

Abstract
Alport syndrome (AS) is an inherited kidney disease that may lead to end-stage renal disease in early adult life. It is a clinically and genetically heterogeneous nephropathy. The possibility of a patient with haematuria or proteinuria being diagnosed as having AS cannot be excluded even if the patient is female or if the family history is unknown. We report a 3-year-old girl with a de novo frameshift mutation, c.3906delA p.(Gly1303Aspfs*17), in the COL4A5 gene. The significance of the electron microscopic study on the glomerular basement membrane must be emphasised because it is the first step towards the diagnosis of AS. Genetic analysis provides the only conclusive diagnosis of AS, by determining the mode of inheritance and prognosis.
Keywords: urinary and genital tract disorders, proteinurea, genetic screening counselling
Background
Alport syndrome (AS) is a hereditary basement membrane disorder characterised by a family history of haematuria, progressive renal dysfunction, hearing loss and ocular abnormalities.1 2 AS is caused by mutations within type Ⅳ collagen genes. There are three patterns of inheritance in AS. About 80% of AS is inherited in an X-linked manner (XLAS) that is caused by mutations in the COL4A5 genes encoding the α5-chain of type Ⅳ collagen. Nearly all affected males develop end-stage renal disease (ESRD) in early adult life, whereas heterozygous women exhibit a wide variability in disease severity, with some affected women exhibiting no haematuria and normal kidney function, and others developing ESRD and deafness.3 About 15% of AS cases are inherited in an autosomal recessive manner (ARAS) caused by mutations in both alleles of the COL4A3/COL4A4 genes encoding the α3/α4 chains of type Ⅳ collagen. The clinical features in women with ARAS are identical to those in men with XLAS.4 Autosomal dominant AS is quite rare and occurs due to heterozygous mutations in either COL4A3 or COL4A4. It is difficult to distinguish between a severely affected woman with XLAS and one with ARAS when there is no consanguinity and no family history of kidney disease.5
Case presentation
A 3-year-old girl with microhaematuria and proteinuria, which was indicated by urinary screening for 3-year-old children, was referred to our hospital. The patient had been a full-term baby (birth weight 2368 g) and the first child of healthy non-consanguineous parents. Her mother has a right solitary kidney. However, there was no family history of haematuria, chronic kidney disease or deafness. During her follow-up, she had persistent microhaematuria and proteinuria. At 4 years of age, she was admitted to our hospital for a renal biopsy. At that time, a urinalysis showed 1+proteinuria (urinary protein/urinary creatinine ratio, 0.85 g/gCr) and 3+haematuria, with the urine sediment containing over 100 red cells per high-power field (HPF). Her blood nitrogen level was 9.6 mg/dL and serum creatinine level was 0.18 mg/dL. Renal ultrasonography was unremarkable. Thirty-six glomeruli were observed under light microscopy (LM); one glomerulus showed segmental sclerosis, but the other glomeruli, tubules and interstitium showed no significant alterations. Immunofluorescence staining for IgG, IgA, IgM, C1q, C3, C4 and fibrinogen showed no significant staining. Electron microscopy (EM) demonstrated irregular thickening with lamellation and segmental thinning of the glomerular basement membrane (GBM) (figures 1 and 2). These results indicated a possibility for the diagnosis of AS despite the absence of family history.
Figure 1.
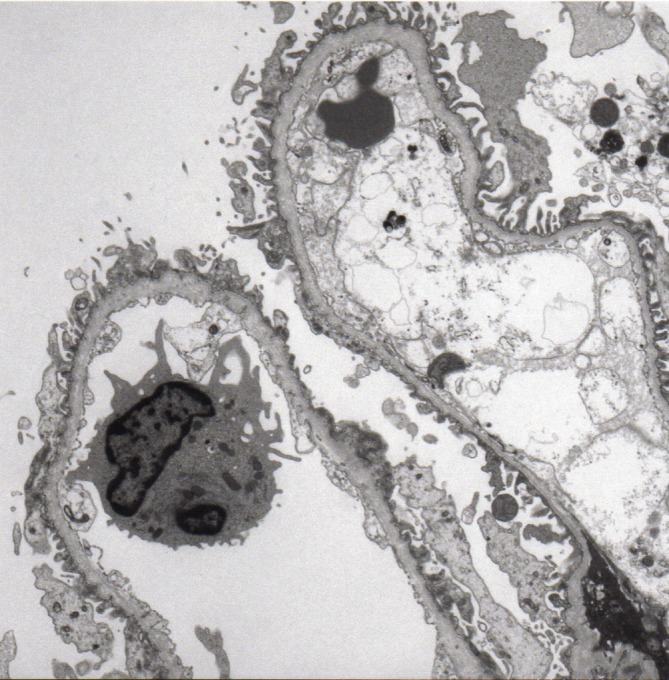
Electron microscopy demonstrated segmental thinning of the glomerular basement membrane. Uranyl acetate-lead citrate, magnification ×3000.
