

Abstract
Inflammation is recently recognized as one of the hallmarks of human cancer. Chronic inflammatory response plays a critical role in cancer development, progression, metastasis, and resistance to chemotherapy. Conversely, the oncogenic aberrations also generate an inflammatory microenvironment, enabling the development and progression of cancer. The molecular mechanisms of action that are responsible for inflammatory cancer and cancer-associated inflammation are not fully understood due to the complex crosstalk between oncogenic and pro-inflammatory genes. However, molecular mediators that regulate both inflammation and cancer, such as NF-κB and STAT have been considered as promising targets for preventing and treating these diseases. Recent works have further demonstrated an important role of oncogenes (e.g., NFAT1, MDM2) and tumor suppressor genes (e.g., p53) in cancer-related inflammation. Natural products that target these molecular mediators have shown anticancer and anti-inflammatory activities in preclinical and clinical studies. Sesquiterpenoids (STs), a class of novel plant-derived secondary metabolites have attracted great interest in recent years because of their diversity in chemical structures and pharmacological activities. At present, we and other investigators have found that dimeric sesquiterpenoids (DSTs) may exert enhanced activity and binding affinity to molecular targets due to the increased number of alkylating centers and improved conformational flexibility and lipophilicity. Here, we focus our discussion on the activities and mechanisms of action of STs and DSTs in treating inflammation and cancer as well as their structure-activity relationships.
Keywords: Cancer, Inflammation, Sesquiterpenoid, MDM2, p53, NF-κB
Introduction
There is an increasing interest in investigation of the relationships between inflammation and cancer. We and others have proposed to dually target molecules involved in both inflammation and cancer as a novel approach to cancer prevention and treatment. Inflammation is the body’s protective response to microbial infection and other environmental stimuli, which usually cause tissue damage. Consequently, infection-triggered inflammatory response causes the destruction of damaged tissues and stimulates its regeneration [1–2]. It has been recognized that acute inflammation contributes to tumor regression while chronic inflammation plays a crucial role in tumor initiation and progression [3–4]. Indeed, more than one fifth of human cancers are associated with chronic and dysregulated inflammation [4]. Chronic inflammation is commonly featured by the persistent activation of immune system, which inevitably results in the accumulation of genetic and epigenetic aberrations, and consequently leads to malignant transformation [4–5]. During this procedure, many molecular mediators, including cytokines [e.g., tumor necrosis factor (TNF)-α and interleukins (ILs)], transcription factors [e.g., nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3)], tumor suppressor genes (e.g., p53) and oncogenes [e.g., murine double minute 2 (MDM2)] are involved in the tumor inflammatory microenvironment; most of the mediators (Fig. 1) have been extensively discussed in recent reviews [5–7]. Here, we focus on the recently uncovered mediators that are involved in inflammation and cancer as well as the changing paradigms in the field.
Fig. 1.
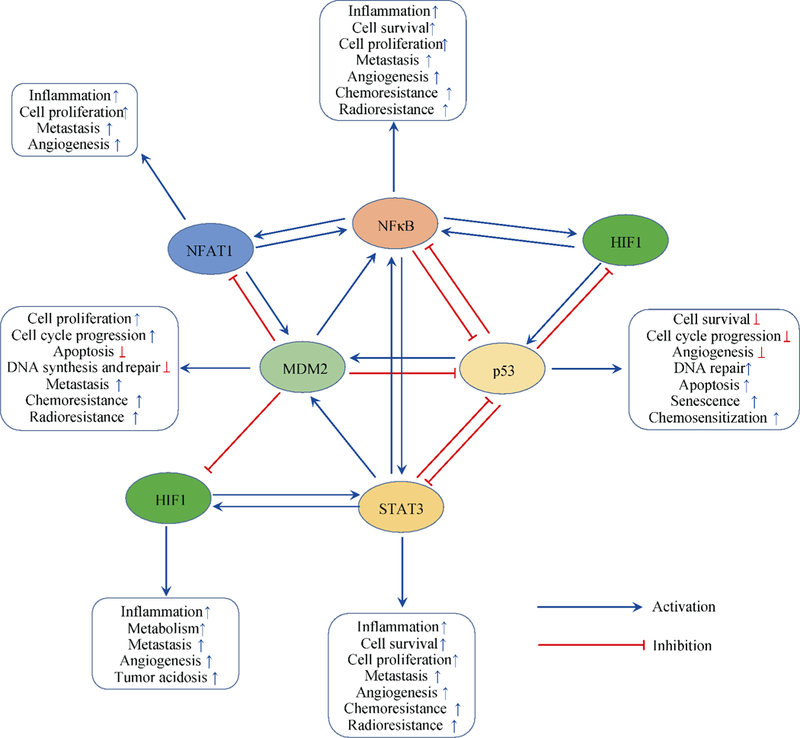
Key mediators of inflammation and cancer that are targeted by STs and DSTs
NF-κB and STAT are well-documented pro-inflammatory transcription factors that play critical roles in inflammation and cancer [8–9]. After activation by pro-inflammatory stimuli, NF-κB regulates the transcription of most of the genes related to inflammation [e.g., TNF, ILs, chemokines, and cyclooxygenase 2 (COX-2)] as well as survival genes in cancer cells [e.g., c-Myc, cyclinDl, and B-cell lymphoma-extra large (Bcl-xl)] [8]. STAT activation contributes to parts of IL-6’s functions and promotes cancer cell proliferation and tumorigenesis [9]. Recent studies have indicated that NF-κB and STAT3 collaboratively control the communication between cancer cells and inflammatory cells [9]. Nuclear factor of activated T-cells (NFAT), which was initially discovered in human T cells, has been demonstrated to play an important role in the regulation of inflammatory response, development and metastasis of cancer, and other biological events in the human body [10–11]. Hypoxia-inducible factor 1 (HIF1) exerts a well-established role in cancer initiation and progression and is essential for the execution of an optimal inflammatory response [12]. The complex cross-talks (Fig. 1) among NF-κB, STAT3, NFAT, and HIF1 in immune cells and tumor cells have yet to be fully comprehended. All of these transcription factors have been demonstrated as promising targets for the treatment of inflammation and cancer.
The MDM2 oncogene and the p53 tumor suppressor gene continue to hold interest as therapeutic targets in human cancers [13–14]. Many MDM2 inhibitors and p53 activators have been developed for the treatment and prevention of human cancers; several of them have currently entered clinical trials [13–16]. Recent studies have shown that MDM2 plays an important role in inflammation and inflammatory diseases via both p53-dependent and p53-independent mechanisms (Fig. 1) [17–21]. MDM2 inhibitors have also been reported to exert potent anti-inflammatory effects in tissue injury by inhibiting NF-κB [17, 21–23]. Because both p53 and NF-κB p65 exert their transactivation activities by interacting with p300/CREB-binding protein (CBP), there is a competitive relationship between p53 and NF-κB p65 [24–26]. Overexpression and activation of either p53 or NF-κB p65 inhibit the activity but not the expression of another protein [27]. Moreover, MDM2 induces NF-κB p65 expression at the transcriptional level, independent of p53 [28–29]. MDM2 directly binds to the first two Sp1-binding sites in the p65 promoter, increases p65 expression, and activates p65-mediated gene expression [28–29]. Recent studies have also uncovered that the nucleotide-binding oligomerization domain (NOD)-like receptors and Src kinase have multiple regulatory roles and have been proposed as molecular targets for treating both chronic inflammation and cancer [30–32].
Natural products are still a major chemical resource for anti-inflammatory and anticancer drug discovery [33–36]. Sesquiterpenoids (STs) are a large class of plant-derived secondary metabolites that exhibit diverse biological activities and have been implicated in traditional medicine against inflammation and cancer [37–38]. This class of natural products show ‘drug-like’ chemical properties, including alkylating center reactivity, lipophilicity, and molecular geometry and electronic features [37–38]. Several STs in clinical trials, including the derivatives of thapsigargin (1), parthenolide (2), and artemisinin (3), have been comprehensively discussed in the recent reviews [37–40]. We have summarized these clinical studies in Table 1. Recent advances have indicated that dimeric sesquiterpenoids (DSTs) exert enhanced anti-inflammatory and anticancer activities due to the increased binding affinity to molecular targets [41–42]. In this review, we will focus our discussion on representative STs and DSTs that have shown potent anti-inflammatory and anticancer activities (Fig. 2 and Table 2) as well as their characterization, molecular target(s), mechanism(s) of action, and structure-activity relationships.
Table 1.
STs and DSTs in human clinical trials
| Compounds | Chemical Structures | Clinical trial phase | Disease | Sponsor | References |
|---|---|---|---|---|---|
| Mipsagargin (G-202) | 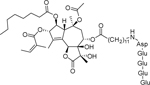 |
Phase 1 | Advanced solid tumors | GenSpera, Inc. | [43] |
| Phase 2 | Chemotherapy-naive metastatic castrate-resistant prostate cancer | GenSpera, Inc. | [44] | ||
| A soluble prodrug of thapsigargin | |||||
| DMAPT (or LC-1) |  |
Phase 1 | Acute myeloid leukemia | Leuchemix Inc. | [45] |
| A water-soluble analog of parthenolide | |||||
| Artesunate | 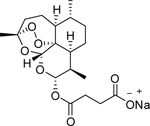 |
Phase 1 | Hepatocellular carcinoma | University Hospital, Ghent | [46] |
| Phase 1 | Solid tumors | Georgetown University | [47] | ||
| Phase 1 | Cervical intraepithelial neoplasia grade 2/3, high-risk HPV (any strain) | Sidney Kimmel Comprehensive Cancer Center |
[48] | ||
| Phase 2 | Colorectal cancer, bowel cancer | St George’s, University of London | [49] | ||
| A semi-synthetic derivative of Artemisinin | Phase 1 | Metastatic breast cancer, locally advanced breast cancer | Heidelberg University | [50] | |
| Artemether | 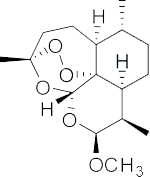 |
Phase 1; Phase 2 | Solid tumors | LondonPharma Ltd. | [51] |
| A semi-synthetic derivative of Artemisinin | |||||
Fig. 2.
