
- 22.Krüger B, Lerch MM, Tessenow W. Direct detection of premature protease activation in living pancreatic acinar cells. Lab Investig J Tech Methods Pathol 1998;78: 763–764. [PubMed] [Google Scholar]
- 23.Sendler M, Beyer G, Mahajan UM, et al. Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology 2015;149:765–776.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diener MK, Hüttner FJ, Kieser M, et al. Partial pancreatoduodenectomy versus duodenum-preserving pancreatic head resection in chronic pancreatitis: the multicentre, randomised, controlled, double-blind Chro-Pac trial. Lancet 2017;390:1027–1037. [DOI] [PubMed] [Google Scholar]
- 25.Wartmann T, Mayerle J, Kähne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 2010;138:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jungermann J, Lerch MM, Weidenbach H, et al. Disassembly of rat pancreatic acinar cell cytoskeleton during supramaximal secretagogue stimulation. Am J Physiol 1995;268:G328–G338. [DOI] [PubMed] [Google Scholar]
- 27.Bidani A, Heming TA. Effects of bafilomycin A1 on functional capabilities of LPS-activated alveolar macrophages. J Leukoc Biol 1995;57:275–281. [DOI] [PubMed] [Google Scholar]
- 28.Hoque R, Mehal WZ. Inflammasomes in pancreatic physiology and disease. Am J Physiol Gastrointest Liver Physiol 2015;308:G643–G651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mareninova OA, Sendler M, Malla SR, et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 2015;1: 678–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest 1997;100:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perides G, Weiss ER, Michael ES, et al. TNF-alpha-dependent regulation of acute pancreatitis severity by Ly-6C(hi) monocytes in mice. J Biol Chem 2011; 286:13327–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofbauer B, Saluja AK, Lerch MM, et al. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol 1998;275: G352–G362. [DOI] [PubMed] [Google Scholar]
- 33.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 2005;5:953–964. [DOI] [PubMed] [Google Scholar]
- 34.Xue J, Sharma V, Hsieh MH, et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun 2015;6:7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Benna J, Hurtado-Nedelec M, Marzaioli V, et al. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev 2016;273: 180–193. [DOI] [PubMed] [Google Scholar]
- 36.Leppkes M, Maueröder C, Hirth S, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun 2016;7:10973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell 2001;12:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louhimo JM, Steer ML, Perides G. Necroptosis is an important severity determinant and potential therapeutic target in experimental severe pancreatitis. Cell Mol Gastroenterol Hepatol 2016;2:519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gea-Sorlí S, Closa D. Role of macrophages in the progression of acute pancreatitis. World J Gastrointest Pharmacol Ther 2010;1:107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levin R, Grinstein S, Canton J. The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev 2016;273:156–179. [DOI] [PubMed] [Google Scholar]
- 41.Vandivier RW, Fadok VA, Hoffmann PR, et al. Elastasemediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest 2002;109:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fadok VA, Bratton DL, Guthrie L, et al. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol 2001; 166:6847–6854. [DOI] [PubMed] [Google Scholar]
- 43.Huynh M-LN, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 2002;109:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffmann PR, Kench JA, Vondracek A, et al. Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. J Immunol 2005;174:1393–1404. [DOI] [PubMed] [Google Scholar]
- 45.Fadok VA, Bratton DL, Rose DM, et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000;405:85–90. [DOI] [PubMed] [Google Scholar]
- 46.Orlowski GM, Colbert JD, Sharma S, et al. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol 2015;195: 1685–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talukdar R, Sareen A, Zhu H, et al. Release of cathepsin B in cytosol causes cell death in acute pancreatitis. Gastroenterology 2016;151:747–758.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung YJ, Isaacs JS, Lee S, et al. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 2003;17:2115–2117. [DOI] [PubMed] [Google Scholar]
- 49.Norman J, Franz M, Messina J, et al. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery 1995;117:648–655. [DOI] [PubMed] [Google Scholar]
- 50.Denham W, Yang J, Fink G, et al. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology 1997;113:1741–1746. [DOI] [PubMed] [Google Scholar]
- 51.Hoque R, Farooq A, Ghani A, et al. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014;146:1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marrache F, Tu SP, Bhagat G, et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 2008;135: 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neuhöfer P, Liang S, Einwächter H, et al. Deletion of IkBa activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A. Gastroenterology 2013; 144:192–201. [DOI] [PubMed] [Google Scholar]
- 54.Li C, Zienkiewicz J, Hawiger J. Interactive sites in the MyD88 Toll/interleukin (IL) 1 receptor domain responsible for coupling to the IL1βeta signaling pathway. J Biol Chem 2005;280:26152–26159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.