Figure 2.
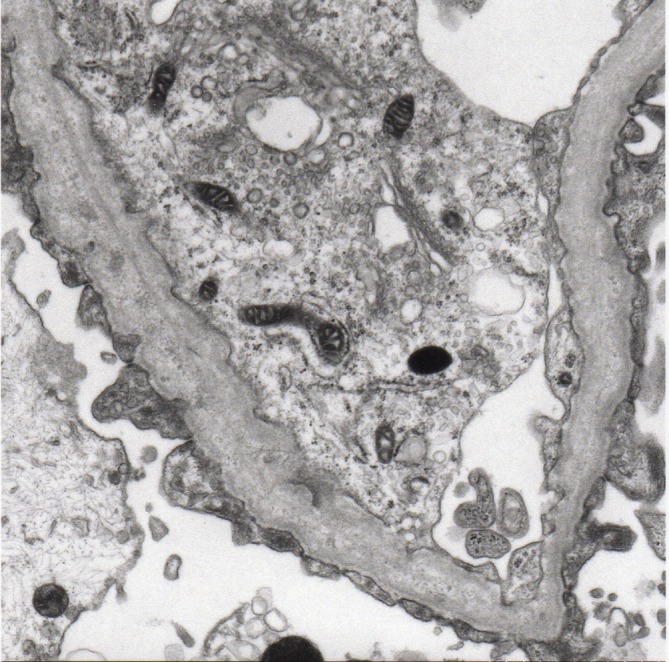
Electron microscopy of the kidney biopsy specimen of the patient demonstrating characteristic features of Alport syndrome. The glomerular basement membrane has focal thickening with lamellation, creating a ‘basket weave’ appearance. Uranyl acetate-lead citrate, magnification ×10 000.
Sequence analysis of COL4A3-5 in the patient and her parents was performed. The study was approved by the Institutional Review Board of Kobe University School of Medicine, and written informed consent was obtained. The analysis revealed that the patient had a heterozygous frameshift mutation (c.3906delA p.(Gly1303Aspfs*17)) in exon 42 of the COL4A5 gene (figure 3), whereas her parents had no mutation. There were no other mutations in COL4A3 or COL4A4 genes.
Figure 3.

Genetic analysis revealed that the patient had a heterozygous frameshift mutation (c.3906delA p.(Gly1303Aspfs*17)) in exon 42, whereas her parents had no mutation.
Treatment
At 4 years and 5 months of age, enalapril maleate was initiated for proteinuria.
Outcome and follow-up
At 5 years of age, she remained well and had no clinically detectable hearing loss or ocular abnormalities. However, a urinalysis revealed 1+proteinuria (urinary protein/urinary creatinine ratio, 0.44 g/gCr) and 3+haematuria, with the urine sediment containing 20–29 red cells per HPF.
Discussion
The patient was a 3-year-old girl with de novo XLAS. Although there was no family history of haematuria, nephropathies or hearing loss, EM findings on renal biopsy specimens indicated GBM abnormalities that are characteristic of AS. Genetic analyses were useful in making a final diagnosis and in distinguishing between de novo XLAS and ARAS.
Although this is a very unusual case of a female child presenting with haematuria and proteinuria at such a young age, the possibility of another disease has been excluded by pathologically and genetically. Pathological findings did not show any other nephritis. We have conducted comprehensive gene analysis for 123 inherited kidney disease causative genes including COL4A3 and COL4A4 genes.