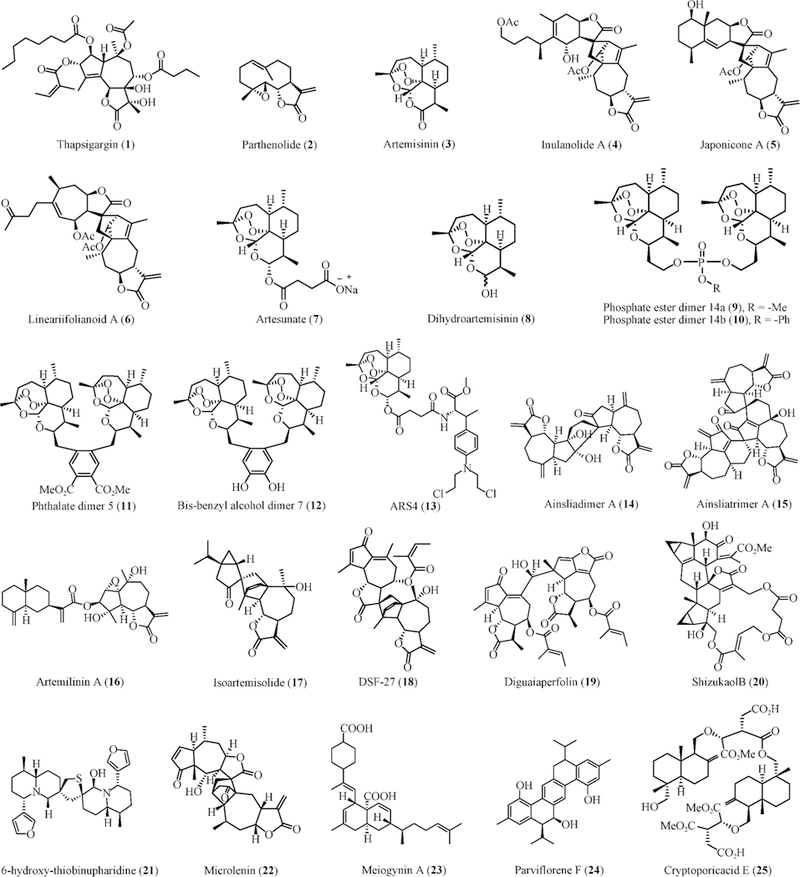
Chemical structures of STs and DSTs with potent anti-inflammatory and anticancer activities
Table 2.
Summary of representative STs and DSTs with anti-inflammatory and anticancer activity and their mechanisms of action
| Compounds | Source | Anti-inflammatory activity |
Anticancer activity | Molecular mechanisms | References |
|---|---|---|---|---|---|
| Inulanolide A (4) |
I. britannica, I. japonica |
Inhibits the production of NO and TNF-α in LPS-stimulated RAW264.7 cells |
Inhibits cell proliferation, arrests cells in G2-M phase, induces cell apoptosis, suppresses cell migration and invasion in breast cancer cells in vitro, independent of p53 Suppresses breast tumor growth and metastasis in vivo, independent of p53 | Inhibits NF-κB activation Inhibits NFAT1-MDM2 pathway, independent of p53 Modulates the expression of proteins related to cell proliferation, cell cycle progression, apoptosis, and DNA damage |
[64, 74] |
| Japonicone A (5) | I. japonica | Inhibits TNF-α-mediated cytotoxicity in L929 cells Inhibits TNF-α-induced endothelial cell activation Reduces TNF-α-induced hepatic injury in d-GalN sensitized mice |
Inhibits cell proliferation, arrests cells in G2-M phase, and induces cell apoptosis in breast cancer cells in vitro, independent of p53 Suppresses breast tumor growth and metastasis in vivo, independent of p53 |
Binds to TNF-α and selectively inhibits the interaction between TNF-α and TNFR1 Inhibits TNF-α-TAK1-IKK-NF-κB signaling axis and TNF-α-induced NF-κB activation and nuclear translocation and downregulates NF-κB target genes Binds to MDM2 and induces MDM2 degradation Inhibits NFAT1 and NFATl-me-diated MDM2 transcription |
[65, 70–73] |
| Lineariifolianoid A (6) | I. lineariifolia | Has no significant effect on TNF-α-mediated L929 cytotoxicity | Inhibits cell proliferation, arrests cells in G2-M phase, induces cell apoptosis, suppresses cell migration and invasion in breast cancer cells in vitro, independent of p53 | Inhibits NFAT1-MDM2 pathway, independent of p53 Modulates the expression of proteins related to cell proliferation, cell cycle progression, apoptosis, and DNA damage |
[68, 75] |
| Artemisinin (3) | A. annua | Exerts anti-inflammatory activity in TPA-induced skin inflammation in mice | Inhibits cancer cell growth and proliferation, arrests cell cycle at G1 phase, and induces cell apoptosis in vitro
Suppresses xenograft tumor growth and sensitizes cancer cells to chemotherapy in vivo |
Inhibits TNF-α-induced phosphorylation and degradation of IκBα, nuclear translocation of NF-κB p65, and expression of NF-κB target genes Reacts with the ferrous atom and produces free radicals or ROS Inhibits MDM2 expression, independent of p53 Modulates the expression of proteins associated with cell cycle progression and apoptosis, independent of p53 |
[78, 89, 92–94] |
| Artesunate (7) | synthetic derivative | Exerts anti-arthritis activity in K/BxN mouse model of rheumatoid arthritis and rat model of Freund’s complete adjuvant-induced arthritis | Induces cancer cell apoptosis | Inhibits nuclear translocation NF-κB and its transcriptional activity Inhibits PI3K/Akt activation Reacts with the ferrous atom and produces free radicals or ROS | [79–82, 89, 90] |
| Dihydroartemisi-nin (8) | synthetic derivative | Inhibits onset of EAE and ameliorates this disease in EAE-inflicted mice Inhibits OVA-induced chronic airway inflammation in mice Protects rats against bleomycine-induced pulmonary fibrosis and alcoholic liver injury |
Inhibits cancer cell growth and proliferation, arrests cell cycle at G1 phase, and induces cell apoptosis in vitro
Suppresses xenograft tumor growth and sensitizes cancer cells to chemotherapy in vivo |
Inhibits mTOR pathway Inhibits phosphorylation of ERK, p38 MAPK and IκBα, and activation of NF-κB Inhibits mRNA and protein expression of TGF-β1, TNF-α, α-SMA, and NF-κB Activates FXR Reacts with the ferrous atom and produces free radicals or ROS Induces transferrin receptor-1 internalization, decreases iron uptake, and disturbs iron homeostasis Inhibits MDM2 expression, independent of p53 Modulates the expression of proteins associated with cell cycle progression and apoptosis |
[83-86, 89,91, 92, 95, 96] |
| Phosphate ester dimer 14a (9) | synthetic derivative | Not reported | Inhibits cancer cell growth and induces apoptosis | Not reported | [97] |
| Phosphate ester dimer 14b (10) | synthetic derivative | Not reported | Inhibits cancer cell growth and induces apoptosis | Not reported | [97] |
| Phthalate dimer 5 (11) | synthetic derivative | Not reported | Inhibits cervical cancer cell growth without apparent cytotoxicity against normal cervical cells | Not reported | [98] |
| Bis-benzyl alcohol dimer 7 (12) | synthetic derivative | Not reported | Inhibits cervical cancer cell growth without apparent cytotoxicity against normal cervical cells | Not reported | [98] |
| ARS4 (13) | synthetic derivative | Not reported | Inhibits cancer cell growth and proliferation, arrests cells in S phase, induces cell apoptosis, and prevents cell migration in ovarian cancer cells in vitro Suppresses xenograft tumor growth and metastasis in vivo | Inhibits MDM2 expression, independent of p53 Modulates the expression of proteins related to cell cycle progression, apoptosis, and EMT |
[100] |
| Ainsliadimer A (14) | A. macrocephala | Inhibits NO production in LPS-stimulated RAW264.7 cells |
Inhibits cancer cell growth in vitro Represses xenograft tumor growth in vivo | Binds to IKKα/β and allosterically inhibits its activities, resulting in inhibition of NF-κB signaling pathway | [102, 104] |
| Ainsliatrimer A (15) | A. fulvioides | Not reported | Inhibits cancer cell growth and induces apoptosis in vitro | Binds to PPARγ and activates its transcriptional activity | [105–107] |
| Artemilinin A (16) | A. argyi | Not reported | Weak cytotoxicity | Not reported | [108] |
| Isoartemisolide (DSF-52, 17) | A. argyi | Inhibits the production of NO, PGE2 and TNF-α in LPS-stimulated BV-2 microglial cells | Weak cytotoxicity | Inhibits NF-κB, JNK/p38 MAPKs, and Jak2/Stat3 pathways | [108, 109] |
| DSF-27 (18) | A. argyi | Inhibits LPS-induced microglial activation and protects neurons from microglia-mediated neuro-inflammatory injury | Not reported | Inhibits the Syk/NF-κB and Jak2/Stat3 signaling pathways | [110] |
| Diguaiaperfolin (19) | E. perfoliatum | Inhibits NO production and iNOS expression in LPS-stimulated RAW264.7 macrophages | Not reported | Not reported | [111] |
| Shizukaol B (20) | C. japonicas | Inhibits ICAM-1/LFA-1-mediated cell aggregation and monocyte adhesion to HUVEC in human promyelocytic HL-60 cells Inhibits the NO production in LPS-stimulated BV-2 cells |
Not reported | Inhibits the expression of ICAM-1, VCAM-1 and E-selectin | [112, 113] |
| 6-hydroxy-thiobi nupharidine (21) | N. pumilum | Inhibits anti-sheep erythrocyte plaque forming cell formation Possesses potent immunosuppressive activity | Inhibits cancer cell growth, induces apoptosis, and inhibits cell invasion in vitro Suppresses lung metastasis of B16 melanoma cell in mice in vivo |
Inhibits NF-κB pathways and induces cleavage of PARP | [114–119] |
| Microlenin (22) | H. microcephalum | Not reported | Induces Ehrlich ascites carcinoma cell death | Inhibits DNA and protein synthesis | [120–122] |
| Meiogynin A(23) | M. cylindrocarpa | Not reported | Induces cancer cell death and apoptosis | Inhibits Bcl-xL/Bak binding and Mcl-1/Bid binding | [123–127] |
| Parviflorene F (24) | C. parviflora | Not reported | Induces cancer cell death and apoptosis | Increases the expression levels of TRAIL-R2 | [128–130] |
| Cryptoporic acid E (25) | C. volvatus | Not reported | Prevents okadaic acid-stimulated tumor promotion in mouse skin Prevents N-Methyl-N-Nitrosourea-induced colon carcinogenesis in rats Prevents 1,2-dimethylhydrazine-caused colon carcinogenesis in mice |
Inhibits superoxide anion radical release | [131, 132] |
Dual Effects of STs and DSTs on Inflammation and Cancer
Inulanolide A, japonicone A, lineariifolianoid A, and their analogs
The Inula plants have a long history of use in traditional Chinese medicine for treating inflammation and cancer [52]. Monomeric STs are characterized as one of the main chemical constituents of Inula plants [52], and have shown chemical diversity [53–55] and excellent pharmacological activities [56–63]. These ST monomers have always been considered as the major bioactive constituents until the discovery of novel DSTs, including inulanolide A (InuA, 4) and its analogs from Inula Britannica [64]. These DSTs have exhibited potent anti-inflammatory activities by inhibiting NF-κB activation and decreasing the production of nitric oxide (NO) and TNF-α in lipopolysaccharide (LPS)-stimulated RAW264.7 cells [64]. Their activities are significantly stronger than the previously identified ST monomers from the same plant [64]. These results have prompted phytochemists to isolate and identify new DSTs with greater biological activities and better drug-like properties, leading to the isolation and characterization of japonicone A (JapA, 5), lineariifolianoid A (LinA, 6) and their analogs [65–69]. All these dimers have exhibited strong inhibitory effects against LPS-induced NO production in RAW264.7 cells [65–69].
Hu et al. have further investigated the molecular mechanisms that are responsible for the anti-inflammatory activities of JapA and characterized this compound as a novel TNF-α antagonist [70]. JapA has been found to directly bind to TNF-α and selectively inhibit the interaction between TNF-α and tumor necrosis factor receptor 1 (TNFR1). However, JapA does not show significant effects on the binding of TNF-α to TNFR2. Consequently, the compound blocks TNF-α-TNFRl-mediated multiple signaling pathways via inhibition of TNF-α-induced NF-κB activation as well as the expression of adhesion molecules [intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1)] and chemokine [monocyte chemoattractant protein-1 (MCP-1)] in the endothelial cells. In the in vivo studies, JapA protects mice from acute hepatitis without compromising the host’s defense against the virus [70].
These DSTs have shown promising anticancer activity in preclinical studies. JapA has been screened in a wide spectrum of human normal and cancer cell lines and exhibits a selective cytotoxicity against human lymphoma cells with IC50 values at sub-micromole levels, but has no apparent effect on normal cells [71]. Li et al. have reported that JapA inhibits the proliferation of lymphoma cells, arrests the cell cycle in G2-M phase, and induces cell apoptosis in vitro, and suppresses the growth and metastasis of lymphoma in vivo. JapA’s anti-lymphoma activities have been attributed to its inhibitory effects on TNF-α-TAK1 (TGF-beta activated kinase 1)-IKK (IκB kinase)-NF-κB signaling axis and TNF-α-induced NF-κB activation and nuclear translocation, which lead to the downregulation of NF-κB target genes that regulate cancer cell growth (cyclin D1 and c-Myc) and apoptosis [B-cell lymphoma 2 (Bcl-2), Bcl-xL, X-linked inhibitor of apoptosis protein (XIAP), TNF receptor-associated factor 2 (TRAF2)] [71].
During our studies on the search for novel dual MDM2 and NFAT1 inhibitors for cancer prevention and therapy, we have identified several STs and DSTs, including JapA, InuA, and LinA [72–75]. JapA has been shown to selectively inhibit breast cancer cell growth without affecting normal breast cells, regardless of p53 status [72]. JapA has also been found to inhibit breast cancer cell proliferation and colony formation, arrest cells in G2-M phase, induce cell apoptosis, and suppress breast tumor growth and metastasis in dose-dependent and p53-independent manners. We have discovered that JapA directly binds to MDM2 protein and induces its autoubiquiti-nation and proteasomal degradation [72]. It has been further found that JapA also inhibits NFATl-mediated MDM2 transcription by inhibiting NFAT1 protein stability and nuclear translocation and disrupting the interaction between NFAT1 DNA binding domain and MDM2 P2 promoter [73]. The importance of NFAT1 and MDM2 for JapA’s anticancer activity has been validated using NFAT1-MDM2 overexpression and knockdown in breast cancer cell lines. InuA and LinA have also exhibited potent anti-breast cancer activity in vitro and in vivo by targeting NFAT1 and MDM2 [74–75].
Artemisinin and its derivatives, dimers, and conjugates
Artemisinin (3) is isolated from the medical plant Artemisia annua L. and identified as a sesquiterpenoid bearing a peroxide bridge [76]. Artemisinin and its derivatives artesunate (7), dihydroartemisinin (8), artemether, and arteether are clinically used antimalarial drugs without host toxicity and have been considered as the most effective drugs in the treatment of cerebral malaria and chloroquine resistant falciparum malaria [77]. In addition to the antimalarial activities, artemisinin and its derivatives have recently been reported to possess anti-inflammatory activity. Wang et al. have demonstrated the anti-inflammatory activity of artemisinin in tet-radecanoylphorbol acetate (TPA)-induced skin inflammation in mice [78]. Artemisinin has further been found to inhibit TNF-α-induced phosphorylation and degradation of inhibitor of kappa B alpha (IκBα) as well as the nuclear translocation of NF-κB p65, resulting in inhibition of the expression of NF-κB target genes [78]. Artemisinin also inhibits reactive oxygen species (ROS) production and phosphorylation of p38 and extracellular signal-regulated kinase (ERK), without affecting the phosphorylation of c-Jun N-terminal kinase (JNK) [78].
Xu et al. have reported that artesunate inhibits the secretion of IL-1β, IL-6, and IL-8 from TNF-α-stimulated human rheumatoid arthritis fibroblast-like synoviocytes (RAFLS) by inhibiting the nuclear translocation of NF-κB and its DNA-binding and transcriptional activities and preventing Akt phosphorylation [79]. The same group has further found that artesunate inhibits the secretion of vascular endothelial growth factor (VEGF) and IL-8 as well as the expression of HIF-1α in RAFLS by inhibiting phosphoinositide 3-kinase (PI3K)/Akt activation [80]. Both studies have indicated that artesunate may have therapeutic potential for human arthritis and have been further validated using the K/BxN mouse model of rheumatoid arthritis and the rat model of Freund’s complete adjuvant-induced arthritis [81–82]. These studies have been comprehensively discussed in recent reviews [40, 77]. Dihydroartemisinin has also recently been observed to inhibit the onset of experimental autoimmune encephalomyelitis (EAE) and ameliorate this disease in EAE-inflicted mice by inhibiting the mammalian target of rapamycin (mTOR) pathway [83]. This compound also suppresses ovalbumin (OVA)-induced chronic airway inflammation in mice by inhibiting ERK, p38 mitogen-activated protein kinase (MAPK) phosphorylation, IκBα phosphorylation, and NF-κB activation [84]. Further studies have indicated that dihydroartemisinin protects rats against bleomycin-induced pulmonary fibrosis and alcoholic liver injury by inhibiting the mRNA and protein expression of transforming growth factor beta 1 (TGF-β1), TNF-α, alpha-smooth muscle actin (α-SMA), and NF-κB and activating farnesoid X receptor (FXR), respectively [85–86].
There is an increasing interest in repurposing artemisinin, artesunate and other derivatives for treating human cancers [reviewed in refs [87–88]]. Accumulating evidence has indicated that artemisinin and its derivatives possess potent in vitro and in vivo activities against various types of human cancer. Because these compounds have shown excellent drug-like properties and safety profiles, repurposing artemisinin and its derivatives as anticancer drugs can significantly reduce the costs during drug development and clinical trials, therefore lowering investment risk and accelerating development process. Currently, artesunate and artemether are under evaluation for their anticancer activity in clinical trials. The detailed information has been summarized in Table 1. Mechanistically, the peroxide bridge of artemisinin and its derivatives is vital for their anticancer activities and can react with the ferrous atom and produce free radicals or ROS, inducing cancer cell apoptosis and oxidative DNA damage [89–91]. Qian et al. have found that dihydroartemisinin induces transferrin receptor-1 internalization, decreases iron uptake, and disturbs iron homeostasis in cancer cells, which are at least partially responsible for the anticancer activity of the compound [92]. Nakase et al. have prepared a series of artemisinin-transferrin conjugates, which exhibit stronger cytotoxicity against prostate cancer cells and induce more cell apoptosis than the parent drug in a transferrin receptor-dependent manner [93]. Artemisinin and its derivatives have been reported to inhibit liver cancer cell growth and proliferation, arrest cell cycle at G1 phase, and induce cell apoptosis in vitro by down-regulating MDM2 expression and modulating the expression of proteins associated with cell cycle progression and apoptosis, independent of p53 [94]. Artemisinin and dihydroartemisinin also inhibit xenograft tumor growth and sensitize liver cancer to the treatment of a chemotherapeutic agent, gemcitabine in vivo [94]. Similar results have been observed in ovarian cancer cells in vitro and in vivo [95]. This group has also demonstrated the potential embryotoxicity of dihydroartemisinin in the embryos of wild-type and TG (flk1: GFP) transgenic zebrafish [96].
Recently, there have been several reports of the potent antitumor activities of artemisinin-derived dimers. Jeyadevan and co-workers have synthesized a series of artemisinin dimers with a different linker group at C-10 [97]. Two phosphate ester dimers of artemisinin, compounds 14a (9) and 14b (10) possess more potent anticancer activity than dihydroar-temisinin and doxorubicin [97]. The same group has further designed and synthesized the second generation trioxane dimers of artemisinin [98]. The lead compounds phthalate dimer 5 (11) and bis-benzyl alcohol dimer 7 (12) selectively induce human cervical cancer cell death without significant cytotoxicity against normal cervical cells [98]. Saikia et al. have designed and synthesized nine artemisinin-derived triazole dimers and evaluated their anticancer activity in leukemia, colon, lung, and liver cancer cells. However, none of them exhibits more significant activities than artemisinin [99].
Interestingly, a recent study has reported the preparation of artemisinin-chemotherapeutic agent conjugates as well as the preclinical assessment of their efficacy and safety in human ovarian cancer models [100]. ARS4 (13), a dihydroar-temisinin-melphalan conjugate exhibits stronger anti-ovarian cancer activity than its parent drugs in vitro and in vivo, without inducing apparent host toxicity. Its anticancer activities have been attributed to ARS4-mediated MDM2 inhibition and its modulatory effects on various proteins that regulate cell cycle progression, apoptosis, and epithelial-mesenchymal transition (EMT), which are consistent with the mechanisms of action of its parent drugs dihydroartemisinin and melphalan [100]. However, further evaluation of these conjugates in more clinically-relevant cancer models is still needed.