In the presence of persistent haematuria and/or proteinuria, renal biopsy is frequently the first investigation performed. However, renal biopsies from AS patients have a widely variable appearance, often with non-specific findings by LM, ranging from a normal appearance to sclerosis. It is difficult to make a diagnosis of AS with LM. Although ultrastructural studies have shown that the fundamental lesion of AS involves the GBM, there are some difficulties encountered while distinguishing between AS and thin basement membrane nephropathy.1 6 In children with AS, thinning of the GBM is the prevalent change. Regardless of age, diffuse attenuation of the GBM is the only pathological finding in 10%–20% of AS cases.1 Thinning of the GBM early in the disease course and characteristic thickening and lamellation of the GBM may be evident later. These findings indicate that a thin GBM is not invariably associated with the benign disease. The lamellation may be patchy, and errors are occasionally made in establishing a diagnosis based on GBM appearance, especially in younger men and women. However, the significance of ultrastructural studies on the GBM must be emphasised because EM findings are the first step to suggest a diagnosis of AS.
There are three presumable reasons that negative family history does not exclude the diagnostic possibility of AS. First, her mother was a ‘carrier’ with no haematuria. Second, approximately 10%–15% of XLAS occur de novo.6 Third, the patient may have had ARAS. It is critical to distinguish between XLAS and ARAS because of the different implications, including the risk of renal failure, for her at-risk family members as well. Female patients of XLAS exhibit extensive phenotypic heterogeneity with respect to the age of onset and rapidity of disease progression, which is not well explained by genotype–phenotype correlations.3 7 The ‘European Community Alport Syndrome Concerted Action’ study indicated that risk factors for developing renal failure include the occurrence and progressive increase in proteinuria and the development of hearing loss.3 Thus, close monitoring of proteinuria and audibility and timely therapy in women with XLAS can delay the progression of the disease. On the contrary, the clinical features in women with ARAS are identical to those in men with XLAS. The recent comprehension is that all women with ARAS develop ESRD at about the same age as do men with XLAS.4 Therefore, it is critical to distinguish between XLAS and ARAS in women with AS in order to institute management. Genetic analysis provides an important means for reliable diagnosis in some clinical situations in which a conclusive diagnosis of AS cannot be established or where it is impossible to determine the mode of transmission, despite careful evaluation of the pedigree. In these situations, genetic analysis has the potential to provide information essential for determining prognosis and guiding genetic counselling.
This case report reflects the importance of recognising the potential for the clinical manifestations of XLAS in women throughout their lifespan and the careful evaluation of ultrastructural studies on the GBM. Hence, genetic analysis is useful for a conclusive diagnosis of AS.
Learning points.
Absence of family history with haematuria, renal failure and hearing loss does not exclude the possibility of diagnosis of Alport syndrome (AS).
Ultrastructural studies on renal biopsy are the first step towards suggesting a diagnosis of AS.
Genetic analysis is a useful tool for establishing a conclusive diagnosis, determining the mode of inheritance and providing information essential for determining prognosis.
Acknowledgments
The authors would like to thank Editage (www.editage.jp) for English language editing.
Footnotes
Contributors: HK and TG contributed to provide medical care for the patient. KN performed the genetic analysis of COL4A5 in the patient. HK also wrote the manuscript with input from all authors.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Gubler MC. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol 2008;4:24–37. 10.1038/ncpneph0671 [DOI] [PubMed] [Google Scholar]
- 2. Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 2013;24:364–75. 10.1681/ASN.2012020148 [DOI] [PubMed] [Google Scholar]
- 3. Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol 2003;14:2603–10. 10.1097/01.ASN.0000090034.71205.74 [DOI] [PubMed] [Google Scholar]
- 4. Storey H, Savige J, Sivakumar V, et al. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol 2013;24:1945–54. 10.1681/ASN.2012100985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Savige J, Colville D, Rheault M, et al. Alport Syndrome in Women and Girls. Clin J Am Soc Nephrol 2016;11:1713–20. 10.2215/CJN.00580116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol 2009;20:1210–5. 10.1681/ASN.2008090984 [DOI] [PubMed] [Google Scholar]
- 7. Rheault MN. Women and alport syndrome. Pediatr Nephrol 2012;27:41–6. 10.1007/s00467-011-1836-7 [DOI] [PMC free article] [PubMed] [Google Scholar]