Ainsliadimer A and ainsliatrimer A
The genus Ainsliaea has been reported to be a rich source of natural STs, especially guaianolides, which have shown diverse biological activities [101–102]. Several Ainsliaea species have been used in traditional Chinese medicine for the treatment of angina and rheumatoid arthritis, such as Ainsliaea macrocephala [101–102]. In a recent study for isolation and characterization of novel bioactive compounds from A. macrocephala, ainsliadimer A (14), a novel guaianolides-type DST with an unprecedented carbon skeleton has been discovered and shown remarkable inhibitory effects on the production of NO [102]. Li et al. have studied the biomimetic total synthesis of ainsliadimer A, which has finally been accomplished in 14 steps [103]. Dong et al. have further investigated its biological activities and have characterized this compound as a potent inhibitor of the NF-κB pathway [104]. They have found that ainsliadimer A selectively binds to IKKα/β via the conserved cysteine 46 residue and allosterically inhibits its activities, resulting in the marked inhibition of NF-κB signaling pathway as well as the induction of cancer cell death in vitro and the repression of xenograft tumor growth in vivo [104].
Another study for identifying novel anticancer natural products from Ainsliaea plants has been performed; a novel trimeric guaianolides, named ainsliatrimer A (15) has been identified from Ainsliaea fulvioides, which has been used in Chinese folk medicine for the treatment of rheumatism, traumatic injuries, edema, stomachache, and anorexia [105]. Ainsliatrimer A and its analogs exhibit potent cytotoxicities against human leukemia (CEM) and colon cancer (LOVO) cell lines with IC50 values in a sub-micromolar range [105]. Li et al. have further accomplished the biomimetic synthesis of ainsliatrimer A and demonstrated that this compound induces cancer cell apoptosis [106]. To further identify the molecular target of ainsliatrimer A, Li et al. have synthesized a biotin-labeled ainsliatrimer A and revealed that peroxisome pro-liferator-activated receptor gamma (PPARγ) is a major cellular target for this compound [107]. Ainsliatrimer A directly binds to PPARγ and activates its transcriptional activity, leading to the expression of PPARγ target gene (e.g., COX-2) and cancer cell death [107].
Others DSTs
As a continuation of work for identifying bioactive constituents from Artemisia plants, a guaianolide-type DST, artemilinin A (16), and a sesquiterpenoid-monoterpenoid dimer, isoartemisolide (also named DSF-52, 17) are isolated from Artemisia argyi [108]. Neither compound shows significant cytotoxicity against cancer cell lines. Isoartemisolide exhibits potent inhibitory effects on the production of NO, pros-taglandin E2 (PGE2) and TNF-α in LPS-stimulated BV-2 microglial cells, which have been attributed to its targeting effects on NF-κB, JNK/p38 MAPKs, and Janus kinase 2 (Jak2)/Stat3 pathways I[108–109]. Another guaianolide-type DST, named DSF-27 (18) from the same medical plant has also shown inhibitory effects on LPS-induced microglial activation, protecting neurons from microglia-mediated neuro-inflammatory injury by inhibiting the spleen tyrosine kinase (Syk)/NF-κB and Jak2/Stat3 signaling pathways [110].
The medicinal plant Eupatorium perfoliatum L. has traditionally been used as an anti-fever, anti-malarial, and anti-inflammatory agent [111]. Mareike and coworkers have evaluated the anti-inflammatory effects of the crude extracts of E. perfoliatum L as well as the defined lead compounds from this plant, including a novel DST, diguaiaperfolin (19). This compound exhibits a strong inhibition of NO production and inducible nitric oxide synthase (iNOS) expression in LPS-stimulated RAW264.7 macrophages without significant cytotoxicity against RAW264.7 macrophages [111]. Another DST, shizukaol B (20) from Chloranthus japonicas has been reported to inhibit ICAM-1/lymphocyte function-associated antigen 1 (LFA-1)-mediated cell aggregation and monocyte adhesion to human umbilical vein endothelial cells (HUVEC) by inhibiting the expression of ICAM-1, VCAM-1, and E-selectin in human promyelocytic HL-60 cells, which suggests that the compound may be a potential anti-atherosclerotic drug [112]. Shizukaol B also possesses potent inhibitory effects on NO production in LPS-stimulated BV-2 cells and may be used as an anti-neuroinflammatory agent as well [113].
The dimeric sesquiterpene thioalkaloid, 6-hydroxythiobi-nupharidine (21) from the rhizome of Nuphar pumilum has been observed to inhibit anti-sheep erythrocyte plaque forming cell formation without causing any apparent cytotoxicity against mouse spleen cells [114]. The same group has further reported that 6-hydroxythiobinupharidine possesses potent immunosuppressive activity [115]. 6-Hydroxythiobinupharidine and its analogs exhibit anti-melanoma activity by inhibiting melanoma cell growth, inducing apoptosis, and inhibiting cell invasion in vitro and suppressing the lung metastasis of B16 melanoma cell in mice in vivo [116–117]. This compound has also been found to exert its anticancer activity by inhibiting NF-κB pathways and inducing cleavage of PARP [118–119].
Microlenin (22), a novel guaianolide-type DST from Helenium microcephalum, has shown excellent anticancer activity in Ehrlich ascites carcinoma cell via inhibition of DNA and protein synthesis [120–122]. Another DST, meiogynin A (23) has been isolated from the bark of Meiogyne cylindrocarpa and identified as a novel inhibitor of Bcl-xL/Bcl-2 antagonist killer 1 (Bak) binding with a Ki value of 10.8 μmol∙L−1 [123–124]. This compound also has inhibitory effects on myeloid cell leukemia-1 (Mcl-1)/BH3 interacting domain death agonist (Bid) binding with a Ki value of 5.2 μmol∙L−1 [125]. The dual targeting effects by meiogynin A result in cancer cell death and apoptosis [123, 126–127]. Parviflorene F (24), an un-symmetrical DST from Curcuma parviflora, has been reported to cause cancer cell death and apoptosis via increasing the expression level of TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2) [128–130]. Cryptoporic acid E (25) from the fungus Cryptoporus volvatus has been characterized as an inhibitor of superoxide anion radical release and shown potent inhibitory effects on okadaic acid-stimulated tumor promotion in mouse skin, N-Methyl-N-Nitrosourea-induced colon carcinogenesis in rats, and 1, 2-dimethylhydrazine-caused colon carcinogenesis in mice [131–132].
Despite a tremendous increase in the identification of novel DSTs from natural source, most of the studies just reported the characterization and initial screening for their anti-inflammatory and anticancer activities [41]. Further detailed investigation is required for determining their in vivo efficacy, toxicity, molecular targets, and mechanisms of action.
Structure–Activity Relationships of STs and DSTs for Their Anti-inflammatory and Anticancer Activities
It has been widely accepted that several chemical properties are critical for the anticancer and anti-inflammatory activities of STs and dimeric DSTs: (1) alkylating centers, (2) oligomerization of sesquiterpenoids, and (3) lipophilicity. All the chemical features (Fig. 3) will be discussed in the following sections.
Fig. 3.
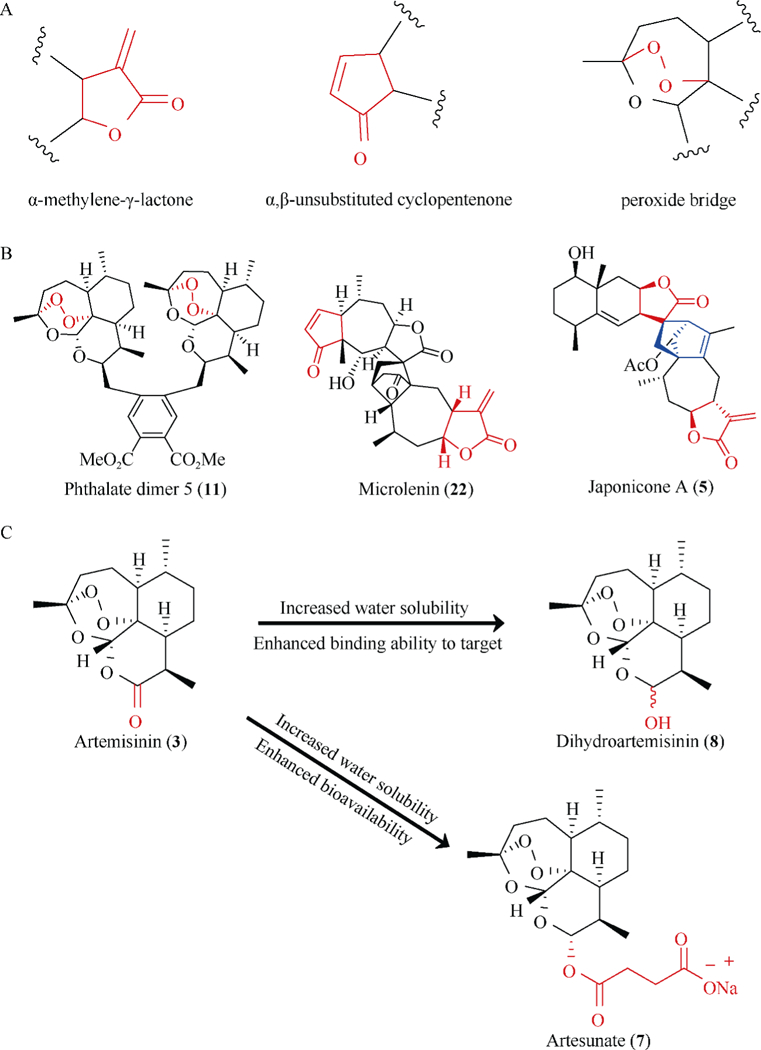
Chemical features of STs and DSTs related to anti-inflammatory and anticancer activities. (A) The common alkylating centers of STs and DSTs are highlighted in red. (B) DSTs often contain more alkylating centers (in red) and a novel linker between the monomers (in blue). (C) The introduction of hydroxyl group and other functional moieties may affect the target binding ability and bioavailability of STs and DSTs
Alkylating centers
The α, β-unsaturated moieties (Fig. 3A), e.g., α-methylene-γ-lactone and α, β-unsubstituted cyclopentenone are typical alkylating centers in STs and DSTs and are critical for the biological activities of this class of compounds [37, 58, 63]. STs and DSTs bearing α, β-unsaturated moieties often exert more potent anticancer and anti-inflammatory activities than the compounds without these functional moieties [58, 63]. Mechanistically, the α, β-unsaturated moiety functions as a reactive Michael acceptor, which forms a covalent bond with biological nucleophiles, especially with the proteins harboring cysteine residues [37, 104]. The formation of stable drug-protein adducts may finally lead to the inactivation of the target proteins by inducing protein conformational changes, inhibiting protein-protein interaction or protein-DNA interaction, blocking the biological functions, and/or promoting their degradation [72–73, 104]. In addition, the peroxide bridge (Fig. 3A) in artemisinin and its analogs and dimers may also be considered as a precursor of an alkylating center [40]. Heme or free iron-induced breaking of the peroxide bridge causes degradation of artemisinin, resulting in the formation of nucleophilic radical metabolites [40]. These metabolites then act as alkylating agents by binding to target proteins containing electrophilic moieties, leading to the inactivation of these target proteins [40]. Moreover, the number of alkylating centers in the STs and DSTs is an important factor for the activities of these compounds. DSTs often contain more than two alkylating centers, causing more potent anticancer and anti-inflammatory activities than their monomers [41–42].
Oligomerization of sesquiterpenoids
Polyvalency effect is a common biological event, which always gives rise to enhanced binding affinity of small ligands to target proteins and increased steric stability of lig- and-receptor complex [42]. We and other investigators have recently discovered that oligomerization, especially dimerization and trimerization of STs can have increased binding affinities that are greater than monomeric STs, leading to enhanced anticancer and anti-inflammatory activities [63, 72–73, 104]. This phenomenon may be attributed to the increased number of alkylating centers as well as the conformational flexibility due to the novel linkage modes between the monomers. Due to the dimerization and trimerization of STs, each dimer and trimer may bear two or three alkylating centers and become bifunctional and trifunctional alkylating agents, e.g., phthalate artemisinin dimer 5 and microlenin (Fig. 3B), often resulting in more than a hundred-fold increase in the binding affinity to a protein target [70, 72, 104]. Furthermore, the linker between the ST monomers is crucial for the conformational flexibility of the oligomers, e.g., JapA (Fig. 3B). More flexible oligomers are often observed to exert more potent activities against inflammation and cancer [41].
Lipophilicity and others
Lipophilicity is a critical factor which controls cell penetrating ability of small molecules [133]. Generally, the presence of lipophilic moieties (e.g., ester group) in STs and DSTs results in enhanced penetration through cell membrane and increased biological activities, whereas the presence of hydrophilic groups (e.g., hydroxyl group) show contrasting results [58, 63, 66]. However, in in vivo studies, high lipophilic STs and DSTs always have very low bioavailability, which finally causes moderate in vivo efficacy (Fig. 3C) [37, 39]. In addition, the biological activities of STs and DSTs are also affected by other factors, including the size and position of lipophilic groups and the number of hydrogen-bond donors and receptors [37, 39]. Many studies have shown that only the addition of the ‘size optimum’ functional groups increases the activity while the presence of oversized or undersized functional groups even reduces the penetrating ability, thereby decreasing their activities [37]. The addition of functional moieties to inappropriate positions may cause steric hindrance to alkylating centers, reducing its binding affinity to the protein target [63, 72–73, 104]. The introduction of hydrophilic groups, e.g., hydroxyl groups to STs and DSTs may not only reduce their penetrating ability, but also increases the number of hydrogen-bond receptors (Fig. 3C). These moieties can form hydrogen-bonds with amino acid residues on target proteins and then enhance the drug-protein binding, leading to enhanced anticancer and anti-inflammatory activities [70, 72, 104].
Discussion and Future Directions
There is increasing evidence demonstrating the correlation between inflammation and cancer [134–135]. The critical mediators of chronic inflammation contribute to cancer initiation and progression, as well as resistance to chemotherapy [6]. Overexpression and activation of oncogenes and mutation and deletion of tumor suppressor genes have recently been observed to play a crucial role in cancer-related inflammation [17–21]. In light of the accumulating reports referring to the critical role of these molecules in inflammation and cancer, the spotlight in this field has been shifted to the identification of pharmacological inhibitors and activators for prevention and treatment of cancer-related and inflammation-related diseases. Natural products hold great promise for anti-inflammatory and anticancer drug discovery because of their excellent chemical properties and promising activities [33–36].
Intense research efforts during recent years have resulted in the discovery of several novel STs and DSTs with potent activities against inflammation and cancer and some of them are undergoing clinical evaluation. STs and DSTs are promising drug candidates for the treatment of inflammation and cancer owing to several favorable features: 1) diverse chemical structures; 2) potent in vitro and in vivo activity; and 3) high binding affinity to molecular targets. However, some drawbacks of this class of compounds limit their development as therapeutics in the clinic. First, most of these STs and DSTs are isolated from plants and/or other natural resources with low yield and content. Second, this class of compounds often has a hydrophobic characteristic, which may result in poor bioavailability and moderate in vivo efficacy. Third, some STs and DSTs have been reported to have low selectivity index and exert cytotoxicity against both normal and cancer cells, which may cause unexpected adverse effects.
To facilitate the transition of STs and DSTs from laboratory to the clinic, the pharmacokinetic properties, especially oral bioavailability should be extensively evaluated and optimized. As summarized in Table 1, most of the STs and DSTs in clinical trials have been optimized for improved water-solubility and bioavailability. Furthermore, the pharmacological and toxicological profiles of STs and DSTs need to be examined, which can help determine the appropriate dose schedules for robust activity but manageable toxicity in the future clinical studies. In addition, optimal drug delivery systems for STs and DSTs can also be utilized to improve the pharmacokinetic profiles and reduce the adverse effects. Because the major molecular targets of STs and DSTs, including NFκB, MDM2, and STAT3 play important roles in chemoresistance, the drug combination studies of STs and DSTs with chemotherapeutic agents and other targeted therapeutics should be carried out in the future. It is known that acquired resistance to therapeutic agents is one of the major clinical challenges in drug development. The STs and DSTs, e.g., JapA and InuA have shown multi-targeting pharmacological properties, which may be developed as effective agents to overwhelm such resistance.
In conclusion, we deem that STs and DSTs represent a novel class of natural products possessing many advantages over other compounds as anticancer and anti-inflammatory agents, However, to facilitate their transition from bench to clinic, further studies are still needed to focus on pharmacological and toxicological profiles, molecular targets and mechanisms of action, biomimetic synthesis and SAR, optimal drug delivery system, and drug combination studies with other therapeutic agents.
Acknowledgements
This work was supported by the National Institutes of Health (NIH/NCI) grant (R01 CA186662). The content is solely the responsibility of the authors, and do not necessarily represent the official views of the National Institutes of Health. The research field in inflammation and cancer is rapidly growing, and we apologize for not being able to cite all the recent publications, due to space limitation. We thank the current and former members of our laboratory and collaborators for their contributions to the publications cited in this review article.
[Research funding] This work was supported by NIH/NCI (Grant R01 CA186662 to R.Z.) and by American Cancer Society (Grant RSG-15–009-01-CDD to W.W.).
Abbreviations
- α-SMA
Alpha-smooth muscle actin
- Bak
Bcl-2 antagonist killer 1
- Bcl-2
B-cell lymphoma-2
- Bcl-xl
B-cell lymphoma-extra large
- Bid
BH3 interacting domain death agonist
- CBP
CREB-binding protein
- COX-2
Cyclooxygenase 2
- DSTs
Dimeric sesquiterpenoids
- EAE
Experimental autoimmune encephalomyelitis
- EMT
Epithelial-mesenchymal transition
- ERK
Extracellular signal–regulated kinase
- FXR
Farnesoid X receptor
- HIF1
Hypoxia-inducible factor 1
- HUVEC
Human umbilical vein endothelial cells
- ICAM-1
Intercellular adhesion molecule 1
- IκBα
Inhibitor of kappa B alpha
- IKK
IκB kinase
- ILs
Interleukins
- iNOS
Inducible nitric oxide synthase
- InuA
Inulanolide A
- Jak2
Janus kinase 2
- JapA
Japonicone A
- JNK
c-Jun N-terminal kinase
- LFA-1
Lymphocyte function-associated antigen 1
- LinA
Lineariifolianoid A
- LPS
Lipopolysaccharide
- MAPK
Mitogen-activated protein kinase
- Mcl-1
Myeloid cell leukemia-1
- MCP-1
Monocyte chemoattractant protein-1
- MDM2
Murine double minute 2
- mTOR
Mammalian target of rapamycin
- NFAT
Nuclear factor of activated T-cells
- NF-κB
Nuclear factor kappa B
- NO
Nitric oxide
- NOD-like
Nucleotide-binding oligomerization domain
- receptors
like receptors
- OVA
Ovalbumin
- PGE2
Prostaglandin E2
- PI3K
Phosphoinositide 3-kinase
- PPARγ
Peroxisome proliferator-activated receptor gamma
- RAFLS
Rheumatoid arthritis fibroblast-like synoviocytes
- ROS
Reactive oxygen species
- STAT
Signal transducer and activator of transcription
- STs
Sesquiterpenoids
- Syk
Spleen tyrosine kinase
- TAK1
TGF-beta activated kinase 1
- TGF-β1
Transforming growth factor beta 1
- TNF-α
Tumor necrosis factor alpha
- TNFR
Tumor necrosis factor receptor
- TPA
Tetradecanoylphorbol acetate
- TRAF2
TNF receptor-associated factor 2
- TRAIL-R2
TNF-related apoptosis-inducing ligand receptor 2
- VCAM-1
Vascular cell adhesion molecule 1
- VEGF
Vascular endothelial growth factor
- XIAP
X-linked inhibitor of apoptosis protein
Biography

Ruiwen ZHANG, M.D., Ph.D., D.A.B.T., FAAAS
Professor ZHANG has a longstanding research interest in the field of translational biomedical research, particularly in translational medicine, cancer therapy and prevention, clinical pharmacology and therapeutics, pharmacogenomics, pharmaceutical and toxicological researches. Thanks to continuously funding from NIH and other governmental and private agencies, Dr. Zhang has maintained a strong research program, making significant contributions to several research fields, including pharmacogenomics of anticancer agents, oncogene and tumor suppressor, molecular targeted cancer therapy, dietary and chemical cancer prevention, drug discovery and development, preclinical and clinical pharmacology, toxicology, and cancer biomarkers. For his outstanding contributions to sciences, Dr. Zhang was elected as a Fellow of American Association for the Advancement of Science (AAAS) in 2009. He has published more than 220 papers, 2 books, and more than 50 invited reviews/book chapters; his publications have been cited more than 10500 times, with an H-index of 55 and an i10-index of 165. He has been invited to give more than 190 presentations in prestigious universities, institutions, and organizations. Dr. Zhang has been a certified toxicologist by the American Board of Toxicology (D.A.B.T.) since 1999 and served on Board of Directors of ABT between 2009 and 2013. He has been an FDA advisory committee member and served as a study section member for NIH, DoD, CDC, FDA, and NIOSH and other international panels. Dr. Zhang is Editor-in-Chief of Current Cancer Drug Targets, Associate Editor-in-Chief of Chinese Journal of Natural Medicines, and also an associate editor, senior editorial board member, or editorial board member of more than 20 scientific journals. He has been an honorary professor of seven universities.
Footnotes
These authors have no conflict of interest to declare.
References
- [1].Pesic M, Greten FR. Inflammation and cancer: tissue regeneration gone awry [J]. Curr Opin Cell Biol, 2016, 43: 55–61. [DOI] [PubMed] [Google Scholar]
- [2].Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil [J]. J Clin Invest, 2015, 125(9): 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents [J]. Nat Rev Clin Oncol, 2015, 12(10): 584–596. [DOI] [PubMed] [Google Scholar]
- [4].Chai EZ, Siveen KS, Shanmugam MK, et al. Analysis of the intricate relationship between chronic inflammation and cancer [J]. Biochem J, 2015, 468(1): 1–15. [DOI] [PubMed] [Google Scholar]
- [5].Fernandes JV, Cobucci RN, Jatoba CA, et al. The role of the mediators of inflammation in cancer development [J]. Pathol Oncol Res, 2015, 21(3): 527–534. [DOI] [PubMed] [Google Scholar]
- [6].Vendramini-Costa DB, Carvalho JE. Molecular link mechanisms between inflammation and cancer [J]. Curr Pharm Des, 2012, 18(26): 3831–3852. [DOI] [PubMed] [Google Scholar]
- [7].Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors [J]. Cancer Immunol Immunother, 2014, 63(1): 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer [J]. Immunol Rev, 2012, 246(1): 379–400. [DOI] [PubMed] [Google Scholar]
- [9].Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling path-ways collaboratively link inflammation to cancer [J]. Protein Cell, 2013, 4(3): 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pan MG, Xiong Y, Chen F. NFAT gene family in inflammation and cancer [J]. Curr Mol Med, 2013, 13(4): 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Qin JJ, Nag S, Wang W, et al. NFAT as cancer target: mission possible? [J]. Biochim Biophys Acta, 2014, 1846(2): 297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Balamurugan K HIF-1 at the crossroads of hypoxia, inflammation, and cancer [J]. Int J Cancer, 2016, 138(5): 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nag S, Qin J, Srivenugopal KS, et al. The MDM2-p53 pathway revisited [J]. J Biomed Res, 2013, 27(4): 254–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qin JJ, Nag S, Voruganti S, et al. Natural product MDM2 inhibitors: anticancer activity and mechanisms of action [J]. Curr Med Chem, 2012, 19(33): 5705–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang B, Golding BT, Hardcastle IR. Small-molecule MDM2-p53 inhibitors: recent advances [J]. Future Med Chem, 2015, 7(5): 631–645. [DOI] [PubMed] [Google Scholar]
- [16].Lv PC, Sun J, Zhu HL. Recent advances of p53-MDM2 small molecule inhibitors (2011-present) [J]. Curr Med Chem, 2015, 22(5): 618–626. [DOI] [PubMed] [Google Scholar]
- [17].Ebrahim M, Mulay SR, Anders HJ, et al. MDM2 beyond cancer: podoptosis, development, inflammation, and tissue regeneration [J]. Histol Histopathol, 2015, 30(11): 1271–1282. [DOI] [PubMed] [Google Scholar]
- [18].Ihling C, Haendeler J, Menzel G, et al. Co-expression of p53 and MDM2 in human atherosclerosis: implications for the regulation of cellularity of atherosclerotic lesions [J]. J Pathol, 1998, 185(3): 303–312. [DOI] [PubMed] [Google Scholar]
- [19].Coindre JM, Hostein I, Maire G, et al. Inflammatory malignant fibrous histiocytomas and dedifferentiated liposarcomas: histological review, genomic profile, and MDM2 and CDK4 status favour a single entity [J]. J Pathol, 2004, 203(3): 822–830. [DOI] [PubMed] [Google Scholar]
- [20].Goodman JE, Hofseth LJ, Hussain SP, et al. Nitric oxide and p53 in cancer-prone chronic inflammation and oxyradical overload disease [J]. Environ Mol Mutagen, 2004, 44(1): 3–9. [DOI] [PubMed] [Google Scholar]
- [21].Mulay SR, Thomasova D, Ryu M, et al. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice [J]. Kidney Int, 2012, 81(12): 1199–1211. [DOI] [PubMed] [Google Scholar]
- [22].Hashimoto T, Ichiki T, Ikeda J, et al. Inhibition of MDM2 attenuates neointimal hyperplasia via suppression of vascular proliferation and inflammation [J]. Cardiovasc Res, 2011, 91(4): 711–719. [DOI] [PubMed] [Google Scholar]
- [23].Thomasova D, Mulay SR, Bruns H, et al. p53-independent roles of MDM2 in NF-kappaB signaling: implications for cancer therapy, wound healing, and autoimmune diseases [J]. Neoplasia, 2012, 14(12): 1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ravi R, Mookerjee B, van Hensbergen Y, et al. p53-mediated repression of nuclear factor-kappaB RelA via the transcriptional integrator p300 [J]. Cancer Res, 1998, 58(20): 4531–4536. [PubMed] [Google Scholar]
- [25].Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53 [J]. Mol Cell Biol, 1999, 19(5): 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wadgaonkar R, Phelps KM, Haque Z, et al. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling [J]. J Biol Chem, 1999, 274(4): 1879–1882. [DOI] [PubMed] [Google Scholar]
- [27].Dey A, Wong ET, Bist P, et al. Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy [J]. Cell Cycle, 2007, 6(17): 2178–2185. [DOI] [PubMed] [Google Scholar]
- [28].Gu L, Findley HW, Zhou M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: a novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia [J]. Blood, 2002, 99(9): 3367–3375. [DOI] [PubMed] [Google Scholar]
- [29].Steinman HA, Burstein E, Lengner C, et al. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis [J]. J Biol Chem, 2004, 279(6): 4877–4886. [DOI] [PubMed] [Google Scholar]
- [30].Zhiyu W, Wang N, Wang Q, et al. The inflammasome: an emerging therapeutic oncotarget for cancer prevention [J]. On-cotarget, 2016, 7(31): 50766–50780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saxena M, Yeretssian G. NOD-like receptors: Master regulators of inflammation and cancer [J]. Front Immunol, 2014, 5: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu ST, Pham H, Pandol SJ, et al. Src as the link between inflammation and cancer [J]. Front Physiol, 2013, 4: 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Safe S, Kasiappan R. Natural products as mechanism-based anticancer agents: Sp transcription factors as targets [J]. Phy-tother Res, 2016, 30(11): 1723–1732. [DOI] [PubMed] [Google Scholar]
- [34].Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014 [J]. J Nat Prod, 2016, 79(3): 629–661. [DOI] [PubMed] [Google Scholar]
- [35].Bishayee A, Sethi G. Bioactive natural products in cancer prevention and therapy: Progress and promise [J]. Semin Cancer Biol, 2016, 40–41: 1–3. [DOI] [PubMed] [Google Scholar]
- [36].Dar KB, Bhat AH, Amin S, et al. Inflammation: A multidimensional insight on natural anti-inflammatory therapeutic compounds [J]. CurrMed Chem, 2016, 23(33): 3775–3800. [DOI] [PubMed] [Google Scholar]
- [37].Ghantous A, Gali-Muhtasib H, Vuorela H, et al. What made sesquiterpene lactones reach cancer clinical trials? [J]. Drug Discov Today, 2010, 15(15–16): 668–678. [DOI] [PubMed] [Google Scholar]
- [38].Chadwick M, Trewin H, Gawthrop F, et al. Sesquiterpenoids lactones: benefits to plants and people [J]. Int J Mol Sci, 2013, 14(6): 12780–12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Merfort I Perspectives on sesquiterpene lactones in inflammation and cancer [J]. Curr Drug Targets, 2011, 12(11): 1560–1573. [DOI] [PubMed] [Google Scholar]
- [40].Shi C, Li H, Yang Y, et al. Anti-inflammatory and immunoregulatory functions of artemisinin and its derivatives [J]. Mediators Inflamm, 2015, 2015: 435713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhan ZJ, Ying YM, Ma LF, et al. Natural disesquiterpenoids [J]. Nat Prod Rep, 2011, 28(3): 594–629. [DOI] [PubMed] [Google Scholar]
- [42].Chow LM, Chan TH. Novel classes of dimer antitumour drug candidates [J]. Curr Pharm Des, 2009, 15(6): 659–674. [DOI] [PubMed] [Google Scholar]
- [43].ClinicalTrials.gov Identifier: NCT01056029 https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [44].EudraCT Number: 2011–002149-36. https://www.clinicaltrialsregister.eu/ctr-search/search, 2017. (accessed 27.02.17).
- [45].Ghantous A, Sinjab A, Herceg Z, et al. Parthenolide: from plant shoots to cancer roots [J]. Drug Discov Today, 2013, 18(17–18): 894–905. [DOI] [PubMed] [Google Scholar]
- [46].ClinicalTrials.gov Identifier: NCT02304289 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [47].ClinicalTrials.gov Identifier: NCT02353026 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [48].ClinicalTrials.gov Identifier: NCT02354534 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [49].ClinicalTrials.gov Identifier: NCT02633098 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [50].ClinicalTrials.gov Identifier: NCT00764036 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [51].ClinicalTrials.gov Identifier: NCT02263950 [EB/OL]. https://clinicaltrials.gov/, 2017. (accessed 27.02.17).
- [52].Wang GW, Qin JJ, Cheng XR, et al. Inula sesquiterpenoids: structural diversity, cytotoxicity and anti-tumor activity [J]. Expert Opin Investig Drugs, 2014, 23(3): 317–345. [DOI] [PubMed] [Google Scholar]
- [53].Hua Y, Qin J, Zhang F, et al. Sesquiterpene lactones from Inula helianthus-aquatica [J]. China J Chin Mater Med, 2012, 37(11): 1586–1589. [PubMed] [Google Scholar]
- [54].Ren J, Qin JJ, Cheng XR, et al. Five new sesquiterpene lactones from Inula hupehensis [J]. Arch Pharm Res, 2013, 36(11): 1319–1325. [DOI] [PubMed] [Google Scholar]
- [55].Cheng XR, Ren J, Wang CH, et al. Hookerolides A-D, the first naturally occurring C-17-pseudoguaianolides from Inula hookeri [J]. Tetrahedron Lett, 2013, 54(15): 1943–1946. [Google Scholar]
- [56].Nie LY, Qin JJ, Huang Y, et al. Sesquiterpenoids from Inula lineariifolia inhibit nitric oxide production [J]. J Nat Prod, 2010, 73(6): 1117–1120. [DOI] [PubMed] [Google Scholar]
- [57].Qin JJ, Jin HZ, Zhu JX, et al. New sesquiterpenes from Inula japonica Thunb. with their inhibitory activities against LPS-induced NO production in RAW264.7 macrophages [J]. Tetrahedron, 2010, 66(48): 9379–9388. [Google Scholar]
- [58].Qin JJ, Zhu JX, Zeng Q, et al. Pseudoguaianolides and guaia-nolides from Inula hupehensis as potential anti-inflammatory agents [J]. J Nat Prod, 2011, 74(9): 1881–1887. [DOI] [PubMed] [Google Scholar]
- [59].Cheng X, Zeng Q, Ren J, et al. Sesquiterpene lactones from Inula falconeri, a plant endemic to the Himalayas, as potential anti-inflammatory agents [J]. Eur J Med Chem, 2011, 46(11): 5408–5415. [DOI] [PubMed] [Google Scholar]
- [60].Zhang SD, Qin JJ, Jin HZ, et al. Sesquiterpenoids from Inula racemosa Hook. f. inhibit nitric oxide production [J]. Planta Med, 2012, 78(2): 166–171. [DOI] [PubMed] [Google Scholar]
- [61].Qin JJ, Zhu JX, Zeng Q, et al. Sesquiterpene lactones from Inula hupehensis inhibit nitric oxide production in RAW264.7 macrophages [J]. Planta Med, 2012, 78(10): 1002–1009. [DOI] [PubMed] [Google Scholar]
- [62].Cheng XR, Zhang SD, Wang CH, et al. Bioactive eudesmane and germacrane derivatives from Inula wissmanniana Hand.-Mazz [J]. Phytochemistry, 2013, 96: 214–222. [DOI] [PubMed] [Google Scholar]
- [63].Qin JJ, Jin HZ, Huang Y, et al. Selective cytotoxicity, inhibition of cell cycle progression, and induction of apoptosis in human breast cancer cells by sesquiterpenoids from Inula lineariifolia Turcz [J]. Eur J Med Chem, 2013, 68: 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jin HZ, Lee D, Lee JH, et al. New sesquiterpene dimers from Inula britannica inhibit NF-kappaB activation and NO and TNF-alpha production in LPS-stimulated RAW264.7 cells [J]. Planta Med, 2006, 72(1): 40–45. [DOI] [PubMed] [Google Scholar]
- [65].Qin JJ, Jin HZ, Fu JJ, et al. Japonicones A-D, bioactive dimeric sesquiterpenes from Inula japonica Thunb [J]. Bioorg Med Chem Lett, 2009, 19(3): 710–713. [DOI] [PubMed] [Google Scholar]
- [66].Qin JJ, Jin HZ, Zhu JX, et al. Japonicones E-L, dimeric sesquiterpene lactones from Inula japonica Thunb [J]. Planta Med, 2010, 76(3): 278–283. [DOI] [PubMed] [Google Scholar]
- [67].Qin JJ, Wang LY, Zhu JX, et al. Neojaponicone A, a bioactive sesquiterpene lactone dimer with an unprecedented carbon skeleton from Inula japonica [J]. Chem Commun (Camb), 2011, 47(4): 1222–1224. [DOI] [PubMed] [Google Scholar]
- [68].Qin JJ, Huang Y, Wang D, et al. Lineariifolianoids A-D, rare unsymmetrical sesquiterpenoid dimers comprised of xanthane and guaiane framework units from Inula lineariifolia [J]. Rsc Advances, 2012, 2(4): 1307–1309. [Google Scholar]
- [69].Zhu JX, Qin JJ, Jin HZ, et al. Japonicones Q-T, four new dimeric sesquiterpene lactones from Inula japonica Thunb. [J]. Fitoterapia, 2013, 84: 40–46. [DOI] [PubMed] [Google Scholar]
- [70].Hu Z, Qin J, Zhang H, et al. Japonicone A antagonizes the activity of TNF-alpha by directly targeting this cytokine and selectively disrupting its interaction with TNF receptor-1 [J]. Biochem Pharmacol, 2012, 84(11): 1482–1491. [DOI] [PubMed] [Google Scholar]
- [71].Li X, Yang X, Liu Y, et al. Japonicone A suppresses growth of Burkitt lymphoma cells through its effect on NF-kappaB [J]. Clin Cancer Res, 2013, 19(11): 2917–2928. [DOI] [PubMed] [Google Scholar]
- [72].Qin JJ, Wang W, Voruganti S, et al. Identification of a new class of natural product MDM2 inhibitor: In vitro and in vivo anti-breast cancer activities and target validation [J]. Oncotarget, 2015, 6(5): 2623–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Qin JJ, Wang W, Voruganti S, et al. Inhibiting NFAT1 for breast cancer therapy: New insights into the mechanism of action of MDM2 inhibitor JapA [J]. Oncotarget, 2015, 6(32): 33106–33119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Qin JJ, Wang W, Sarkar S, et al. Inulanolide A as a new dual inhibitor of NFAT1-MDM2 pathway for breast cancer therapy [J]. Oncotarget, 2016, 7(22): 32566–32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Qin JJ, Sarkar S, Voruganti S, et al. Identification of lineariifo-lianoid A as a novel dual NFAT1 and MDM2 inhibitor for human cancer therapy [J]. J Biomed Res, 2016, 30(4): 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tu Y Artemisinin-A gift from traditional Chinese medicine to the world (Nobel Lecture) [J]. Angew Chem Int Ed Engl, 2016, 55(35): 10210–10226. [DOI] [PubMed] [Google Scholar]
- [77].Kong LY, Tan RX. Artemisinin, a miracle of traditional Chinese medicine [J]. Nat Prod Rep, 2015, 32(12): 1617–1621. [DOI] [PubMed] [Google Scholar]
- [78].Wang KS, Li J, Wang Z, et al. Artemisinin inhibits inflammatory response via regulating NF-kappaB and MAPK signaling pathways [J]. Immunopharmacol Immunotoxicol, 2016: 1–9. [DOI] [PubMed] [Google Scholar]
- [79].Xu H, He Y, Yang X, et al. Anti-malarial agent artesunate inhibits TNF-alpha-induced production of proinflammatory cytokines via inhibition of NF-kappaB and PI3 kinase/Akt signal pathway in human rheumatoid arthritis fibroblast-like synoviocytes [J]. Rheumatology (Oxford), 2007, 46(6): 920–926. [DOI] [PubMed] [Google Scholar]
- [80].He Y, Fan J, Lin H, et al. The anti-malaria agent artesunate inhibits expression of vascular endothelial growth factor and hypoxia-inducible factor-1alpha in human rheumatoid arthritis fibroblast-like synoviocyte [J]. Rheumatol Int, 2011, 31(1): 53–60. [DOI] [PubMed] [Google Scholar]
- [81].Hou L, Block KE, Huang H. Artesunate abolishes germinal center B cells and inhibits autoimmune arthritis [J]. PLoS One, 2014, 9(8): e104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Guruprasad B, Chaudhary P, Choedon T, et al. Artesunate ameliorates functional limitations in Freund’s complete adjuvant-induced monoarthritis in rat by maintaining oxidative homeostasis and inhibiting COX-2 expression [J]. Inflammation, 2015, 38(3): 1028–1035. [DOI] [PubMed] [Google Scholar]
- [83].Zhao YG, Wang Y, Guo Z, et al. Dihydroartemisinin ameliorates inflammatory disease by its reciprocal effects on Th and regulatory T cell function via modulating the mammalian target of rapamycin pathway [J]. J Immunol, 2012, 189(9): 4417–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wei M, Xie X, Chu X, et al. Dihydroartemisinin suppresses ovalbumin-induced airway inflammation in a mouse allergic asthma model [J]. Immunopharmacol Immunotoxicol, 2013, 35(3): 382–389. [DOI] [PubMed] [Google Scholar]
- [85].Yang D, Yuan W, Lv C, et al. Dihydroartemisinin supresses inflammation and fibrosis in bleomycine-induced pulmonary fibrosis in rats [J]. Int J Clin Exp Pathol, 2015, 8(2): 1270–1281. [PMC free article] [PubMed] [Google Scholar]
- [86].Xu W, Lu C, Yao L, et al. Dihydroartemisinin protects against alcoholic liver injury through alleviating hepatocyte steatosis in a farnesoid X receptor-dependent manner [J]. Toxicol Appl Pharmacol, 2016, 315: 23–34. [DOI] [PubMed] [Google Scholar]
- [87].Das AK. Anticancer effect of antimalarial artemisinin compounds [J]. Ann Med Health Sci Res, 2015, 5(2): 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Augustin Y, Krishna S, Kumar D, et al. The wisdom of crowds and the repurposing of artesunate as an anticancer drug [J]. Ecancermedicalscience, 2015, 9: ed50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mercer AE, Copple IM, Maggs JL, et al. The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials [J]. J Biol Chem, 2011, 286(2): 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Berdelle N, Nikolova T, Quiros S, et al. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells [J]. Mol Cancer Ther, 2011, 10(12): 2224–2233. [DOI] [PubMed] [Google Scholar]
- [91].Kong R, Jia G, Cheng ZX, et al. Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in pancreatic cancer cells via ROS-mediated up-regulation of death receptor 5 [J]. PLoS One, 2012, 7(5): e37222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ba Q, Zhou N, Duan J, et al. Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1 [J]. PLoS One, 2012, 7(8): e42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nakase I, Gallis B, Takatani-Nakase T, et al. Transferrin receptor-dependent cytotoxicity of artemisinin-transferrin conjugates on prostate cancer cells and induction of apoptosis [J]. Cancer Lett, 2009, 274(2): 290–298. [DOI] [PubMed] [Google Scholar]
- [94].Hou J, Wang D, Zhang R, et al. Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action [J]. Clin Cancer Res, 2008, 14(17): 5519–5530. [DOI] [PubMed] [Google Scholar]
- [95].Chen T, Li M, Zhang R, et al. Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy [J]. J Cell Mol Med, 2009, 13(7): 1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ba Q, Duan J, Tian JQ, et al. Dihydroartemisinin promotes angiogenesis during the early embryonic development of zebrafish [J]. Acta Pharmacol Sin, 2013, 34(8): 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Jeyadevan JP, Bray PG, Chadwick J, et al. Antimalarial and antitumor evaluation of novel C-10 non-acetal dimers of 10beta-(2-hydroxyethyl)deoxoartemisinin [J]. J Med Chem, 2004, 47(5): 1290–1298. [DOI] [PubMed] [Google Scholar]
- [98].Paik IH, Xie S, Shapiro TA, et al. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity [J]. J Med Chem, 2006, 49(9): 2731–2734. [DOI] [PubMed] [Google Scholar]
- [99].Saikia B, Saikia PP, Goswami A, et al. Synthesis of a novel series of 1,2,3-triazole-containing artemisinin dimers with potent anticancer activity involving huisgen 1,3-dipolar cycloaddition reaction [J]. Synthesis-Stuttgart, 2011(19): 3173–3179. [Google Scholar]
- [100].Li X, Zhou Y, Liu Y, et al. Preclinical efficacy and safety assessment of artemisinin-chemotherapeutic agent conjugates for ovarian cancer [J]. EBioMedicine, 2016, 14: 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Li C, Jones AX, Lei X. Synthesis and mode of action of oligomeric sesquiterpene lactones [J]. Nat Prod Rep, 2016, 33(5): 602–611. [DOI] [PubMed] [Google Scholar]
- [102].Wu ZJ, Xu XK, Shen YH, et al. Ainsliadimer A, a new sesquiterpene lactone dimer with an unusual carbon skeleton from Ainsliaea macrocephala [J]. Org Lett, 2008, 10(12): 2397–2400. [DOI] [PubMed] [Google Scholar]
- [103].Li C, Yu X, Lei X. A biomimetic total synthesis of (+)-ainsliadimer A [J]. Org Lett, 2010, 12(19): 4284–4287. [DOI] [PubMed] [Google Scholar]
- [104].Dong T, Li C, Wang X, et al. Ainsliadimer A selectively inhibits IKKalpha/beta by covalently binding a conserved cysteine [J]. Nat Commun, 2015, 6: 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wang Y, Shen YH, Jin HZ, et al. Ainsliatrimers A and B, the first two guaianolide trimers from Ainsliaea fulvioides [J]. Org Lett, 2008, 10(24): 5517–5520. [DOI] [PubMed] [Google Scholar]
- [106].Li C, Dong T, Dian LY, et al. Biomimetic syntheses and structural elucidation of the apoptosis-inducing sesquiterpenoid trimers: (–)-ainsliatrimers A and B [J]. Chem Sci, 2013, 4(3): 1163–1167. [Google Scholar]
- [107].Li C, Dong T, Li Q, et al. Probing the anticancer mechanism of (–)-ainsliatrimer A through diverted total synthesis and bioorthogonal ligation [J]. Angew Chem Int Ed Engl, 2014, 53(45): 12111–12115. [DOI] [PubMed] [Google Scholar]
- [108].Wang S, Li J, Sun J, et al. NO inhibitory guaianolide-derived terpenoids from Artemisia argyi [J]. Fitoterapia, 2013, 85: 169–175. [DOI] [PubMed] [Google Scholar]
- [109].Zeng KW, Wang S, Dong X, et al. Sesquiterpene dimer (DSF-52) from Artemisia argyi inhibits microglia-mediated neuroinflammation via suppression of NF-kappaB, JNK/p38 MAPKs and Jak2/Stat3 signaling pathways [J]. Phytomedicine, 2014, 21(3): 298–306. [DOI] [PubMed] [Google Scholar]
- [110].Zeng KW, Wang S, Dong X, et al. Sesquiterpene dimmer (DSF-27) inhibits the release of neuroinflammatory mediators from microglia by targeting spleen tyrosine kinase (Syk) and Janus kinase 2 (Jak2): Two major non-receptor tyrosine signaling proteins involved in inflammatory events [J]. Toxicol Appl Pharmacol, 2014, 275(3): 244–256. [DOI] [PubMed] [Google Scholar]
- [111].Maas M, Deters AM, Hensel A. Anti-inflammatory activity of Eupatorium perfoliatum L. extracts, eupafolin, and dimeric guaianolide via iNOS inhibitory activity and modulation of inflammation-related cytokines and chemokines [J]. J Ethno-pharmacol, 2011, 137(1): 371–381. [DOI] [PubMed] [Google Scholar]
- [112].Kwon OE, Lee HS, Lee SW, et al. Dimeric sesquiterpenoids isolated from Chloranthus japonicus inhibited the expression of cell adhesion molecules [J]. J Ethnopharmacol, 2006, 104(1–2): 270–277. [DOI] [PubMed] [Google Scholar]
- [113].Wang LJ, Xiong J, Liu ST, et al. Sesquiterpenoids from Chloranthus henryi and their anti-neuroinflammatory activities [J]. Chem Biodivers, 2014, 11(6): 919–928. [DOI] [PubMed] [Google Scholar]
- [114].Yamahara J, Shimoda H, Matsuda H, et al. Potent immunosuppressive principles, dimeric sesquiterpene thioalkaloids, isolated from nupharis rhizoma, the rhizoma of Nuphar pumilum (nymphaeaceae): structure-requirement of nuphar-alkaloid for immunosuppressive activity [J]. Biol Pharm Bull, 1996, 19(9): 1241–1243. [DOI] [PubMed] [Google Scholar]
- [115].Matsuda H, Shimoda H, Yoshikawa M. Dimeric sesquiterpene thioalkaloids with potent immunosuppressive activity from the rhizome of Nuphar pumilum: structural requirements of nuphar alkaloids for immunosuppressive activity [J]. Bioorg Med Chem, 2001, 9(4): 1031–1035. [DOI] [PubMed] [Google Scholar]
- [116].Matsuda H, Morikawa T, Oda M, et al. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum [J]. Bioorg Med Chem Lett, 2003, 13(24): 4445–4449. [DOI] [PubMed] [Google Scholar]
- [117].Matsuda H, Yoshida K, Miyagawa K, et al. Nuphar alkaloids with immediately apoptosis-inducing activity from Nuphar pumilum and their structural requirements for the activity [J]. Bioorg Med Chem Lett, 2006, 16(6): 1567–1573. [DOI] [PubMed] [Google Scholar]
- [118].Ozer J, Eisner N, Ostrozhenkova E, et al. Nuphar lutea thioal-kaloids inhibit the nuclear factor kappaB pathway, potentiate apoptosis and are synergistic with cisplatin and etoposide [J]. Cancer Biol Ther, 2009, 8(19): 1860–1868. [DOI] [PubMed] [Google Scholar]
- [119].Korotkov A, Li H, Chapman CW, et al. Total syntheses and biological evaluation of both enantiomers of several hydroxylated dimeric nuphar alkaloids [J]. Angew Chem Int Ed Engl, 2015, 54(36): 10604–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Imakura Y, Lee KH, Sims D, et al. Antitumor agents XXVIII: Structural elucidation of the novel antitumor sesquiterpene lactone, microlenin, from Helenium microcephalum [J]. J Pharm Sci, 1978, 67(9): 1228–1232. [DOI] [PubMed] [Google Scholar]
- [121].Imakura Y, Lee KH, Sims D, et al. Antitumor agents XXXVI: Structural elucidation of sesquiterpene lactones microhelenins-A, B, and C, microlenin acetate, and plenolin from Helenium microcephalum [J]. J Pharm Sci, 1980, 69(9): 1044–1049. [DOI] [PubMed] [Google Scholar]
- [122].Hall IH, Lee KH, Imakura Y, et al. Antitumor agents LXIII: the effects of microlenin on nucleic acid and protein syntheses of Ehrlich ascites cells [J]. J Pharm Sci, 1983, 72(9): 1008–1011. [DOI] [PubMed] [Google Scholar]
- [123].Litaudon M, Bousserouel H, Awang K, et al. A dimeric ses-quiterpenoid from a Malaysian Meiogyne as a new inhibitor of Bcl-xL/BakBH3 domain peptide interaction [J]. J Nat Prod, 2009, 72(3): 480–483. [DOI] [PubMed] [Google Scholar]
- [124].Fotsop DF, Roussi F, Leverrier A, et al. Biomimetic total synthesis of meiogynin A, an inhibitor of Bcl-xL and Bak interaction [J]. J Org Chem, 2010, 75(21): 7412–7415. [DOI] [PubMed] [Google Scholar]
- [125].Desrat S, Remeur C, Geny C, et al. From meiogynin A to the synthesis of dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins [J]. Chem Commun (Camb), 2014, 50(62): 8593–8596. [DOI] [PubMed] [Google Scholar]
- [126].Desrat S, Pujals A, Colas C, et al. Pro-apoptotic meiogynin A derivatives that target Bcl-xL and Mcl-1 [J]. Bioorg Med Chem Lett, 2014, 24(21): 5086–5088. [DOI] [PubMed] [Google Scholar]
- [127].Desrat S, Remeur C, Roussi F. Development of an efficient route toward meiogynin A-inspired dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins [J]. Org Biomol Chem, 2015, 13(19): 5520–5531. [DOI] [PubMed] [Google Scholar]
- [128].Toume K, Takahashi M, Yamaguchi K, et al. Parviflorenes B-F, novel cytotoxic unsymmetrical sesquiterpene-dimers with three backbone skeletons front Curcuma parviflora [J]. Tetrahedron, 2004, 60(48): 10817–10824. [Google Scholar]
- [129].Toume K, Sato M, Koyano T, et al. Cytotoxic dimeric ses-quiterpenoids from Curcuma parviflora: isolation of three new parviflorenes and absolute stereochemistry of parviflorenes A, B, D, F, and G [J]. Tetrahedron, 2005, 61(28): 6700–6706. [Google Scholar]
- [130].Ohtsuki T, Tamaki M, Toume K, et al. A novel sesquiterpenoid dimer parviflorene F induces apoptosis by up-regulating the expression of TRAIL-R2 and a caspase-dependent mechanism [J]. Bioorg Med Chem, 2008, 16(4): 1756–1763. [DOI] [PubMed] [Google Scholar]
- [131].Matsunaga S, Furuya-Suguri H, Nishiwaki S, et al. Differential effects of cryptoporic acids D and E, inhibitors of superoxide anion radical release, on tumor promotion of okadaic acid in mouse skin [J]. Carcinogenesis, 1991, 12(6): 1129–1131. [DOI] [PubMed] [Google Scholar]
- [132].Narisawa T, Fukaura Y, Kotanagi H, et al. Inhibitory effect of cryptoporic acid E, a product from fungus Cryptoporus volvatus, on colon carcinogenesis induced with N-methyl-N-nitrosourea in rats and with 1,2-dimethylhydrazine in mice [J]. Jpn J Cancer Res, 1992, 83(8): 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bergstrom CA, Yazdanian M. Lipophilicity in drug development: Too much or not enough? [J]. AAPS J, 2016, 18(5): 1095–1100. [DOI] [PubMed] [Google Scholar]
- [134].Kundu JK, Surh YJ. Emerging avenues linking inflammation and cancer [J]. Free Radic Biol Med, 2012, 52(9): 2013–2037. [DOI] [PubMed] [Google Scholar]
- [135].Guven Maiorov E, Keskin O, Gursoy A, et al. The structural network of inflammation and cancer: merits and challenges [J]. Semin Cancer Biol, 2013, 23(4): 243–251. [DOI] [PubMed] [Google Scholar]